

Abstract
Osteoarthritis (OA) and post-traumatic OA (PTOA) are caused by an imbalance in catabolic and anabolic processes in articular cartilage and pro-inflammatory changes throughout the joint, leading to joint degeneration and pain. We examined whether interleukin-6 (IL-6) signaling contributed to cartilage degradation and pain in PTOA. Genetic ablation of Il6 in male mice decreased PTOA-associated cartilage catabolism, innervation of the knee joint, and nociceptive signaling without improving PTOA-associated subchondral bone sclerosis or chondrocyte apoptosis. These effects were not observed in female Il6−/− mice. Compared to wild-type mice, the activation of the IL-6 downstream mediators STAT3 and ERK was reduced in the knees and dorsal root ganglia (DRG) of male Il6−/− mice after knee injury. Janus kinases (JAKs) were critical for STAT and ERK signaling in cartilage catabolism and DRG pain signaling in tissue explants. Whereas STAT3 signaling was important for cartilage catabolism, ERK signaling mediated neurite outgrowth and the activation of nociceptive neurons. These data demonstrate that IL-6 mediates both cartilage degradation and pain associated with PTOA in a sex-specific manner and identify tissue-specific contributions of downstream effectors of IL-6 signaling, which are potential therapeutic targets for disease-modifying OA drugs.
Introduction
Osteoarthritis (OA) is the most prevalent joint disorder and a leading cause of disability, to which multiple risk factors contribute, including aging, sex, increased body mass index (BMI), and prior joint injury (1–3). The prevalence of OA varies by age and sex. A higher prevalence of OA is found in males as compared to females prior to the age of 50 years; however, OA is more prevalent in females after menopause than males (4). It is estimated that 9.6% of men and 18.0% of women older than 60 years of age have symptomatic OA worldwide (5). Post-traumatic OA (PTOA), a specific type of OA caused by injury, accounts for approximately 12% of symptomatic OA (6). Pain is a dominant symptom of OA and PTOA (referred to here collectively as OA/PTOA) and serves as a key criterion in the diagnosis and treatment (7). Clinically, women generally experience more pain and worse knee OA symptom than males (8–10). Overall, OA/PTOA accounts for ~35% of patients with chronic pain and affects nearly 32.5 million people (1 in every 10 adults), which translates to an estimated $65.5 billion of annual medical costs in the US alone (11). Despite the extensive socioeconomic burden of OA/PTOA, no effective disease-modifying osteoarthritis drugs (DMOADs) are yet available for OA/PTOA–associated joint degeneration and pain management. Current treatments, such as topical capsaicin, acetaminophen, NSAIDs, and corticosteroid injections, either have limited efficacy or variable negative side effects (12, 13).
OA/PTOA is not simply a disease affecting structural alterations to the articular cartilages, but rather a disease affecting the entire joint, which can involve the ligaments, meniscus, nerves, subchondral bone, synovial membrane, and periarticular muscles (14, 15). The physical manifestations of OA/PTOA, such as articular cartilage and meniscus degeneration, synovial hyperplasia, osteophyte formation, and subchondral bone sclerosis, are caused by an imbalance in anabolic and catabolic responses, as well as proinflammatory changes (16, 17). Factors involved in OA/PTOA initiation and progression include proinflammatory cytokines [tumor necrosis factor (TNF), interleukin-1β (IL-1β), IL-6, and IL-17] and matrix-degrading enzymes, such as the metalloproteases MMP-1, MMP-3, MMP-9, MMP-13, ADAMTS-4, ADAMTS-5 (18). In addition to structural changes to the joint, the pathology of OA/PTOA–associated pain has also been under investigation. As in all types of chronic pain, OA/PTOA–associated pain is the dynamic result of a complex interaction between local tissue damage and inflammation, peripheral and central sensitization, and the brain (19). TNFα, IL-1β, IL-6 (20–22), C-C Motif Chemokine Ligand 2 (CCL2) (23, 24), and the neurotrophic factor, nerve growth factor (NGF) (25, 26) are all reported as being correlated with OA/PTOA–associated pain. Because IL-6 is both a major predictor of structural changes in OA/PTOA (27) and has been associated with inflammatory pain in other contexts (28), it may serve as a critical effector connecting OA/PTOA–associated joint changes and pain.
IL-6, as an inflammatory cytokine, has been implicated in both OA/PTOA cartilage degeneration and unrelated conditions of inflammatory pain. IL-6 signals by binding to the IL-6–specific membrane receptor mIL-6R, known as ‘classical signaling’, or by binding to the soluble form of this receptor (sIL-6R), known as ‘trans-signaling’. Both interactions activate the transducer glycoprotein 130 (gp130) and induce a cascade of Janus kinase (JAK) protein phosphorylations, which further leads to the phosphorylation and activation of Signal transducer and activator of transcription (STAT) and/or Mitogen-activated protein kinase (MAPK) proteins (29, 30). An increase in IL-6 has been identified in the articular cartilage (31), synovial fluid (32), and infrapatellar fat pad (33) in human OA cohort studies, and a large scale, long-term follow-up (15-years) study identified increased amounts of serum IL-6 as an important predictor of radiographic knee OA (27). In murine OA studies, the role of IL-6 has proven to be complex. It has been reported that aged Il6−/− males, but not aged females, exhibit higher incidences and more substantial cartilage damage when compared with age-matched controls (34). Conversely, genetic ablation of Il6 following joint injury in mice (35, 36) has also been associated with decreased cartilage degeneration, whereas intraarticular injections of recombinant IL-6 protein induce cartilage destruction (37). Our previous work also demonstrated that phosphorylated STAT3, a downstream mediator of IL-6 signaling, is increased within cartilage and synovium following a joint injury (38). Other work has indicated that treatment with Stattic (a STAT3 inhibitor) by oral gavage to injured mice reduces cartilage degradation during the development of PTOA in vivo (39). Based on current evidence, IL-6 displays variable roles in age-related OA and PTOA.
In addition to its role as a cytokine during immune responses, IL-6 signaling also functions in the nervous system, where IL-6 and IL-6R are produced by neurons in sympathetic and sensory ganglia (40). IL-6 has been linked to chronic pain through the induction of the pain-activated cation channel TRPV1 (41, 42) and induces neurite outgrowth in the PC12 neuronal cell line in vitro, similar to effects caused by NGF (40, 43). Although no direct functional role has been identified between IL-6 and OA pain specifically, several clinical studies have demonstrated a correlation between pain in OA patients and higher concentrations of IL-6 in synovial fluid (44–47). Based on these data, we hypothesize that IL-6 may be involved not only in PTOA-associated cartilage degeneration but also in PTOA-associated pain.
Although accumulated evidence indicates a contribution of IL-6 to cartilage degradation in PTOA progression, there is little evidence regarding the link between IL-6 and PTOA-associated pain or the downstream molecular mechanisms responsible for the IL-6 signaling contribution to cartilage degeneration and/or pain. Here, we utilized the destabilization of the medial meniscus (DMM) surgical model of PTOA with conventional Il6−/− and control mice of both sexes to study the function and mechanisms of IL-6 signaling in both cartilage degeneration and pain signaling within nociceptive neurons associated with the knee joint. IL-6 mediated both PTOA-associated cartilage degradation and pain only in males and identified specific roles for downstream mediators of IL-6 signaling within chondrocytes and nociceptive neurons. Collectively, these findings have important implications for therapeutic targets of disease-modifying osteoarthritis drugs (DMOADs) and for sex-specific considerations in the treatment of OA patients.
Results
Genetic ablation of Il6 alleviates PTOA-associated cartilage catabolism in male mice
To determine the necessity of IL-6 in PTOA-associated cartilage degeneration and pain, we first performed both sham and a modified DMM surgery on conventional Il6 knockout (Il6 −/−) and wild-type (WT) control male mice at 4 months of age, followed by assessments of joint cartilage degeneration and pain at 4, 8, and 12 weeks post-injury (wpi) (fig. S1A). Similar to prior studies, our modified DMM surgery induced a progressive OA phenotype in WT male mice, as shown by histological assessments and confirmed with a standard histological scoring system for murine OA – the Osteoarthritis Research Society International (OARSI) score (OARSI scoring) from 4 to 12 wpi (fig. S1, B and C). To identify any innate differences between the two mouse strains, we first characterized WT and Il6 −/− mice with sham injury first and did not detect any significant difference in cartilage (fig. S2, A to C), subchondral bone (fig. S2D), or pain sensation at either the molecular or behavioral level (fig. S2, E to F). These data enabled us to compare the two mouse strains directly in subsequent experiments.
In Il6 −/− mice, DMM surgery induced minor OA phenotypes with a reduction in Safranin-O (cartilage) staining and/or small fibrillations at the cartilage surface, similar to that seen in WT mice at 4 weeks post-DMM. However, at 8 and 12 weeks after DMM injury, Il6 −/− mice showed significantly less cartilage degradation than WT mice, which exhibited severe clefting and cartilage erosions at both time points (Fig. 1A). This is evidenced by the significantly lower OARSI scores in Il6 −/− mice at 8 and 12 weeks post-DMM when compared to WT controls (Fig. 1B). Further, OARSI scores of Il6 −/− mice following DMM were noted as similar to WT sham controls at these later timepoints, indicating that genetic ablation of Il6 is capable of attenuating cartilage degradation in PTOA for prolonged periods of time in male mice.
Fig. 1. Loss of IL-6 reduces cartilage degradation in male mice with PTOA.
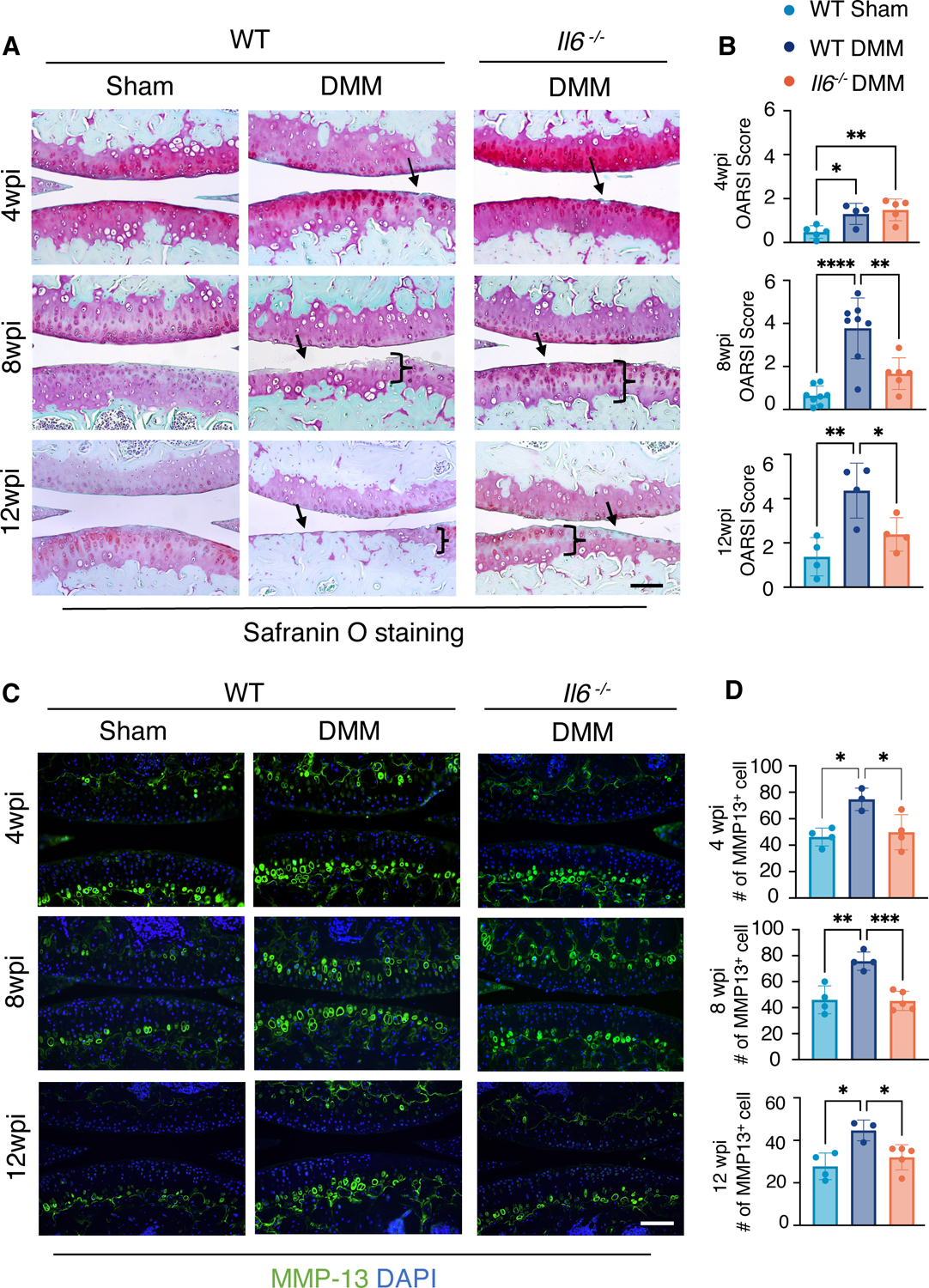
(A and B) Safranin-O staining of knee joint sections (A) and OARSI scores quantifying the cartilage phenotypes (B) in WT and Il6−/− male mice at 4, 8, and 12 weeks after sham or DMM surgery. N ≥ 5 mice per condition (4wpi); N≥ 6 mice per condition (8wpi); N≥ 4 mice per condition (12wpi). wpi, weeks post-injury. Scale bar, 100 μm. (C) Immunofluorescence staining for MMP-13 in knee sections from WT or Il6−/− male mice at 4, 8, and 12 weeks after sham or DMM surgery. DAPI indicates nuclei. Scale bar, 100 μm. (D) Quantification of the number of cells with MMP-13 staining. Green fluorescence encircling cells was used to identify individual MMP-13+ chondrocytes. N≥ 3 mice per condition. One-way ANOVA. All quantitative data represent mean ± SD. * p<0.05, ** ≤p<0.01, *** ≤p<0.001, **** p<0.0001.
