

Abstract
The immature phenotype of embryonic stem cell-derived cardiomyocytes (ESC-CMs) limits their application. However, the molecular mechanisms of cardiomyocyte maturation remain largely unexplored. This study found that overexpression of long noncoding RNA (lncRNA)-Cmarr, which was highly expressed in cardiomyocytes, promoted the maturation change and physiological maturation of mouse ESC-CMs (mESC-CMs). Moreover, transplantation of cardiac patch overexpressing Cmarr exhibited better retention of mESC-CMs, reduced infarct area by enhancing vascular density in the host heart, and improved cardiac function in mice after myocardial infarction. Mechanism studies identified that Cmarr acted as a competitive endogenous RNA to impede the repression of miR-540-3p on Dtna expression and promoted the binding of the dystrophin-glycoprotein complex (DGC) and yes-associated protein (YAP), which in turn reduced the proportion of nuclear YAP and the expression of YAP target genes. Therefore, this study revealed the function and mechanism of Cmarr in promoting cardiomyocyte maturation and provided a lncRNA that can be used as a functional factor in the construction of cardiac patches for the treatment of myocardial infarction.
Keywords: MT:Noncoding RNAs, Cmarr, miR-540-3p, Dtna, DGC, cardiomyocyte maturation, myocardial infarction, cardiac patch
Graphical abstract
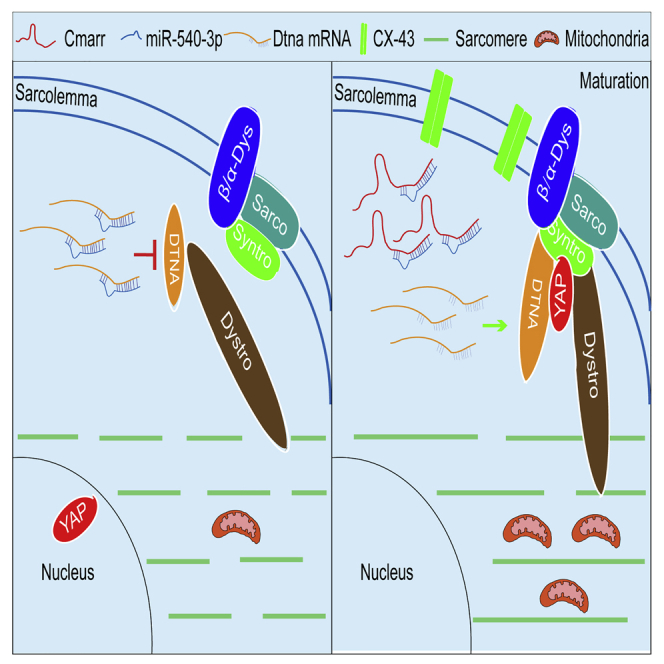
Kang and colleagues described that lncRNA Cmarr competed with miR-540-3p and released its inhibition on Dtna expression, facilitating the DGC-YAP complex formation and promoting the maturation of mESC-CMs. They also showed that the Cmarr-engineered patch recovered the impaired function of MI heart and provided a functional lncRNA in cardiomyocyte maturation and MI treatment.
Introduction
Unlimited differentiation potential of pluripotent stem cells (PSCs) into cardiomyocytes in vitro provides the possibility of developing cell transplantation therapies for injured hearts.1,2 However, PSC-derived cardiomyocytes (PSC-CMs) have a fetal-like phenotype including molecular, structural, and functional properties, which is one of the hurdles that should be overcome for better application of PSC-CMs.3,4 Nowadays, researchers have shown that the mature cardiomyocytes generated from PSCs formed better grafts in infarcted rat hearts, suggesting that the maturation status of transplanted PSC-CMs might affect the cardiac function recovery of infarcted hearts.5 Accumulating evidence revealed the maturation drivers were valuable to the development of PSC-CM therapies. Transplantation of PSC-CMs overexpressing Igf2 significantly improved cardiac function in mice after myocardial infarction (MI).6 Similarly, overexpression of N-cadherin in the transplanted cardiomyocytes also promoted the functional integration and augmented the heart function of MI mice.7 It follows that pursuing the molecular mechanisms of PSC-CM maturation is of great importance.
Long noncoding RNAs (lncRNAs) are a group of RNAs that are longer than 200 nucleotides and have no coding potential.8 Recently, several studies have shown that lncRNAs played a vital role not only in cardiac development and function but also in the differentiation of embryonic stem cells (ESCs) into cardiomyocytes. lncRNAs Fendrr and Upperhand were indispensable for heart development.9,10 Mhrt and H19 protected the heart from pathological remodeling.11,12 lncRNA Bvht and lincRNA-1405 regulated the differentiation of ESCs to cardiomyocytes.1,13 In addition, Touma et al., reported that mutual regulation of lncRNA-PPP1R1B and TCAP might drive cardiomyocyte maturation.14 These studies raise our curiosity about how heart-rich lncRNAs regulate cardiomyocyte maturation.
The dystrophin-glycoprotein complex (DGC) is formed by dystrophin associating with dystrobrevin and various glycoproteins and plays a critical role in mature cardiomyocytes. DGC functions in stabilizing the sarcolemma, linking the actin cytoskeleton to the extracellular matrix, and transporting force to the extracellular matrix,15, 16, 17 suggesting that the formation of DGC contributes to the maturation of cardiomyocytes. Recent studies have shown that the crosstalk between DGC and the Hippo signaling pathway controls the switch between proliferation and maturation in cardiomyocytes. Activation of the Hippo signaling pathway could phosphorylate its core effector yes-associated protein (YAP), which reduced the nuclear distribution of YAP and the expression level of its downstream cell cycle-related genes.17,18 The assembly of DGC could interact with YAP in the cytoplasm and promote cardiomyocyte maturation.19 However, the molecular mechanisms for the assembly and regulation of DGC during cardiomyocyte maturation remain unclear.
Here, our study showed that a heart-rich lncRNA, Cmarr, upregulated during cardiac development, facilitated the maturation transition of mESC-CMs in vitro and in the engineered cardiac patch, which was beneficial to the cardiac function recovery of MI heart. Further studies indicated that Cmarr specifically interacted with miR-540-3p, impeding the repression of Dtna (a key component of DGC) expression by miR-540-3p, and thus served as a regulator of the DGC-YAP complex and cardiomyocyte maturation.
Results
Cmarr promotes the maturation change of mESC-CMs
Maturation of cardiomyocytes is achieved gradually during heart development. To address the potential molecules associated with cardiomyocyte maturation, we collected the hearts at different stages (Embryonic day 10.5 (E10.5), E14.5, E17.5, neonatal, and adult) and performed RNA sequencing (RNA-seq) to investigate the whole heart transcriptome at distinct stages. qPCR assay verified that the β-isoform to α-isoform transition of the myosin heavy chain (β-MHC to α-MHC) occurred with the development of heart (Figure S1A). Similarly, the expression of the mature gene Kcnj2 gradually increased, whereas the expression of the fetal gene Tgf significantly decreased at the indicated stages of heart development (Figure S1B). Gene set enrichment analysis (GSEA) further confirmed that the expression of cell cycle-related genes was negatively correlated with heart development (Figure S1C). Subsequently, we found that 65 lncRNAs were persistently upregulated during heart development (Figure 1A). Cardiomyocytes maintained their proliferative potential within 1 week after birth and rapidly reached maturation characteristics. We focused on the 26 candidate lncRNAs that were highly expressed in the adult heart and significantly upregulated during the fetal-to-adult transition (Figure 1B). Then, we analyzed the lncRNA profiles in cardiomyocytes (GSE41538) and fibroblasts (GSE103870)20,21 and identified that 68 lncRNAs were highly expressed in cardiomyocytes (Figure S2A). We cross-analyzed these 68 cardiomyocyte-expressed lncRNAs and the 26 candidate lncRNAs and found only one lncRNA (Xloc_010735, AK084656),22,23 termed cardiomyocyte maturation-related RNA (Cmarr), was highly expressed in cardiomyocytes and potentially associated with cardiac maturation (Figures S2B and S2C). qPCR assay confirmed that Cmarr was highly expressed in the heart tissue and significantly upregulated during heart development (Figures 1C and 1D). Rapid amplification of cDNA ends (RACE) assay showed that the length of Cmarr was 870 nt, which consisted of two exons (Figure 1E). Coding potential analysis24 showed that Cmarr had a low coding probability, similar to Xist (Figure S2D). We further performed the polysome profile assay followed by qPCR and found that polysome were highly enriched on the Gapdh mRNA, whereas there was no polysome enriched on the Cmarr, indicating that Cmarr was indeed a noncoding transcript (Figure S2E). Cmarr was also upregulated in the mESC-CMs, neonatal cardiomyocytes, and adult cardiomyocytes along with the increased maturity as indicated by a decreased ratio of Ki67-positive cardiomyocytes and increased expression levels of maturation genes (α/β-MHC and Kcnj2) (Figures 1F and 1G). These results suggested that Cmarr may be associated with cardiomyocyte maturation.
Figure 1.

