

Abstract
Fe–N-heterocyclic carbene (NHC) complexes attract increasing attention as photosensitisers and photoredox catalysts. Such applications generally rely on sufficiently long excited state lifetimes and efficient bimolecular quenching, which leads to there being few examples of successful usage of Fe–NHC complexes to date. Here, we have employed [Fe(iii)(btz)3]3+ (btz = (3,3′-dimethyl-1,1′-bis(p-tolyl)-4,4′-bis(1,2,3-triazol-5-ylidene))) in the addition of alkyl halides to alkenes and alkynes via visible light-mediated atom transfer radical addition (ATRA). Unlike other Fe–NHC complexes, [Fe(iii/ii)(btz)3]3+/2+ benefits from sizable charge transfer excited state lifetimes ≥0.1 ns in both oxidation states, and the Fe(iii) 2LMCT and Fe(ii) 3MLCT states are strong oxidants and reductants, respectively. The combined reactivity of both excited states enables efficient one-electron reduction of the alkyl halide substrate under green light irradiation. The two-photon mechanism proceeds via reductive quenching of the Fe(iii) 2LMCT state by a sacrificial electron donor and subsequent excitation of the Fe(ii) product to its highly reducing 3MLCT state. This route is shown to be more efficient than the alternative, where oxidative quenching of the less reducing Fe(iii) 2LMCT state by the alkyl halide drives the reaction, in the absence of a sacrificial electron donor.
An iron complex with N-heterocyclic carbene ligands engages in efficient photoredox catalysis via excited state electron transfer reactions of its Fe(ii) and Fe(iii) oxidation states.
Introduction
In recent years the field of visible light-mediated photoredox catalysis based on single-photon excitation of light-harvesting molecules has gained increasingly more recognition as a highly useful tool1,2 towards facilitating greener methods of conducting chemical transformations.3,4 While a large number of such reactions is efficiently catalysed by a range of organic photosensitisers (PS),1,5 particular focus has also been given to transition metal complexes4,6 as catalysts. This is largely due to their strong visible light absorption and favourable reversible redox chemistry in both the ground and excited states (ESs), making them highly suitable candidates for photoredox catalysts (PCs). To date, the field is strongly dominated by complexes of ruthenium(ii),7–11 iridium(iii)10,12–14 and copper(i).15–20 However, especially the former two metals are scarce,21 expensive, and their complexes have absorption maxima within the blue or violet region of the visible spectrum, warranting comparatively high-energy irradiation.8,12,22 Therefore, there is an ongoing demand for environmentally benign and inexpensive catalysts with absorption maxima at longer wavelengths.23
Photosensitisers based on Earth-abundant iron – while exhibiting many of the aforementioned beneficial characteristics – notoriously suffer from short excited state lifetimes due to low lying metal-centred states.24,25 Many reports of photoredox catalysis involving iron catalysts26–28 rely on intramolecular ligand-to-metal charge-transfer (LMCT) with the substrate coordinated as a ligand. Meanwhile, efficient intermolecular charge-transfer (CT)29,30 involving the ESs of iron photosensitisers had been considered inaccessible31 until recently. To date, there are few substantiated examples of bimolecular quenching of iron CT states being used in photoredox catalysis as shown on the example of one dehalogenation reaction and one cyclisation reaction, where the involvement of CT states of iron has been firmly established.32,33
These photocatalytic reactions were enabled by the emergence of Fe–NHC-complexes29,34–36 (NHC = N-heterocyclic carbene) with excited state lifetimes approaching and exceeding the nanosecond threshold, which has brought forth the first examples of intermolecular ES quenching of CT states in an iron PS, [Fe(phtmeimb)2]PF6 (phtmeimb = tris(3-methylimidazolin-2-ylidene)(phenyl)borate).29,30,37 The possibility for their application was recently further explored by Aydogan et al.,32,33 where sizeable cage escape yields (CEYs) and corresponding quantum efficiencies with common trialkylamine donors were observed. However, these comparatively high CEYs were dependant on the use of halogenated solvents, thereby potentially restricting the scope of possible applications.
Furthermore, inherent thermodynamic restrictions of traditional single-photon driven photocatalysis pose an issue that requires addressing to date.38,39 As a result, multi-photon processes have been given progressively more consideration for photoredox reactions. The energy limitations imposed by visible light-photons can hereby be overcome by combining the energies of at least two photons in the same catalytic turnover, producing stronger oxidants and reductants, making previously unattainable transformations accessible.38–41 Such a reaction mode is comparable to the Z-scheme in natural and artificial photosynthesis.42 Additionally, undesired side reactions caused by irradiation at shorter wavelengths2 can be circumvented by using light with lower photon energy, while still providing a sufficient thermodynamic driving force. However, examples of applications of such a two-photon mechanism in photoredox catalysis of organic reactions using metal complexes are very scarce.43,44
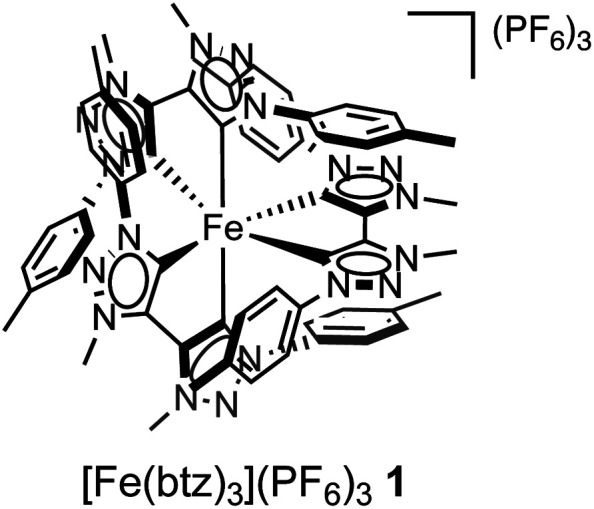
Here, we present the first case of an iron-based PS, [Fe(iii)(btz)3](PF6)31 (btz = 3,3′-dimethyl-1,1′-bis(p-tolyl)-4,4′-bis(1,2,3-triazol-5-ylidene))36 (Fig. 1), driving a photoredox catalysis reaction via two consecutive excited state electron transfer reactions, generally referred to as consecutive photo-induced electron transfer (conPET).38,45–47 By virtue of the reasonably long-lived CT states of both the Fe(iii) and Fe(ii) oxidation states of 1,36,55 this complex could be applied as photoredox catalyst (PC) in a benchmark photoredox reaction, the atom transfer radical addition (ATRA) reaction (Schemes 1 and 2). Herein, we investigated the addition of alkyl halides to alkenes and alkynes, which led to a range of synthetically valuable,48 difunctionalised products being obtained. Catalytic efficiencies were comparable to results obtained by traditional ruthenium-based ATRA catalysis,10 while using green light for excitation.
Fig. 1. Structure of the photoredox catalyst [Fe(btz)3](PF6)31 used in this study.
Scheme 1. Optimised reaction conditions for the ATRA reaction using 1 in the reductive quenching (RQ) route.

Scheme 2. Optimised reaction conditions for the ATRA reaction using 1 in the oxidative quenching (OQ) route.

Results and discussion
Initial testing, optimisation & substrate scope
Reductive quenching route
Investigations of the photocatalytic activity of [Fe(iii)(btz)3](PF6)31 in the ATRA reaction were conducted using a modified system based on a method previously established by Stephenson and co-workers for perfluorous tagging of alkenes and alkynes with the archetypal [Ru(bpy)3]2+.10 Complex 1 was chosen as PC due to the relatively long-lived CT states of the Fe(iii)-state as well as its Fe(ii)-congener 2.36,55 Both oxidation states exhibit absorption bands in the green light region,36 enabling the consecutive absorption of two photons in one catalytic turnover. If operative, such a mechanism should give access to the more strongly reducing 3MLCT (metal-to-ligand charge-transfer) excited state of the Fe(ii)-form (approx. −1.6 to −1.8 V vs. Fc). Otherwise, the Fe(ii) ground state generated in the reductive quenching step could be anticipated to be insufficiently reducing (−0.58 V vs. Fc)36 for the activation of the perfluoroalkyl halide.
