

Abstract
The discovery of N6-methyldeoxyadenine (6mA) across eukaryotes created excitement for additional epigenetic mechanisms. However, some studies have highlighted confounding factors, challenging the prevalence of 6mA in eukaryotes. We developed a metagenomic method to quantitatively deconvolve 6mA events from a gDNA sample into species of interest, genomic regions, and sources of contamination. Applying this method, we observed high-resolution 6mA deposition in two protozoa. We found that commensal/soil bacteria explained the vast majority of 6mA in insect and plant samples. We found no evidence of high 6mA in Drosophila, Arabidopsis, or human. Plasmids used for genetic manipulation even from Dam methyltransferase mutant Escherichia coli, could carry abundant 6mA, confounding the evaluation of candidate 6mA methyltransferases and demethylases. This work advocates for a re-assessment of 6mA in eukaryotes.
One Sentence Summary:
To help clarify DNA adenine methylation in eukaryotes, we developed a method and report findings with broad implications.
For decades, N6-methyldeoxyadenine (6mA) is known to be widespread in prokaryotes regulating DNA replication, repair and transcription (1–3). Recently, 6mA has been reported to be also prevalent in eukaryotes. Unlike the generally high abundance of 6mA in bacteria, 6mA/A levels in eukaryotic organisms vary over several orders of magnitudes (4–13). A few unicellular organisms have very high 6mA/A levels: 0.4% in Chlamydomonas reinhardtii (4), 0.66% in Tetrahymena thermophila (5) and up to 2.8% in early-diverging fungi (6). In contrast, 6mA/A levels reported in multicellular eukaryotes are much lower: ~0.1% to ~0.0001%, or undetectable (8, 10–12, 14, 15). Nevertheless, important functions have been assigned to 6mA in eukaryotes, suggesting additional epigenetic mechanisms in basic biology and human diseases (11). However, other studies have cast doubt on the existence and levels of 6mA in eukaryotic DNA (15–19). For example, liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) can reliably quantify 6mA with high sensitivity, but it cannot discriminate eukaryotic 6mA from bacterial 6mA contamination (16, 20). Unique metabolically generated stable isotope labeling can address this limitation of LC-MS/MS (17, 18); however, it can only be used in cultured cells. Anti-6mA antibody-based dot blotting is commonly used to estimate 6mA levels (4, 5, 7, 9–12), but it cannot rule out bacterial contamination. In addition, anti-6mA antibody-based DNA immuno-precipitation sequencing (DIP-seq) is often used for 6mA mapping (7, 8, 10, 13, 21), but it can be confounded by 6mA-independent factors such as DNA secondary structures (20) and RNA contamination (15). Restriction enzyme-based 6mA analyses are constrained by their limited recognition motifs (4, 22). Single molecule real time (SMRT) sequencing (23) and nanopore sequencing (24) provide opportunities for directly mapping 6mA events (3, 25, 26), but the existing methods are mainly for mapping 6mA in prokaryotes and protozoa with high 6mA abundance (3, 14, 26–29). For eukaryotes with low 6mA abundance, these methods are prone to make many false positive calls due to low sensitivity (14–16).
The lack of a reliable technology that accurately quantify 6mA/A levels in eukaryotic genomes motivated us to develop a method, named 6mASCOPE, for quantitative 6mA deconvolution (Fig. 1). The method, based on a short insert SMRT library design (Fig. 1A), examines all DNA molecules sequenced in a gDNA sample, separates the total sequences into different sources, and quantitatively deconvolves the total 6mA events into each of the sources (Fig. 1B). We first validated our method over a wide range of 6mA/A levels, from 10−6 to 10−1, and then examined a number of eukaryotes.
Fig. 1. Overview of 6mASCOPE for quantitative 6mA deconvolution.
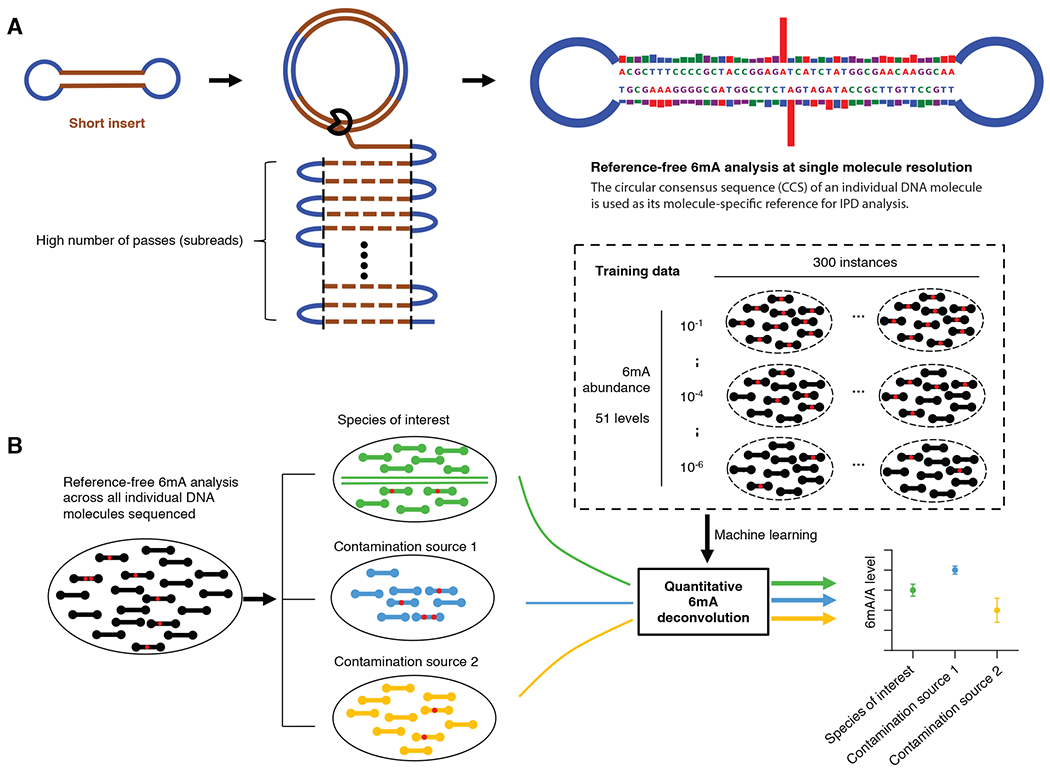
(A) Reference-free 6mA analysis of single molecules. Each molecule (short insert) is sequenced for a large number of passes (subreads). The subreads are combined to a circular consensus sequence (CCS), serving as the molecule-specific reference for in silico IPD estimation, and provide repeated measures of IPD values for 6mA analysis (Methods). Blue segment: SMRT adapter. (B) After single molecule 6mA analysis (a red dot indicates a 6mA event), CCSs (black rods) from a sequenced gDNA sample are separated into the eukaryotic genome (green) and contamination sources (blue and yellow). The 6mA/A levels of each species (or genomic region) are estimated using a machine learning model trained across a wide range of 6mA abundance, with defined confidence intervals.