Cmarr promotes the maturation change of mESC-CMs
(A and B) Heatmap showing the expression landscape of 65 upregulated lncRNAs (A) and the selected 26 lncRNAs (B) in mouse embryonic heart (E10.5, E14.5, and E17.5), neonatal heart, and adult heart. (C and D) qPCR assay for the relative expression of Cmarr in adult tissues (heart, muscle, thymus, testis, liver, lung, spleen, kidney, and intestinum tenue) (C) and developing heart (E10.5, E14.5, E17.5, neonatal, and adult) of mouse (D). (E) RACE assay for identifying Cmarr transcript in adult heart of mouse. (F) Ki67 immunostaining (red) in the mESCs (mESC-CMs [D10]), neonatal heart, and adult heart. Cardiomyocytes and cell nuclei were stained with α-ACTININ (green) and Hoechst 33342 (HO.33342, blue), respectively. Scale bar: 25 μm. (G) qPCR assay for the ratio of α/β-MHC and the expression levels of Kcnj2 and Cmarr in mESC-CMs and neonatal and adult hearts. (H) qPCR assay to verify the overexpression efficiency of Cmarr in mESC-CMs (Cmarr OE). (I) Ki67 immunostaining and statistics of Ki67+ cardiomyocytes in Ctrl and Cmarr OE groups. Scale bar: 25 μm. (J) Quantification of cell area and cell perimeter after Cmarr overexpression. Scale bar: 25 μm. (K) qPCR assay for the ratio of α/β-MHC (left) and the expression levels of maturation-related genes (α-MHC, Kcnj2, and Gja1) after Cmarr overexpression. (L) Representative immunostaining images of CX-43 in Ctrl and Cmarr OE mESC-CMs. Scale bar: 25 μm. Data are presented as the mean ± SEM (n = 3). The statistical significance is performed according to Student’s t-tests (unpaired two-tailed). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus Ctrl.
Then, Cmarr was overexpressed in the mESC-CMs (Cmarr OE) (Figure 1H), and the results showed that overexpression of Cmarr reduced the ratio of Ki67+ mESC-CMs and increased the cell area and perimeter of mESC-CMs (Figures 1I and 1J), as well as a significant increase in the ratio of α/β-MHC and expression levels of cardiac maturation markers (α-MHC, Kcnj2, and Gja1) (Figure 1K). Similarly, the gap junction protein CX-43 obviously appeared on the cytomembrane of the mESC-CMs after overexpressing Cmarr (Figure 1L). To further explore whether knockdown of Cmarr could blunt the maturation process of mESC-CMs, we cultured the mESC-CMs on the Matrigel-coated plate with or without shCmarr virus and the results showed that knockdown of Cmarr resulted in decreased expression levels of cardiomyocyte maturation markers (α-MHC, Kcnj2, and Gja1) and calcium handling genes (Ryr2, Atp2a2, and Slc8a1) (Figures S3A–S3C), as well as an increased proportion of Ki67+ mESC-CMs (Figure S3D). Together, these results indicated that Cmarr is critical for promoting the maturation changes of mESC-CMs.
Overexpression of Cmarr promotes the physiological maturation of mESC-CMs
To determine whether the maturation of structure and gene expression was accompanied by changes in physiological phenotype, we first measured the action potentials (APs) of mESC-CMs after overexpression of Cmarr by whole-cell patch clamp experiment (Figure 2A). The results showed that the Cmarr OE mESC-CMs exhibited a more negative resting membrane potential and prolonged AP duration at 90% of the repolarization (APD 90) compared with control (Ctrl) mESC-CMs (Figures 2B and 2C), indicating that overexpression of Cmarr improved the electrophysiology properties of mESC-CMs. We then investigated the calcium handling capacity of mESC-CMs after overexpression of Cmarr. Our findings showed that the transient amplitude, maximum upstroke velocity, and maximum decay velocity of the Cmarr OE mESC-CMs were higher than those of Ctrl mESC-CMs as detected by the fluorescent Ca2+ dye Cal-520 acetoxymethyl ester assay (Figures 2D–2G). Similarly, compared with the Ctrl cells, the expression levels of calcium handling-related genes (Ryr2, Atp2a2, and Slc8a1) were upregulated in the Cmarr OE cells (Figure 2H). Next, we detected the mitochondrial content by immunostaining of the mitochondrial marker Tom20, and the results showed that the number of mitochondria was increased in the mESC-CMs after overexpression of Cmarr (Figure 2I). We further measured the oxygen consumption rate (OCR) of mESC-CMs to exogenous fatty acids (palmitate) after overexpression of Cmarr and found that Cmarr OE mESC-CMs exhibited a higher basal and maximal OCR in response to exogenous palmitate (Figures 2J and 2K), indicating that overexpression of Cmarr enhanced mitochondrial maturation of mESC-CMs. These data demonstrated that overexpression of Cmarr improves the physiological characteristics of mESC-CMs.
Figure 2.

Overexpression of Cmarr promotes the physiological maturation of mESC-CMs
(A) Representative action potential recordings in Ctrl and Cmarr OE mESC-CMs. (B and C) Statistics of resting membrane potentials (RMPs) (B) and action potential duration to 90% (APD90) (C) in the Ctrl and Cmarr OE mESC-CMs. n = 9 for each group. (D) Representative images of Ca2+ imaging analysis in Ctrl and Cmarr OE mESC-CMs. (E–G) Statistics of calcium transient amplitudes (E), upstroke velocities (F), and decay velocities (G) in Ctrl and Cmarr OE mESC-CMs. n = 15 for each group. (H) qPCR assay for the relative expression levels of calcium handling-related genes (Ryr2, Atp2a2, and Slc8a1) after Cmarr overexpression. (I) Tom20 immunostaining (left) and statistics for relative fluorescence intensity (right) in Ctrl and Cmarr OE mESC-CMs. Scale bar: 25 μm. (J) Oxygen consumption rate (OCR) of Ctrl (left) and Cmarr OE (right) mESC-CMs after addition of exogenous palmitate. (K) Statistics of basal and maximum OCR in response to exogenous fatty acids (FAs). n = 4 for each group. The statistical significance is performed according to Student’s t-tests (unpaired two-tailed). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus Ctrl.
Cmarr induces maturation of transplanted mESC-CMs in extracellular matrix patch and improves the cardiac function of MI mice
Previous literature has shown that Ti2C-cryogel treatment or overexpression of N-cadherin promotes functional maturation of PSC-derived cardiomyocytes, which have a conductive effect on enhancing the reparative potency of cardiomyocytes after transplantation into infarcted hearts,7,25 suggesting that promoting the maturation properties of PSC-CMs will be beneficial for the treatment of MI. Considering the effect of Cmarr in promoting mESC-CM maturation in vitro, we further investigated the therapeutic efficacy of Cmarr-overexpressed mESC-CMs in treating MI. Since the low retention and poor survival of PSC-CM grafts via direct intramyocardial injection or intravascular infusion, a cardiac patch formed by decellularized cardiac extracellular matrix (ECM) could achieve effective cell delivery.26,27 The mESC-CMs were seeded onto the piece of ECM at a density of 1 × 104 cells/mm2 to construct the cardiac patches (Figures 3A and 3B), and infected with Fuw-puro and Fuw-Cmarr lentivirus, respectively. After cultured for 1 week in vitro, the ratio of α/β-MHC and the expression levels of Kcnj2 and Gja1 were upregulated in the Cmarr-overexpressed cardiac patch (Cmarr patch) (Figures 3C and 3D). Next, we examined whether the Cmarr patch played a therapeutic role in mice after MI. A total of 44 mice were used for MI surgery and 41 mice survived after surgery (survival rate of almost 93%). The echocardiography analysis was employed for the survival mice and there was no significant difference in heart function between MI, MI + Ctrl patch, and MI + Cmarr patch groups at day 3 after MI (Figure 3E). MI mice with an ejection fraction lower than 40% (MI, eight mice; MI + Ctrl patch, eight mice; and MI + Cmarr patch; eight mice) and sham mice (eight mice) were used for further analysis. Two weeks after transplantation, echocardiography was performed to evaluate the recovery of cardiac function, and our results showed a significant increase in left-ventricular ejection fraction (LVEF) and fraction shortening in the Cmarr patch group (Figure 3F). In addition, histological analysis showed that the average infarct size of the Cmarr patch group was smaller than that of the MI and Ctrl patch groups (Figure 3G). These results indicated that the cardiac patches generated by Cmarr-overexpressed mESC-CMs could effectively reduce the infarct area and restore the cardiac function of MI hearts.
Figure 3.