In PTOA, articular cartilage degradation is generally the result of an imbalance in cartilage anabolism and catabolism (remodeling) and chondrocyte cell death. To understand how removal of Il6 affects cartilage degradation in PTOA, we first examined the catabolic protein, MMP-13, using immunofluorescent (IF) staining on knee joint sections from Il6 −/− and WT control mice at 4, 8, and 12 weeks post-DMM. MMP-13 was induced by DMM in WT mice at all time points, but there were significantly fewer MMP-13+ chondrocytes in joint cartilages from Il6 −/− mice as compared to tissues from WT mice, indicating a reduction in cartilage catabolism in Il6 −/− mice from early to late timepoints following DMM injury (Fig. 1, C and D). Similar to the OARSI scores, the numbers of MMP-13+ chondrocytes in joint cartilages from Il6 −/− mice following DMM were comparable to WT sham controls. Antibodies against Aggrecan (ACAN) and its degradation product, Aggrecan C-terminal neoepitope (NITEGE), were also used for immunostaining and further characterization the cartilage phenotypes. These data demonstrated that Il6 deletion alleviated the reduction in ACAN (anabolism) and the increase in NITEGE (catabolism) caused by DMM injury (fig. S3, A and B). To examine whether the reduced cartilage degradation observed in Il6 −/− knee joints following DMM occurs through altered chondrocyte survival, we performed TUNEL staining on knee joint sections from Il6 −/− and WT control mice at 4, 8, and 12 weeks post-DMM. At 4 weeks post-DMM, in both WT and Il6 −/− mice, there was a minor increase in apoptosis within the articular cartilage after injury (fig. S4); however, by 8 wpi there was a significant increase in apoptosis in the superficial and deep zones of the articular cartilage in both WT and Il6 −/− mice as compared to sham control. We observed no difference between the DMM injury groups of WT and Il6 −/− mice (fig. S4). Collectively, these observations indicate that IL-6 promotes cartilage degradation in PTOA in male mice by stimulating cartilage catabolism mediated by MMP-13 and Aggrecanase, not by altering chondrocyte cell death.
The loss of IL-6 reduces pain in male mice with PTOA
Because pain is a prominent symptom of OA and IL-6 is known to be involved in various settings of pain, we measured local knee pain in both WT and Il6 −/− male mice following DMM injury or sham surgery using a pressure application measurement (PAM) device. The PAM device measures the precise maximum force that a mouse can withstand at the knee joint prior to vocalization and/or limb withdrawal (48, 49). A low paw withdrawal threshold indicates a higher pain sensitivity. In WT mice at 1 week post-injury, there was significant acute pain caused by the surgery in both sham and DMM-injured mice. In the sham group, surgical pain declined over time and returned to baseline by 8 wpi (Fig. 2A). On the contrary, local knee pain in WT DMM mice was significantly enhanced compared to the sham group from 4 to 8 wpi, indicating DMM injury induced chronic knee pain in WT male mice (Fig. 2A). However, Il6 −/− male mice with DMM injury exhibited significantly less pain from 2 to 8 wpi compared to WT DMM injured mice and demonstrated a complete recovery and return to baseline over this time (Fig. 2A).
Fig. 2. Loss of IL-6 reduces pain in male mice with PTOA.
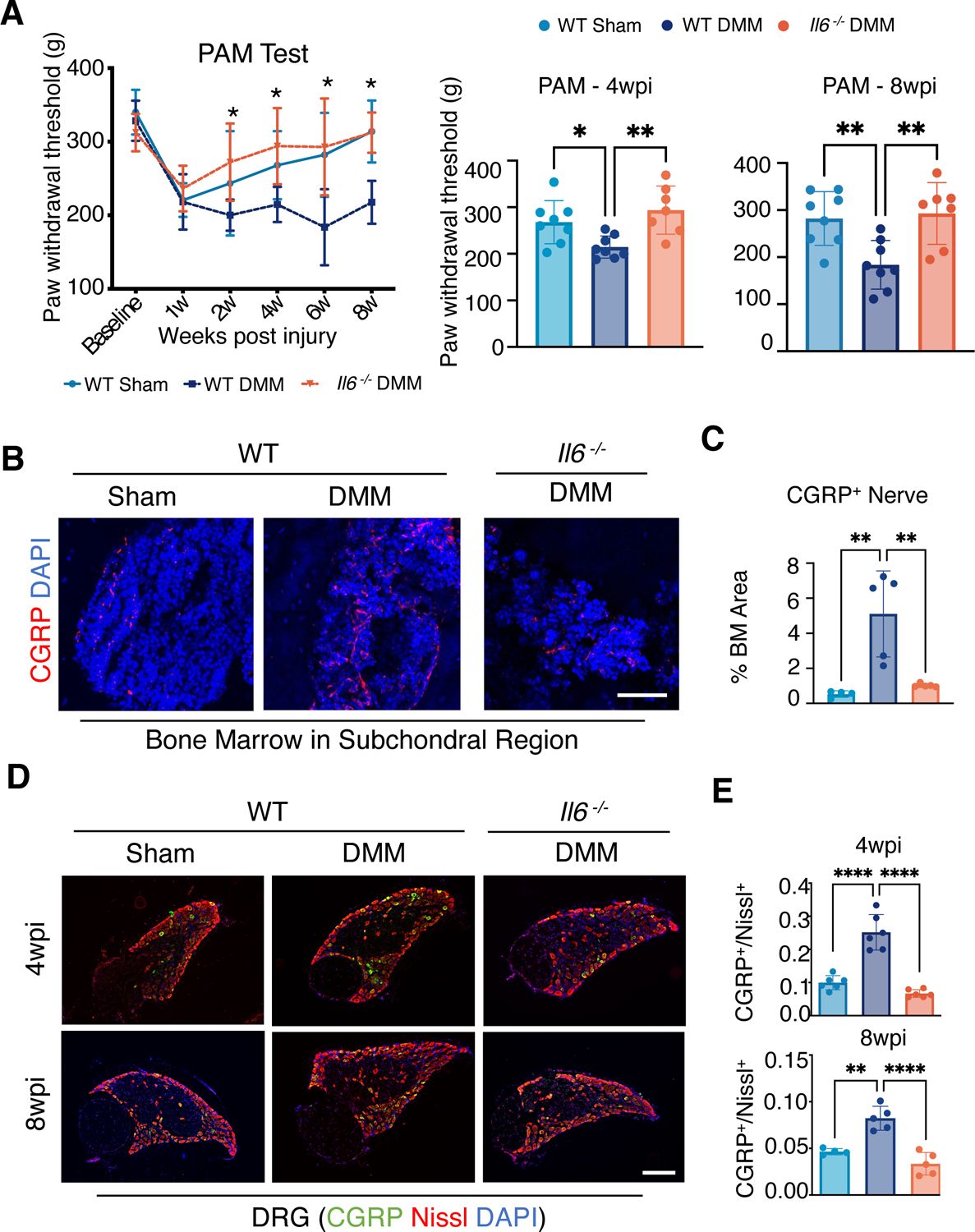
(A) Behavioral tests for knee pain (mechanical hyperalgesia) in WT and Il6−/− male mice at the indicated times after sham or DMM surgery. Paw withdrawal threshold was measured with a PAM device with pressure applied to the knee joint. Bar graphs show data at 4 and 8 wpi. N≥ 8 mice per condition. Two-way ANOVA with Bonferroni’s post-hoc test. Data represent mean ± SEM. * indicates p<0.05 between WT DMM condition and Il6−/− DMM condition. (B and C) Immunofluorescence (IF) staining (B) and quantification (C) showing innervation by CGRP+ neurons at subchondral bone marrow of the medial compartment. DAPI indicates nuclei. Scale bar, 50 μm. N≥ 4 mice per condition. One-way ANOVA. Data represent mean ± SD. ** p<0.01. (D) IF staining showing CGRP in L3–5 DRG from WT and Il6−/− mice after sham or DMM surgery at 4 and 8 wpi. Nissl counterstain labels all neurons, and DAPI indicates nuclei. Scale bar, 200 μm. (E) Quantification of the ratio of CGRP+ neurons to the total number of neurons. N≥ 4 mice per condition. One-way ANOVA. Data represent mean ± SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To investigate the underlying mechanisms that may account for the reduced pain response observed in Il6 −/− male mice following DMM or sham injury, we first characterized the subchondral bone and bone marrow innervation in the sham and DMM-injured knee joints of WT and Il6−/− male mice by immunostaining joint sections for the pain-associated molecule, Calcitonin gene-related peptide (CGRP). CGRP is a neurotransmitter of the nociceptive sensory C-fibers, which are unmyelinated sensory neurons with a low-threshold response to injury or damage in pain pathways (50, 51), and higher expression of CGRP in osteochondral junctions of OA conditions have been found in both human and murine OA pain studies (52, 53). We observed an enhancement of CGRP+ innervation within subchondral regions in WT mice, but not in Il6 −/− mice, following DMM injury (Fig. 2, B and C). To assess molecular markers of pain, we measured the number of CGRP+ neurons within specific dorsal root ganglia (DRG), which are clusters of nociceptive sensory neurons that transmit pain signals from the peripheral to the central nervous system. Specifically, Lumbar 3–5 (L3–L5) DRG correspond to the peripheral nerves innervating knee joint tissues, including the synovium, ligaments, osteochondral junction, subchondral marrow, and meniscus. We visualized all neurons within a DRG by performing Nissl staining (NeuroTrace) on DRG tissue sections while simultaneously staining for CGRP, thus enabling us to measure the proportion of activated pain-sensing neurons within those DRG. A higher ratio of CGRP+ cells were observed in the DRG of WT mice following DMM injury as compared to WT sham mice at 4 and 8 weeks following surgery. In contrast, DRG sections from Il6 −/− DMM mice had CGRP+ cell ratios similar to those of sham-operated WT mice at both early (4 wpi) and late (8 wpi) timepoints (Fig. 2, D and E). These data are consistent with the PAM behavioral pain tests, suggesting an early rescue of the pain response caused by DMM injury in male mice by deleting Il6. It is noteworthy that pain measures, including both behavioral and molecular readouts on DRG sections, were rescued in Il6 −/− male mice at a timepoint when cartilage damage was minimal and remained comparable between in Il6 −/− and WT control mice following DMM injury. These data suggest a potential uncoupling of pain and overt cartilage degradation in PTOA; however, IL-6 appears to play a substantial role in both PTOA-associated cartilage degeneration and pain in male mice.