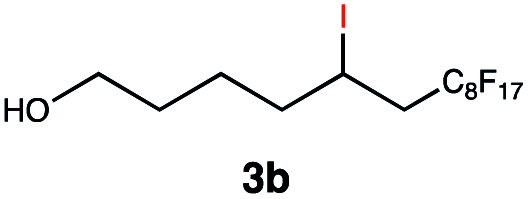
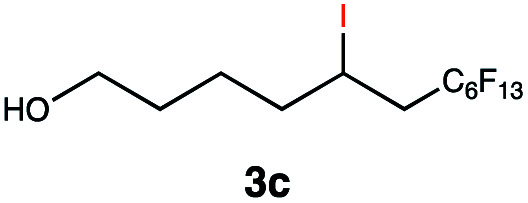
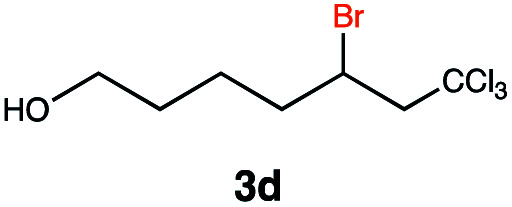
Different sacrificial electron donors were compared (Table S2†), but due to its matching redox properties, inexpensive and readily available triethylamine (TEA) (1 equiv.) was primarily used with our PC 1. Under irradiation at 530 nm using a 3.03 W LED array the combination of PC 1 and TEA afforded product 3b of the model reaction between 5-hexen-1-ol 3a and C8F17I, with full conversion being reached within 8 min. Therefore, all further optimisations were conducted with this sacrificial reductant, resulting in a reliable protocol using mild, atom efficient reaction conditions with a low catalyst loading of 0.5 mol%, and only a substoichiometric amount (0.34 equiv.) of sacrificial electron donor needed to achieve full conversion in the model system within 10 min. A 4 : 3 mixture of acetonitrile and methanol10 was chosen as the reaction solvent affording good solubility of the reactants (Tables S3 and S4†). The possibility of using other alkyl halides was also investigated (Table S5†), however the addition of perfluoroalkyl halides as well as CBrCl3 were most successful. The wider applicability of our procedure in terms of choice of alkene/alkyne was investigated by expanding the scope of substrates for the addition of perfluoroalkyl iodides and CBrCl3 (Table 1) using the optimised conditions (Scheme 1).
Scope of the visible light-mediated ATRA reaction through reductive (RQ) and oxidative (OQ) quenching of 1a,b.
| Entry | Substrate | Route | Time/min | Product | Yieldc/(%) |
|---|---|---|---|---|---|
| 1 |

|
RQ | 10 |
|
91 |
| OQ | 40 | 92 | |||
| 2 | RQ | 10 |
|
93 | |
| OQ | 40 | 93 | |||
| 3 | RQ | 60 |
|
43 | |
| OQ | 40 | 35 | |||
| 4 |
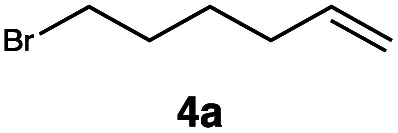
|
RQ | 10 |
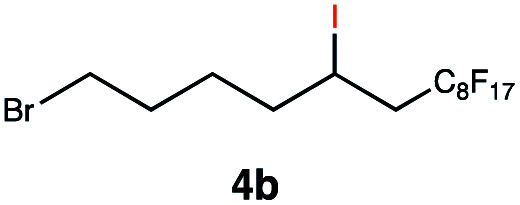
|
90 |
| OQ | 40 | 94 | |||
| 5 |
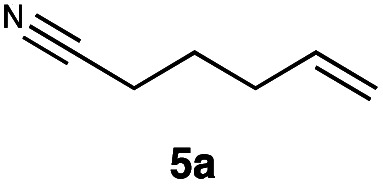
|
RQ | 40 |
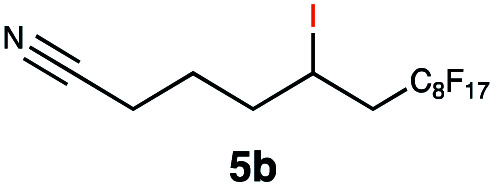
|
76 |
| OQ | 40 | 57 | |||
| 6 |
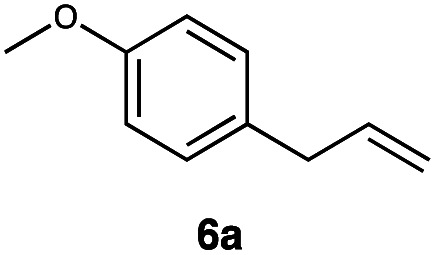
|
RQ | 40 |
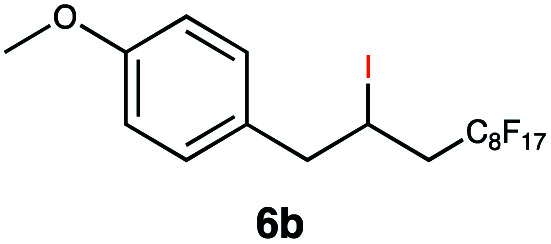
|
58 |
| OQ | 80 | 74 | |||
| 7 |
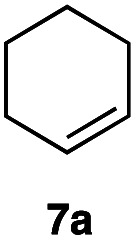
|
RQ | 15 |
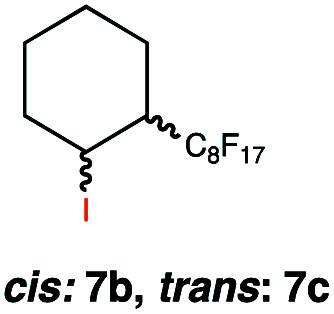
|
22 (cis : trans 17 : 5) |
| OQ | 40 | 32 (cis : trans 25 : 7) | |||
| 8 |
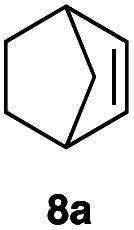
|
RQ | 15 |
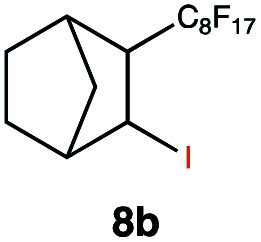
|
80 |
| OQ | 40 | 91 | |||
| 9 |
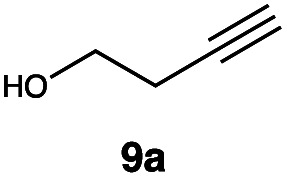
|
RQ | 20 |
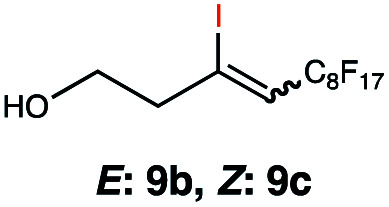
|
62 (E : Z 42 : 20) |
| OQ | 80 | 81 (E : Z 65 : 16) | |||
| 10 |
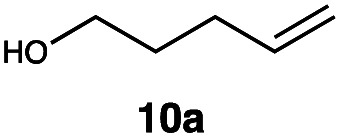
|
RQ | 10 |
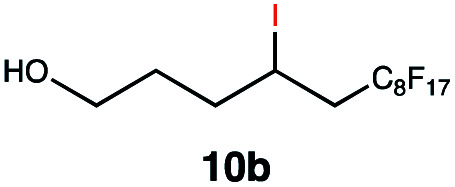
|
93 |
| OQ | 40 | 89 | |||
| 11 |
|
RQ | 30 |
|
43 |
| OQ | 40 | 87 | |||
| RQ | 30 |
|
26 | ||
| 12 |
|
RQ | 15 |
|
98 |
| OQ | 40 | 97 | |||
| 13 |
|
RQ | 30 |
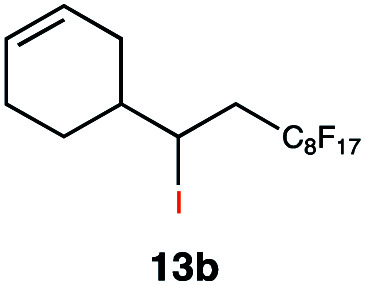
|
63 |
| 14d |
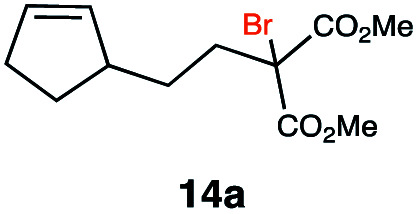
|
RQ | 45 |
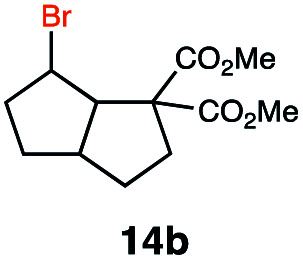
|
49 |
| OQ | 40 | 31 | |||
| 15 |
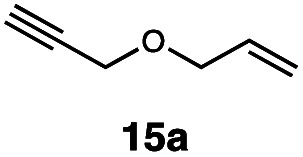
|
RQ | 150 |
|
51 (E : Z 25 : 26) |
| OQ | 150 | 49 (E : Z 25 : 24) |
Reaction conditions: RQ: 0.5 mol% (1), TEA (0.34 equiv.),  (1.33 equiv.), 4 : 3 MeCN : MeOH.