Cmarr induces the maturation of transplanted mESC-CMs in extracellular matrix patch and improves the cardiac function of MI mice
(A) Schematic illustration of the cardiac patch. (B) Representative image of a decellularized mouse heart. (C and D) qPCR assay for the expression level of Cmarr (C), the ratio of α/β-MHC, and the expression levels of Kcnj2 and Gja1 (D) in Ctrl and Cmarr patch. n = 3 for each group. (E and F) Echocardiography analysis of ejection fraction and fraction shortening in sham (n = 8), MI (n = 8), MI + Ctrl patch (n = 8), and MI + Cmarr patch (n = 8) at 3 days (E) and 2 weeks (F) post-MI. (G) Representative images of Masson staining of dissected hearts (left) and the quantification of infarct size (right) in each group 2 weeks post-MI. Scale bar: 500 μm. (H) Immunostaining of GFP (green), CTNT (red), and Hoechst 33342 (blue) in the heart 2 weeks post-MI. The white dashed line represents the interface between the patch and myocardium. Scale bar: 25 μm. (I) TUNEL assay for apoptotic cells in the cardiac patch 2 weeks post-MI. Statistics of TUNEL+ cells are shown (right). n = 8 for each group. The white dashed line represents the interface between the patch and myocardium. Scale bar: 100 μm. (J) Immunostaining of vWF and α-SMA in the border zone of infarcted hearts 2 weeks after transplantation and vascular density was measured by calculating the number of vWF+ structures in each group. n = 8 for each group. Scale bar: 100 μm. The statistical significance is performed according to one-way analysis of variance (ANOVA) followed by Tukey’s post hoc. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus MI. #p < 0.05, ##p < 0.01, and ###p < 0.001 versus MI + Ctrl patch.
Consistently, we found that the Cmarr patch exhibited better retention of transplanted mESC-CMs and the ratio of apoptotic cells (TUNEL+) was significantly decreased in the Cmarr patch group (Figures 3H and 3I). Previous studies have shown that transplantation of cardiac patches with PSC-CMs promotes angiogenesis in the border zone of infarction.27,28 We then investigated the effect of transplantation of cardiac patch overexpressing Cmarr on angiogenesis in mice after MI by von Willebrand Factor (vWF) and α-smooth muscle actin (α-SMA) immunostaining, and the results showed that the vascular density was increased in the Cmarr patch group (Figure 3J). Besides, the results of H&E staining showed that the cardiac patch was closely adhered and structurally integrated to the host myocardium and no significant insulating tissue was found in the junction 2 weeks after transplantation, indicating that the transplanted cells in the cardiac patch might be coupled to the host myocardium and achieve function recovery of the MI hearts (Figure S4A). In addition, there was only negligible CD3+ T cell infiltration in the transplanted patch, suggesting that transplanted cells were immunologically compatible with the host tissues (Figure S4B). Together, our findings suggested that overexpression of Cmarr may contribute to the survival of transplanted mESC-CMs in cardiac patches and the angiogenesis in the host hearts, ultimately exhibiting better therapeutic outcomes.
Cmarr counteracts miR-540-3p to regulate cardiomyocyte maturation
The mechanism of lncRNA action is largely determined by its location in the cell. Cmarr was primarily distributed in the cytoplasm of cardiomyocytes (Figure 4A), which indicated a possible function by competing with microRNAs (miRNAs). By aligning the sequence of Cmarr with the miRNA library,29 we revealed that 25 miRNAs potentially interacted with Cmarr (Figure S5A). Cross-analysis of these candidate miRNAs and miRNAs differentially expressed during heart development (Figures S5B–S5D),30 we identified two miRNAs (miR-540-3p and miR-541-5p), both of which were downregulated during heart development and might interact with Cmarr (Figure 4B). The luciferase reporter assay showed that only miR-540-3p mimics significantly reduced the activity of Cmarr luciferase reporter (Figure 4C). Correspondingly, we found the negligible effect of miR-540-3p on the luciferase activity of Cmarr mut (mutated in miR-540-3p target site) (Figures 4D and 4E). Further, RNA pull-down assay with MS2-binding protein (MS2 bp), which specifically binds to RNA containing MS2 sequences (MS2bs), showed that miR-540-3p was enriched by MS2bs-Cmarr RNA, but not MS2bs-Cmarr mut RNA (Figure 4F), indicating that Cmarr could interact with miR-540-3p. Our results also showed that inhibition of miR-540-3p significantly reduced the ratio of Ki67+ cells (Figure S6A) and increased the cell area and perimeter of mESC-CMs (Figure S6B). Furthermore, inhibition of miR-540-3p also increased the expression levels of cardiomyocyte maturation markers and the distribution of CX-43 on the cytomembrane (Figures S6C and S6D), indicating that inhibition of miR-540-3p mimicked the overexpression of Cmarr and promoted the maturation changes of mESC-CMs. Further, our results showed that overexpression of Cmarr mut failed to promote the maturation of mESC-CMs (Figures 4G–4M). Together, these results indicated that Cmarr promotes cardiomyocyte maturation by competing with miR-540-3p.
Figure 4.

Cmarr counteracts miR-540-3p to regulate cardiomyocyte maturation
(A) qPCR assay for the distribution of Cmarr in the cytoplasm and nucleus of mESC-CMs. Xist and Gapdh were used as the nucleus and cytoplasm control (Ctrl), respectively. (B) Venn diagram showing predicted Cmarr-binding miRNAs during heart development (up) and heatmap showing the expression pattern of miR-540-3p and miR-541-5p during heart development (down). (C) Luciferase reporter assay for pGL3-Cmarr reporter activity after cotransfection with Ctrl mimics, miR-540-3p mimics, or miR-541-5p mimics. The luciferase signal was normalized to the Renilla signal. (D) Targeting sequences of miR-540-3p in Cmarr and the mutant sequences in Cmarr mut. (E) Luciferase reporter assay for pGL3-Cmarr mut (mutation of miR-540-3p target site) reporter activity after cotransfection with Ctrl mimics or miR-540-3p mimics. (F) MS2bp-YFP RNA pull-down assay for the enrichment of miR-540-3p in MS2bs-Luci, MS2bs-Cmarr, or MS2bs-Cmarr mut. (G and H) Quantification of Ki67+ cell ratio (G), and cell area and perimeter (H) after overexpression of Cmarr or Cmarr mut. (I) qPCR assay for the expression levels of maturation-related genes (α-MHC, Kcnj2, and Gja1) after overexpression of Cmarr or Cmarr mut. (J) Representative immunostaining images of CX-43 in Ctrl, Cmarr OE, and Cmarr mut OE mESC-CMs. Scale bar: 25 μm. (K) Statistics of calcium transient amplitudes in Ctrl, Cmarr OE, and Cmarr mut OE mESC-CMs, n = 9 for each group. (L) qPCR assay for the relative expression levels of calcium handling-related genes (Ryr2, Atp2a2, and Slc8a1) in Ctrl, Cmarr OE, and Cmarr mut OE mESC-CMs. (M) Tom20 immunostaining (left) and statistics for relative fluorescence intensity (right). Scale bar: 25 μm. Data are presented as the mean ± SEM. The statistical significance is performed according to Student’s t-tests (unpaired two-tailed) (Figures 4C and 4E) or one-way ANOVA followed by Tukey’s post hoc (Figures 4F–4I and 4K–4M). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus Ctrl. #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Cmarr OE.
miR-540-3p directly represses Dtna expression and impedes the DGC-YAP interaction
miRNAs generally play their roles by binding to the 3′ UTR of target mRNAs and downregulating their expression. To further explore the underlying mechanism of Cmarr/miR-540-3p on mESC-CM maturation, we focused on pursuing the downstream targets of miR-540-3p. Target screening analysis using miRWalk2.0 hinted that 1,257 genes might be the targets of miR-540-3p. Cross-analysis between the predicted targets and genes positively correlated with heart development revealed that 18 genes might be the potential targets of miR-540-3p during heart development (Figures 5A and 5B). We then identified Dtna, which encoded an intracellular component of DGC,15 as a potential target of miR-540-3p (Figure 5C). As expected, our results showed that miR-540-3p mimics significantly reduced the activity of the Dtna 3′ UTR luciferase reporter, but not the Dtna 3′ UTR mut (mutation in miR-540-3p binding site) luciferase reporter (Figures 5D and 5E). Similarly, the expression level of DTNA was negatively regulated by miR-540-3p (Figure 5F). In cardiomyocytes, the DGC binds to YAP and impedes its transfer from the cytoplasm to the nucleus, thus facilitating the maturation transition of cardiomyocytes.17,19 We found that knockdown of Dtna weakened the interaction of the DGC component with YAP and increased the percentage of nuclear YAP (Figures 5G–5J). Further, we detected the expression of downstream target genes of YAP, and our findings showed that knockdown of Dtna upregulated the expression levels of YAP target genes (Figure 5K). These results demonstrated that Dtna is the target of miR-540-3p and is required for the formation of the DGC-YAP complex.
Figure 5.

miR-540-3p directly represses Dtna expression and impedes the DGC-YAP interaction
(A) Venn diagram showing the miR-540-3p target genes upregulated during heart development. (B) Heatmap showing the expression landscape of overlapping genes during heart development. (C) qPCR assay for the expression level of Dtna during heart development. (D) Targeting sequences of miR-540-3p in Dtna 3′ UTR and the mutant sequences in Dtna 3′ UTR mut. (E) Luciferase reporter assay for the activity of pGL3-Dtna 3′ UTR and pGL3-Dtna 3′ UTR mut (mutation of miR-540-3p target site) reporters after cotransfection with Ctrl or miR-540-3p mimics. (F) Western blot assay for the expression of DTNA after transfection with miR-540-3p mimics (left) or miR-540-3p inhibitor (miR-540-3p inhi) (right). Ctrl mimics and Ctrl inhibitor (Ctrl inhi) were used as the corresponding Ctrl, and GAPDH was used as a loading Ctrl. (G) Western blot assay for the expression of DTNA in Ctrl (Plko_ctrl) and Dtna knockdown (Plko_shDtna) mESC-CMs. (H) Co-IP assay for the interaction of SYNTROPHIN-DYSTROPHIN (left) and SYNTROPHIN-YAP (right) after Dtna knockdown. (I) Representative immunostaining images of YAP after Dtna knockdown. Scale bar: 25 μm. (J) The protein level of YAP was derived from nuclear and cytoplasmic extraction of Plko_ctrl and Plko_shDtna mESC-CMs, respectively. H3 and TUBULIN were used as the nuclear and cytoplasmic Ctrl, respectively. (K) qPCR assay for the expression levels of YAP target genes in Plko_ctrl and Plko_shDtna mESC-CMs. Data are presented as the mean ± SEM (n = 3). The statistical significance is performed according to Student’s t-tests (unpaired two-tailed). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus Plko_ctrl.