Because OA is a disease of the whole joint and OA progression and pain have been associated with aberrant subchondral bone remodeling and innervation (53, 54), we used micro computed tomography (microCT) to evaluate the subchondral bone in WT and Il6−/− knees following DMM or sham surgery. At 8 weeks post-DMM injury, we observed subchondral bone sclerosis in both WT and Il6 −/− male mice (fig. S5, A and B), suggesting that removal of Il6 did not attenuate subchondral sclerosi, and that the reduced PTOA-associated pain and cartilage degradation in Il6−/− male mice did not occur due to corrections in subchondral bone remodeling.
Multiple downstream mediators of IL-6 signaling are activated in joint tissues and peripheral nerves following joint injury in male mice
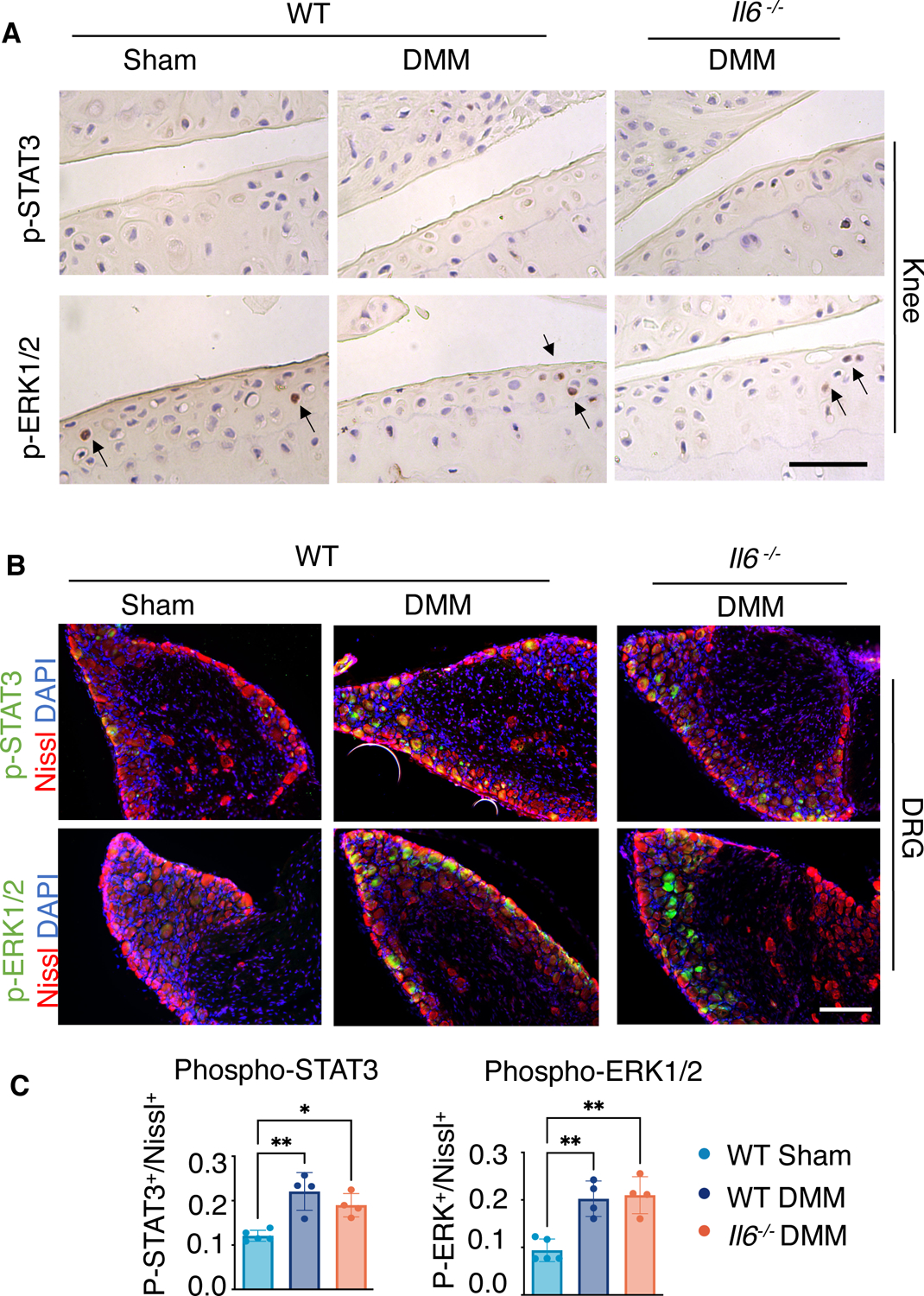
To further understand the molecular mechanisms by which IL-6 signaling may promote cartilage degradation and pain in PTOA of male mice, we examined changes in activation of STAT3 and extracellular-regulated kinase (ERK), both of which are downstream mediators of IL-6 signaling, in both cartilage and L3 DRG at 8 weeks post-DMM injury. Within the knees of WT male mice, we observed a significant increase in phosphorylated STAT3 (p-STAT3) and phosphorylated ERK1 and ERK2 (p-ERK1/2) within cartilage and synovial tissues following injury (Fig. 3A). Distant from the injury site at the knee, DRG associated with L3 vertebrae also showed an increase in p-STAT3 and p-ERK1/2 in injured male mice (Fig. 3B). In Il6 −/− male mice, whose cartilage degradation and pain were attenuated compared to WT mice (Fig. 1A and 2A), the number of cells with activated p-STAT3 and p-ERK1/2 following DMM injury were reduced in both knee and L3 DRG to similar amounts as observed in sham controls (Fig. 3B). These data indicate that the activation of downstream IL-6 signaling mechanisms within knee joint tissues and their associated DRG neurons may play essential roles in PTOA-associated cartilage degeneration and pain in male mice. Notably, IL-6 signaling through MAPKs, such as ERK, has been shown to induce neurite outgrowth in PC12 cells (55), and is also strongly implicated in the pathogenesis of pain (56).
Fig. 3. IL-6 downstream signaling within joint cartilage and DRGs of WT and Il6−/− male mice after DMM injury.
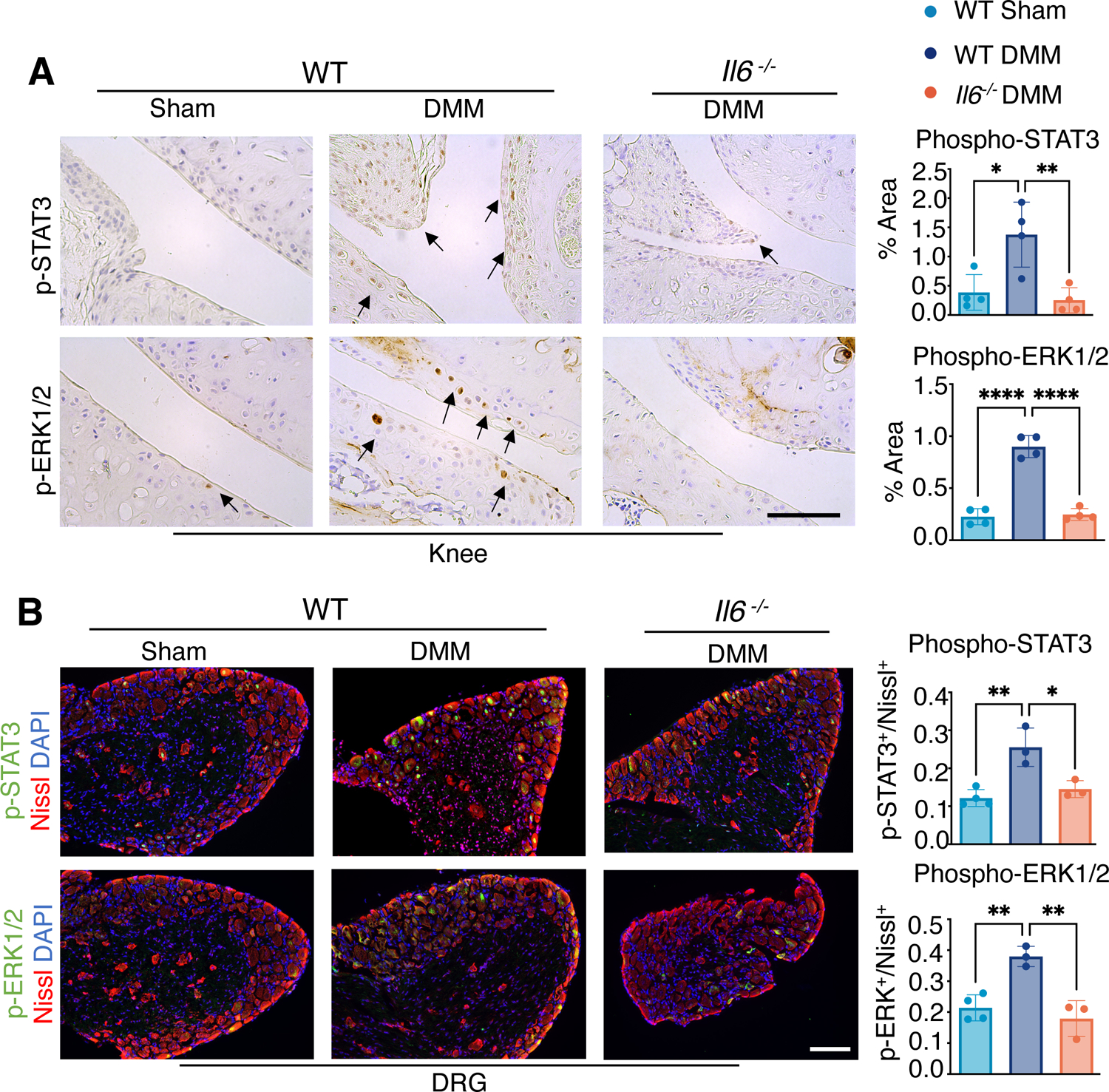
(A) Immunohistochemistry (IHC) showing phosphorylated STAT3 (p-STAT3) and ERK1/2 (p-ERK1/2) in knee joints of WT and Il6−/− mice after sham or DMM surgery at 8 wpi. Scale bar, 100 μm. The ratio of p-STAT3+ or p-ERK1/2+ staining area to total examined cartilage area was quantified. N≥ 4 mice per condition. (B) Immunofluorescence staining for p-STAT3 or p-ERK1/2 in L3 DRG from WT and Il6−/− mice after sham or DMM surgery at 8 wpi. Nissl labels all neurons, and DAPI labels nuclei. Scale bar, 100 μm. The ratio of p-STAT3+ or p-ERK1/2+ neurons to total neurons was quantified. N≥ 3 mice per condition. One-way ANOVA. Data represent mean ± SD. * p<0.05, ** p<0.01.
Il6 deletion in female mice does not affect PTOA-associated cartilage degradation or pain
In addition to the results evaluated in male mice as described above, we also examined joint cartilage and pain phenotypes associated with PTOA in female mice. As previously shown, female mice exhibit a slower and blunted cartilage catabolic response to DMM injury as compared to male mice (57). We did not observe significant cartilage degradation in WT female mice subjected to DMM surgery until 12-wpi, which was still only characterized by small cartilage fibrillations and vertical clefts at the surface of the cartilage, producing an OARSI score of 2 on average (Fig. 4, A and B; fig. S6, A and B). Histology and OARSI scores of knee sections from Il6−/− female mice indicated a similar PTOA cartilage phenotype as compared to WT female mice following DMM injury, indicating that removal of Il6 in females did not provide chondroprotection at this stage (Fig. 4, A and B). As for pain, both WT and Il6 −/− female mice exhibited persistent knee pain (fig. S6C). At 12 weeks post-DMM, when the PTOA-associated cartilage phenotypes were visible, Il6 −/− female mice continued to show enhanced knee pain when compared with sham female mice (Fig. 4C). To evaluate innervation of the subchondral bone marrow, we again performed CGRP immunostaining of knee joints from females at 12 weeks following DMM injury. Unlike male mice, Il6 −/− female mice showed persistent CGRP staining in subchondral marrow regions following DMM injury (Fig. 4, D and E), and the numbers of CGRP+ cells in DRG neurons remained high (Fig. 4, F and G).