(1.33 equiv.), 4 : 3 MeCN : MeOH.
Reaction conditions: OQ: 1.5 mol% (1),  (1.33 equiv.), MeCN.
(1.33 equiv.), MeCN.
Isolated yield (%) after purification via silica gel column chromatography.
Intramolecular reaction. No external halide source was added.
This protocol was applied to a wide scope of substrates (Table 1), where excellent yields were afforded mainly for the transformation of terminal alkenes (entries 1–5 & 9–12). When using cyclohexene as a starting material (entry 7), comparatively low yields were observed. In contrast, for norbornene, another compound harbouring an internal double bond, excellent yields were obtained (entry 8). The increased reactivity for the latter can likely be attributed to its ring strain.
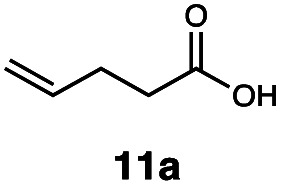
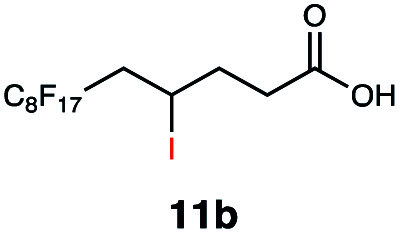
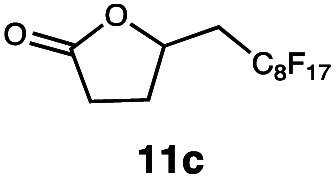
Carbonyl functionalities such as ketones and carboxylic acids were well-tolerated (entries 11 & 12), although the latter gave a mixture of the desired product 11b and the corresponding lactone 11c as a result of the basic conditions. Slightly diminished yields were noted for 5b (entry 5), which could be explained by quenching of the excited state of 1 by the low-lying LUMO of the nitrile-group present in substrate 5a, although no corresponding products were isolated. Furthermore, incompatibility of our system with Michael acceptors, such as methyl-vinyl ketone and methyl acrylate, was noted (Table S6†).
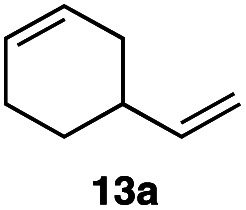
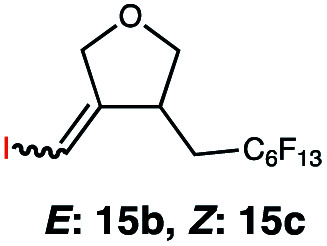
In order to investigate the extent to which addition to terminal alkenes might be favoured over internal double bonds, the reaction of C8F17I with 4-vinyl-1-cyclohexene was studied (entry 13). Although addition to both available double bonds was observed, 13b was obtained as the main product with a yield of 63%, resulting from preferential addition to the external double bond. The starting material 13a was not fully converted and other products were present in trace amounts according to 1H NMR spectroscopy, further highlighting the impact of steric hindrance. To probe the chemoselectivity of the reaction with regard to alkene- and alkyne-functionalities, perfluorohexyl iodide was added to 3-(allyloxy)-1-propyne 15a, affording 15b and 15c as the sole products (entry 15). Besides strongly supporting the presence of radicals, this further served to illustrate the preferential addition of the initially generated perfluoroalkyl radical to the alkene functionality over an alkyne moiety within the same molecule.
Control experiments were conducted using different iron compounds such as FeBr2 and [Fe(bpy)3](PF6)2 (Fig. S1 and S2†) as well as FeBr3, showing the necessity of 1 for the reactions to proceed efficiently, if at all. Furthermore, the absence of PC or light resulted in no conversion being observed.
With [Ru(bpy)3]Cl2 as PC, the reductive route resulted in full conversion within 4 min (compare 1: 8 min) under our standard conditions using green light (Table S1†).
Oxidative quenching route
An alternative reaction mode based on oxidative quenching of the excited PC 1 by perfluoroalkyl halides could omit the use of sacrificial reagents, thereby increasing the overall atom efficiency. Such a reaction route has previously been reported for the visible light-mediated ATRA reaction with the archetypal [Ru(bpy)3]2+ as PC.10 Since the 2LMCT state of 1* is a similarly strong reductant (Eo(IV/*III) = −1.0 V vs. Fc36) as the 3MLCT excited state of [Ru(bpy)3]2+, (E1/2(III/*II) = −1.2 V vs. Fc in acetonitrile36) we also pursued experiments with PC 1 in the absence of sacrificial electron donors.
Preliminary probing of the catalytic activity gave promising results and upon optimisation of the reaction conditions (Tables S7–S9†), a reliable procedure for the ATRA reaction via oxidative quenching (OQ) of 1 was established (Scheme 2), which afforded the desired ATRA product with full conversion within 40 min using 1.5 mol% of the PC. Acetonitrile was used as reaction solvent as opposed to the binary mixture with methanol (4 : 3) since the latter failed to give full conversion of the model substrate 3a.
This protocol was thereafter applied to the same substrate scope as in the RQ route, affording similar results in terms of yield and selectivity (Table 1). A decrease in yield was noted for the conversion of 5a compared to the RQ reaction (entry 5), whereas the conversion of 6a (entry 6) as well as the alkyne-substrate 9a resulted in a higher yield being obtained (entry 9). Unsurprisingly, for the ATRA reaction of the carboxylic acid 11a with C8F17I, no formation of the lactone 11c as a side-product was observed, which is explained by the lack of base under these reaction conditions. Additionally, no conversion of Michael acceptors (Table S12†) was observed in this route either.
Similar control experiments as for the RQ route were also performed for the OQ route, where the use of different iron compounds resulted in no conversion of the model substrate 3a (Table S11†), indicating that 1 is required to drive the reaction. Absence of any PC or light again resulted in none of product 3b being formed, further showcasing that irradiation of 1 is essential. Direct comparison with [Ru(bpy)3]Cl2 as PC, using the same reaction conditions and green light irradiation, revealed that the reaction stops at only 34% conversion in 40 min (Table S10†), whereas >99% conversion was obtained within the same duration using the Fe-PC 1 (Table S9†). In comparison to the archetypal noble metal photosensitiser, the Fe-PC gives comparable or superior results under green light irradiation for both routes, due to the fact that lower energy photons are required for its excitation.