Dtna is a functional target of Cmarr/miR-540-3p interaction
To confirm whether Cmarr impeded the inhibition of Dtna expression by miR-540-3p, we introduced Cmarr lentiviruses into miR-540-3p-overexpressed mESC-CMs (Figure S7A). We found that overexpression of Cmarr increased the expression level of DTNA in the miR-540-3p-overexpressed mESC-CMs, suggesting that Cmarr indirectly increased the expression of DTNA through endogenous competition with miR-540-3p (Figure S7B). Next, to confirm that the interaction between Cmarr and miR-540-3p regulated mESC-CM maturation by affecting the expression of Dtna and the translocation of YAP, we detected the expression of Dtna and the cell distribution of YAP in Cmarr OE and Cmarr mut OE mESC-CMs, respectively. Our results showed that overexpression of Cmarr significantly increased the expression level of DTNA (Figure 6A), and decreased the percentage of nuclear YAP (Figure 6B). However, overexpression of Cmarr mut, which relieved its interaction with miR-540-3p, did not affect the expression of DTNA and the distribution of YAP (Figures 6A and 6B). To further validate that Cmarr/miR-540-3p targeted Dtna to regulate the maturation of mESC-CMs, we infected Cmarr OE cells with shDtna lentiviruses (Cmarr_shDtna) (Figure 6C). Our results showed that knockdown of Dtna significantly rescued the reduction of nuclear YAP and YAP target gene expression (Figures 6D and 6E) and impeded the promotion effect of mESC-CM maturation by Cmarr overexpression (Figures 6F–6K). Besides, the expression of DTNA was positively correlated with the maturation of human ESC (hESC)-CMs (1 year versus 20 days) and heart (adult versus fetal) (GSE62913) (Figures S8A and S8B),31 suggesting that DTNA might also be involved in the maturation process of human cardiomyocytes. Together, these results demonstrated that Dtna is a functional target of Cmarr/miR-540-3p interaction in the process of mESC-CM maturation.
Figure 6.

Dtna is a functional target of Cmarr/miR-540-3p interaction
(A) Western blot assay for the expression of DTNA in Ctrl, Cmarr OE, and Cmarr mut OE mESC-CMs (n = 4). (B) The protein level of YAP was derived from nuclear and cytoplasmic extraction of Ctrl, Cmarr OE, and Cmarr mut OE mESC-CMs. H3 and TUBULIN were used as the nuclear and cytoplasmic Ctrl, respectively. (C) Expression levels of DTNA in Ctrl (Ctrl_plko), Cmarr overexpressed (Cmarr OE_plko), and Cmarr overexpressed with Dtna interference (Cmarr OE_shDtna) mESCs (n = 4). (D) The protein level of YAP derived from nuclear and cytoplasmic extraction of Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs, respectively. (E) qPCR assay for the expression levels of YAP target genes in Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs. (F and G) Quantification of Ki67+ cell ratio (F), cell area and perimeter (G) in Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs. (H) qPCR assay for the expression levels of maturation-related genes (α-MHC, Kcnj2, and Gja1) in Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs. (I) Representative immunostaining images of CX-43 in Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs. Scale bar: 25 μm. (J) qPCR assay for the relative expression levels of calcium handling-related genes in Ctrl_plko, Cmarr OE_plko, and Cmarr OE_shDtna mESC-CMs. (K) Tom20 immunostaining (left) and statistics for relative fluorescence intensity (right). Scale bar: 25 μm. Data are presented as the mean ± SEM from at least three independent experiments. The statistical significance was performed according to one-way ANOVA followed by Tukey’s post hoc. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus Ctrl_plko. #p < 0.05, ##p < 0.01, and ###p < 0.001 versus Cmarr OE_plko.
Discussion
PSC-CMs can be generated in vitro in high yield and purity,2 but the PSC-CMs remain immature, thus limiting their potential applications in cardiac disease modeling, drug discovery, and cell therapy.32 Although multiple approaches, such as mechanical stress, electrical stimulation, substrate stiffness changes, and endothelial cell coculture,33 can induce PSC-CMs to a more mature phenotype, maturation remains hampered due to the complicated regulatory networks and unidentified factors in cardiomyocyte maturation.34 lncRNAs act as epigenetic regulators with higher specificity than coding genes and are participants in diverse biological processes such as myocardial differentiation and cardiac diseases.1,11, 12, 13 Recently, the expression profiles of several lncRNAs have been uncovered during the postnatal period of heart development,14 but the roles and mechanisms of lncRNAs in cardiomyocyte maturation remain largely unknown. In this study, we decoded the lncRNA profiles of embryonic, neonatal, and adult hearts and identified that lncRNA Cmarr, which was highly expressed in the heart, promoted the maturation transition, and the physiological phenotype change of mESC-CMs served as a molecular driver of mESC-CM maturation.
lncRNAs can execute their functions through diverse modes, especially by functioning as competitive endogenous RNAs (ceRNAs) of specific miRNAs. Cytoplasm-distributed lncRNAs fine-tune their targeted miRNAs as intrinsic regulators to modulate biological function. lncRNA-ROR, which acts as a ceRNA of miR-145, affects the differentiation potential of ESCs.35 HBL1 interacts with miR-1 to regulate cardiomyocyte differentiation.36 lncRNA CHRF can regulate cardiac hypertrophy by targeting miR-489.37 Currently, a few miRNAs, including Let-7 and miR-200c, have been reported to modulate cardiomyocyte maturation,31,38 but there is no evidence about the mutual interaction of miRNA and intrinsic lncRNA in cardiac maturation. Our results showed that inhibition of miR-540-3p facilitated the maturation changes of mESC-CMs and that Cmarr interacted with miR-540-3p as a ceRNA and antagonized its effect on downstream genes during cardiomyocyte maturation. These findings revealed an interaction model between lncRNA and miRNA during cardiomyocyte maturation and developed the understanding of the mechanisms involved in cardiomyocyte maturation.
The DGC consists of a core subcomplex (dystrophin-dystroglycan), an intracellular subcomplex (dystrobrevin-syntrophin), and a transmembrane subcomplex (sarcoglycan-sarcospan) that connects the ECM to the intracellular cytoskeleton and transmits the extracellular signal to regulate the maturation of cardiomyocytes.39,40 Deletion of DGC components dystrophin or sarcoglycan impairs mechanical stress transmission, increases sarcolemma permeability, and leads to cardiomyopathy.39,41, 42, 43 Deficiency of the intracellular component syntrophin blocks extracellular signal transduction and structural maturation of cardiomyocytes.39 Downregulation or mutation of α-dystrobrevin (coded by the gene Dtna) is also associated with cardiomyopathy and congenital heart defects.40,43,44 Besides, deficiency of the dystrophin or sarcoglycan of the DGC complex significantly leads to mitochondrial dysfunction and metabolic impairment, presented by decreased mitochondrial oxidative phosphorylation, enhanced reactive oxygen species (ROS) generation, and disorganized mitochondrial network.45 Recent studies have shown that the ECM component agrin impairs the assembly of DGC, decreases the expression of mitochondrial marker Tom20, and reduces the maturation of cardiomyocytes.19 Our study showed that Dtna was the downstream target of the Cmarr/miR-540-3p axis and knockdown of Dtna impaired the integrity of DGC, which uncovered a precise mechanism for the regulation of Dtna in cardiomyocyte maturation. Cytoplasmic YAP is reported to facilitate the formation of DGC.19 Activation of Hippo signaling also promotes the formation of DGC-YAP complex in the cytoplasm of cardiomyocytes and facilitates the transition from proliferation to maturation,17,19 whereas nuclear YAP cooperates with HIF-1α to impair the transition of glycolysis to oxidative phosphorylation and metabolic maturation in neonatal rat cardiomyocytes.46,47 We also found that knockdown of Dtna impeded the DGC-YAP assembly, increased the YAP nuclear localization, and decreased the Tom20 expression, thus reducing the maturation of mESC-CMs. Together, our study complements the mechanisms of Dtna expression and the DGC-YAP formation by Cmarr/miR-540-3p in cardiomyocyte maturation.