Fig. 4. Cartilage and pain phenotypes in WT and Il6−/− female mice at 12 weeks after DMM injury.
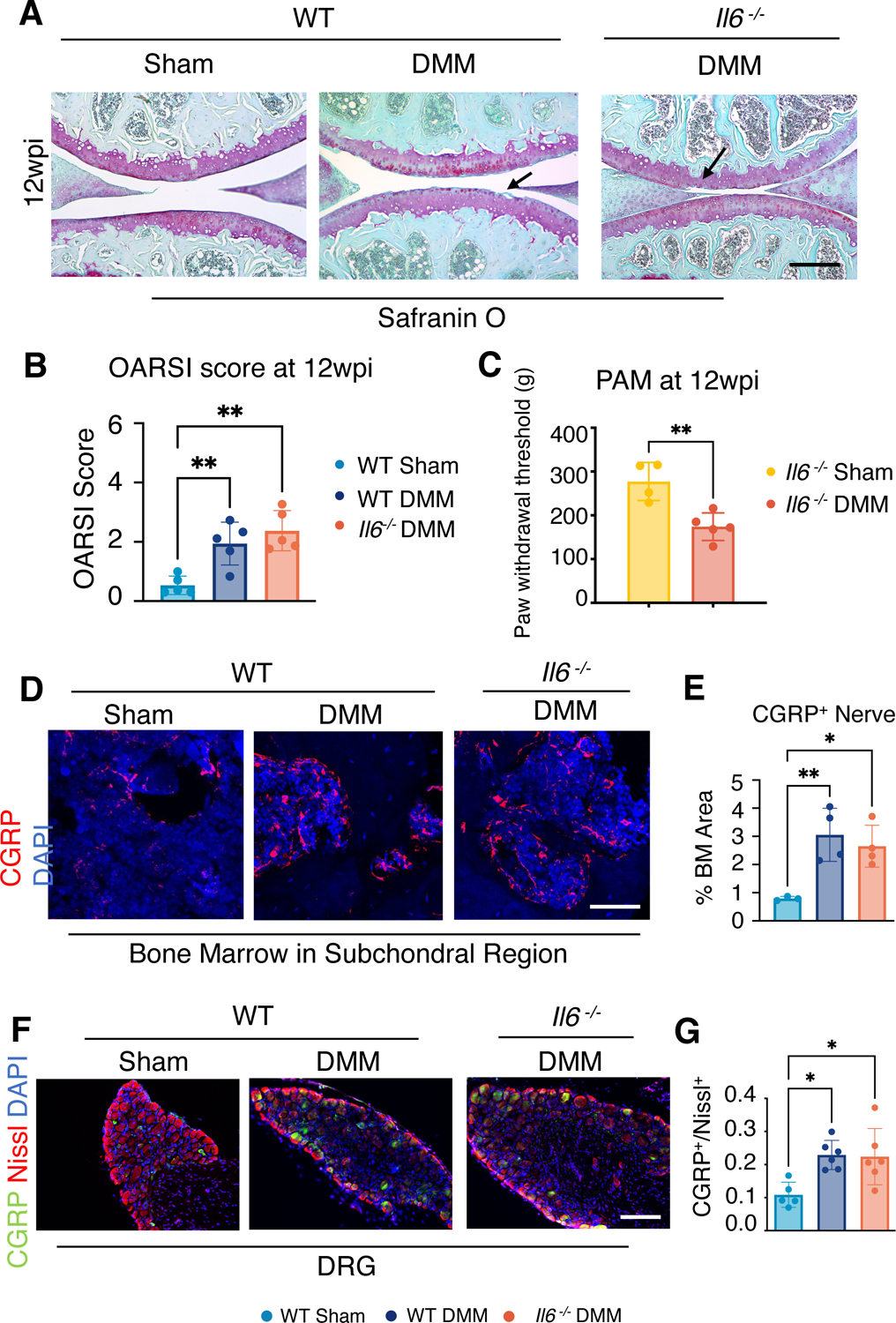
(A) Safranin-O staining of knee joint sections from WT and Il6−/− female mice at wpi. N≥ 5 mice per condition. Scale bar, 200 μm. (B) OARSI scores of female mice at 12 wpi. N≥ 5 mice per condition. (C) PAM assessment of knee pain in DMM and sham-operated Il6−/− female mice at 12 wpi. N≥ 4 mice per condition. Unpaired t-test. Data represent mean ± SD. (D and E) Immunofluorescence (IF) staining (D) and quantification (E) showing innervation by CGRP+ neurons at subchondral bone marrow at 12 wpi in WT and Il6−/− female mice. Scale bar, 50 μm. N≥ 3 mice per condition. One-way ANOVA. Data represent mean ± SD. (F) IF staining for CGRP in L3 DRG from WT and Il6−/− female mice after sham or DMM surgery at 12 wpi. Nissl labels all neurons, and DAPI labels nuclei. N≥ 5 mice per condition; Scale bar, 100 μm. (G) Quantification of the ratio of CGRP+ neurons to total neurons. One-way ANOVA. Data represent mean ± SD. * p<0.05, ** p<0.01.
We also assessed activation of the IL-6 signaling mediators, STAT3 and ERK1/2, in both joint cartilages and DRG in females. We observed no difference in p-STAT3 or p-ERK1/2 in the joint cartilages of WT or Il6 −/− female mice following DMM (Fig. 5A). In contrast, DMM injury increased p-STAT3 and p-ERK1/2 in the DRG neurons of both WT and Il6−/− female mice (Fig. 5, B and C). The failure to rescue PTOA-associated cartilage degradation and pain in female mice by the genetic ablation of Il6, as well as the differential signaling responses observed between male and female mice following DMM injury, suggests that at least some of the molecular mechanisms regulating PTOA-associated cartilage degeneration and pain differ between males and females.
Fig. 5. IL-6 downstream signaling within joint cartilage and DRGs of WT and Il6−/− female mice at 12 weeks after DMM injury.
(A) Immunohistochemistry showing phosphorylated STAT3 (p-STAT3) and ERK1/2 (p-ERK1/2) in knee joints of WT and Il6−/− female mice after sham or DMM surgery at 12 wpi. N≥ 5 mice per condition; Scale bar, 50 μm. (B) Immunofluorescence staining for p-STAT3 or p-ERK1/2 in L3 DRG from WT and Il6−/− female mice after sham or DMM surgery at 12 wpi. Nissl labels all neurons, and DAPI labels nuclei. N≥ 4 mice per condition. Scale bar, 100 μm. (C) Quantification of the ratio of p-STAT3+ or p-ERK1/2+ neurons to total neurons. One-way ANOVA. Data represent mean ± SD. * p<0.05, ** p<0.01.
Differential responses to IL-6 in cartilage and DRG explants between male and female mice
To better understand the specific effects of IL-6 signaling within cartilage tissue and DRG neurons of both male and female WT mice, we isolated femoral cartilage and L3 DRGs and cultured the explants with or without recombinant IL-6 protein (rIL-6) ex vivo. IL-6 treatment decreased ACAN staining (anabolism) in cartilage explants from males but not in females; however, it increased MMP-13 staining (catabolism) in cartilage explants from both males and females (fig. S7A). This observation demonstrated that IL-6 can promote cartilage degeneration similar to that observed in OA in tissue explants ex vivo with limited sex differences. We also treated DRG explants from both male and female WT mice with rIL-6. We observed that the DRG explants from female mice were more prone to the induction of neurite outgrowths in control conditions as compared to male DRG explants and that rIL-6 treatment induced greater neurite outgrowth in male DRG as compared to female DRG (fig. S7B). Consistent with these results, the numbers of CGRP+ neurons were higher in DRG explants from females than in those from males under control conditions (Fig. S7C). Additionally, male DRG explants exhibited a significant increase in the number of CGRP+ neurons following rIL-6 treatment; however, female DRG showed no such increase in CGRP positivity in response to rIL-6 (Fig. S7C).
JAK and ERK inhibitors reduce IL-6–induced cartilage catabolism and pain-associated cellular responses
To investigate which downstream mediators of IL-6 signaling drove catabolic and anabolic cartilage responses, we first tested the effects of specific JAK, STAT, and ERK inhibitors on rIL-6–induced cartilage degeneration using femoral cartilage explants from 2-month-old male mice and ATDC5 chondrogenic cells that are co-treated with rIL-6 (Fig. 6, A and B; fig. S8A). In both cartilage explants and ATDC5 cells, inhibition of JAK signaling with Ruxolitinib alleviated the rIL-6–mediated reduction in ACAN accumulation and Acan expression (anabolism) and rescued the rIL-6–mediated induction of Mmp13 and MMP13 (catabolism) (Fig. 6, A and B). The STAT3 inhibitor Stattic had similar effects as Ruxolitinib in blocking rIL-6–induced increases in Mmp13 and MMP13, but it enhanced rIL-6–mediated suppression of Acan and ACAN (Fig. 6, A and B). The ERK1/2 inhibitor U0126 did not counteract the rIL-6–induced decreases in Acan and ACAN and had only a modest dampening effect on rIL-6–induced increases in Mmp13 and MMP13 (Fig. 6, A and B). Based on these in vitro experiments, JAK signaling, upstream of STAT3 and ERK signaling, appears to be a key effector of IL-6–induced cartilage degeneration through decreasing anabolism and increasing catabolism. Whereas inhibition of STAT3 reduced IL-6–induced cartilage catabolism, it did not rescue the negative effects on cartilage anabolism and actually worsened anabolic suppression in ATDC5 cells (Fig. 6, A and B). This effect may be due to an increase in phosphorylation pf the MAPK kinase P38, which was not observed following JAK or ERK inhibition (fig. S8B). Collectively, these data indicate that STAT3 or ERK inhibition alone may be insufficient to maintain healthy cartilage following the pathologic activation of IL-6 signaling in chondrocytes.
Fig. 6. IL-6 induces OA-associated molecular and cellular phenotypes in vitro and ex vivo.