While the OQ route has the general advantage of not requiring a sacrificial reductant, it is noteworthy that a much lower catalyst loading is needed for the reaction to go to completion in considerably shorter time in case of the reductive route.
Furthermore, the longevity of PC 1 is much better under RQ reaction conditions, as shown by experiments, wherein considerable catalytic activity was maintained over multiple cycles after addition of fresh starting materials (Fig. S10 and S11†).
Mechanistic investigations
Reductive quenching route and consecutive photo-induced electron transfer
Following the successful application of 1 as PC for the visible light-mediated ATRA reaction under conditions that would allow for reductive quenching, we sought to elucidate the reaction mechanism of this route. Based on the mechanistic investigations described below, we propose a catalytic cycle involving excitation of both the Fe(iii)- and Fe(ii)-ground states (Scheme 3).
Scheme 3. Proposed reaction mechanism for the visible light-mediated ATRA reaction via reductive quenching of the excited state of [Fe(btz)3](PF6)31.

The visible light-induced formation of radical species catalysed by 1 in the reductive quenching route was demonstrated through trapping experiments using TEMPO (= (2,2,6,6-tetramethylpiperidin-1-yl)oxyl) (Fig. S5†), which resulted in no product being formed. Additionally to the results obtained by conversion of substrate 15a (Table 1, entry 15), which also provided evidence for radical formation in the reaction, the radical ring-closing reaction of diethyl 2,2-diallylmalonate (Scheme 4a) and ring-opening of trans-(2-vinylcyclopropyl)methanol 17 (Scheme 4b) were successfully shown.
Scheme 4. Radical (a) ring-closing of diethyl 2,2-diallylmalonate and (b) ring-opening of trans-(2-vinylcyclopropyl)methanol via the reductive quenching route of the visible light-mediated ATRA reaction with 1.

Having established that the reaction must involve the generation of perfluoroalkyl radicals as a consequence of a visible light-induced process, the first step of the proposed mechanism, the excitation and subsequent reductive quenching of [Fe(iii)(btz)3]3+1 to produce [Fe(ii)(btz)3]2+2, was investigated.
UV-vis absorption spectroscopy (Fig. S6†) of 1 in the presence of the sacrificial reductant TEA and under visible light irradiation showed the expected formation of 2 (Scheme 3), presumably by reductive quenching of the 2LMCT state of the photoexcited *[Fe(iii)(btz)3]3+1*via single electron transfer. Emission quenching data corroborated that the short-lived ES of 1* (τ0 = 94 ps in acetonitrile : methanol (4 : 3) (Table 2 and Fig. S13†), 100 ps in acetonitrile36) is quenched with reasonable efficiency by 0.5 M TEA (ηq = 31%, Table S16†) corresponding to an essentially diffusion controlled bimolecular rate constant kq (Table 2, see Fig. S15† for quenching studies and Stern–Volmer plot). Transient absorption spectroscopy furthermore confirmed that the quenching occurs by reductive electron transfer producing stable 2 (Fig. 2) with a quantum yield ηqηce of about 9% (Table S16†). Notably, the lower yield of quenching implied by the short lifetime is balanced by a cage escape yield ηce > 20% that largely exceeds the very moderate values previously found for the quenching of the 2LMCT state of [Fe(iii)(phtmeimb)2]+ in acetonitrile.30,37
Excited state reactions in the cycle shown in Scheme 3 (KSV = Stern–Volmer quenching rate, kq = bimolecular rate constant, ηq = quenching yield, ηce = cage escape yield, ηqηce = quantum yield)a.
| PS (τ0/ps) | |||||
|---|---|---|---|---|---|
| [Fe(iii)(btz)3]3+, 1 (94) | [Fe(ii)(btz)3]2+, 2 (330) | ||||
| K SV/M−1 | k q/109 M−1 s−1 | η q ([Q]/M) | η ce | η q η ce ([Q]/M) | |
| TEA | 0.81b | 8.4 | 0.31 (0.50) | 0.29c | 0 (≤0.50)d |
| C8F17I | n.a.e | n.a. | 0 (≤0.45)f | n.a. | 0.18c (0.45)g,h |
| 5-Hexen-1-ol | n.a. | n.a. | 0 (≤0.14)f | n.a. | 0 (≤0.14)d |
In MeCN : MeOH (4 : 3).
Stern–Volmer constant from steady state emission quenching.
±20%.
No detectable products in ns-transient absorption.
Not applicable.
No emission quenching up to indicated quencher concentration.
From ns-transient absorption.
η ce ≥ 0.3 with ηq(0.45 M) ≤ 0.6 (based on kq ≤ 2 × 1010 M−1 s−1).
Fig. 2. Absorption changes upon laser flash excitation (490 nm) of [Fe(iii)(btz)3](PF6)31 in MeCN : MeOH (4 : 3) with TEA (0.29 M) ( ) and difference spectrum between [Fe(iii)(btz)3](PF6)31 and [Fe(ii)(btz)3](PF6)22 (2–1, ▬).
) and difference spectrum between [Fe(iii)(btz)3](PF6)31 and [Fe(ii)(btz)3](PF6)22 (2–1, ▬).

In case of conventional single-photon excitation-based photoredox catalysis, the reduction of C8F17I by 2 would constitute the next step in the mechanism, regenerating the GS of 1 and initiating the ATRA reaction. In our case, the reduced PC 2 is evidently not sufficiently reducing (−0.58 V vs. Fc)36 in its ground state as shown by the stability of isolated 2 towards C8F17I under exclusion of light. The conversion of 2 to 1 observed upon exposure of a solution of 2 and C8F17I to ambient light (Fig. S17†) suggested however a photo-induced oxidation of 2*. Despite its rather low ES state energy (1–1.2 eV estimated from the lowest energy absorption band assuming ET ≈ 2/3ES), the non-emissive 3MLCT excited state of 2 should still be a strong reductant (−1.6 to −1.8 V vs. Fc), owing to the low potential of the Fe(iii/ii)-couple (−0.58 V vs. Fc).36 It is thereby not only much more reducing than the ground state of 2 but also compared to the more energetic (2.2 eV) 2LMCT excited state of 1 (−1.0 V vs. Fc).36 The 3MLCT state of the Fe(ii)-oxidation state also features an even longer excited state lifetime (τ0 = 330 ps, (Table 2 and Fig. S14†) in acetonitrile : methanol (4 : 3)) compared to the 2LMCT state of its Fe(iii)-congener that could facilitate reasonably efficient quenching by the perfluoroalkyl halide.
To investigate this further, 2 was formed in situ in the presence of TEA and the model substrate 3a under green light irradiation without C8F17I present (Fig. S6† for absorption spectra). However, subsequent addition of the alkyl halide to the sample and storage in the dark (Fig. 4) resulted in no product being formed, thus supporting that the reaction cannot be driven by the GS of 2. Therefore, the sample was irradiated at 700 nm, where exclusively 2 but not 1 exhibits notable absorption (Fig. S12†) as to exclude formation of 1*. According to UV-vis absorption spectroscopy, complex 1 was restored in the process and no further reaction could occur due to the lack of absorption of 1 at this wavelength. By 1H NMR spectroscopic analysis, it was shown that 5% of the starting material had been converted during this “single turnover” with only 0.5 mol% of PC present, providing strong support for an underlying radical chain propagation pathway (see Fig. S90–S95† for NMR spectra). This result is further consistent with the fact that substoichiometric amounts of the sacrificial reductant are sufficient to drive the reaction to completion as complementary formation of ATRA products via quenching of 1* by C8F17I could be ruled out in the acetonitrile : methanol (4 : 3) solvent mixture (vide infra).