Transplantation of PSC-CMs with higher maturation has been shown to improve cardiac function in MI hearts. Overexpression of Igf2 in PSC-CMs promotes the structural maturation of PSC-CMs through suppressing Igf1R/INSR signaling, and transplantation of PSC-CMs overexpressing Igf2 significantly improves the heart function in mice after MI.6 Similarly, transplantation of PSC-CMs with high N-cadherin expression level exhibit higher maturation and better retention, ultimately improving the heart function of MI mice.7 However, several studies point out that adult mature cardiomyocytes fail to survive after transplantation due to their poor proliferative capacity,48,49 implying that exogenous cardiomyocytes should tune the survival and functional performance to a better balance for the treatment of MI. Cardiac patch acts as a promising method for the treatment of MI that could promote the maturation of PSC-CMs and improve the survival rate of transplanted cardiomyocytes in the hearts after MI.25,27,28,50,51 Previous studies have shown that culturing in Ti2C-8-cryogel promotes the structural and functional maturation of PSC-CMs. Transplantation of Ti2C-8-cryogel cardiac patch exhibits a higher survival rate of transplanted cardiomyocytes, significantly decreases the infarct area, and improves the cardiac function of MI hearts,25 linking the maturation of transplanted PSC-CMs to the repair efficacy of cardiac patch in the treatment of MI. Our study showed that Cmarr promoted the maturation of mESC-CMs in the cardiac patch, and transplantation of cardiac patch overexpressing Cmarr exhibited better retention and lower apoptosis ratio of transplanted mESC-CMs, and significantly promoted the vascularization in the border zone of infarction, reduced infarct size, and improved cardiac function of MI hearts, suggesting that combination of cardiomyocyte maturation drivers with engineered cardiac patch could provide a method for cellular therapy of MI.
In summary, our study indicated that heart-rich Cmarr competed with miR-540-3p and impeded the inhibition of Dtna expression by miR-540-3p, which facilitated the formation of the DGC-YAP complex, thereby facilitating the maturation transition of mESC-CMs. Furthermore, our study demonstrated that transplantation of cardiac patch overexpressing Cmarr significantly reduced the infarct area and improved the function of MI heart. Thus, our study elucidated the function and mechanism of Cmarr in promoting cardiomyocyte maturation, developed our understanding of epigenetic regulation in cardiomyocyte maturation, and provided a functional lncRNA for the construction of an engineered cardiac patch for the treatment of MI.
Materials and methods
Experimental animals
C57BL/6 male mice (6–8 weeks old) were purchased from Shanghai Laboratory Animal Center (SLAC), Shanghai. Related animal experiments were carried out and approved by the Institutional Animal Care and Use Committee of Tongji University (TJLAC-020-122).
Plasmids
Sequences of Cmarr and mutant Cmarr were cloned into the Fuw lentiviral vector (Fuw-Cmarr and Fuw-Cmarr mut, respectively). cDNA of puromycin was also cloned into the Fuw lentiviral vector, which was used as a negative Ctrl. The shRNA targeting Dtna or Cmarr was cloned into the plko lentiviral vector, and the plko vector with scramble RNA was used as a negative Ctrl. The T2A-puromycin donor was cloned into the pLB vector (TIANGEN). The sgRNA targeting the end of the Slc8a1 gene was cloned into the pX330 vector (Addgene).
For the luciferase reporter assay, sequences of Cmarr, Cmarr mutant (Cmarr mut), Dtna 3′ UTR, and Dtna 3′ UTR mutant (Dtna 3′ UTR mut) were cloned into the pGL3 vector (Promega). For the miRNA pull-down assay, sequences of Renilla, Cmarr, and Cmarr mutant were cloned into the pMS2 vector. All primers are listed in Table S1.
Generation of Slc8a1-T2A-Puro mESCs
The small guide RNA (sgRNA) targeting the end of gene Slc8a1 was designed from http://crispr.mit.edu/. This sgRNA was inserted into the pX330 vector. T2A-puromycin-resistant gene was inserted into the pLB vector (TIANGEN). 5 μg of pX330-sgRNA and 10 μg of the donor were mixed with the P3 primary cell solution (Lonza V4XP-3-32). These plasmids were synchronously electroplated into wild-type 46C mESCs using the CG-104 program of the 4D Nucleofector system according to the manufacturer’s instructions. Primers are listed in Table S1.
Cell culture
mESCs (wild-type 46C and Slc8a1-T2A-Puro 46C) were cultured in mESC medium (DMEM [Invitrogen], 15% FBS [Invitrogen], 2 mM nonessential amino acids [Invitrogen], 2 mM L-GlutaMax [Invitrogen], 1 mM sodium pyruvate [Invitrogen], 0.1 mM β-mercaptoethanol [Sigma], and 1 × leukemia inhibitory factor (LIF) [Millipore]) on the feeder layer. 293FT and NIH3T3 cells were cultured in DMEM supplemented with 10% FBS and 2 mM nonessential amino acids. mESC-CMs were cultured in cardiac myocyte medium (ScienCell) supplemented with 5% FBS, 1× cardiac myocyte growth supplement (CMGS), and 1× penicillin/streptomycin (PS) solution.
Cardiomyocyte differentiation
Cardiomyocyte differentiation was performed as previously described.1 Briefly, mESCs were maintained on the feeder layer in serum-free medium (SFM) for two passages (DMEM/Neurobasal [v/v = 1:1], 0.5 × N2 supplement [GIBCO], 0.5 × B27 supplement [GIBCO], 2 mM L-GlutaMax, and 1.5 × 10−4 M monothioglycerol with 10 ng/mL hBMP4 [R&D] and 1 × LIF). Then, mESCs were dissociated and suspended in the cardiomyocyte differentiation medium (DMEM/DMEM-F12 (v/v = 1:1), 1 × N2 supplement, 1 × B27 supplement, 2 mM L-GlutaMax, and 4.5 × 10−4 M monothioglycerol) with 25 ng/mL L-ascorbic acid and cultured for 2 days at a density of 5 × 104 cells/mL in 6-cm Petri dishes. Embryoid bodies (EBs) were subsequently dissociated and resuspended in the cardiomyocyte differentiation medium containing 0.2 ng/mL hBMP4, 5 ng/mL hActivin A, 5 ng/mL vascular endothelial growth factor (VEGF), and 25 ng/mL L-ascorbic acid for 40 h at a density of 5 × 104 cells/mL. The reconstituted EBs were dissociated and attached to 12-well plates (pre-coated with gelatin) in the Stem Cell Pro-34 medium supplemented with 5 ng/mL VEGF, 10 ng/mL basic fibroblast growth factor (bFGF), 25 ng/mL fibroblast growth factor 10 (FGF10), and 25 ng/mL L-ascorbic acid at a density of 2.5 × 105 cells/well. Contracted cardiomyocytes were observed after another 3 days of culture. Puromycin (1 ng/mL) was added on day 7 of cardiomyocyte differentiation to eliminate non-cardiomyocytes, and mESC-CMs were dissociated for further studies.
Cytoplasmic and nuclear fractionation
Cytoplasmic and nuclear RNA was isolated as previously described.52 mESC-CMs were dissociated, washed twice with ice-cold PBS, and centrifuged. The cell pellets were incubated in 200 μL of lysis buffer A (10 mM Tris [pH 8.0], 140 mM NaCl, 1.5 mM MgCl2, 0.5% Nonidet P-40) on ice for 5 min. Then, the cell lysate was centrifuged (1,000 × g for 3 min) at 4°C. The supernatant containing the cytoplasmic fraction was added to 1 mL of RNAiso Plus (TaKaRa) to extract cytoplasmic RNA. The nuclear pellets were washed twice with 200 μL of lysis buffer A and once with lysis buffer A containing 1% Tween-40 and 0.5% deoxycholic acid. Finally, 1 mL of RNAiso Plus was added to the nuclear pellets to extract nuclear RNA.
Dual-luciferase assay
Dual-luciferase assay was performed as previously described.53 Dtna 3′ UTR, Dtna 3′ UTR mutant, Cmarr, and Cmarr mutant sequences were cloned into the pGL3 reporter vector (Promega), respectively. NIH3T3 cells grown in 24-well plates (3 × 104 cells/well) were transfected with 200 ng of pGL3 reporter, 10 ng of Renilla luciferase vector (pRL-Tk, Promega), and 50 nM mimics (Ctrl, miR-540-3p, or miR-541-5p) using X-treme GENE HP DNA Transfection Reagent (Roche). Cells were lysed 48 h later with the lysis buffer. Firefly and Renilla luciferase activities were detected by the M5 plate reader (SpectraMax). The firefly signal was normalized to the Renilla signal. Ctrl mimics were used as a negative Ctrl.
Co-immunoprecipitation
Co-immunoprecipitation (co-IP) assay was performed as previously described.1 Briefly, protein A + G beads were incubated with 3 μg of anti-syntrophin antibody overnight at 4°C. Next day, mESC-CMs (5 million) grown in a 10-cm dish were dissociated and lysed with 400 μL of lysis buffer on ice for 30 min (lysis I [40 mM Tris, pH 7.4; 450 mM NaCl; 1% Triton X-100; 20% glycerol; and 1 mM EDTA]; lysis II [40 mM Tris, pH 7.4; 10 mM NaCl; 1% Triton X-100; 20% glycerol] (v/v) = 1:2). The cell lysate was sonicated and incubated with anti-syntrophin-coated beads for 6 h at 4°C. After discarding the supernatant, the complex was washed five times with 400 μL of lysis buffer, then 50 μL of 1× SDS buffer was added to the complex and heated at 95°C for 10 min.