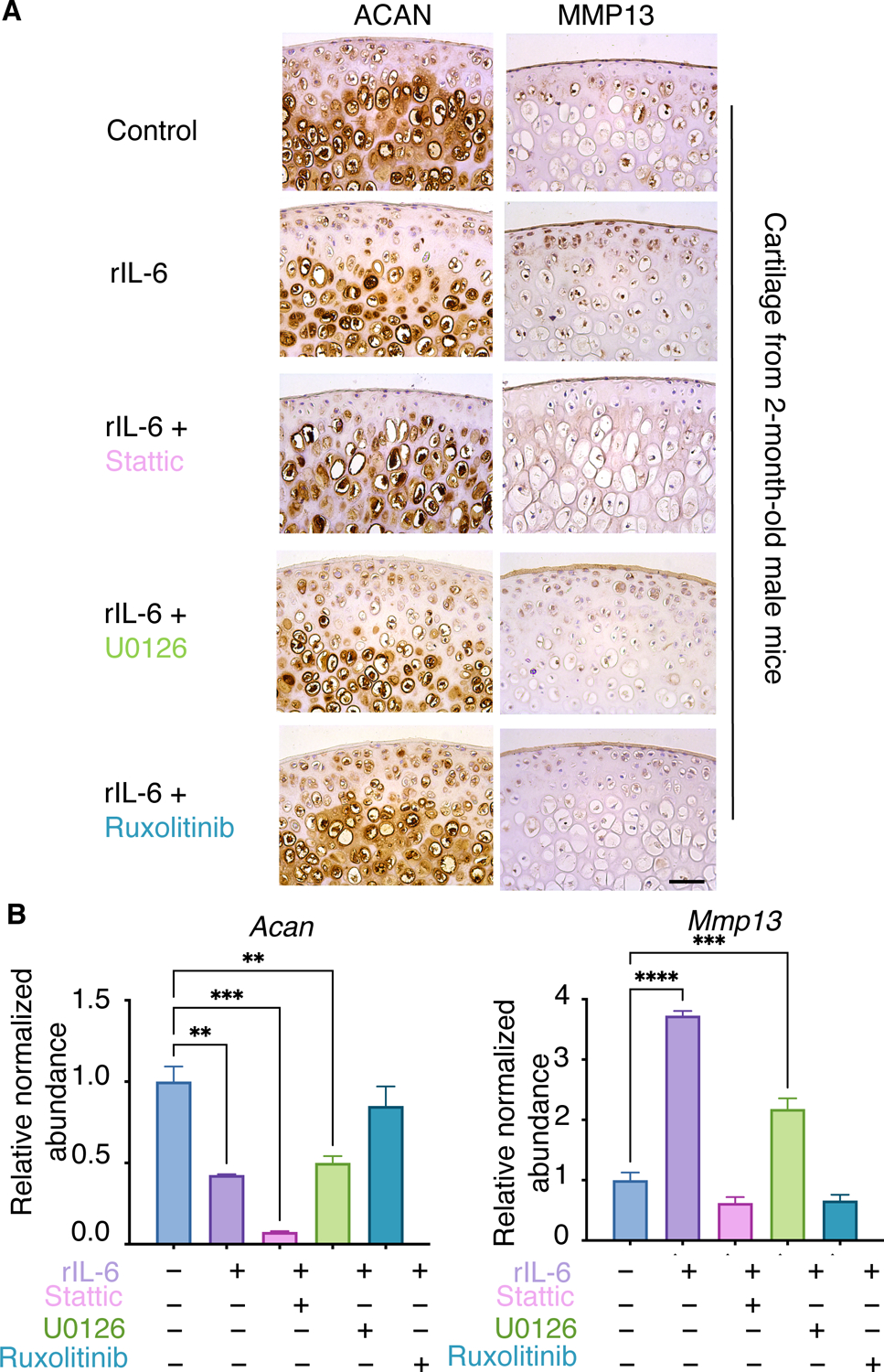
(A) Femoral cartilage explants from 2-month-old male mice were cultured with rIL-6 plus or minus the indicated inhibitors. N=3 mice per condition. Scale bar, 50 μm. (B) qPCR of ATDC5 cells cultured with DMSO (–) or rIL-6 plus or minus inhibitors. N≥ 3 independent experiments per condition. Data represent mean ± SEM. Only the statistical significance by one-way ANOVA between DMSO group and treatment groups is shown in the graph. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To understand the response of peripheral nerves to IL-6 signaling and its downstream mediators, we harvested L3–L5 DRGs from 2-month-old male mice and cultured them as either intact DRG explants (Fig. 7, A to C) or disassociated primary neuron cultures (fig. S9) and treated them with rIL-6 or recombinant NGF (rNGF). NGF is a well-established neurotrophic factor that stimulates neurite outgrowth and induces pain-related factors such as the neuropeptide CGRP (58). Consistent with results described above (fig. S7B), rIL-6 stimulated neurite outgrowth in DRG explants and isolated primary neuron cultures similarly to rNGF (Fig. 7, A and B and fig. S9). In addition to neurite outgrowth, rIL-6 stimulation also significantly increased expression of the pain-associated factors Cgrp and C-C motif chemokine receptor 2 (Ccr2) in DRG explants (Fig. 7C). Similar to the experiments with ATDC5 cells, we evaluated the efficacy of JAK, STAT, and ERK inhibitors in blocking rIL-6–induced neurite outgrowth and the expression of pain-associated factors in both DRG explant and primary DRG neuron cultures (Fig. 7. A to C; fig. S8, A and C, and S9). Again, inhibition of JAK signaling with Ruxolitinib attenuated both neurite outgrowth and the increase in pain-associated factors induced by rIL-6. However, unlike what we observed in chondrocytes, inhibition of ERK1/2, but not STAT3, rescued the rIL-6–induced pathologic effects on DRG neurons and the induction of pain-associated factors. Although treatment with U0126 reduced neurite outgrowth as well as the expression of Cgrp and Ccr2 induced by rIL-6, treatment with Stattic exhibited minimal effects on neurite outgrowth while exacerbating the rIL-6–induced expression of Cgrp and Ccr2. These data suggest that inhibition of STAT3 would have no effect or potentially agonistic effects on PTOA-associated pain, whereas JAK and ERK inhibition may be a relevant approach to treating IL-6–induced or PTOA-associated pain.
Fig. 7. IL-6 induces pain-associated factors in male DRG.
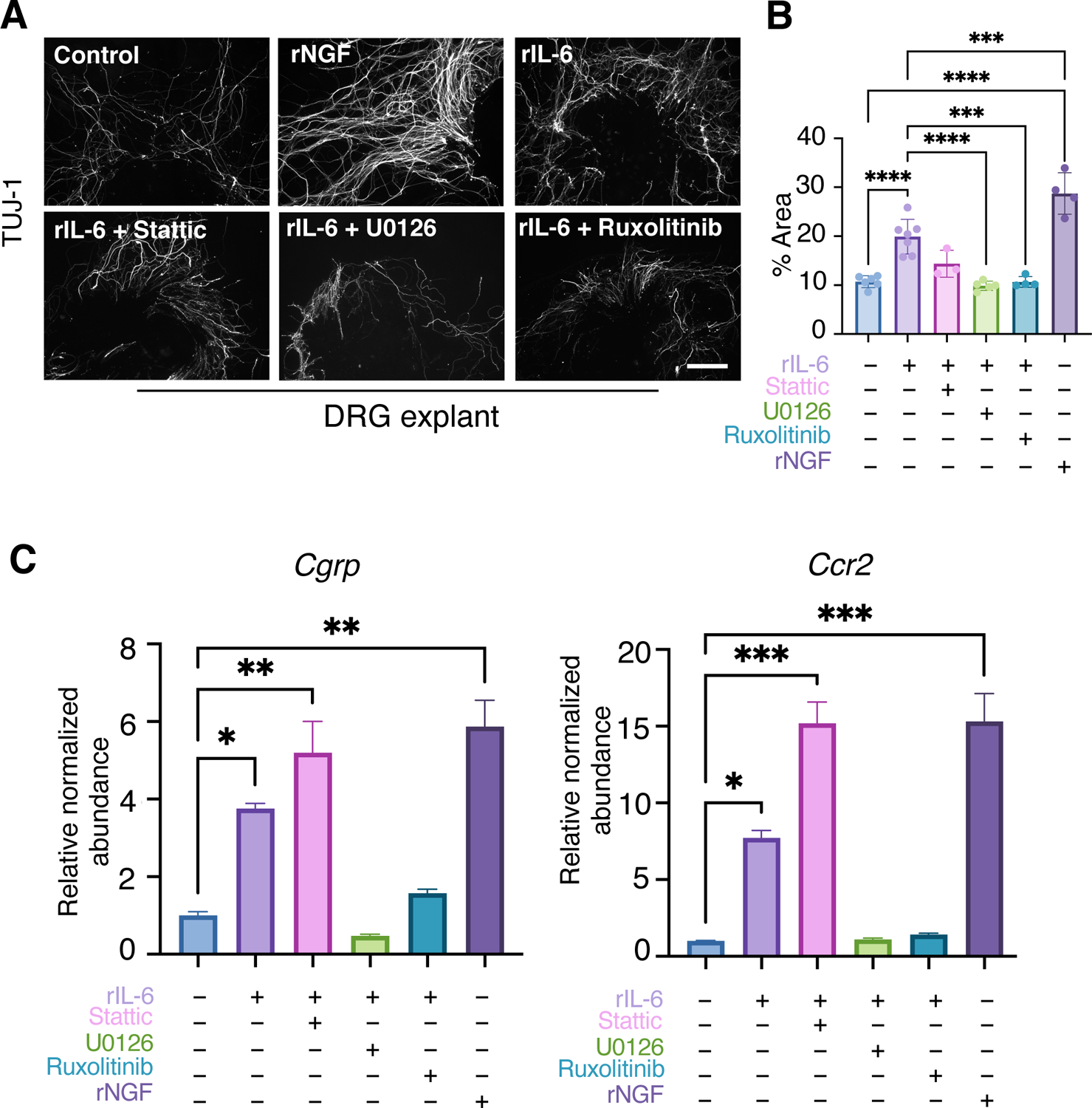
(A) Immunofluorescence staining for TUJ-1 to visualize neurites in L3–L5 DRG from male mice cultured with rNGF, rIL-6, or rIL-6 plus the indicated inhibitors. N≥3 explants per condition. Scale bar, 200 μm. (B) Quantification of the percentage of TUJ-1 staining area over the total examined area. Data represent mean ± SD. Only the statistical significance by one-way ANOVA between DMSO group and treatment groups as well as rIL-6 and treatment groups are shown. (C) qPCR of DRG neurons from male mice treated with DMSO (–) or rIL-6 plus or minus inhibitors as indicated. N≥ 3 explants per condition. Data represent mean ± SEM. Only the statistical significance by one-way ANOVA between DMSO group and treatment groups is shown. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Discussion
Here, we identified a critical role for IL-6 signaling in mediating PTOA-associated cartilage degeneration and pain in male mice. Genetic ablation of Il6 reduced cartilage degradation through the attenuation of cartilage catabolism without affecting chondrocyte cell death. Additionally, deletion of Il6 reduced PTOA-associated pain responses in male mice, likely through a decrease in knee joint innervation and a reduction in pain mediators within knee-associated DRG neurons, even though subchondral bone sclerosis persisted in the mutants. This observation indicates that innervation of the subchondral bone and subchondral bone sclerosis are separable events during PTOA. Because deletion of Il6 reduced the amount of activated STAT3 and ERK1/2 in DRG following joint injury, cytokine-induced innervation and pain signaling likely propagates the PTOA-associated pain phenotype, which is separate from the subchondral bone sclerosis phenotype. We further demonstrated the differential effects that downstream mediators of IL-6 signaling play in both cartilage and DRG. JAK signaling was identified as a critical mediator of IL-6–induced cartilage catabolism and pain signaling in nociceptive neurons. STAT3 signaling was also a potent driver of cartilage catabolism; however, its inhibition had negative effects on cartilage anabolism and further exacerbated pain-associated signaling in DRG neurons, suggesting that STAT3 inhibitors may not be an appropriate DMOAD. ERK signaling was essential for IL-6–induced neurite outgrowth and pain signaling in DRG neurons; however, inhibition of ERK1/2 exerted only mildly suppressive effects on cartilage catabolism. Collectively, this study provides new insights into IL-6 and its downstream signaling effectors as potential therapeutic targets in the treatment of PTOA-associated cartilage degeneration and pain (Fig. 8) and indicates that IL-6 inhibition may have sex-specific effects in the treatment of PTOA.
Fig. 8. Schematic model of IL-6 signaling in male PTOA.