Fig. 4. Wavelength-switching experiment for the visible light-mediated ATRA reaction of C8F17I with 5-hexen-1-ol to form 3b using 1 as PC and TEA as sacrificial reductant (see ESI† for details regarding the set-up) as support for the involvement of conPET; absorption spectra normalised to 1. (A) A solution of 1 (0.357 mM), 5-hexen-1-ol (71.4 mM), TEA (23.6 mM) & mesitylene (15 μL, internal standard) in acetonitrile : methanol (4 : 3) was irradiated at 525 nm for 25 min; absorption spectrum of the reaction solution containing 1 (red line). (B) Absorption spectrum of the reaction solution containing in situ generated 2 (brown line); addition of C8F17I (95 mM). (C) Reaction solution after addition of the perfluoroalkyl halide and irradiation at 700 nm for 60 min; 5% NMR yield of product 3b. (D) Reaction solution after addition of perfluoroalkyl halide and storing in the dark for 1 h; no formation of 3b.

The proposed mechanism was further supported by emission quenching data, which gave no indication of a reaction between 1* and the perfluoroalkyl halide occurring in acetonitrile : methanol (4 : 3) (Table 2). Formation of perfluoroalkyl radicals by oxidative quenching could thus be specifically excluded under these conditions. The proposed reactivity of 2* towards the alkyl halide was directly evidenced by transient absorption spectroscopy (Fig. 3). The observed absorption changes after excitation with a ns-laser pulse unambiguously show the irreversible transformation of Fe(ii) to Fe(iii) via oxidative quenching by C8F17I that proceeds with a quantum yield ηqηce of about 18% (Table S16†). Under the conditions of the ATRA experiments, the strongly reducing α-amino alkyl radical (E° = −1.4 (ref. 49) to −2.1 V (ref. 50 and 51) vs. Fc) formed from the alkylamine donor in the reductive quenching step could also be expected to contribute to the reduction of the alkyl halide. However, any contribution from the donor radical would be a side reaction as only the oxidative quenching of 2* ensures the necessary regeneration of the Fe(iii)-state to close the catalytic cycle.
Fig. 3. Absorption changes 50 ns after laser flash excitation (650 nm) of [Fe(ii)(btz)3](PF6)22 in MeCN : MeOH (4 : 3) with C8F17I (0.45 M) ( ) and difference spectrum between [Fe(ii)(btz)3](PF6)22 and [Fe(iii)(btz)3](PF6)31 (1–2, ▬).
) and difference spectrum between [Fe(ii)(btz)3](PF6)22 and [Fe(iii)(btz)3](PF6)31 (1–2, ▬).

The radical obtained in the oxidative quenching step can attack the alkene or alkyne resulting in the formation of an alkyl- or alkenyl-radical respectively. Alternatively, the secondary alkyl radical could be oxidised to form a carbocation via reductive quenching of 1* followed by addition of the halide ion. However, the latter pathway is less likely to be operative with regard to the concentration required for significant quenching of the short-lived 1* in competition with the large concentration of TEA present. With the relevant concentrations, any competing quenching by the alkene component could also be excluded for either oxidation state of the PC and no excited state reaction of 2* with the amine could be observed, which further supports the suggested mechanism (Scheme 3). Importantly, the oxidative quenching of 2* does not only provide a pathway for the reduction of the alkyl halide but also ensures the necessary regeneration of the Fe(iii)-state, which is in agreement with our mechanistic proposal.
Further evidence for the radical chain propagation mechanism was provided by determination of the quantum yield Φ of the model reaction for the reductive cycle using a method developed by Pitre et al.56 (for calculations see Tables S14 and S15,† for absorption spectra see Fig. S8†). Here, a value of Φ = 6 was afforded after accounting for the fact that two photons are proposed to be involved in one catalytic turnover, which coincided well with the observations made in the wavelength-switching experiment.
Overall, the benefits of this mechanism based on the reductive route can be attributed to the formation of *[Fe(ii)(btz)3]2+2* as a sufficiently strong reductant for the reduction of C8F17I. While α-amino alkyl radicals might also contribute to this reaction, it is the oxidative quenching of 2* that enables catalytic turnover.
The ES electron transfer reactivity of both 1 and its Fe(ii) congener 2 had so far been unexplored and the kinetics of the quenching reactions as well as the cage escape yields of the electron transfer products needed to be investigated. Through our findings, we were able to show that a conPET mechanism, where the excited states of both the Fe(iii)- and the Fe(ii)-species are involved in product formation, was operative here. This is in line with the pronounced absorption of the reduced PC at the irradiation wavelength (530 nm, Fig. 4 and S12†), which should facilitate formation of 2* during the reaction. With 2* being formed by consecutive absorption of two photons, the reductive route constitutes the first example of a conPET being directly observed and utilised in a reaction catalysed by an Earth-abundant transition-metal complex.
Additionally, to our knowledge, a reaction driven by bimolecular oxidative quenching of the 3MLCT state of an Fe(ii)-PC has only a single, very recent precedence in the form of cross coupling reactions via aryl radicals that employ a novel Fe(ii)-PS with doubly cyclometalated phenylphenanthroline ligands.52
Oxidative quenching route
In addition to the RQ route, we also investigated the underlying mechanism of the visible light-mediated ATRA reaction in the absence of electron donors. Based on the findings of these studies (see below), we propose that the reaction proceeds via oxidative quenching of 1*, as shown in Scheme 5.
Scheme 5. Proposed reaction mechanism for the visible light-mediated ATRA reaction via oxidative quenching of the excited state of [Fe(btz)3](PF6)31.

As in case of the RQ route, addition of TEMPO to the reaction inhibited product formation (Table S13†). Along with the formation of cyclised product both for substrate 15a (Table 1, entry 15) and diethyl 2,2-diallylmalonate (Scheme 6a) as well as the radical ring-opening of trans-(2-vinylcyclopropyl)methanol (Scheme 6b), this provided clear evidence for the generation of perfluoroalkyl radicals, which occurred only under irradiation.
Scheme 6. Radical (a) ring-closing of diethyl 2,2-diallylmalonate and (b) ring-opening of trans-(2-vinylcyclopropyl)methanol via the oxidative quenching route of the visible light-mediated ATRA reaction with 1.

Therefore, in the absence of the amine as electron donor, the reaction(s) leading to the reduction of the perfluoroalkyl halide can be attributed to the oxidative quenching of 1*, as neither α-amino alkyl radicals nor the stronger excited state reductant 2* are available. This would then lead to formation of the oxidized PC 1′, here formally denoted as [Fe(iv)(btz)3]4+. However, as the one-electron oxidised state of 1 has not been unambiguously characterised, it could also correspond to the product of ligand oxidation, [Fe(iii)(btz)2(btz+)]4+.36 Tracing the progress of the reaction by UV-vis absorption spectroscopy did also not reveal any distinct intermediate species being formed (Fig. S7†).
While we had observed that in acetonitrile : methanol (4 : 3), aforementioned bimolecular quenching of 1* by C8F17I does not take place (Table 2, see Fig. S16† for quenching studies and Stern–Volmer plot), we were however able to observe weak emission quenching of the 2LMCT state by C8F17I in neat acetonitrile despite the lower solubility of the perfluoroalkyl halide in the latter solvent (Table 3). This coincided well with the fact that the ATRA reaction by OQ only proceeded efficiently when using neat acetonitrile as reaction solvent.
Excited state reactions in the cycle shown in Scheme 5 (KSV = Stern–Volmer quenching rate, kq = bimolecular rate constant, ηq = quenching yield, ηce = cage escape yield)a.
| PS (τ0/ps) | ||||
|---|---|---|---|---|
| [Fe(iii)(btz)3]3+, 1 (100) | ||||
| K SV/M−1 | k q/109 M−1 s−1 | η q ([Q]/M) | η ce | |
| C8F17I | 0.43b | 4.3 | 0.09 (0.12) | <0.15c |
| 5-Hexen-1-ol | n.a.d | n.a. | 0 (≤0.14)e | n.a. |
In MeCN.