Immunostaining
Immunostaining assay was performed as previously described.54,55 mESC-CMs cultured on a 12-mm coverslip were fixed with 4% paraformaldehyde for 15 min at 4°C. For the immunostaining of CX-43, cells were first fixed with ice-cold methyl alcohol at −20°C for 15 min, followed by 4% paraformaldehyde for 15 min at 4°C. Next, the cells were washed three times with PBS and blocked with PBS supplemented with 10% donkey serum and 0.1% Triton X-100 for 1 h at room temperature. Then, the cells were incubated with the corresponding antibodies overnight at 4°C. Next day, the cells were incubated with fluorescent secondary antibodies and Hoechst 33342 (nuclear staining) for 1 h at room temperature. Immunostaining images were captured by a Nikon ECLIPSE Ti-S microscope. Cell area, cell perimeter, and fluorescence intensity were quantified using ImageJ software (https://imagej.nih.gov/ij/). The antibodies are listed in Table S2.
MS2bp-YFP RNA pull-down assay
RNA pull-down assay was performed as previously described.56 Briefly, 2 million 293FT cells grown in a 6-cm dish were transfected with 3 μg of MS2bp-YFP fusion plasmid, 500 nM miR-540-3p mimics, and 3 μg of MS2bs-Cmarr or MS2bs-Cmarr mut plasmid using X-treme GENE HP DNA Transfection Reagent (Roche). The cells were lysed 48 h later with the RNA immunoprecipitation (RIP) buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES [pH = 7.0], 0.5% NP-40, and 1 mM dithiothreitol). Anti-GFP (Abcam) (able to recognize MS2bp-YFP fusion protein) was used to immunoprecipitate the RNA-protein complex. The complex was treated with RNAiso Plus (TaKaRa) to purify miRNA.
Ca2+ imaging analysis
The contractile mESC-CMs were incubated with Tyrode’s solution containing Cal-520 and 0.02% Pluronic F-127 (AAT Bioquest) for 20 min at 37°C. The cells were washed with warm medium. The spontaneous Ca2+ transients were recorded with a Leica confocal microscope in line-scan (X-T) mode. Ca2+ images were analyzed using MATLAB software (MathWorks).28
ECM construction of mouse heart
To prepare the cardiac ECM, the decellularization of the mouse heart was performed as previously described.27,57,58 Briefly, the heart (derived from 8-week-old male mice) was frozen at −80°C for 30 min and perfused with deionized water for about 30 min until it became white and hypertrophied. Then, the white heart was perfused with 1% SDS solution and 1% Triton solution until it became completely translucent. Finally, the decellularized heart was washed with PBS (with PS solution, 100 U/mL) for 2 h. The decellularized heart was soaked in PBS solution and placed at 4°C.
Generation of cardiac patch
The generation of the cardiac patch was performed as previously described.27,59 Briefly, the ECM was cut into pieces and placed into the 12-well plate with the endocardial side up. Then the mESC-CMs were seeded onto the ECM at a density of 1 × 104 cells/mm2. After 1 week of culture, the cardiac patch was transplanted to the top of the MI zone through a fibrin-based method.
The MI mice construction and echocardiography analysis
The construction of the MI model was performed as previously described.60 The adult mice were anesthetized with 2% isoflurane and a small incision was made in the left chest. The third intercostal space was exposed and a small hole was created. The middle of the rib was pushed and the hemostatic clamp was simultaneously held slightly open to cause the heart to pop out. Then, the left main descending coronary artery (LCA) was sutured and ligated with a 7-0 silk suture. Next, the heart was placed back into the chest, and the skin cut was sutured and ligated with 5-0 silk suture immediately. The mice used in our study were intraperitoneally injected with cyclosporine A (Selleck, S2286, 5 mg/kg/d) for seven consecutive days from the day before to the sixth day of MI to inhibit immune rejection. Echocardiography analysis of these mice was performed 14 days after transplantation (Visual Sonics Vevo 2100 system equipped with a 40-MHz 550 s probe).
Histology and immunostaining of heart slides
The mouse heart was fixed with 4% paraformaldehyde solution at 4°C for 24 h and dehydrated with a sucrose gradient. The heart below the ligation site to the apex were sectioned at a thickness of 8 μm and sampled at 500-μm intervals for the Masson trichrome (Yeasen) staining.
For immunofluorescence staining, the slides were blocked in PBS supplemented with 10% donkey serum and 0.1% Triton X-100 for 1 h at room temperature. Then, the slides were incubated with the corresponding antibodies overnight at 4°C. Next day, the slides were incubated with fluorescent secondary antibodies and Hoechst 33342 (nuclear staining) for 1 h at room temperature. TUNEL staining was performed by a one-step TUNEL Apoptosis Assay Kit (Beyotime, C1088) according to the manufacturer's instructions. Three views were randomly selected from each slide to quantify the number of vWF or TUNEL-positive cells. Infarct size was quantified as the average of five sections through the following formula: infarct size = [infarct perimeter (infarct epicardium + infarct endocardium) × 100]/left ventricle perimeter (left ventricle epicardium + left ventricle endocardium).
Recording of APs
The APs of mESC-CMs were performed as previously described.61,62 Briefly, mESC-CMs were attached to the coverslip at a density of 3 × 104 cells/cm2. The next day, a coverslip was put into the recording chamber filled with warm Tyrode’s solution. The APs were recorded by the EPC-10 amplifier (Heka Electronics) in current-clamp mode. The internal solution was 0.1 mM NaGTP, 1 mM MgCl2, 1 mM EGTA, 5 mM MgATP, 5 mM Na2-phosphocreatine, 10 mM HEPES, 20 mM KCl, and 110 mM K-aspartate (pH = 7.3, adjusted with KOH). Data were analyzed by CAPA software.
Mitochondrial function analysis
The Seahorse XF96 extracellular flux analyzer combined with Seahorse XF Palmitate-BSA fatty acid β-oxidation (FAO) substrate kit (Agilent) was used to assess the mitochondrial oxidation capacity of mESC-CMs in response to exogenous fatty acid (palmitate).31,63 Briefly, mESC-CMs were seeded onto Seahorse plates at a density of 3 × 105 cells/well for 3 days. BSA Ctrl (33 μM) or palmitate: BSA (200:33 μM) was provided to mESC-CMs to measure baseline OCR followed by sequential automatic injection of 10 μM oligomycin (Sigma-Aldrich), 2 μM carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (Sigma- Aldrich), and 10 μM antimycin A (Sigma-Aldrich) to analyze the OCR. The OCR was normalized to the number of cells. The basal/maximal change of the OCR due to exogenous fatty acids was defined as basal/maximal palmitate-BSA rate minus basal/maximal BSA rate.
qRT-PCR
Total RNA was extracted using RNAiso Plus (TaKaRa). Reverse transcription of 500 ng of total RNA was performed using a PrimeScript RT reagent kit (TaKaRa). qPCR was performed on the Mx3000 instrument (Agilent) using the SYBR Kit (Bio-Rad). Gene expression was normalized to Gapdh mRNA.54 qPCR primers are listed in Table S3.
Western blot
Cells were dissociated, washed twice with ice-cold PBS, and lysed in RIPA buffer. The total protein concentration was detected by a BCA kit (Thermo). Equal amounts of cell lysates were loaded onto the SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Blots were blocked in 3% BSA (Ameresco) and then probed with the following primary antibodies: anti-DTNA (Abcam, 1:2,000), anti-H3 (Cell Signaling Technology, 1:2,000), anti-TUBULIN (Cell Signaling Technology, 1:2,000), anti-YAP (Cell Signaling Technology, 1:2,000), anti-α-SYNTROPHIN (Santa-Cruz 1:1,000), anti-DYSPROTHIN (1:1,000), and anti-GAPDH (Bioworld, 1:2,000). GAPDH was used as a loading Ctrl. The grayscale analysis of protein bands was performed with ImageJ software.1 The antibodies are listed in Table S4.
Polysome profile analysis
Polysome profile analysis was performed as previously described.23 Briefly, cells were treated with 100 μg/mL cyclohexanone (CHX) (Selleck, S7418) for 10 min at 37°C. Then the cells were lysed with 1 mL of polysome extraction buffer (20 mM Tris-HCl, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40, 100 μg/mL CHX, 1× protease inhibitor, and RNase inhibitor) on ice for 10 min and centrifuged at 13,000 rpm for 10 min. The supernatant was transferred to the top of a 13.2-mL centrifuge tube (Beckman Coulter, 331372) containing a 10%–50% sucrose gradient solution. Then the solution was centrifuged at 39,000 rpm for 90 min at 4°C. The solution was collected into 12 tubes and RNA was extracted from each tube. Enrichment of polysomes on the Gapdh mRNA or Cmarr was analyzed by qRT-PCR.