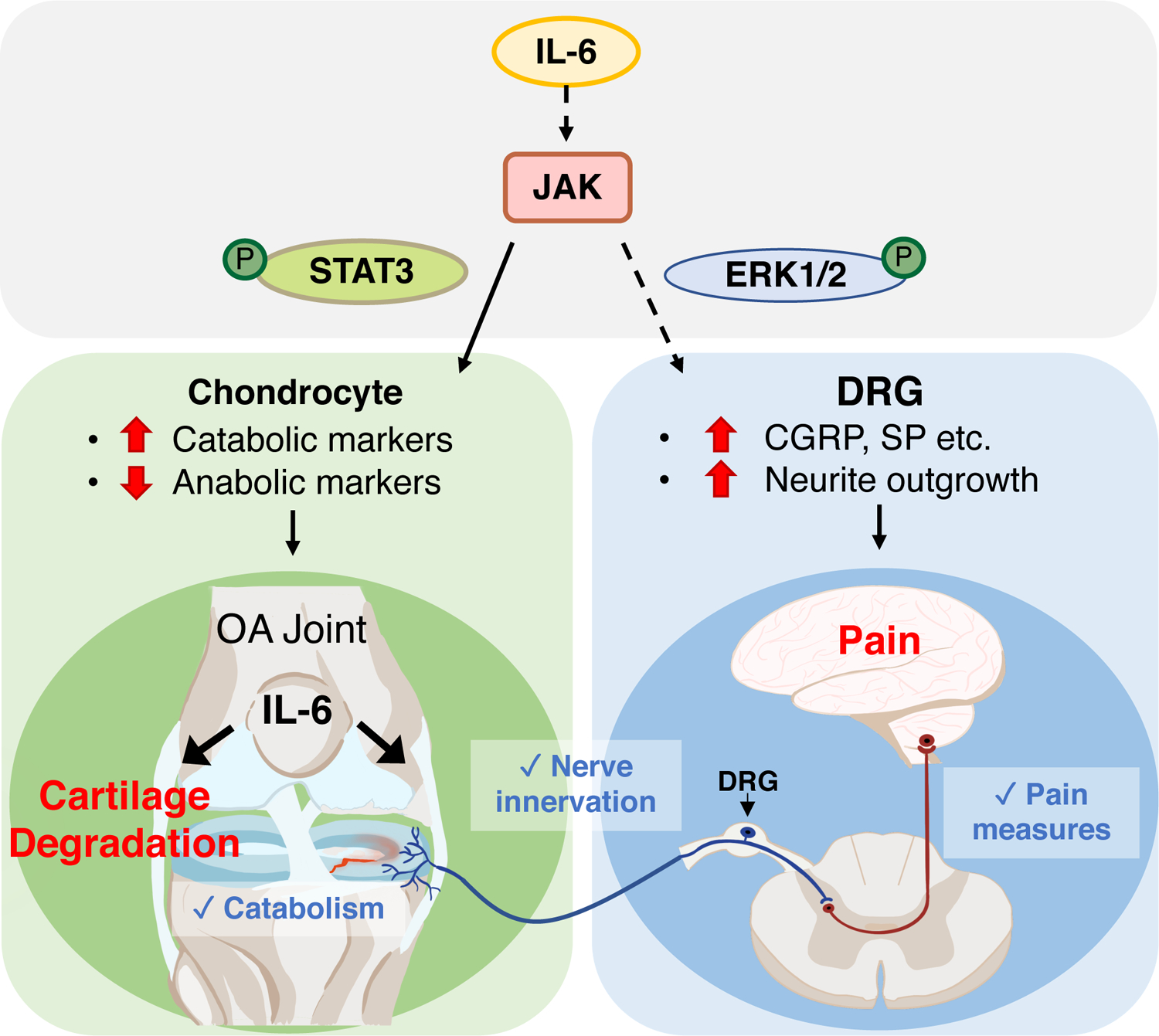
Genetic ablation of Il6 in male mice attenuated PTOA-associated cartilage catabolism and pain. IL-6 promoted cartilage catabolism and inhibited cartilage anabolism through JAK and its downstream effector STAT3, as demonstrated by pharmacological intervention. Genetic ablation of Il6 in male mice reduced pain sensation, innervation of the knee joint, and nociceptive signaling through JAK and ERK signaling, which was important for neurite outgrowth and pain signaling in DRG neurons.
The potential cellular or tissue sources of IL-6 in the context of OA/PTOA have been explored in multiple studies. In OA/PTOA knee joints, IL-6 can be secreted from chondrocytes, osteoblasts, meniscal cells, infrapatellar fat, synovial fibroblasts, and subchondral bone mesenchymal stromal cells (33, 59–65). Specifically, senescent cells and the senescence-associated secretory phenotype (SASP) within the joint environment have been found to be highly correlated with OA progression and pain (66–68) and a source of IL-6 as well (69). Besides these cell types within the knee joint, immune cells such as macrophages, monocytes, T cells, and B cells can infiltrate the joint after injury, releasing cytokines (including IL-6) and chemokines (70). In addition, IL-6 has been recognized as an important component of the neuroinflammatory cascade pathway across Schwann cells to neurons and microglia (71). Our data highlighted here from Il6 −/− mice do not delineate if one or more of these potential cellular sources of IL-6 are important in promoting PTOA-associated cartilage degeneration and pain in male mice, but rather establishes a proof-of-concept that IL-6 inhibition may serve as a sex-specific therapeutic strategy irrespective of the cellular source.
Although we focused on the exacerbating role of IL-6 in injury-induced OA, a study by de Hooge et al. reported that aged Il6 −/− males have higher incidences and more extensive cartilage damage when compared with aged WT controls (34). Our results demonstrated that Il6 deletion in male mice attenuated cartilage degradation and pain in injury-induced OA, but the previous study demonstrated that Il6 deletion accelerates age-induced OA (29). These opposing effects indicate a potential dual role for IL-6 in conditions of aging and acute injury–induced OA. Consistent with our data showing insensitivity of injury-induced OA to Il6 deletion in female mice, the study by de Hooge et al. also indicates that aged Il6−/− female mice do not show substantial cartilage damage similar to aged male Il6−/− mice (29), which further confirms that Il6 deletion lacks major joint-associated effects in female mice.
Much of the current OA-related research using mice are based on the in vivo analysis of PTOA in male mice because female mice are in general more refractory to PTOA-associated cartilage degradation (57). In our study, we examined both male and female mice and observed that both sexes exhibited similar pain responses following DMM injury as measured by the PAM device, even though WT female mice developed slower cartilage degeneration phenotypes in PTOA. Unlike the situation in male mice, genetic ablation of Il6 in female mice did not alter the course of either PTOA-associated cartilage degeneration or pain through the activation of STAT3 and ERK1/2 in DRG neurons over the course of the 12-week assessments. These findings bring to the forefront three important points: 1) PTOA-associated chronic pain may be discordant with severe cartilage degeneration, because chronic pain was observed in both male and female mice at timepoints (early and late) in which visible cartilage effects of PTOA are absent or extremely minor; 2) Il6 deletion in DMM-injured female mice did not alleviate pain or reduce the amounts of activated STAT3 and ERK1/2, thereby indicating that the pain response in female mice lacking IL-6 may be driven by other JAK-STAT–activating cytokines associated with PTOA; and 3) removal or inhibition of IL-6 signaling was only effective in the prevention of PTOA-associated cartilage degeneration and pain in males and explants from males, which suggests that the underlying mechanisms regulating PTOA phenotypic progression may differ between males and females at the molecular level (at least in mice).
To address the potential for differential responses of male and female mice to IL-6 signaling in relevant joint-associated tissues, we assessed the cell and tissue autonomy of how both male and female cartilage and DRG explants responded to rIL-6. Cartilage explants from both sexes responded in a similar manner by increasing MMP13 (catabolism) following rIL-6 treatment, suggesting that at this concentration and duration of rIL-6 treatments the cartilage is capable of undergoing catabolic changes in response to IL-6 signaling irrespective of sex. However, ACAN (anabolism) decreased only in the cartilage explants from males, but not females. This may indicate different sensitivities of anabolism and catabolism between males and females in response to rIL-6 treatment. Furthermore, IL-6 signaling may not be activated to the same degree in females as compared to males to elicit prominent cartilage degradation in vivo, which is consistent with our findings showing a lack of change in p-STAT3 and p-ERK1/2 in vivo within the articular cartilage of female mice at 12-weeks post DMM. In addition, we demonstrated that female DRG have a greater cell- and tissue- autonomous potential to develop neurite outgrowths at baseline that are comparable to male DRG stimulated with rIL-6, and that female DRG cannot be further stimulated to produce additional neurite outgrowths by rIL-6 treatment. A similar effect is also observed in CGRP expression in male and female DRG in control and rIL-6 treatments. These data are consistent with female tissues being sensitive to numerous pain-associated cytokines and suggests that removal of Il6 could be insufficient to alter PTOA-associated pain responses in female mice. Prior work in murine and cell models have also exposed the relationship between sex hormones and IL-6, which demonstrated that estrogen is a negative regulator of Il6 (72) and that higher amounts of estrogen receptor reduce Il6 expression (73). Because females produce higher amounts of estrogen and estrogen receptors than males, females in general may have a more blunted effect to IL-6 signaling activation, such as the decreased severity of joint cartilage degradation following DMM injury observed in females as compared to male mice. This hypothesis also matches the clinical observation that females after menopause, who have lost estrogen protection, are more prone to OA and display more severe OA symptom than males (8). Alternatively, females may have alternate pain signaling mechanisms to overcome the reduced effects of IL-6 signaling, for instance through other pro-inflammatory cytokines acting through similar downstream effectors as IL-6. In humans, IL-6 serum amounts are similar between males and females with OA; however, females exhibit higher serum IL-6 reactivity to evoked acute pain as compared to men with advanced knee OA (74). Further investigations are needed to identify underlying mechanisms of the sex differences in response to IL-6 signaling, IL-6–modulating drugs, and their effects on PTOA-associated cartilage degeneration and pain.
Most clinical studies on IL-6 and OA mainly focus on using IL-6 as a biomarker for OA, and no direct causal link has been demonstrated between IL-6 and OA pain; however, IL-6 signaling modulators have been applied to the treatment of other arthritis conditions in recent years. Tocilizumab, an antibody against the IL-6 receptor, is effective in treating rheumatoid arthritis (RA) and entered the market as a key disease-modifying anti-rheumatic drug (DMARD). Given that there are important differences in the pathological mechanisms between RA and OA, the clinical results for their use in RA do not directly indicate their potential usefulness for treating OA. A recent double-blinded, randomized controlled trial (NCT02477059) demonstrated that tocilizumab did not substantially reduce pain or improve function in patients with hand OA, and subsequently generated more adverse events such as infections and neutropenia within the treatment group (75). This result is unexpected in lieu of our findings; however, we suspect that hand OA is likely under the control of different signaling mechanisms as compared to PTOA of the knee due the different mechanical loading and movement between hand and knee (76). Further, signaling differences are known to exist between the membrane-bound and soluble forms of the IL-6 receptor, and therefore general targeting of the IL-6 receptor may have unintended consequences. Thus, a more specific downstream effector of the IL-6 receptor may be an even more appealing drug target, providing better effectiveness with fewer side effects. Small molecule JAK inhibitors are the newest class of investigational drugs for treating RA and have proven efficacious in early pain relief and in limiting the progression of structural damage in RA (77). However, their use and effectiveness in OA or PTOA have not been reported with clinical trials. Our study provides new insights into the potential importance and application of JAK inhibition and their potential effects on cartilage degradation and pain in PTOA.
We demonstrated that STAT3 and ERK signaling regulate specific aspects of cartilage degradation and pain downstream of IL-6 signaling. Although studies have shown the STAT3 inhibitor, Stattic, to be effective in alleviating cartilage degradation in DMM-induced OA (39), our data indicates that STAT3 inhibition may exacerbate IL-6–induced suppression of cartilage anabolic genes and further potentiate the expression of pain mediators. Therefore, drugs solely targeting STAT3 may not be the most effective therapeutic approach for treating PTOA-associated cartilage degeneration and pain. In opposition to STAT3 inhibition, ERK1/2 inhibition using U0126 significantly reduced the expression of pain mediators induced by IL-6 signaling but had limited efficacy in reducing IL-6–mediated cartilage catabolism. Therefore, the most effective approaches to treating both PTOA-associated cartilage catabolism and pain may lie in the dual inhibition of STAT3 and ERK signaling simultaneously, or alternatively the use of JAK inhibitors.
In conclusion, our findings demonstrated that IL-6 signaling mediated both cartilage degradation and pain in the PTOA of male, but not female, mice. This study further defined the differential effects of downstream mediators of IL-6 signaling in both chondrocytes and DRG neurons, which has provided critical insights into not only the most relevant therapeutic targets of DMOADs, but also the population that would stand most to benefit from their use.