From steady state emission quenching.
Estimated from absence of detectable ns-transient absorption.
Not applicable.
No emission quenching up to indicated quencher concentration.
No products of the quenching reaction could be detected by transient absorption spectroscopy. This result was however anticipated, in view of the low yield of quenching. Considering the absence of low-lying electronically excited states in alkyl halides and their poor electron donor ability, quenching of 1* by C8F17I due to energy transfer or reductive electron transfer appear very unlikely. Furthermore, control experiments (Table S10†) confirmed that no ATRA products were detected in the dark or in the absence of the PC. In combination with the radical trapping experiments described above, these results strongly indicate that the observed quenching of the 2LMCT state of 1* by C8F17I can be attributed to oxidative electron transfer.
For the reduction of C8F17I a potential of −1.70 V vs. Fc (−1.32 V vs. SCE) has been cited previously.10 Peak potentials from irreversible electrochemistry of alkyl halides are however no suitable proxy of their thermodynamic reduction potential.53 This is evident from literature reports10 that describe perfluorous tagging via oxidative quenching of [Ru(bpy)3]Cl2 (Eo(III/II*) = −1.19 V vs. Fc in acetonitrile54) by C8F17I using DMSO as reaction solvent, as well as from our previously mentioned control experiments for the oxidative quenching using [Ru(bpy)3]2+.
We therefore suggest that the less efficient reactions in the absence of TEA are initiated by the rather inefficient quenching of 1* (Eo(IV/*III) = −1.0 V vs. Fc) with C8F17I to give the oxidized PC 1′ and a perfluoroalkyl radical, which could attack the alkene or alkyne. This is also in line with our observation that the PC exhibits substantially reduced longevity in the OQ route, as 1′ is known to not be very stable based on the irreversible oxidative voltammetry of 1.36 The proposed mechanism would thus constitute the first case of photoredox catalysis based on bimolecular oxidative quenching of the 2LMCT state of an Fe–NHC-complex.
Following the oxidative quenching, two mechanistic pathways to the desired ATRA product are possible, one of which is based on radical chain propagation while the other includes generation and subsequent trapping of an alkyl/alkenyl-radical.10 Determination of the quantum yield (Φ = 4.5, for calculations see Tables S14 and S15,† for absorption spectra see Fig. S9†) showed that, as in the RQ route, radical chain propagation must occur in the OQ route, once the initial perfluoroalkyl radical has been generated. Otherwise, said radical might reduce 1′ while restoring 1, which in the absence of an added electron donor is the most likely process to close the catalytic cycle. Partially, the product of the ATRA reaction would consequently be formed through the trapping of the resulting carbocation by the halide ion.
Conclusions
We have demonstrated that the Fe–NHC complex [Fe(iii)(btz)3]3+ can drive photoredox reactions by consecutive excitation of both the Fe(iii)- and Fe(ii)-ground state using the same irradiation wavelength. The two generated excited states (2LMCT and 3MLCT respectively) are shown to be sufficiently long-lived to engage in efficient bimolecular quenching. The utility of such an unprecedented reaction mode for iron complexes was illustrated in the well-explored ATRA reaction. Here, the formation of C–C and C-halide bonds for a range of over 10 common substrate types, exhibiting different functional groups and structures, was catalysed, resulting in synthetically useful, difunctionalised products.
Despite the lower photon energy required for excitation of the Fe-PC, efficiencies for this benchmark photoredox reaction are comparable to those obtained by commonly used noble metal catalysts thereby showcasing the usefulness of iron photosensitisers as a viable greener alternative to more common noble-metal PCs.
In the absence of an electron donor, the process has to be initiated by oxidative quenching. Efficiency and durability of the PC are somewhat limited by the relatively moderate reducing power of the 2LMCT state of the Fe(iii)-PC and the limited stability of the resulting Fe(iv) state. A more advantageous situation arises in the presence of an electron donor that leads to reductive quenching of the PC and its subsequent photoexcitation. The resulting 3MLCT state of the Fe(ii)-PC exhibits both the necessary lifetime and reducing power to ensure efficient reduction of the alkyl halide, C8F17I. Among PRC reactions catalysed by complexes of Earth-abundant transition metals, a mechanism involving two conPET events from charge transfer states within the same catalytic cycle has no precedence. Our example thus demonstrates for the first time that also with Fe–NHC-type PCs, two-photon mechanisms can be a viable approach to thermodynamically demanding PRC reactions.
Generally, our findings establish that even Fe–NHC complexes with sub-ns lifetimes of their charge transfer excited states can serve as efficient PCs in PRC applications, particularly where the excited state reaction products escape geminate recombination with sizeable yield in the reaction solvent. Therefore, already the currently available Fe–NHC complexes can be considered attractive alternatives to the established noble metal PCs for a variety of visible light induced organic reactions. Prospective developments should also enable even more demanding applications in e.g., solar to chemical energy conversion.
Experimental section
General procedure for the visible light-mediated photocatalytic ATRA reaction
Photochemical reactions were conducted in a TAK120 AC photoreactor purchased from HK Testsysteme GmbH. A 6 mL clear glass crimp top vial equipped with a magnetic stir bar was charged with the photocatalyst (PC) (0.5–1.5 mol%, 1.25–3.8 μmol), alkene (0.25 mmol, 1.0 equiv.), alkyl halide (0.33 mmol, 1.33 equiv.) and solvent (3.5 mL). For the reductive quenching (RQ) route a reductant (0.085–0.25 mmol, 0.34–1 equiv.) was furthermore added to the reaction mixture.
After sealing the vial using an aluminium cap with a septum, the reaction solution was flushed with argon for 5 min unless otherwise stated. The sample was then irradiated in the photoreactor at 530 nm (3.03 W per slot).
Condition & scope screenings were performed in deuterated solvents and NMR yields were determined via1H NMR-spectroscopy by integration against the internal standard mesitylene (30 μL per sample).
Reactions, wherein the products were isolated, were conducted in non-deuterated solvents & without addition of an internal standard. Isolation was performed by removal of the solvent in vacuo and subsequent purification by silica gel column chromatography affording elemental analysis pure products.
Visible light-mediated ATRA reaction of alkyl halides to alkenes & alkynes via reductive quenching of [Fe(btz)3](PF6)31 – procedure A
A 6 mL clear glass crimp top vial equipped with a magnetic stir bar was charged with [Fe(btz)3](PF6)31 (0.00125 mmol, 0.005 equiv.), alkene (0.25 mmol, 1.0 equiv.), alkyl halide (0.33 mmol, 1.3 equiv.), MeOH : MeCN (3 : 4, 3.5 mL) as well as TEA (0.085 mmol, 0.34 equiv.). After sealing the vial using an aluminium cap with a septum, the reaction solution was flushed with Argon (g) for 5 min. The sample was then irradiated at 530 nm (3.03 W per slot) in the photoreactor under vigorous stirring until completion or until the reaction ceased to progress further. The solvent was then removed under reduced pressure and the product was isolated by silica gel column chromatography using the indicated eluent system.
For more facile product characterisation multiple batches were prepared in parallel and isolated together in some instances, the individual number of which is indicated for the respective product (see ESI†).
Visible light-mediated ATRA reaction of alkyl halides to alkenes & alkynes via reductive quenching of [Fe(btz)3](PF6)31 – procedure B
A 6 mL clear glass crimp top vial equipped with a magnetic stir bar was charged with [Fe(btz)3](PF6)31 (3.8 μmol, 0.015 equiv.), alkene (0.25 mmol, 1.0 equiv.), alkyl halide (0.33 mmol, 1.3 equiv.) and MeCN (3.5 mL). After sealing the vial using an aluminium cap with a septum, the reaction solution was flushed with argon (g) for 5 min. The sample was then irradiated at 530 nm (3.03 W per slot) in the photoreactor under vigorous stirring for the indicated duration (40 or 80 min). The solvent was then removed under reduced pressure and the product was isolated by silica gel column chromatography using the indicated eluent system.