Results
A method for quantitative 6mA deconvolution
Existing SMRT sequencing-based methods for modification detection require a reference genome, as they compare the inter-pulse duration (IPD) associated with a base of interest in the native DNA to the expected IPD value estimated based on the base and its flanking DNA sequence in the reference genome provided, calculating an IPD ratio (25, 29, 30). Within this design, only those sequencing reads that map to the provided reference genome are analyzed for 6mA, ignoring potential bacterial contamination, carrying abundant 6mA events, beyond the eukaryotic reference genome.
To solve this problem, we took a metagenomic approach not limited to the eukaryotic species of interest. First, in contrast to existing methods that depends on a reference genome for IPD analysis, we took a reference-free approach by using the circular consensus sequence (CCS, a feature of SMRT sequencing for error correction) of an individual DNA molecule as its molecule-specific reference for IPD analysis (23, 25) (Fig. 1A; Methods), thus examining all the sequenced genetic contents for 6mA analysis. We designed relatively short SMRT insert libraries (200~400bp) (Fig. S1A; Methods; Supplementary Text) so that each DNA molecule could be sequenced for a large number of passes (mean: 272X; median: 181X; Fig. 1A and Fig. S1B), which facilitated a CCS base calling accuracy of >99.84% (Phred Score 28; Methods; Supplementary Text, Fig. S2) and enabled reliable IPD analysis on single molecules (Fig. 2A, B). We then used a metagenomic approach to map the CCS reads to a comprehensive collection of genomes (Methods) and performed 6mA quantification (described below) separately for each subgroup of genetic contents in a gDNA sample: species of interest, genomic regions of interest, and sources of contamination.
Fig. 2. 6mASCOPE method evaluation.
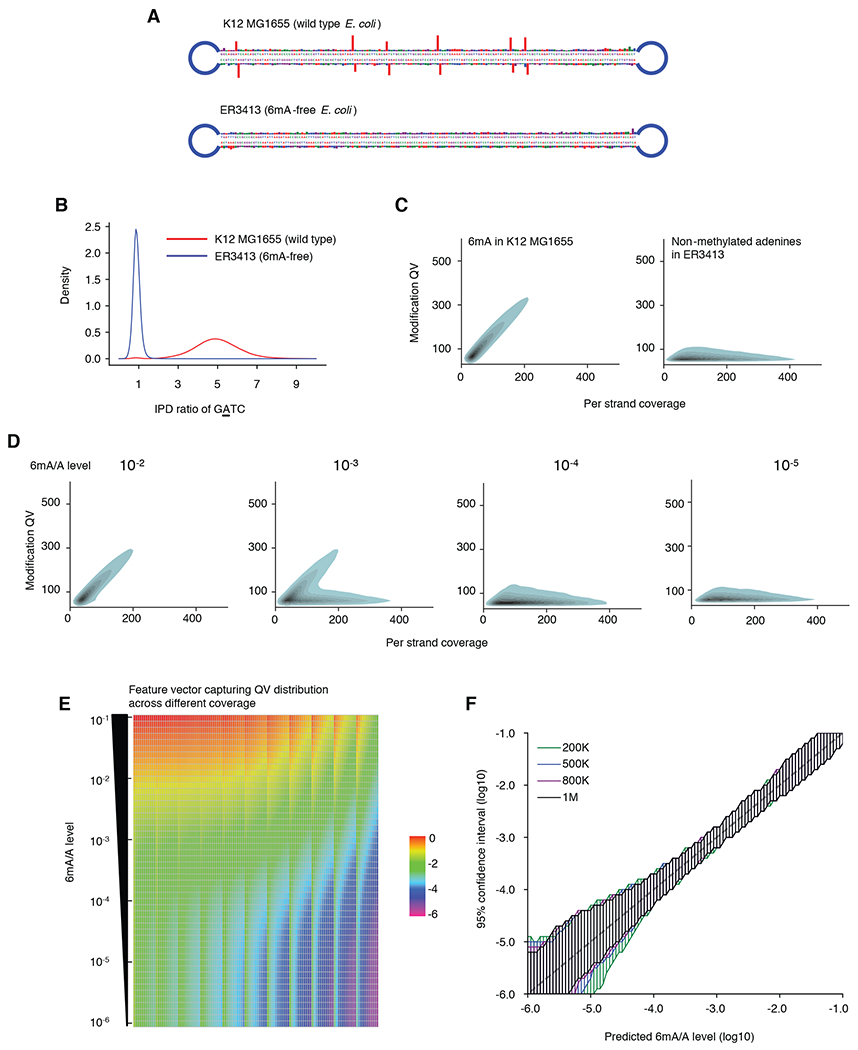
(A) IPD ratios on illustrative molecules from E. coli wild type strain K12 MG1655 and 6mA-free strain ER3413. Blue segment: SMRT adapter. (B) IPD ratio of adenines on GATC motif in E. coli K12 MG1655 and ER3413. 6mA events have IPD ratios ~5 while non-methylated adenines have IPD ratios ~1. (C) Modification Quality values (QVs) of 6mA linearly (slope ~1.7) deviate from the non-methylated adenines with better separation at high CCS passes. For illustration, kernel density estimation of adenines with QV 50 is shown. Left, 6mA in GATC, GCACNNNNNNGTT and AACNNNNNNTGC from E. coli K12 MG1655. Right, non-methylated adenines in E. coli ER3413. (D) QV distribution varies across different 6mA/A levels. Same legend as in (C). (E) Feature vectors used for machine learning model training. Rows: 51 6mA/A levels (10−1 to 10−6) are constructed by mixing negative and positive controls in silico at different ratios. Each column represents the percentage (averaged across 300 replicates, log10 transformed) of adenines over a number of slopes across CCS passes 20-240x, divided into 11 bins (Methods). (F) For each 6mA quantification (x-axis), 6mASCOPE also provides the 95% confidence interval (y-axis) (Methods). Colors represent the number of CCS reads used for 6mA quantification.
The current standard method to detect 6mA from SMRT sequencing is based on a defined cut off on a modification quality value (QV; essentially a transformed p value; Methods) (3, 28, 31). Because QV varies dramatically over sequencing depth or number of CCS passes on individual molecules (Fig. 2C) (28, 30), a fixed cutoff can create false positive 6mA calls, especially from genomic regions with high sequencing depth (e.g., mitochondrial genomes). We built on a critical observation of linear increase (slope ~1.7 for 6mA events) of QV over CCS passes (better separation from non-methylated adenine’s at higher coverages, Figs. 2C & D) and developed a machine learning model for 6mA quantification from QV values calculated in the reference-free single molecule IPD analysis. The core idea was to train the machine learning model across a wide range of 6mA/A levels (training datasets described below), and use the model to predict 6mA/A levels of newly sequenced gDNA samples based on the collective QV distribution, instead of an arbitrary QV cutoff (Fig. 2D; Methods).