Materials and Methods
Mice
Interleukin-6 knockout mice (Il6 −/−) in the C57BL/6J background were used in this study and obtained from The Jackson Laboratory. C57BL/6J mice were used as wildtype (WT) controls and also obtained from The Jackson Laboratory. All surgeries were performed at 4 months of age. Mice were harvested at 4-weeks (5-month-old), 8-weeks (6-month-old) and 12-weeks (7-month-old) post injury. Details are included in table S1. The numbers of mice used in each experiment are indicated in data file S1. All animal experiments were conducted in accordance with policies and regulations of the NIH and approved by the Duke University Institutional Animal Care and Use Committee (IACUC) (reference number for approval: A068-20-03).
Modified destabilized medial meniscus (DMM) OA model and surgical procedures
Destabilization of the medial meniscus was used as our model of post-traumatic OA (PTOA) in mice. We followed the well-established surgical method of destabilized medial meniscus with a slight modification.(36, 78, 79) Briefly, the left hind-limbs were shaved and wiped with iodine and 70% ethanol to sanitize. After exposing thee left knee, the medial collateral ligament was transected. Next, the medial meniscus was destabilized from its anterior-medial tibial attachment using a 25G needle, and put back to its original location. The skin incision was sutured with 5–0 monofilament Nylon after the surgery. Sham surgeries were performed on separate animals with incisions made on the skin to expose the knee, however neither the ligament transection nor the meniscus detachment were performed.
Histology
Knees were collected at 4-, 8- and 12-wpi, and fixed with 4% PFA for two days at 4°C. Samples were decalcified with 14% EDTA for 10–14 days, and processed for paraffin embedding. The medial compartment of the knee joints was sectioned at 5-micron thickness. Sections were collected at the point of meniscus separation, and stopped when the meniscus were no longer present in tissue sections, which resulted in about 60 sections per sample. We then performed immunohistochemistry (IHC) or immunofluorescence (IF), and evaluated cartilage degradation and other morphological changes by Safranin O staining with fast green counterstaining by following established protocols(80). Specifically, sections were de-paraffinized, and unmasked for antigen with EDTA (pH =8) for phospho-STAT3 IHC, and citrate buffer for phospho-ERK1/2 IHC at 95°C. MMP-13 IF staining were performed with paraffin slides as described previously(81). Cell apoptosis was examined utilizing a TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) kit (Roche). In brief, after de-paraffinization, sections were subjected to antigen retrieval with proteinase K (10ug/ml) for 20 mins at room temperature. After washes with PBS, sections were incubated in a humidified chamber with the TUNEL reaction mix for 60 minutes at 37°C, followed by coverslipping and imaging. Methods for nerve staining in the knee was adapted from prior work(82). The 50-micron thick sections were obtained from frozen embedded knee tissues by performing tape transfer. After bleaching with 3% H2O2, tissue sections were unmasked by 0.3% Triton X-100 in PBS for 1 hour and incubated with CGRP antibody (1:1400) overnight. On day 2, after blocking with secondary antibody (AlexFluor 594) the sections were imaged with Zeiss 780 inverted confocal microscope. Fiji was used to perform quantifications, where the ratio of area of positive staining to the area of region of interest (ROI) were calculated, and IHC quantification was performed according to a published protocol (83).
In addition to knee joint tissues, DRG associated with L3–L5 vertebrae were harvested at the same time for frozen sections. To prepare frozen sections, DRGs were harvested and processed through a gradient of sucrose from 15% to 30% for 2 days before embedding in O.C.T. For IF staining, DRG samples were sectioned at 12-micron thickness. To unmask antigen, citrate buffer was used for p-STAT3 staining at sub-boiling temperature, and methanol was used for p-ERK1/2 staining at −20°C. Nissl substance in neurons were stained by NeuroTrace 530/615 red fluorescent (Invitrogen), which labeled all neurons. Quantifications were performed by calculating the ratio of the number of CGRP+ neurons (green) to the total number of neurons (stained by NeuroTrace in red and having large nuclei with DAPI in blue). The ratio of CGRP+ neurons over the total number of neurons is calculated to account for the fact that different numbers of total neurons are obtained in different DRG sections. DRG explant cultures were adapted from established protocols(84). For TUJ-1 staining on DRG explants, DRGs were fix with 3.7% formaldehyde for 30 minutes, and then incubated with TUJ-1 antibody as a 1:2000 dilution in PBS buffer with 0.3% Triton-100 and 10% normal goat serum overnight at 4°C. After incubation with appropriate secondary antibody diluted in PBS, sections were coverslipped with DAPI and prepared for imaging. For RNA and protein extraction, DRG were snap frozen in liquid nitrogen and stored at −80 degrees. (See tables S2 and S3 for the details of antibodies and reagents).
Osteoarthritis Research Society International (OARSI) osteoarthritis cartilage histopathology assessment (OARSI Score)
OARSI scoring is a standard histological scoring system for murine OA(85, 86). In our experiments, five sections at different depths, approximately 50um apart, were randomly selected for each animal. Based on Safranin-O stained sections, cartilage changes were scored blindly from 0 to 6 by at least three individuals. Every data point in the OARSI score panels represent one animal, which is an average score from three graders on five individual sections per animal. A score of 0–2, represents only minor changes observed within the articular cartilage, while scores of 3–6 indicate increasing areas of cartilage erosion across the articular surface.
MicroCT analysis
MicroCT analysis were performed on knees from WT control and Il6 −/− mice prior to decalcification using a VivaCT 80 scanner with 55-kVp source (Scanco USA) as previously described(87). Quantification of microCT data were calculated for the subchondral region, which we defined as 0.1mm region above proximal growth plate of the tibia extending to the articular cartilage. Bone volume/total volume (BV/TV) at the region of interest is used as primary outcome parameter.
Behavioral testing
For pain measurements, behavioral tests were conducted before harvesting tissue at 1-, 2-, 4-, 6-, and 8-weeks post-injury (Sham/DMM) using the Pressure Application Measurement (PAM) device to assess mechanical hyperalgesia(88–91). Animals were adapted to the test environment with stable room temperature and humidity for two days before the baseline testing. In PAM tests, a gradually increasing force was applied across the knee joint of the mouse which is lightly but securely held, until the animal provides an indication of pain or discomfort. The peak force applied within the 5 second maximum test duration is recorded by the device connecting to force sensor worn on the operator’s finger and is displayed in grams as the paw withdrawal threshold.
Cell and Ex vivo cartilage and DRG explant cultures
ATDC5 cells (RIKEN BioResource Center) were maintained and cultured as previously described(38). In short, ATDC5 cells were maintained in complete media DMEM/F-12 (1:1) medium with addition of 10% FBS and 1% Penicillin/Streptomycin. For experimental purposes, ATDC5 cells were differentiated in complete media supplemented with insulin, transferrin, and sodium selenite (ITS) for 7 to 14-days. Before treatment with rIL-6 protein or inhibitors, differentiated ATDC5 chondrogenic cells were washed with PBS once, and the indicated treatments were added to the media for 48 hours. A concentration of 50ng/ml recombinant IL-6 (rIL-6) protein was applied to the culture; the JAK inhibitor, Ruxolitinib (Selleckchem), was used at a final concentration of 0.5uM; the STAT3 inhibitor, Stattic (Sigma), was used at a final concentration of 20uM; and the ERK1/2 inhibitor, U0126 (Sigma), was used at a final concentration of 10uM.
For cartilage explant culture, femoral cartilages were harvested from three-week-old (fig. S7A) and 2-month-old mice (Fig. 6) and cultured in the same media as ATDC5 cells (DMEM/F-12 medium + 5% FBS + 1% Penicillin/Streptomycin + ITS) with 100ng/ml rIL-6 for 7 days. Ruxolitinib was used at a final concentration of 10uM; Stattic was used at a final concentration of 20uM; and U0126 was used at a final concentration of 20uM.
DRG explant cultures are adapted from the protocol described previously(84). Briefly, following DRG harvest from 1 month old WT mice, the intact DRGs were placed in a 35mm Petri dish containing 3 mL of ice-cold serum free Neurobasal-A media with 1% Penicillin/Streptomycin. Each individual DRG was cleaned and trimmed of excess fibers and connective tissues under a surgical microscope and was placed in a new petri dish containing ice-cold media. All steps after this were performed using aseptic technique in a Class II biosafety cabinet. DRG were washed with sterile HBSS solution twice and plated in PDL pre-coated plates. Neurobasal medium supplemented with 2% v/v B-27 Supplement were gently added to cover and maintain the entire explant at 37°C and 5% CO2 in a cell culture incubator. Isolated primary neurons from DRG were disassociated and cultured according to a published protocol (92). A concentration of 50ng/ml rIL-6 and 50ng/ml recombinant NGF (rNGF) were added to the DRG explant or isolated neuron culture. Ruxolitinib was used at a final concentration of 10uM; Stattic was used at a final concentration of 20uM; and U0126 was used at a final concentration of 10uM. DRG explants were cultured with treatment for 2 days, 4 days, or 10 days for neurite outgrowth assays and tissue section staining, and 24 hours for isolated neuron cultures.
Transcript and protein analyses
ATDC5 cells and DRGs were collected in RLT buffer and TRIzol respectively for RNA extraction, and RIPA lysis buffer (radioimmunoprecipitation assay (RIPA) buffer plus protease inhibitor and phosphatase inhibitors) for protein extraction. A motorized tissue grinder was used to homogenize DRG in order to extract RNA or protein. RNA was extracted with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions, and RNA concentrations were measured using a NanoDrop 1000 spectrophotometer. After RNA extraction, cDNA were synthesized and real-time qPCR analysis were performed for the chondrogenic related genes (Acan and Mmp13), several pain mediators (Cgrp and Ccr2) and a housekeeping gene, Gapdh (see table S4 for sequences). For Western analysis, 30ug of protein were loaded to each well, and underwent electrophoresis through 4–20% gradient Tris-Glycine gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes with a Trans-Blot Turbo Transfer System (Bio-Rad). IL-6 downstream signaling activation was monitored using primary antibodies against STAT3, p-STAT3 (Tyr705), ERK1/2, p-ERK1/2 (Thr202/Tyr204), p38 MAPK, and pp38 MAPK (Thr180/Tyr182), which were incubated overnight in 1:1000 dilution in 5% nonfat milk dissolved in 1x TBST. After blotting with appropriate secondary antibodies, gels were developed using the Pierce ECL Western Blotting Substrate or SuperSignal West Femto Maximum Sensitivity Substrate depending on the intensity and were imaged using a Chemidoc system (Bio-Rad).
Statistical analysis
Sample size was determined based on the preliminary experiments and prior studies with a consideration in the variations of the sample. Slide selection and quantifications were done in a randomized and blinded manner. Statistical significance and p-values were determined using one-way ANOVA for OARSI scores, quantification of IF stainings, and BV/TV values from our microCT analyses. Two-way ANOVA with Bonferroni’s post-hoc test were used for behavioral analysis, and unpaired two-tailed t-tests were utilized for qPCR results (data file S1).
Supplementary Material
Acknowledgements:
We would like to thank Dr. Yun Gu, Dr. Deepika Sharma, and the Duke Light Microscopy Core Facility for technical and equipment support.
Funding:
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institute of Health (NIH) under the award numbers R01AR071722 and R01AR063071 to M.J.H and Duke University Anesthesiology Research Fund to R-R.J.
Footnotes
Competing Interests: The authors declare that they have no competing interests.