UV-vis absorption and emission spectroscopy
UV-vis absorption spectra were recorded on a Probe Drum Lab-in-a-box spectrometer. Steady-state emission measurements were performed on a Fluorolog-3 (Horiba) fluorimeter. Emission spectra were background subtracted and corrected for the wavelength dependent instrument response.
All samples were prepared in a solvent mixture of acetonitrile (spectroscopic grade Uvasol®, ≥99.9%, Merck) and methanol (ACS spectroscopic grade, ≥99.9%, Merck) (volume ratio 4 : 3) or in neat acetonitrile with absorptions of around 0.0755 ± 0.005 at the excitation wavelength (502 or 525 nm).
Transient absorption spectroscopy
Nanosecond transient absorption measurements were obtained with LP920-S (Edinburgh Instruments) or LKS.60 (Applied Photophysics) laser flash photolysis spectrometers. Samples were excited at 465 or 650 nm (for Fe(ii)) and 490 nm (for Fe(iii)) with 8–10 ns pulses (about 18 mJ per pulse) provided by a frequency tripled Q-switched Nd:YAG laser (EKSPLA NT342B or Quantel, BrilliantB) combined with an optical parametric oscillator (OPO). All measurements were performed at right angle in a 10 × 10 mm quartz cuvette with samples deaerated by purging with Ar and an absorption of around 0.5 at the excitation wavelength. Solutions of [Ru(bpy)3]2+ in MeCN : MeOH (4 : 3) were used as actinometer.
Femtosecond transient absorption measurements were performed on a Newport TAS system with a Coherent Libra Ti:sapphire amplifier (1.5 mJ, 3 kHz, 800 nm, fwhm 40 fs). Excitation wavelengths of 550 nm (for Fe(iii)) and 450 nm (for Fe(ii)) were generated by optical parametric amplifiers (TOPAS-Prime and NIRUVVIS, Light Conversion) with the pump power adjusted to about 3 mW on the 1 mm cuvette. The white light supercontinuum probe light was generated in a CaF2 crystal (Crystran) and was detected with a silicon diode array (Newport). Correction for spectral chirp and data fitting was performed with Surface Xplorer v4.
Author contributions
A. I. performed large parts of the experimental work, including initial testing, optimisations and substrate scope for the reductive quenching route, planned and carried out mechanistic investigations such as radical trapping and wavelength-switching experiments as well as chemical actinometry, conceived the proposed mechanisms, wrote major parts of the entire manuscript and coordinated the writing.
J. S. performed large parts of the experimental work, including initial testing, optimisations and substrate scope for the oxidative quenching route, planned and carried out mechanistic investigations such as radical trapping and radical clock experiments, conceived the proposed mechanisms, contributed to writing of the first draft and to reviewing of the manuscript.
C. J. performed all measurements based on transient absorption and emission spectroscopy, determined excited state lifetimes, rate constants and yields of excited state reactions, and contributed to the mechanistic interpretation and to the writing of the manuscript.
L. H. M. d. G. performed testing and optimisations in the initial stage of the project and contributed to the writing the manuscript.
S. K. contributed to experiment design and to reviewing of the manuscript.
R. L. devised the spectroscopic reactivity studies, guided the mechanistic interpretation and made major contributions to the writing of the manuscript.
K. W. conceived and planned the research and contributed to the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank Carl-Johan Wallentin for initial discussions and Jens Uhlig for providing us with a custom-built sample holder for irradiation with LED bars. We would like to thank the Swedish Foundation for Strategic Research (SSF, EM16-0067) as well as the Knut and Alice Wallenberg (KAW, 2018.0074) Foundation for financial support. KW acknowledges support from the Swedish Research Council (VR, 2020-03207), the Swedish Energy Agency (Energimyndigheten), the LMK Foundation and the Sten K Johnson Foundation. SK acknowledges support from Wenner-Gren Stiftelserna. RL acknowledges financial support by the Swedish Research Council (VR, 2020-05058).
Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02122f
References
- Marzo L. Pagire S. K. Reiser O. König B. Angew. Chem., Int. Ed. Engl. 2018;57:10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Narayanam J. M. Stephenson C. R. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Yoon T. P. Ischay M. A. Du J. Nat. Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- Prier C. K. Rankic D. A. MacMillan D. W. C. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicewicz D. A. Nguyen T. M. ACS Catal. 2013;4:355–360. doi: 10.1021/cs400956a. [DOI] [Google Scholar]
- Twilton J. Le C. Zhang P. Shaw M. H. Evans R. W. MacMillan D. W. C. Nat. Rev. Chem. 2017;1:0052. doi: 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Hedstrand D. M. Kruizinga W. H. Kellogg R. M. Tetrahedron Lett. 1978;19:1255–1258. doi: 10.1016/S0040-4039(01)94515-0. [DOI] [Google Scholar]
- Lytle F. E. Hercules D. M. J. Am. Chem. Soc. 2002;91:253–257. doi: 10.1021/ja01030a006. [DOI] [Google Scholar]
- Ischay M. A. Anzovino M. E. Du J. Yoon T. P. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- Wallentin C.-J. Nguyen J. D. Finkbeiner P. Stephenson C. R. J. J. Am. Chem. Soc. 2012;134:8875–8884. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]
- Ischay M. A. Lu Z. Yoon T. P. J. Am. Chem. Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeian K. Djurovich P. I. Garces F. O. Carlson G. Watts R. J. Inorg. Chem. 1991;30:1685–1687. doi: 10.1021/ic00008a003. [DOI] [Google Scholar]
- Nguyen J. D. Tucker J. W. Konieczynska M. D. Stephenson C. R. J. J. Am. Chem. Soc. 2011;133:4160–4163. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras J. A. Mills I. N. Transue W. J. Bernhard S. J. Am. Chem. Soc. 2016;138:9460–9472. doi: 10.1021/jacs.6b03246. [DOI] [PubMed] [Google Scholar]
- Mitani M. Kato I. Koyama K. J. Am. Chem. Soc. 1983;105:6719–6721. doi: 10.1021/ja00360a033. [DOI] [Google Scholar]
- Pirtsch M. Paria S. Matsuno T. Isobe H. Reiser O. Chem.–Eur. J. 2012;18:7336–7340. doi: 10.1002/chem.201200967. [DOI] [PubMed] [Google Scholar]
- Bagal D. B. Kachkovskyi G. Knorn M. Rawner T. Bhanage B. M. Reiser O. Angew. Chem., Int. Ed. Engl. 2015;54:6999–7002. doi: 10.1002/anie.201501880. [DOI] [PubMed] [Google Scholar]
- Pintauer T. Chem. Pap. 2016;70:22–42. [Google Scholar]