We constructed high quality benchmark datasets for the machine learning model training. For 6mA negative controls, we used HEK-WGA (whole genome amplification of HEK-293 cell gDNA, 6mA/A level < 10−6 by ultra-high performance liquid chromatography tandem mass spectrometry, UHPLC-MS/MS), HEK293 (native gDNA, 6mA/A level <10−6 by UHPLC-MS/MS), and HEK-WGA-MsssI (CpG sites in vitro methylated using a 5mC methyltransferase, MsssI), with the latter two representing the influence of 5mC events on IPD (16, 25) (Methods). These samples were each methylated in vitro using three bacterial 6mA methyltransferases (Dam: GATC; TaqI: TCGA; and EcoRI: GAATTC) to create three positive controls: HEK-WGA-3M, HEK293-3M, HEK-WGA-MsssI-3M (Fig. S3). By mixing negative and positive controls in silico at different ratios, we created a wide range of 6mA/A levels (10−1 to 10−6) for the model training (Methods; Fig. 2E). Using Leave-One-Out cross validation, we compared several models (Fig. S4) and selected Random Forest. Our model showed reliable quantification of 6mA/A levels with defined 95% confidence intervals (CIs; Figs. 2F, S5; Methods). CI depends on both 6mA/A level and number of CCS reads (Fig. 2F, Fig. S5B, Supplementary Text), which facilitated dataset-specific CI estimation along with 6mA quantification.
In contrast to existing methods (Table S1), 6mASCOPE takes a metagenomic approach and specifically quantifies 6mA events in eukaryotic genomes over contamination, because CCS reads, grouped by species (or specific genomic regions), are separately quantified for 6mA/A levels. For validation, we applied 6mASCOPE on a series of in vitro mixed E. coli, H. pylori and S. cerevisiae samples with a wide range of 6mA/A levels (10−2 to 10−6 by UHPLC-MS/MS) and found that 6mASCOPE reliably deconvolved different sources into expected ratios along with stable 6mA quantification (Fig. S6).
High-resolution insights of 6mA deposition in two protozoans
Although previous studies reported enrichment of 6mA events in the linkers near transcription start sites (TSS) in two protozoans, C. reinhardtii and T. thermophila (4, 5), it remains unclear which specific regions within the linkers are enriched for 6mA events. We SMRT sequenced both organisms and obtained 862,205 and 975,050 CCS reads for single molecule 6mA analysis, respectively (Methods, Table S2). We first reproduced that 6mA has a periodical pattern inversely correlated with nucleosomes near TSSs (Fig. S7, Methods). Next, by dividing genomic regions between nucleosome dyad and the middle of each nucleosome linker into ten bins (Methods) and quantifying 6mA/A levels in each bin using 6mASCOPE, we found that 6mA was enriched at the nucleosome-linker boundaries in C. reinhardtii (Fig. 3A, D) instead of at the middle of the linkers as previously reported. In contrast, 6mA/A levels of T. thermophila increased from the nucleosome boundaries to the middle of linkers (Fig. 3A, E; S8). We further used 6mASCOPE to examine the enrichment of 6mA across different motifs: for C. reinhardtii, we confirmed that 6mA is enriched in the VATB motif (Fig. 3B; V = A, C or G; B = C, G or T) and essentially absent in non-VATB motifs; for T. thermophila, although 6mA was reported to be enriched across NATN motif (5), our 6mASCOPE analysis revealed that VATB sites have a 2~3 fold higher 6mA/A level than TATN and NATA sites (Fig. 3C).
Fig. 3. 6mASCOPE discovers high resolution 6mA deposition in C. reinhardtii and T. Thermophila.
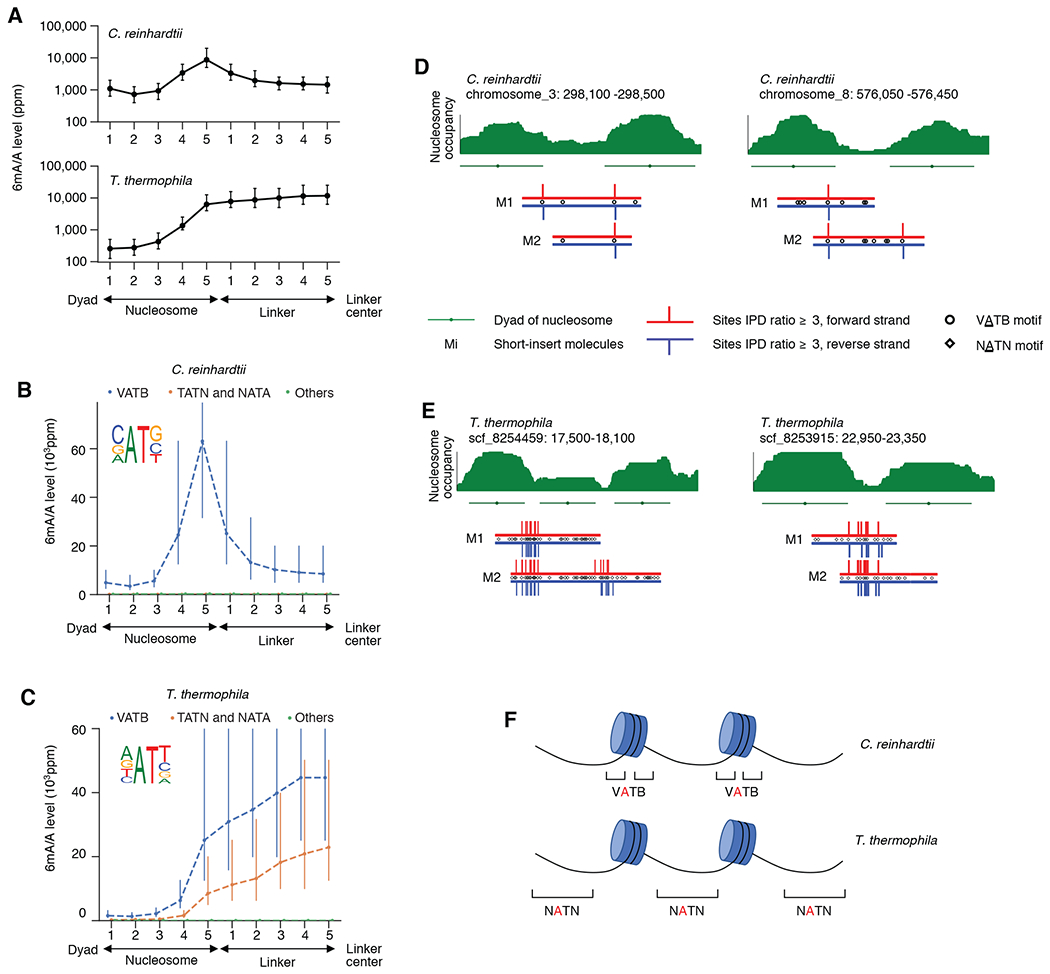
(A) 6mA deposition relative to nucleosomes and linkers in C. reinhardtii and T. thermophila. Genomic regions between the nucleosome dyad and the linker center are divided into ten bins (x-axis) across the genome. 6mA/A level (y-axis) was quantified with 6mASCOPE. Error bars: 95% CIs. (B) 6mA is enriched in VATB motif at nucleosome-linker boundaries in C. reinhardtii. Adenines in each bin are divided into three groups: VATB, TATN/NATA, and others. x- and y-axes are the same as in (A). Error bars: 95% CIs. (C) 6mA is enriched across the NATN motif at linkers in T. thermophila. Same legend as in (B). (D) and (E), Illustrative examples of 6mA enrichment in C. reinhardtii (D) and T. thermophila (E). Nucleosome occupancy (green stack) is based on MNase-seq data (Methods). Nucleosomes (green lines) and dyads (green dots) are determined by iNPS(v1.2.2). SMRT CCS reads (Mi) are shown with red (forward strand) and blue (reverse strand) lines. IPD ratios 3 are shown. (F) Schematic of 6mA enrichment at the nucleosome-linker boundaries in C. reinhardtii, and the gradual 6mA increase from nucleosome boundaries to linker centers in T. thermophila.