Data and Materials Availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Hunter DJ, Bierma-Zeinstra S, Osteoarthritis. Lancet 393, 1745–1759 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Lespasio MJ et al. , Knee Osteoarthritis: A Primer. Perm J 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace IJ et al. , Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc Natl Acad Sci U S A 114, 9332–9336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nevitt MC, Felson DT, Sex hormones and the risk of osteoarthritis in women: epidemiological evidence. Ann Rheum Dis 55, 673–676 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolf AD, Pfleger B, Burden of major musculoskeletal conditions. Bull World Health Organ 81, 646–656 (2003). [PMC free article] [PubMed] [Google Scholar]
- 6.Brown TD, Johnston RC, Saltzman CL, Marsh JL, Buckwalter JA, Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma 20, 739–744 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Neogi T, The epidemiology and impact of pain in osteoarthritis. Osteoarthritis Cartilage 21, 1145–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elbaz A et al. , Sex and body mass index correlate with Western Ontario and McMaster Universities Osteoarthritis Index and quality of life scores in knee osteoarthritis. Arch Phys Med Rehabil 92, 1618–1623 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Tonelli SM, Rakel BA, Cooper NA, Angstom WL, Sluka KA, Women with knee osteoarthritis have more pain and poorer function than men, but similar physical activity prior to total knee replacement. Biol Sex Differ 2, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass N et al. , Examining sex differences in knee pain: the multicenter osteoarthritis study. Osteoarthr Cartilage 22, 1100–1106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.S W-C. Hochberg MC, Cisternas MG, US Bone and Joint Initiative. The Burden of Musculoskeletal Diseases in the United States (BMUS). (2019).
- 12.Kolasinski SL et al. , 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Rheumatol 72, 220–233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloppenburg M, Berenbaum F, Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthritis Cartilage 28, 242–248 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Brandt KD, Radin EL, Dieppe PA, van de Putte L, Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis 65, 1261–1264 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeser RF, Goldring SR, Scanzello CR, Goldring MB, Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 64, 1697–1707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felson DT, Clinical practice. Osteoarthritis of the knee. N Engl J Med 354, 841–848 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Loeser RF, Collins JA, Diekman BO, Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 12, 412–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Bosch MHJ, van Lent P, van der Kraan PM, Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis Cartilage 10.1016/j.joca.2020.01.016 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Malfait AM, Schnitzer TJ, Towards a mechanism-based approach to pain management in osteoarthritis. Nat Rev Rheumatol 9, 654–664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenn D, Richter F, Schaible HG, Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: an inflammatory mechanism of joint pain. Arthritis Rheum 56, 351–359 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Schaible HG et al. , The role of proinflammatory cytokines in the generation and maintenance of joint pain. Ann N Y Acad Sci 1193, 60–69 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Schaible HG, Nociceptive neurons detect cytokines in arthritis. Arthritis Res Ther 16, 470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zarebska JM et al. , CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthr Cartilage 25, 406–412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller RE et al. , CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 109, 20602–20607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pecchi E et al. , Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther 16, R16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller RE, Block JA, Malfait AM, Nerve growth factor blockade for the management of osteoarthritis pain: what can we learn from clinical trials and preclinical models? Curr Opin Rheumatol 29, 110–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livshits G et al. , Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: The Chingford Study. Arthritis Rheum 60, 2037–2045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller RE, Miller RJ, Malfait AM, Osteoarthritis joint pain: the cytokine connection. Cytokine 70, 185–193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baran P et al. , The balance of interleukin (IL)-6, IL-6.soluble IL-6 receptor (sIL-6R), and IL-6.sIL-6R.sgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem 293, 6762–6775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunter CA, Jones SA, IL-6 as a keystone cytokine in health and disease. Nat Immunol 16, 448–457 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Qu XQ, Wang WJ, Tang SS, Liu Y, Wang JL, Correlation between interleukin-6 expression in articular cartilage bone and osteoarthritis. Genet Mol Res 14, 14189–14195 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Vuolteenaho K et al. , High Synovial Fluid Interleukin-6 Levels Are Associated with Increased Matrix Metalloproteinase Levels and Severe Radiographic Changes in Osteoarthritis Patients. Osteoarthr Cartilage 25, S92–S93 (2017). [Google Scholar]
- 33.Distel E et al. , The infrapatellar fat pad in knee osteoarthritis: an important source of interleukin-6 and its soluble receptor. Arthritis Rheum 60, 3374–3377 (2009). [DOI] [PubMed] [Google Scholar]
- 34.de Hooge AS et al. , Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage 13, 66–73 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Little CB, Hunter DJ, Post-traumatic osteoarthritis: from mouse models to clinical trials. Nat Rev Rheumatol 9, 485–497 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Culley KL et al. , Mouse models of osteoarthritis: surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol Biol 1226, 143–173 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Ryu JH et al. , Interleukin-6 plays an essential role in hypoxia-inducible factor 2alpha-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 63, 2732–2743 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Liu Z et al. , A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci Signal 8, ra71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latourte A et al. , Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis 76, 748–755 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Erta M, Quintana A, Hidalgo J, Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci 8, 1254–1266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andratsch M et al. , A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci 29, 13473–13483 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svensson CI, Interleukin-6: a local pain trigger? Arthritis Res Ther 12, 145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satoh T et al. , Induction of neuronal differentiation in PC12 cells by B-cell stimulatory factor 2/interleukin 6. Mol Cell Biol 8, 3546–3549 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Penninx BW et al. , Inflammatory markers and physical function among older adults with knee osteoarthritis. J Rheumatol 31, 2027–2031 (2004). [PubMed] [Google Scholar]
- 45.Leung YY, Huebner JL, Haaland B, Wong SBS, Kraus VB, Synovial fluid pro-inflammatory profile differs according to the characteristics of knee pain. Osteoarthritis Cartilage 25, 1420–1427 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Radojčić MR et al. , Biomarker of extracellular matrix remodelling C1M and proinflammatory cytokine interleukin 6 are related to synovitis and pain in end-stage knee osteoarthritis patients. Pain 158, 1254–1263 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Eitner A et al. , Pain sensation in human osteoarthritic knee joints is strongly enhanced by diabetes mellitus. Pain 158, 1743–1753 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Miller RE et al. , Visualization of Peripheral Neuron Sensitization in a Surgical Mouse Model of Osteoarthritis by In Vivo Calcium Imaging. Arthritis Rheumatol 70, 88–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malfait AM, Little CB, McDougall JJ, A commentary on modelling osteoarthritis pain in small animals. Osteoarthritis Cartilage 21, 1316–1326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Assas BM, Pennock JI, Miyan JA, Calcitonin gene-related peptide is a key neurotransmitter in the neuro-immune axis. Front Neurosci 8, 23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thakur M, Dickenson AH, Baron R, Osteoarthritis pain: nociceptive or neuropathic? Nat Rev Rheumatol 10, 374–380 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Aso K et al. , Contribution of nerves within osteochondral channels to osteoarthritis knee pain in humans and rats. Osteoarthritis Cartilage 28, 1245–1254 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Zhu S et al. , Subchondral bone osteoclasts induce sensory innervation and osteoarthritis pain. J Clin Invest 129, 1076–1093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Y, Chen X, Wang S, Jing Y, Su J, Subchondral bone microenvironment in osteoarthritis and pain. Bone Res 9, 20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bongartz H, Seiss EA, Bock J, Schaper F, Glucocorticoids attenuate interleukin-6-induced c-Fos and Egr1 expression and impair neuritogenesis in PC12 cells. J Neurochem 157, 532–549 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Ji RR, Gereau R. W. t., Malcangio M, Strichartz GR, MAP kinase and pain. Brain Res Rev 60, 135–148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma HL et al. , Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage 15, 695–700 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Drubin DG, Feinstein SC, Shooter EM, Kirschner MW, Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol 101, 1799–1807 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greene MA, Loeser RF, Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage 23, 1966–1971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiegertjes R, van de Loo FAJ, Blaney Davidson EN, A roadmap to target interleukin-6 in osteoarthritis. Rheumatology (Oxford) 59, 2681–2694 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guerne PA, Zuraw BL, Vaughan JH, Carson DA, Lotz M, Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest 83, 585–592 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guerne PA, Carson DA, Lotz M, IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J Immunol 144, 499–505 (1990). [PubMed] [Google Scholar]
- 63.Ishimi Y et al. , IL-6 is produced by osteoblasts and induces bone resorption. J Immunol 145, 3297–3303 (1990). [PubMed] [Google Scholar]
- 64.Wu X et al. , Interleukin-6 from subchondral bone mesenchymal stem cells contributes to the pathological phenotypes of experimental osteoarthritis. Am J Transl Res 10, 1143–1154 (2018). [PMC free article] [PubMed] [Google Scholar]
- 65.Sakao K et al. , Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J Bone Miner Metab 27, 412–423 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Jeon OH, David N, Campisi J, Elisseeff JH, Senescent cells and osteoarthritis: a painful connection. J Clin Invest 128, 1229–1237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCulloch K, Litherland GJ, Rai TS, Cellular senescence in osteoarthritis pathology. Aging Cell 16, 210–218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu W et al. , Senescent Tissue-Resident Mesenchymal Stromal Cells Are an Internal Source of Inflammation in Human Osteoarthritic Cartilage. Front Cell Dev Biol 9, 725071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vinatier C, Domínguez E, Guicheux J, Caramés B, Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Frontiers in physiology 9, 706–706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haseeb A, Haqqi TM, Immunopathogenesis of osteoarthritis. Clin Immunol 146, 185–196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu Z et al. , CNTF-STAT3-IL-6 Axis Mediates Neuroinflammatory Cascade across Schwann Cell-Neuron-Microglia. Cell Rep 31, 107657 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Kassem M, Harris SA, Spelsberg TC, Riggs BL, Estrogen inhibits interleukin-6 production and gene expression in a human osteoblastic cell line with high levels of estrogen receptors. J Bone Miner Res 11, 193–199 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Galien R, Garcia T, Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res 25, 2424–2429 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mun CJ et al. , Sex Differences in Interleukin-6 Responses Over Time Following Laboratory Pain Testing Among Patients With Knee Osteoarthritis. The journal of pain 21, 731–741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Richette P et al. , Efficacy of tocilizumab in patients with hand osteoarthritis: double blind, randomised, placebo-controlled, multicentre trial. Ann Rheum Dis 10.1136/annrheumdis-2020-218547 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Leung GJ, Rainsford KD, Kean WF, Osteoarthritis of the hand I: aetiology and pathogenesis, risk factors, investigation and diagnosis. Journal of Pharmacy and Pharmacology 66, 339–346 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Harrington R, Al Nokhatha SA, Conway R, JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J Inflamm Res 13, 519–531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glasson SS, Blanchet TJ, Morris EA, The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15, 1061–1069 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Sophocleous A, Huesa C, Osteoarthritis Mouse Model of Destabilization of the Medial Meniscus. Methods Mol Biol 1914, 281–293 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Hilton MJ, Springer Science+Business Media, Skeletal development and repair : methods and protocols, Springer protocols, (Humana Press, New York, 2014), pp. x, 328 pages. [Google Scholar]
- 81.Haseeb A et al. , SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc Natl Acad Sci U S A 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Z et al. , Fracture repair requires TrkA signaling by skeletal sensory nerves. J Clin Invest 129, 5137–5150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Crowe AR, Yue W, Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio-protocol 9, e3465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fornaro M, Sharthiya H, Tiwari V, Adult Mouse DRG Explant and Dissociated Cell Models to Investigate Neuroplasticity and Responses to Environmental Insults Including Viral Infection. J Vis Exp 10.3791/56757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Glasson SS, Chambers MG, Van Den Berg WB, Little CB, The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18 Suppl 3, S17–23 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Pritzker KP et al. , Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14, 13–29 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Wang C et al. , NOTCH signaling in skeletal progenitors is critical for fracture repair. J Clin Invest 126, 1471–1481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barton NJ et al. , Pressure application measurement (PAM): a novel behavioural technique for measuring hypersensitivity in a rat model of joint pain. J Neurosci Methods 163, 67–75 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Leuchtweis J, Imhof AK, Montechiaro F, Schaible HG, Boettger MK, Validation of the digital pressure application measurement (PAM) device for detection of primary mechanical hyperalgesia in rat and mouse antigen-induced knee joint arthritis. Methods Find Exp Clin Pharmacol 32, 575–583 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Miller RE, Block JA, Malfait AM, What is new in pain modification in osteoarthritis? Rheumatology (Oxford) 10.1093/rheumatology/kex522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K et al. , PD-1 blockade inhibits osteoclast formation and murine bone cancer pain. J Clin Invest 130, 3603–3620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee S.-i., Levine J, Isolation and Growth of Adult Mouse Dorsal Root Ganglia Neurons. Bio-protocol 5, e1601 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.