RNA-seq analysis
Paired-end RNA-seq was performed for developing mouse hearts (E10.5, E14.5, E17.5, neonatal, and adult). RNA-seq reads of development hearts were aligned to the mm9 UCSC reference gene GTF using TopHat2 with the default parameters. Transcript assembly and differential expression analysis with the mm9 UCSC reference gene GTF and the reference genome GSE52313 were performed through Cufflinks2 and Cuffdiff2 with the default parameters, respectively. For lncRNA analysis, transcripts were filtered for a minimum length of 200 bp, and the protein coding potential was evaluated using the Coding-Non-Coding Index (CNCI). Upregulated noncoding RNAs were sorted by the changes in fragments per kilobase per million mapped reads (FPKM) during heart development (E14.5 > E10.5, E17.5 > E14.5, Neonatal > E17.5, adult > neonatal, adult/E10.5 > 10, and neonatal/E17.5 > 1.5) (Tables S5 and S6). Upregulated genes were sorted by the changes in FPKM during heart development (E14.5 > E10.5, E17.5 > E14.5, neonatal > E17.5, adult > neonatal, adult/E10.5 > 10) (Table S7). For miRNA analysis, miRNA sequencing (miRNA-seq) data were analyzed using CAP-miRSeq.30 Up- and downregulated miRNAs were sorted by changes in mature miRNA expression (read counts) during heart development (upregulated miRNAs: adult/E10.5 > 1.5, adult/E14.5 > 1.5, adult/E17.5 > 1.5, neonatal/E10.5 > 1.5, neonatal/14.5 > 1.5 [Table S8]. downregulated miRNAs: adult/E10.5 < 1.5, adult/E14.5 < 1.5, adult/E17.5 < 1.5, neonatal/E10.5 < 1.5, neonatal/14.5 < 1.5 [Table S9]).
Statistical analysis
Data in this study are presented as the mean ± SEM (at least three independent experiments). Statistical significance was performed according to Student’s t-tests (unpaired two-tailed) for two groups and according to one-way analysis of variance (ANOVA) followed by Tukey’s post hoc for more than two groups (GraphPad Prism software): ∗/# indicates p < 0.05, ∗∗/## indicates p < 0.01, and ∗∗∗/### indicates p < 0.001.
Data availability
The RNA-seq data produced in this study are available in the following databases: Gene Expression Omnibus (GEO) under accession number GSE158202.
Acknowledgments
This study was supported by grants from the National Key R&D Program of China (2021YFA1100400, 2018YFA0800100, and 2017YFA0103700), the National Natural Science Foundation of China (31830059, 31970599, 32170575, 32000605, and 31721003), the Shanghai Rising-Star Program (20QA1409600), the Fundamental Research Funds for the Central Universities (22120220104), and the Shanghai Municipal Medical and Health Discipline Construction Projects (2017ZZ02015), and the project was funded by China Postdoctoral Science Foundation (2019M661609).
Author contributions
J.K., Y.W., and X.G. planned and performed experiments and wrote the manuscript. Y.W., X.G., T.H., K.F., Y.X., Y.C., and P.Z. performed experiments and collected the data. Y.W., X.G., Y.X., Y.Y., X.W., and H.Y. performed data analysis. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.07.022.
Supplemental information
References
- 1.Guo X., Xu Y., Wang Z., Wu Y., Chen J., Wang G., Lu C., Jia W., Xi J., Zhu S., et al. A linc1405/eomes complex promotes cardiac mesoderm specification and cardiogenesis. Cell Stem Cell. 2018;22:893–908.e6. doi: 10.1016/j.stem.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Kattman S.J., Witty A.D., Gagliardi M., Dubois N.C., Niapour M., Hotta A., Ellis J., Keller G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Kadota S., Pabon L., Reinecke H., Murry C.E. In vivo maturation of human induced pluripotent stem cell-derived cardiomyocytes in neonatal and adult rat hearts. Stem Cell Rep. 2017;8:278–289. doi: 10.1016/j.stemcr.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uosaki H., Cahan P., Lee D.I., Wang S., Miyamoto M., Fernandez L., Kass D.A., Kwon C. Transcriptional landscape of cardiomyocyte maturation. Cell Rep. 2015;13:1705–1716. doi: 10.1016/j.celrep.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funakoshi S., Fernandes I., Mastikhina O., Wilkinson D., Tran T., Dhahri W., Mazine A., Yang D., Burnett B., Lee J., et al. Generation of mature compact ventricular cardiomyocytes from human pluripotent stem cells. Nat. Commun. 2021;12:3155. doi: 10.1038/s41467-021-23329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sui Y., Zhang W., Tang T., Gao L., Cao T., Zhu H., You Q., Yu B., Yang T. Insulin-like growth factor-II overexpression accelerates parthenogenetic stem cell differentiation into cardiomyocytes and improves cardiac function after acute myocardial infarction in mice. Stem Cell Res. Ther. 2020;11:86. doi: 10.1186/s13287-020-1575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou X., Zhao M., Fan C., Fast V.G., Valarmathi M.T., Zhu W., Zhang J. N-cadherin overexpression enhances the reparative potency of human-induced pluripotent stem cell-derived cardiac myocytes in infarcted mouse hearts. Cardiovasc. Res. 2020;116:671–685. doi: 10.1093/cvr/cvz179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marques A.C., Ponting C.P. Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol. 2009;10:R124. doi: 10.1186/gb-2009-10-11-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M., Herrmann B.G. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson K.M., Anderson D.M., McAnally J.R., Shelton J.M., Bassel-Duby R., Olson E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature. 2016;539:433–436. doi: 10.1038/nature20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han P., Li W., Lin C.H., Yang J., Shang C., Nuernberg S.T., Jin K.K., Xu W., Lin C.Y., Lin C.J., et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L., An X., Li Z., Song Y., Li L., Zuo S., Liu N., Yang G., Wang H., Cheng X., et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc. Res. 2016;111:56–65. doi: 10.1093/cvr/cvw078. [DOI] [PubMed] [Google Scholar]
- 13.Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touma M., Kang X., Zhao Y., Cass A.A., Gao F., Biniwale R., Coppola G., Xiao X., Reemtsen B., Wang Y. Decoding the long noncoding RNA during cardiac maturation: a roadmap for functional discovery. Circ. Cardiovasc. Genet. 2016;9:395–407. doi: 10.1161/CIRCGENETICS.115.001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehmsen J., Poon E., Davies K. The dystrophin-associated protein complex. J. Cell Sci. 2002;115:2801–2803. doi: 10.1242/jcs.115.14.2801. [DOI] [PubMed] [Google Scholar]
- 16.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morikawa Y., Heallen T., Leach J., Xiao Y., Martin J.F. Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature. 2017;547:227–231. doi: 10.1038/nature22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leach J.P., Heallen T., Zhang M., Rahmani M., Morikawa Y., Hill M.C., Segura A., Willerson J.T., Martin J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260–264. doi: 10.1038/nature24045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassat E., Mutlak Y.E., Genzelinakh A., Shadrin I.Y., Baruch Umansky K., Yifa O., Kain D., Rajchman D., Leach J., Riabov Bassat D., et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–184. doi: 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eulalio A., Mano M., Dal Ferro M., Zentilin L., Sinagra G., Zacchigna S., Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 21.Mullin N.K., Mallipeddi N.V., Hamburg-Shields E., Ibarra B., Khalil A.M., Atit R.P. Wnt/beta-catenin signaling pathway regulates specific lncRNAs that impact dermal fibroblasts and skin fibrosis. Front. Genet. 2017;8:183. doi: 10.3389/fgene.2017.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ounzain S., Micheletti R., Beckmann T., Schroen B., Alexanian M., Pezzuto I., Crippa S., Nemir M., Sarre A., Johnson R., et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur. Heart J. 2015;36:353–368a. doi: 10.1093/eurheartj/ehu180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyu Y., Jia W., Wu Y., Zhao X., Xia Y., Guo X., Kang J. Cpmer: a new conserved eEF1A2-binding partner that regulates Eomes translation and cardiomyocyte differentiation. Stem Cell Rep. 2022;17:1154–1169. doi: 10.1016/j.stemcr.2022.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang Y.J., Yang D.C., Kong L., Hou M., Meng Y.Q., Wei L., Gao G. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017;45:W12–W16. doi: 10.1093/nar/gkx428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye G., Wen Z., Wen F., Song X., Wang L., Li C., He Y., Prakash S., Qiu X. Mussel-inspired conductive Ti2C-cryogel promotes functional maturation of cardiomyocytes and enhances repair of myocardial infarction. Theranostics. 2020;10:2047–2066. doi: 10.7150/thno.38876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godier-Furnémont A.F.G., Martens T.P., Koeckert M.S., Wan L., Parks J., Arai K., Zhang G., Hudson B., Homma S., Vunjak-Novakovic G. Composite scaffold provides a cell delivery platform for cardiovascular repair. Proc. Natl. Acad. Sci. U. S. A. 2011;108:7974–7979. doi: 10.1073/pnas.1104619108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q., Yang H., Bai A., Jiang W., Li X., Wang X., Mao Y., Lu C., Qian R., Guo F., et al. Functional engineered human cardiac patches prepared from nature's platform improve heart function after acute myocardial infarction. Biomaterials. 2016;105:52–65. doi: 10.1016/j.biomaterials.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 28.Yang H., Wei L., Liu C., Zhong W., Li B., Chen Y., Han R., Zhuang J., Qu J., Tao H., et al. Engineering human ventricular heart tissue based on macroporous iron oxide scaffolds. Acta Biomater. 2019;88:540–553. doi: 10.1016/j.actbio.2019.02.024. [DOI] [PubMed] [Google Scholar]