6mA from commensal bacteria contribute to most 6mA events in insect and plant samples
A previous study quantified 6mA in D. melanogaster using UHPLC-MS/MS and reported that 6mA/A reaches the peak level of ~700ppm (parts per million) in ~0.75h embryos and falls ~10ppm at later stages such as adult tissues (8). We first collected the fly embryo sample at ~0.75h and got 674,650 SMRT CCS reads for single molecule 6mA analysis (Table S2). Despite strict measures to avoid contamination (Methods), we found that while 96.12% of the CCS reads map to the D. melanogaster genome reference, 3.88% of the CCS reads map to a few microbes (Fig. 4A). Specifically, the contamination reads came from Saccharomyces cerevisiae (1.65%), the major food source of Drosophila (32), and two genera of bacteria, Acetobacter (0.86%) and Lactobacillus (0.23%), the main gut commensal bacteria of D. melanogaster (33). We separately quantified 6mA/A levels in the D. melanogaster genome and in each contamination source and found that the level of 6mA/A in total gDNA was 100 ppm (CI: 50-200, consistent with the ~121ppm UHPLC-MS/MS estimate), 2 ppm in D. melanogaster (CI: 1-10), 2 ppm in Saccharomyces (CI:1-10), 5,495 ppm in Acetobacter (, CI: 3,162-10,000), 977 ppm in Lactobacillus (CI: 501-1,995), and 7,413 ppm in Others (including additional bacteria genera and un-annotated sequences, Methods, CI: 3,981-12,589;) (Fig. 4B, Fig. S9). Importantly, despite their relatively low abundance (3.88%), bacteria contributed to most of the 6mA events in the total gDNA (Fig. 4C). In Acetobacter, we observed a high confidence bacterial 6mA motif (GANTC)(Fig. 4B), consistent with REBASE database (34). The 6mA/A level of 2 ppm (CI: 1-10ppm) estimated for D. melanogaster, in contrast with the ~700ppm previously reported, only explains 1.44% of the total 6mA events in the gDNA sample (Fig. 4C; considering taxonomy abundances). We next applied 6mASCOPE to examine a D. melanogaster adult sample (whole animal), which showed very different microbiome composition with extremely low bacteria contamination, yet still no evidence of high 6mA/A level in Drosophila (Fig. S10). We also reanalyzed the 6mA DIP-seq data from a previous D. melanogaster study (8) and found reads that map to multiple bacterial genomes. It is also worth noting that N4-methylcytosine (4mC), another form of DNA methylation prevalent in bacteria, was also detected in CCS reads from Acetobacter enriched at GTAC sites (Fig. S11), a motif previously reported in Acetobacter (34). This highlights that 4mC analysis for eukaryotic organisms also should be cautiously examined for possible bacterial contamination.
Fig. 4. 6mASCOPE analyses show that commensal bacteria contribute to the vast majority of 6mA events in insect and plant samples.
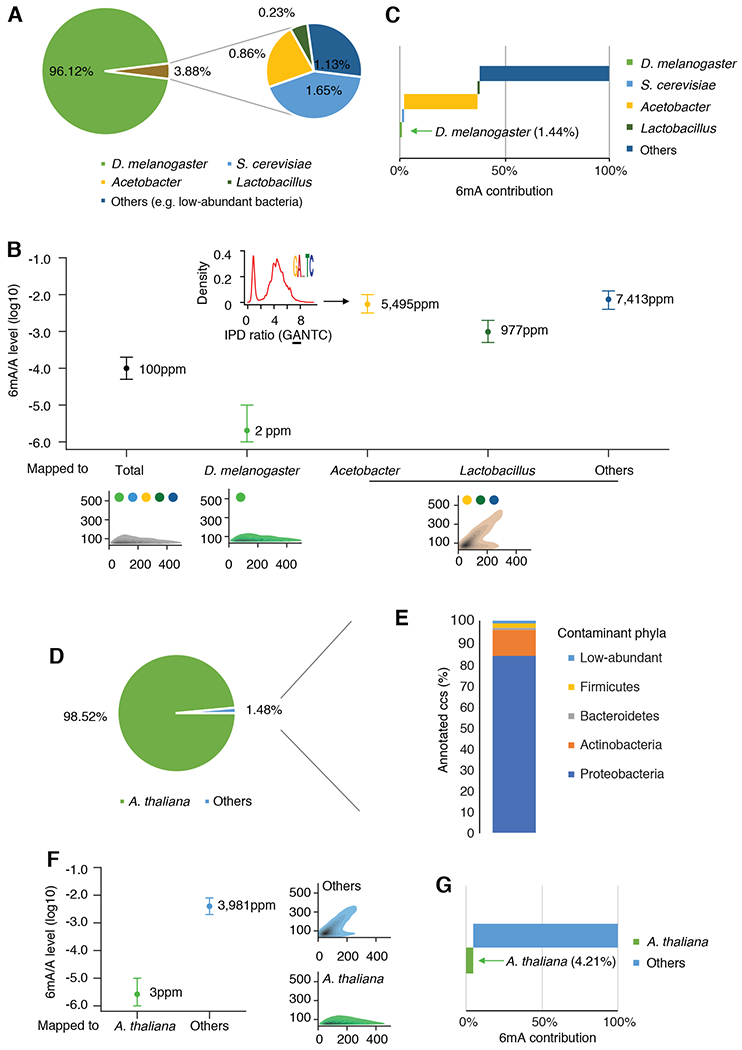
(A) Taxonomic compositions (%) in the D. melanogaster embryo ~0.75h gDNA sample. CCS reads mapped to Acetobacter or Lactobacillus are summarized by genus. (B) 6mA quantification of the D. melanogaster genome and contaminations. For each subgroup, 6mA/A levels are quantified by 6mASCOPE (error bars: 95% CIs). QV distributions are shown at bottom (color dots: species/genus). 6mA/A level of S. cerevisiae is further examined with additional sequencing (Fig. S9). CCS reads from Acetobacter, Lactobacillus and Others (e.g. low-abundant bacteria) are grouped together due to low CCS read counts within each subgroup and CIs are defined based on 8,000 CCS reads. Arrow denotes the density of IPD ratios in GANTC motif in Acetobacter. (C) 6mA contribution (%) from each subgroup in the D. melanogaster embryo sample. (D & E) Taxonomic compositions (%) in the A. thaliana 21-day seedling gDNA sample. The CCS reads in subgroup “Others” (D) are taxonomy classified with Kraken2. Main classes of Proteobacteria are shown in Fig. S12. (F) 6mA quantification of the A. thaliana genome and the contamination (Others). Same legend as in (B). (G) 6mA contribution (%) from each subgroup in the A. thaliana seedling sample.