- Rawner T. Lutsker E. Kaiser C. A. Reiser O. Angew. Chem., Int. Ed. Engl. 2018;8:3950–3956. [Google Scholar]
- Hossain A. Engl S. Lutsker E. Reiser O. ACS Catal. 2019;9:1103–1109. doi: 10.1021/acscatal.8b04188. [DOI] [Google Scholar]
- Bozic-Weber B. Constable E. C. Housecroft C. E. Coord. Chem. Rev. 2013;257:3089–3106. doi: 10.1016/j.ccr.2013.05.019. [DOI] [Google Scholar]
- Day J. I. Teegardin K. Weaver J. Chan J. Org. Process Res. Dev. 2016;20:1156–1163. doi: 10.1021/acs.oprd.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. B. Wenger O. S. Chem.–Eur. J. 2018;24:2039–2058. doi: 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- Liu Y. Persson P. Sundström V. Wärnmark K. Acc. Chem. Res. 2016;49:1477–1485. doi: 10.1021/acs.accounts.6b00186. [DOI] [PubMed] [Google Scholar]
- McCusker J. K. Science. 2019;363:484–488. doi: 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Li Z. Wang X. Xia S. Jin J. Org. Lett. 2019;21:4259–4265. doi: 10.1021/acs.orglett.9b01439. [DOI] [PubMed] [Google Scholar]
- Feng G. Wang X. Jin J. Eur. J. Org. Chem. 2019;2019:6728–6732. doi: 10.1002/ejoc.201901381. [DOI] [Google Scholar]
- Xia S. Hu K. Lei C. Jin J. Org. Lett. 2020;22:1385–1389. doi: 10.1021/acs.orglett.0c00002. [DOI] [PubMed] [Google Scholar]
- Kjaer K. S. Kaul N. Prakash O. Chábera P. Rosemann N. W. Honarfar A. Gordivska O. Fredin L. A. Bergquist K. E. Häggström L. Ericsson T. Lindh L. Yartsev A. Styring S. Huang P. Uhlig J. Bendix J. Strand D. Sundström V. Persson P. Lomoth R. Wärnmark K. Science. 2019;363:249–253. doi: 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Rosemann N. W. Chábera P. Prakash O. Kaufhold S. Wärnmark K. Yartsev A. Persson P. J. Am. Chem. Soc. 2020;142:8565–8569. doi: 10.1021/jacs.0c00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M. D. McCusker J. K. J. Am. Chem. Soc. 2020;142:16229–16233. doi: 10.1021/jacs.0c08389. [DOI] [PubMed] [Google Scholar]
- Aydogan A. Bangle R. E. Cadranel A. Turlington M. D. Conroy D. T. Cauet E. Singleton M. L. Meyer G. J. Sampaio R. N. Elias B. Troian-Gautier L. J. Am. Chem. Soc. 2021;143:15661–15673. doi: 10.1021/jacs.1c06081. [DOI] [PubMed] [Google Scholar]
- Aydogan A. Bangle R. E. De Kreijger S. Dickenson J. C. Singleton M. L. Cauet E. Cadranel A. Meyer G. J. Elias B. Sampaio R. N. Troian-Gautier L. Catal.: Sci. Technol. 2021;11:8037–8051. doi: 10.1039/D1CY01771C. [DOI] [Google Scholar]
- Liu Y. Harlang T. Canton S. E. Chábera P. Suarez-Alcantara K. Fleckhaus A. Vithanage D. A. Göransson E. Corani A. Lomoth R. Sundström V. Wärnmark K. Chem. Commun. 2013;49:6412–6414. doi: 10.1039/C3CC43833C. [DOI] [PubMed] [Google Scholar]
- Harlang T. C. Liu Y. Gordivska O. Fredin L. A. Ponseca Jr C. S. Huang P. Chábera P. Kjaer K. S. Mateos H. Uhlig J. Lomoth R. Wallenberg R. Styring S. Persson P. Sundström V. Wärnmark K. Nat. Chem. 2015;7:883–889. doi: 10.1038/nchem.2365. [DOI] [PubMed] [Google Scholar]
- Chábera P. Liu Y. Prakash O. Thyrhaug E. Nahhas A. E. Honarfar A. Essen S. Fredin L. A. Harlang T. C. Kjaer K. S. Handrup K. Ericson F. Tatsuno H. Morgan K. Schnadt J. Häggström L. Ericsson T. Sobkowiak A. Lidin S. Huang P. Styring S. Uhlig J. Bendix J. Lomoth R. Sundström V. Persson P. Wärnmark K. Nature. 2017;543:695–699. doi: 10.1038/nature21430. [DOI] [PubMed] [Google Scholar]
- Schwarz J. Ilic A. Johnson C. Lomoth R. Wärnmark K. Chem. Commun. 2022;58:5351–5354. doi: 10.1039/D2CC01016J. [DOI] [PubMed] [Google Scholar]
- Ghosh I. Ghosh T. Bardagi J. I. König B. Science. 2014;346:725–728. doi: 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]
- Glaser F. Kerzig C. Wenger O. S. Angew. Chem., Int. Ed. Engl. 2020;59:10266–10284. doi: 10.1002/anie.201915762. [DOI] [PubMed] [Google Scholar]
- MacKenzie I. A. Wang L. Onuska N. P. R. Williams O. F. Begam K. Moran A. M. Dunietz B. D. Nicewicz D. A. Nature. 2020;580:76–80. doi: 10.1038/s41586-020-2131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targos K. Williams O. P. Wickens Z. K. J. Am. Chem. Soc. 2021;143:4125–4132. doi: 10.1021/jacs.1c00399. [DOI] [PubMed] [Google Scholar]
- Wang Y. Suzuki H. Xie J. Tomita O. Martin D. J. Higashi M. Kong D. Abe R. Tang J. Chem. Rev. 2018;118:5201–5241. doi: 10.1021/acs.chemrev.7b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y. Yang Q. Schelter E. J. Angew. Chem., Int. Ed. Engl. 2018;57:10999–11003. doi: 10.1002/anie.201804022. [DOI] [PubMed] [Google Scholar]
- Giedyk M. Narobe R. Weiß S. Touraud D. Kunz W. König B. Nat. Catal. 2019;3:40–47. doi: 10.1038/s41929-019-0369-5. [DOI] [Google Scholar]
- Haimerl J. Ghosh I. König B. Vogelsang J. Lupton J. M. Chem. Sci. 2019;10:681–687. doi: 10.1039/C8SC03860K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graml A. Nevesely T. Jan Kutta R. Cibulka R. König B. Nat. Commun. 2020;11:3174. doi: 10.1038/s41467-020-16909-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl F. Bergwinkl S. Allacher C. Dick B. Chem.–Eur. J. 2020;26:7946–7954. doi: 10.1002/chem.201905167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T. Cheung C. W. Hu X. Angew. Chem., Int. Ed. Engl. 2014;53:4910–4914. doi: 10.1002/anie.201402511. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya K. Das P. K. J. Phys. Chem. 2002;90:3987–3993. doi: 10.1021/j100408a032. [DOI] [Google Scholar]
- Wayner D. D. M. McPhee D. J. Griller D. J. Am. Chem. Soc. 2002;110:132–137. doi: 10.1021/ja00209a021. [DOI] [Google Scholar]
- Lai R. Y. Bard A. J. J. Phys. Chem. A. 2003;107:3335–3340. doi: 10.1021/jp026743j. [DOI] [Google Scholar]
- Leis W. Arguello Cordero M. A. Lochbrunner S. Schubert H. Berkefeld A. J. Am. Chem. Soc. 2022;144:1169–1173. doi: 10.1021/jacs.1c13083. [DOI] [PubMed] [Google Scholar]
- Andrieux C. P. Gelis L. Medebielle M. Pinson J. Saveant J. M. J. Am. Chem. Soc. 2002;112:3509–3520. doi: 10.1021/ja00165a040. [DOI] [Google Scholar]
- Bock C. R. Connor J. A. Gutierrez A. R. Meyer T. J. Whitten D. G. Sullivan B. P. Nagle J. K. J. Am. Chem. Soc. 1979;101:4815–4824. doi: 10.1021/ja00511a007. [DOI] [Google Scholar]
- Chábera P. Kjaer K. S. Prakash O. Honarfar A. Liu Y. Fredin L. A. Harlang T. C. B. Lidin S. Uhlig J. Sundström V. Lomoth R. Persson P. Wärnmark K. J. Phys. Chem. Lett. 2018;9(3):459–463. doi: 10.1021/acs.jpclett.7b02962. [DOI] [PubMed] [Google Scholar]
- Pitre S. McTiernan C. Vine W. DiPucchio R. Grenier M. Scaiano J. C. Sci. Rep. 2015;5:16397. doi: 10.1038/srep16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.