- 29.Chang T.H., Huang H.Y., Hsu J.B.K., Weng S.L., Horng J.T., Huang H.D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinf. 2013;14(Suppl 2):S4. doi: 10.1186/1471-2105-14-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Z., Evans J., Bhagwate A., Middha S., Bockol M., Yan H., Kocher J.P. CAP-miRSeq: a comprehensive analysis pipeline for microRNA sequencing data. BMC Genom. 2014;15:423. doi: 10.1186/1471-2164-15-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuppusamy K.T., Jones D.C., Sperber H., Madan A., Fischer K.A., Rodriguez M.L., Pabon L., Zhu W.Z., Tulloch N.L., Yang X., et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA. 2015;112:E2785–E2794. doi: 10.1073/pnas.1424042112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan F., Lee A.S., Liang P., Sanchez-Freire V., Nguyen P.K., Wang L., Han L., Yen M., Wang Y., Sun N., et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee D.S., Chen J.H., Lundy D.J., Liu C.H., Hwang S.M., Pabon L., Shieh R.C., Chen C.C., Wu S.N., Yan Y.T., et al. Defined MicroRNAs induce aspects of maturation in mouse and human embryonic-stem-cell-derived cardiomyocytes. Cell Rep. 2015;12:1960–1967. doi: 10.1016/j.celrep.2015.08.042. [DOI] [PubMed] [Google Scholar]
- 34.Karbassi E., Fenix A., Marchiano S., Muraoka N., Nakamura K., Yang X., Murry C.E. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020;17:341–359. doi: 10.1038/s41569-019-0331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., Xu Z., Jiang J., Xu C., Kang J., Xiao L., Wu M., Xiong J., Guo X., Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell. 2013;25:69–80. doi: 10.1016/j.devcel.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Liu J., Li Y., Lin B., Sheng Y., Yang L. HBL1 is a human long noncoding RNA that modulates cardiomyocyte development from pluripotent stem cells by counteracting MIR1. Dev. Cell. 2017;43:333–348.e5. doi: 10.1016/j.devcel.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 38.Poon E.N.Y., Hao B., Guan D., Jun Li M., Lu J., Yang Y., Wu B., Wu S.C.M., Webb S.E., Liang Y., et al. Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovasc. Res. 2018;114:894–906. doi: 10.1093/cvr/cvy019. [DOI] [PubMed] [Google Scholar]
- 39.Bhat H.F., Mir S.S., Dar K.B., Bhat Z.F., Shah R.A., Ganai N.A. ABC of multifaceted dystrophin glycoprotein complex (DGC) J. Cell. Physiol. 2018;233:5142–5159. doi: 10.1002/jcp.25982. [DOI] [PubMed] [Google Scholar]
- 40.Grady R.M., Grange R.W., Lau K.S., Maimone M.M., Nichol M.C., Stull J.T., Sanes J.R. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- 41.Lapidos K.A., Kakkar R., McNally E.M. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 42.Danialou G., Comtois A.S., Dudley R., Karpati G., Vincent G., Des Rosiers C., Petrof B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001;15:1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 43.Badorff C., Knowlton K.U. Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: from bench to bedside. Med. Microbiol. Immunol. 2004;193:121–126. doi: 10.1007/s00430-003-0189-7. [DOI] [PubMed] [Google Scholar]
- 44.Strakova J., Dean J.D., Sharpe K.M., Meyers T.A., Odom G.L., Townsend D. Dystrobrevin increases dystrophin's binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. J. Mol. Cell. Cardiol. 2014;76:106–115. doi: 10.1016/j.yjmcc.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ignatieva E., Smolina N., Kostareva A., Dmitrieva R. Skeletal muscle mitochondria dysfunction in genetic neuromuscular disorders with cardiac phenotype. Int. J. Mol. Sci. 2021;22:7349. doi: 10.3390/ijms22147349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu D., Linders A., Yamak A., Correia C., Kijlstra J.D., Garakani A., Xiao L., Milan D.J., van der Meer P., Serra M., et al. Metabolic maturation of human pluripotent stem cell-derived cardiomyocytes by inhibition of HIF1alpha and LDHA. Circ. Res. 2018;123:1066–1079. doi: 10.1161/CIRCRESAHA.118.313249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kashihara T., Mukai R., Oka S.I., Zhai P., Nakada Y., Yang Z., Mizushima W., Nakahara T., Warren J.S., Abdellatif M., Sadoshima J. YAP mediates compensatory cardiac hypertrophy through aerobic glycolysis in response to pressure overload. J. Clin. Invest. 2022;132:e150595. doi: 10.1172/JCI150595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reinecke H., Zhang M., Bartosek T., Murry C.E. Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation. 1999;100:193–202. doi: 10.1161/01.cir.100.2.193. [DOI] [PubMed] [Google Scholar]
- 49.Pushp P., Nogueira D.E.S., Rodrigues C.A.V., Ferreira F.C., Cabral J.M.S., Gupta M.K. A concise review on induced pluripotent stem cell-derived cardiomyocytes for personalized regenerative medicine. Stem Cell Rev. Rep. 2021;17:748–776. doi: 10.1007/s12015-020-10061-2. [DOI] [PubMed] [Google Scholar]
- 50.Gao L., Gregorich Z.R., Zhu W., Mattapally S., Oduk Y., Lou X., Kannappan R., Borovjagin A.V., Walcott G.P., Pollard A.E., et al. Large cardiac muscle patches engineered from human induced-pluripotent stem cell-derived cardiac cells improve recovery from myocardial infarction in swine. Circulation. 2018;137:1712–1730. doi: 10.1161/CIRCULATIONAHA.117.030785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng L., Hu Q., Wang X., Mansoor A., Lee J., Feygin J., Zhang G., Suntharalingam P., Boozer S., Mhashilkar A., et al. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation. 2007;115:1866–1875. doi: 10.1161/CIRCULATIONAHA.106.659730. [DOI] [PubMed] [Google Scholar]
- 52.Hwang H.W., Wentzel E.A., Mendell J.T. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 53.Guo X., Liu Q., Wang G., Zhu S., Gao L., Hong W., Chen Y., Wu M., Liu H., Jiang C., Kang J. microRNA-29b is a novel mediator of Sox2 function in the regulation of somatic cell reprogramming. Cell Res. 2013;23:142–156. doi: 10.1038/cr.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xi J., Wu Y., Li G., Ma L., Feng K., Guo X., Jia W., Wang G., Yang G., Li P., Kang J. Mir-29b mediates the neural tube versus neural crest fate decision during embryonic stem cell neural differentiation. Stem Cell Rep. 2017;9:571–586. doi: 10.1016/j.stemcr.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu Y., Xi J., Wang G., Guo Z., Sun Q., Lu C., Ma L., Wu Y., Jia W., Zhu S., et al. PAUPAR and PAX6 sequentially regulate human embryonic stem cell cortical differentiation. Nucleic Acids Res. 2021;49:1935–1950. doi: 10.1093/nar/gkab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., De S., Huarte M., Zhan M., Becker K.G., Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012;47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu T.Y., Lin B., Kim J., Sullivan M., Tobita K., Salama G., Yang L. Repopulation of decellularized mouse heart with human induced pluripotent stem cell-derived cardiovascular progenitor cells. Nat. Commun. 2013;4:2307. doi: 10.1038/ncomms3307. [DOI] [PubMed] [Google Scholar]
- 58.Li B., Yang H., Wang X., Zhan Y., Sheng W., Cai H., Xin H., Liang Q., Zhou P., Lu C., et al. Engineering human ventricular heart muscles based on a highly efficient system for purification of human pluripotent stem cell-derived ventricular cardiomyocytes. Stem Cell Res. Ther. 2017;8:202. doi: 10.1186/s13287-017-0651-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yasui H., Lee J.K., Yoshida A., Yokoyama T., Nakanishi H., Miwa K., Naito A.T., Oka T., Akazawa H., Nakai J., et al. Excitation propagation in three-dimensional engineered hearts using decellularized extracellular matrix. Biomaterials. 2014;35:7839–7850. doi: 10.1016/j.biomaterials.2014.05.080. [DOI] [PubMed] [Google Scholar]
- 60.Gao E., Lei Y.H., Shang X., Huang Z.M., Zuo L., Boucher M., Fan Q., Chuprun J.K., Ma X.L., Koch W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010;107:1445–1453. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang P., Huang J.J., Ou-Yang K.F., Liang H., Li M.L., Wang Y.J., Yang H.T. Minimal contribution of IP3R2 in cardiac differentiation and derived ventricular-like myocytes from human embryonic stem cells. Acta Pharmacol. Sin. 2020;41:1576–1586. doi: 10.1038/s41401-020-00528-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poon E.N.Y., Luo X.L., Webb S.E., Yan B., Zhao R., Wu S.C.M., Yang Y., Zhang P., Bai H., Shao J., et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020;30:626–629. doi: 10.1038/s41422-020-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Horikoshi Y., Yan Y., Terashvili M., Wells C., Horikoshi H., Fujita S., Bosnjak Z.J., Bai X. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells. 2019;8 doi: 10.3390/cells8091095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data produced in this study are available in the following databases: Gene Expression Omnibus (GEO) under accession number GSE158202.