In addition to insects, we hypothesized that soil bacteria can confound 6mA analysis in plants. We applied 6mASCOPE to A. thaliana 21-day-old seedlings (Methods), which was reported as having ~2500ppm 6mA/A by LC-MS/MS (9). Among the total 535,030 SMRT CCS reads for single molecule 6mA analysis, 98.52% can be mapped to the A. thaliana genome (Fig. 4D). Among the other 1.48% (subgroup “Others”), 24.12% can be annotated and classified (using Kraken2) into several phyla: Proteobacteria (Fig. S12), Actinobacteria, Bacteroidetes , Firmicutes. These phyla and classes (Fig. 4E, Fig. S12) are consistent with A. thaliana root microbiome (35). Using 6mASCOPE, we separately quantified 6mA/A levels for A. thaliana (3ppm, CI 1-10ppm) and Others (3,981ppm, CI 1,995-7,943), and found that CCS reads mapped to A. thaliana only contribute to 4.21% of the total 6mA events in the total gDNA sample (Fig. 4F, G). Consistently, 6mASCOPE analysis of the A. thaliana 21-day-old root sample also demonstrated significant microbiome contamination (greater than the seedlings) with a smaller contribution from A. thaliana to the total 6mA events (Fig. S13).
6mASCOPE finds no evidence of high abundance of 6mA in the human cells examined
We next examined the abundance of 6mA in human cells and tissues. We chose to investigate peripheral blood mononuclear cells (PBMC), which are composed of 70-90% lymphocytes (36) because lymphocytes have been shown to have a high 6mA/A level of ~0.051% (510ppm) (12). We also collected two glioblastoma brain tissue samples because glioblastoma stem cell and primary glioblastoma were reported with a 6mA/A level of ~1000 ppm by dot blotting and mass spectrometry (11).
We obtained 570,283, 247,700, and 280,763 SMRT CCS reads from the PBMC sample and the two glioblastomas brain tissues, respectively, for single molecule 6mA analysis; 99.53%, 99.88%, and 99.86% of CCS reads could be mapped to the human reference indicating highly pure samples. The 6mA/A levels estimated by 6mASCOPE in glioblastoma samples were ~10−6 , with 3ppm (CI: 1-16ppm) for Glioblastoma-1 and 2ppm (CI: 1-13ppm) for Glioblastoma-2 (Fig. 5A; Methods). This level was comparable to the negative controls with extremely low 6mA/A levels: HEK-WGA (1ppm, 1-6ppm) and native HEK293 (1ppm, 1-6ppm), when the confidence intervals were taken into consideration. In the PBMC sample, the 6mA/A level estimation of 17ppm (CI: 4-63ppm) by 6mASCOPE is consistent with the measurements of UHPLC-MS/MS (Fig. 5A). These data suggested that 6mA, if present in glioblastoma and PBMC, were either significantly lower than the reported levels in the recent studies (Glioblastoma ~1000 ppm; Lymphocytes: ~510 ppm), or 6mA/A level may be highly variable between different samples of the same cell type, tissue or a specific disease. Motif enrichment analysis did not support a reliable motif in these samples (Fig. S14).
Fig. 5.
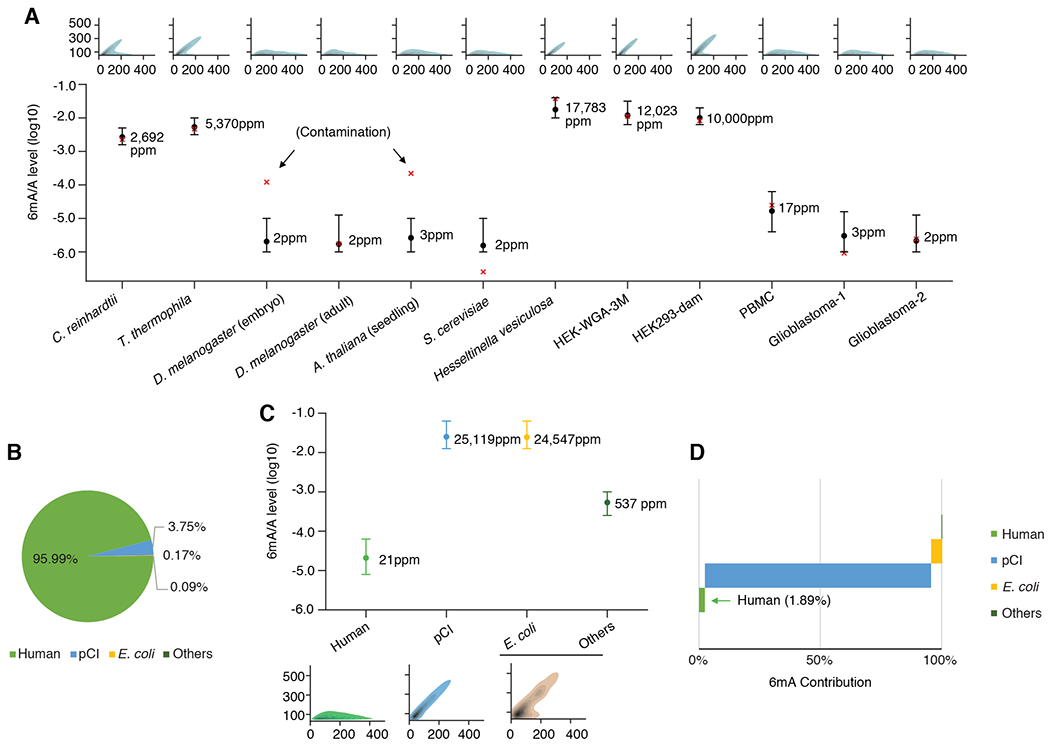
6mASCOPE based quantitative deconvolution across multiple human gDNA samples. (A) 6mA/A levels on the genome of interest quantified by 6mASCOPE (error bars: 95% CIs). 6mA/A level in S. cerevisiae is consistent with independent UHPLC-MS/MS measurement (0.3ppm, lower than the minimum 6mA/A level used in 6mASCOPE training dataset). Except for D. melanogaster embryo and A. thaliana gDNA samples (both are contaminated by bacteria), 6mA/A levels by 6mASCOPE are consistent with UHPLC-MS/MS (red cross). For all samples except HEK-WGA-3M and HEK293-dam, the UHPLC-MS/MS is performed independently using the same batch of gDNA samples. For HEK-WGA-3M and HEK293-dam, the UHPLC-MS/MS estimates are mimicked: nearly all the expected motif(s) are methylated in vitro by the methyltransferase(s). For each gDNA sample, QV distribution is shown on the top. (B) Sources (%) of CCS reads in the HEK-pCI sample (transfection of an empty pCI plasmid into HEK 293 cells). (C) 6mA quantification (%) of different sources in HEK-pCI ; same legend as in (A). CCS reads from E. coli and Others are grouped together and their CIs are determined based on 8,000 CCS reads. (D) 6mA contribution (%) from the subgroups in the HEK-pCI sample.
Across all the samples examined in this study, we observed largely consistent 6mA/A level estimates between 6mASCOPE and UHPLC-MS/MS (Fig. 5A) except the D. melanogaster embryo and A. thaliana samples, for which the much higher 6mA/A estimates by UHPLC-MS/MS were due to bacterial contamination (Fig. 4), highlighting the capability and reliability of 6mASCOPE. In addition to 6mA quantification of individual species, our method was also able to quantify 6mA/A levels in specific genomic regions of interest. Previous studies have reported enrichment of 6mA in mitochondrial DNA (mtDNA) (12, 13, 21, 37) and in young full-length LINE-1 elements (L1s) (10, 11, 21). For mtDNA, 6mASCOPE did not find significant 6mA enrichment in the 7,205 CCS reads from the HEK293 sample that map to mtDNA, in comparison to a negative control (targeted mitochondrial genome amplification, 10−5.72, CI: 10−6.00 to 10−4.90; Fig. S15). For L1 elements, although 6mASCOPE appeared to suggest a higher 6mA/A level in the young full-length L1s than older L1s, a further comparison with a WGA negative control did not support 6mA enrichment in young L1 elements (Fig. S16), highlighting the importance of using negative controls to capture possible uncharacterized biases (14, 38). This result was consistent with our previous study of human lymphoblastoid cells, in which increased IPD patterns exist not only in A’s but also C’s, G’s, and T’s, of young L1 elements, which suggested confounding factors such as secondary structure (14).
Plasmids used for genetic manipulation can carry confounding bacterial-origin 6mA
Genetic manipulation is commonly used in epigenetic research to characterize putative methyltransferases and demethylases. E. coli is often used as hosts for plasmid selection and expansion. As a result, the plasmids can contain 6mA events written by bacterial methyltransferase(s) and confound 6mA study in eukaryotic cells.
To illustrate this, we transfected an empty pCI plasmid vector from E. coli into HEK 293 cells following the standard Lipofection-based protocol (Methods). Total gDNA harvested at 72 hrs post transfection was SMRT sequenced and analyzed using 6mASCOPE. Among the 741,558 CCS reads, 95.99% could be mapped to the human genome, while 3.75% came from the pCI vector (Fig. 5B), and the remaining 0.26% CCS reads (Fig. 5B) also include reads that map to the E. coli genome (Methods), implying possible carryover of gDNA from E. coli to the HEK293 during transfection. By separately quantifying the 6mA/A level in each subgroup, pCI showed a high 6mA/A level of 10−1.60 (25,119 ppm), about the same as E. coli (Fig. 5C). Considering its abundance, pCI contributed to 93.91% of the total 6mA events in this post-transfection HEK293 total gDNA (Fig. 5C, D). This cautioned that genetic manipulation experiments involving plasmids may confound the characterization of putative 6mA methyltransferases and demethylases. While the use of methylation-free bacteria as the host for plasmid preparation can avoid this type of contamination, it is worth noting that the Dam methyltransferase mutant E.coli, previously used in a few studies (7, 37), still has substantial 6mA events because of the remaining 6mA methyltransferase hsdM (2, 28) (Fig. S17 based on 6mASCOPE analysis). So, we suggest the use of E.coli strains with both Dam and hsdM deleted as the plasmid host.
Discussion
This study did not mean to exclude the potential presence of authentic, high levels of 6mA/A in multicellular eukaryotes in certain samples that we did not examine here. However, we do advocate for a re-assessment of 6mA across eukaryotic genomes using 6mASCOPE to quantitatively estimate the confounding impact of bacterial contamination. To facilitate the broad use of 6mASCOPE, we have released a detailed experimental protocol and an automated software package (39).
We also stressed the possibility of plasmid 6mA contamination, even from Dam methyltransferase mutant E. coll, during genetic manipulation, and suggested that it may have confounded previous characterization of 6mA enzymes. Lipofection or electroporation, which transfect plasmid DNA directly into the target cells, are more likely to introduce contamination, while lentiviral transduction, would be less affected if the original plasmids were completely removed during viral packaging.
Although this study was focused on 6mA, a similar need also applies to 4mC detection. The analysis of the D. melanogaster embryo sample not only discovered bacterial-origin 6mA events but also bacterial-origin 4mC events. This suggests that similar caution is needed when studying 4mC given that recent studies have attempted to call 4mC sites from eukaryotes using SMRT sequencing (40), despite it being prone for false positive calls (16), especially given the lack of evidence for 4mC in mouse even using ultrasensitive UHPLC-MS/MS (19). More broadly, this study also helps guide rigorous technological development for the detection of other forms of rare DNA and RNA modifications.
We would also like to highlight a few limitations of this study. First, the focus of 6mASCOPE is more about quantitatively deconvolving the global 6mA/A level into different species and genomic regions of interests, rather than mapping specific 6mA events in a particular genome. We prioritized this focus because the most controversial 6mA findings to date were those reporting high 6mA/A levels in multicellular eukaryotes. The precise mapping of specific 6mA events in a particular genome would require deeper SMRT sequencing and can be pursued in future work. Second, for reliable data interpretation, it is important to combine the 6mA/A levels estimated by 6mASCOPE with their confidence intervals which depend on sequencing depth. However, even with a large number of CCS reads, 6mASCOPE does not precisely differentiate 6mA/A levels below 10ppm because the confidence interval includes 1ppm, which is the lowest 6mA/A level in our training dataset (Fig. 2F; Supplementary Text). Third, two recent studies reported that ribo-m6A on mRNA can be a source of 6mA on DNA via the nucleotide-salvage pathway (17, 18). 6mA events that are misincorporated via this pathway cannot be distinguished from other 6mA events by SMRT sequencing or 6mASCOPE, and isotope labeling coupled with LC/MS-MS is needed instead (17). Fourth, for each gDNA sample, the CCS reads analyzed by 6mASCOPE only represent the DNA molecules that were sequenced by SMRT sequencing. Although SMRT DNA polymerases can effectively sequence through diverse genomic regions with very complex secondary structures (41), it might miss some DNA molecules with certain unknown properties. Last, although 6mASCOPE enables quantitative 6mA deconvolution, it could be confounded by other DNA modifications that indirectly influence SMRT DNA polymerase kinetics of adenines or flanking bases (3, 25, 30), so we suggest to combine LC/MS-MS with 6mASCOPE for joint 6mA quantification and deconvolution of eukaryotic gDNA samples as performed in this study.
Supplementary Material
Acknowledgements
We thank P. Hegemann lab, Humboldt University of Berlin, for the C. reinhardtii strains. We thank H.D. Madhani lab, The University of California, San Francisco, for the S. cerevisiae strain. We thank F. Wang and B. Yao, Emory University, for the D. melanogaster embryos and adults. We thank J. Dong lab, Rutgers University, for the A. thaliana strains. We thank J. Mo, Chinese Academy of Science, for helping with UHPLC-MS/MS analysis. We thank members from the Fang lab for helpful discussions. We thank computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Funding:
Supported by Icahn Institute for Genomics and Multiscale Biology (G.F.), the National Institutes of Health grants R35 GM139655 (G.F.), R01 HG011095 (G.F.) and R56 AG071291 (G.F.). Irma T. Hirschl/Monique Weill-Caulier Trust (G.F.), and Nash Family Foundation (G.F.). UHPLC-MS/MS analyses of 6mA were supported by Strategic Priority Research Program of the Chinese Academy of Sciences XDPB2004 and National Natural Science Foundation of China 22021003 (H.W.).
Footnotes
Computing interests: The authors declare no competing financial interests.
Data and materials availability:
All sequencing data generated in this study have been-submitted to NCBI with accession number PRJNA667898. The software supporting all proposed methods is available along with a tutorial at Zenodo (39) and at GitHub http://www.github.com/fanglab/6mASCOPE.
References and Notes:
- 1.Sánchez-Romero MA, Casadesús J, The bacterial epigenome. Nat. Rev. Microbiol 18, 7–20 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE, Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol. 30, 1232–1239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaulaurier J, Schadt EE, Fang G, Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet 20, 157–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Dore LC, Weng X, Ji Q, Mets L, He C, N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 161, 879–892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Chen X, Sheng Y, Liu Y, Gao S, N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res. 45 (2017), doi: 10.1093/nar/gkx883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K, Haridas S, Kuo A, Salamov A, Ahrendt SR, Lau R, Bowen BP, Lipzen A, Sullivan W, Andreopoulos BB, Clum A, Lindquist E, Daum C, Northen TR, Kunde-Ramamoorthy G, Schmitz RJ, Gryganskyi A, Culley D, Magnuson J, James TY, O’Malley MA, Stajich JE, Spatafora JW, Visel A, Grigoriev IV, Widespread adenine N6-methylation of active genes in fungi. Nat Genet (2017), doi: 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- 7.Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y, DNA Methylation on N6-Adenine in C. elegans. Cell. 161, 868–878 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, Li C, Liu B, Luo Y, Zhu Y, Zhang N, He S, He C, Wang H, Chen D, N6-methyladenine DNA modification in Drosophila. Cell. 161, 893–906 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Liang Z, Shen L, Cui X, Bao S, Geng Y, Yu G, Liang F, Xie S, Lu T, Gu X, Yu H, DNA N6-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 45, 406–416.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, Tackett A, Wang G, Hon LS, Fang G, Swenberg JA, Xiao AZ, DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature. 532, 329–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q, Yu Y, Wang P, Wang Y, Gorkin DU, Zhang C, Dowiak AV, Lin K, Zeng C, Sui Y, Kim LJY, Miller TE, Jiang L, Lee CH, Huang Z, Fang X, Zhai K, Mack SC, Sander M, Bao S, Kerstetter-Fogle AE, Sloan AE, Xiao AZ, Rich JN, N6-methyladenine DNA Modification in Glioblastoma. Cell. 175, 1228–1243.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Xiao C, Zhu S, He M, Chen D, Zhang Q, Chen Y, Yu G, Liu J, Xie SQ, Luo F, Liang Z, Wang DP, Bo XC, Gu XF, Wang K, Yan GR, N 6 -Methyladenine DNA Modification in the Human Genome. Mol. Cell 71, 306–318.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Hao Z, Wu T, Cui X, Zhu P, Tan C, Dou X, Hsu K-W, Lin Y-T, Peng P-H, Zhang L-S, Gao Y, Hu L, Sun H-L, Zhu A, Liu J, Wu K-J, He C, N6-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol. Cell, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu S, Beaulaurier J, Deikus G, Wu TP, Strahl M, Hao Z, Luo G, Gregory JA, Chess A, He C, Xiao A, Sebra R, Schadt EE, Fang G, Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 28, 1067–1078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Douvlataniotis K, Bensberg M, Lentini A, Gylemo B, Nestor CE, No evidence for DNA N6-methyladenine in mammals. Sci. Adv 6, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brown ZK, Boulias K, Wang J, Wang SY, O’Brown NM, Hao Z, Shibuya H, Fady PE, Shi Y, He C, Megason SG, Liu T, Greer EL, Sources of artifact in measurements of 6mA and 4mC abundance in eukaryotic genomic DNA. BMC Genomics. 20, 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musheev MU, Baumgärtner A, Krebs L, Niehrs C, The origin of genomic N 6-methyl-deoxyadenosine in mammalian cells. Nat. Chem. Biol 16, 630–634 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Lai W, Li Y, Chen S, Liu B, Zhang N, Mo J, Lyu C, Zheng J, Du YR, Jiang G, Xu GL, Wang H, N6-methyladenine is incorporated into mammalian genome by DNA polymerase. Cell Res., 3–6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schiffers S, Ebert C, Rahimoff R, Kosmatchev O, Steinbacher J, Bohne AV, Spada F, Michalakis S, Nickelsen J, Müller M, Carell T, Quantitative LC–MS Provides No Evidence for m 6 dA or m 4 dC in the Genome of Mouse Embryonic Stem Cells and Tissues. Angew. Chemie - Int. Ed 56, 11268–11271 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Lentini A, Lagerwall C, Vikingsson S, Mjoseng HK, Douvlataniotis K, Vogt H, Green H, Meehan RR, Benson M, Nestor CE, A reassessment of DNA-immunoprecipitation-based genomic profiling. Nat. Methods 15, 499–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh CWQWQ, Goh YT, Toh JDWDW, Neo SP, Ng SBB, Gunaratne J, Gao Y-GG, Quake SR, Burkholder WFF, Goh WSSSS, Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. 46, 11659–11670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo GZ, Wang F, Weng X, Chen K, Hao Z, Yu M, Deng X, Liu J, He C, Characterization of eukaryotic DNA N(6)-methyladenine by a highly sensitive restriction enzyme-assisted sequencing. Nat Commun. 7, 11301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, Töpfer A, Alonge M, Mahmoud M, Qian Y, Chin C-S, Phillippy AM, Schatz MC, Myers G, DePristo MA, Ruan J, Marschall T, Sedlazeck FJ, Zook JM, Li H, Koren S, Carroll A, Rank DR, Hunkapiller MW, Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deamer D, Akeson M, Branton D, Three decades of nanopore sequencing. Nat. Biotechnol 34, 518–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW, Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tourancheau A, Mead EA, Zhang XS, Fang G, Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 18, 491–498 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaulaurier J, Zhu S, Deikus G, Mogno I, Zhang XS, Davis-Richardson A, Canepa R, Triplett EW, Faith JJ, Sebra R, Schadt EE, Fang G, Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat Biotechnol. 36, 61–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, Posfai J, Singh K, Visel A, Wetmore K, Zhao Z, Rubin EM, Korlach J, Pennacchio LA, Roberts RJ, The Epigenomic Landscape of Prokaryotes. PLoS Genet. 12, 1–28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beaulaurier J, Zhang XS, Zhu S, Sebra R, Rosenbluh C, Deikus G, Shen N, Munera D, Waldor MK, Chess A, Blaser MJ, Schadt EE, Fang G, Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes. Nat Commun. 6, 7438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A, Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 23, 129–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliveira PH, Ribis JW, Garrett EM, Trzilova D, Kim A, Sekulovic O, Mead EA, Pak T, Zhu S, Deikus G, Touchon M, Lewis-Sandari M, Beckford C, Zeitouni NE, Altman DR, Webster E, Oussenko I, Bunyavanich S, Aggarwal AK, Bashir A, Patel G, Wallach F, Hamula C, Huprikar S, Schadt EE, Sebra R, van Bakel H, Kasarskis A, Tamayo R, Shen A, Fang G, Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat. Microbiol 5, 166–180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murgier J, Everaerts C, Farine JP, Ferveur JF, Live yeast in juvenile diet induces species-specific effects on Drosophila adult behaviour and fitness. Sci. Rep 9, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee WJ, Brey PT, How microbiomes influence metazoan development: Insights from history and drosophila modeling of gut-microbe interactions. Annu. Rev. Cell Dev. Biol 29, 571–592 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Roberts RJ, Vincze T, Posfai J, Macelis D, REBASE-a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelbrektson A, Kunin V, Engelbrektson A, Kunin V, Glavina del Rio T, Hugenholtz P, Tringe SG, Defining the core Arabidopsis thaliana root microbiome. Nature. 488, 86–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corkum CP, Ings DP, Burgess C, Karwowska S, Kroll W, Michalak TI, Immune cell subsets and their gene expression profiles from human PBMC isolated by Vacutainer Cell Preparation Tube (CPT™) and standard density gradient. BMC Immunol. 16, 1–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma C, Niu R, Huang T, Shao LW, Peng Y, Ding W, Wang Y, Jia G, He C, Li CY, He A, Liu Y, N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell Biol 21 (2019), , doi: 10.1038/s41556-018-0238-5. [DOI] [PubMed] [Google Scholar]
- 38.Guiblet WM, Cremona MA, Cechova M, Harris RS, Kejnovská I, Kejnovsky E, Eckert K, Chiaromonte F, Makova KD, Long-read sequencing technology indicates genome-wide effects of non-B DNA on polymerization speed and error rate. Genome Res. 28, 1767–1778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong Y, Cao L, Deikus G, Fan Y, Mead E, Lai W, Zhang Y, Yong R, Sebra R, Wang H, Zhang X-S, Fang G, Code and processed data for: Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution (version 1.0) (2021), doi: 10.5281/ZENODO.5659041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye P, Luan Y, Chen K, Liu Y, Xiao C, Xie Z, MethSMRT: An integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 45 (2017), doi: 10.1093/nar/gkw950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ, Sequencing the unsequenceable: Expanded CGG-repeat alleles of the fragile X gene. Genome Res. 23, 121–128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao B, Li Y, Wang Z, Chen L, Poidevin M, Zhang C, Lin L, Wang F, Bao H, Jiao B, Lim J, Cheng Y, Huang L, Phillips BL, Xu T, Duan R, Moberg KH, Wu H, Jin P, Active N 6 - Methyladenine Demethylation by DMAD Regulates Gene Expression by Coordinating with Polycomb Protein in Neurons. Mol. Cell 71, 848–857.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bian C, Guo X, Zhang Y, Wang L, Xu T, DeLong A, Dong J, Protein phosphatase 2A promotes stomatal development by stabilizing SPEECHLESS in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A 117, 13127–13137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bulanenkova S, Snezhkov E, Nikolaev L, Sverdlov E, Identification and mapping of open chromatin regions within a 140 kb polygenic locus of human chromosome 19 using E. coli Dam methylase. Genetica. 130, 83–92 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Zhang XS, Blaser MJ, Natural transformation of an engineered helicobacter pylori strain deficient in type II restriction endonucleases. J. Bacteriol 194, 3407–3416 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fulton TM, Chunwongse J, Tanksley SD, Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Report 13, 207–209 (1995). [Google Scholar]
- 47.Li H, Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10 (2009), doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W, Liu Y, Zhu S, Green CD, Wei G, Han JDJ, Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data. Nat. Commun 5 (2014), doi: 10.1038/ncomms5909. [DOI] [PubMed] [Google Scholar]
- 50.Gomes RJ, de F. Borges M, de F. Rosa M, Castro-Gómez RJH, Spinosa WA, Acetic acid bacteria in the food industry: Systematics, characteristics and applications. Food Technol. Biotechnol 56, 139–151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey TL, Johnson J, Grant CE, Noble WS, The MEME Suite. Nucleic Acids Res. 43, W39–W49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babushok DV, Kazazian HH, Progress in understanding the biology of the human mutagen LINE-1. Hum. Mutat 28, 527–539 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Castro-Diaz N, Ecco G, Coluccio A, Kapopoulou A, Yazdanpanah B, Friedli M, Duc J, Jang SM, Turelli P, Trono D, Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev. 28, 1397–1409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai W, Lyu C, Wang H, Vertical Ultrafiltration-Facilitated DNA Digestion for Rapid and Sensitive UHPLC-MS/MS Detection of DNA Modifications. Anal. Chem 90, 6859–6866 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Anton BP, Mongodin EF, Agrawal S, Fomenkov A, Byrd DR, Roberts RJ, Raleigh EA, Complete genome sequence of ER2796, a DNA methyltransferase-deficient strain of Escherichia coli K-12. PLoS One. 10, 1–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.