

Abstract
The selective inhibition of NaV1.7 is a promising strategy for developing novel analgesic agents with fewer adverse effects. Although the potent selective inhibition of NaV1.7 has been recently achieved, multiple NaV1.7 inhibitors failed in clinical development. In this review, the relationship between preclinical in vivo efficacy and NaV1.7 coverage among three types of voltage-gated sodium channel (VGSC) inhibitors, namely conventional VGSC inhibitors, sulphonamides and acyl sulphonamides, is discussed. By demonstrating the PK/PD discrepancy of preclinical studies versus in vivo models and clinical results, the potential reasons behind the disconnect between preclinical results and clinical outcomes are discussed together with strategies for developing ideal analgesic agents.
The relationship between preclinical in vivo efficacy and NaV1.7 coverage among three types of VGSC inhibitors is discussed.
1. Introduction
Pain sensation is a critical signal for preventing dangerous signs, and consecutive instances of pain are often problematic. Chronic pain is one of the most typical symptoms reported by patients, and it sometimes has devastating consequences, resulting in a huge economic burden on the health care system.1 To alleviate such devastating conditions, various analgesic agents are utilised in clinical settings.2–6 Typical analgesic agents are listed in Table 1. Opioids are highly efficacious agents for the treatment of pain disorders, but their abuse potential is a critical concern.4 Although non-steroidal anti-inflammatory drugs (NSAIDs) are useful for treating inflammatory pain, their maximum efficacy and duration of efficacy are limited. It was reported that the prolonged use of NSAIDs was associated with gastrointestinal (GI) and cardiovascular (CV) adverse effects.5 Gabapentinoids are specific ligands for the α2δ subunit of voltage-gated Ca2+ channels, and they are widely used in the first-line treatment of neuropathic pain (NP). However, central nervous system (CNS) adverse effects such as dizziness, somnolence or abuse potential, together with narrow safety margins, should be considered in the use of gabapentinoids.6 Although voltage-gated sodium channel (VGSC, NaV) inhibitors are also efficacious for various pain disorders, CNS and CV adverse effects often limit their usage.7 Thus, the unmet medical needs in the treatment of pain disorders remain considerably high, and many analgesic drug candidates have been extensively developed.2,3
The targets for analgesics.
| Target | Target location | Example | Challenges |
|---|---|---|---|
| Opioid | CNS | Morphine | Abuse potential |
| Fentanyl | |||
| Inhibition of prostaglandin synthesis (NSAID) | Peripheral tissue | Aspirin | Limited efficacy |
| Diclofenac | GI and CV adverse effects | ||
| Loxoprofen | |||
| Gabapentinoid (α2δ ligand) | CNS | Gabapentin | CNS adverse effects |
| Pregabalin | |||
| Mirogabalin | |||
| VGSC inhibitor | PNS and CNS | Lidocaine | CNS and CV adverse effects |
| Mexiletine |
VGSCs regulate neuronal signals in the CNS and peripheral nervous system (PNS). Since the discovery of such fundamental roles of VGSCs, their modulation has been studied extensively for the treatment of various disorders, including epilepsy and pain. In 2006, the discovery of loss-of-function mutations of the NaV1.7 gene (SCN9A) in humans accelerated such efforts. A hereditary loss-of-function mutation of SCN9A leads to a rare genetic condition called congenital insensitivity to pain (CIP), which is characterised by the inability to perceive physical pain.8 Although patients with CIP exhibit a normal phenotype similar to that of healthy individuals, they experience anosmia, which is a lack of olfactory function.9 Conversely, the excessive expression of SCN9A leads to hereditary pain disorders, such as paroxysmal extreme pain disorder (PEPD) and inherited erythromelalgia (IEM).10 Gain-of-function variants of SCN9A are also associated with itch conditions in humans.11 The fundamental role in pain signalling by NaV1.7 has been reported in rodents. Although global deletion of SCN9A is lethal in mice, genetic and animal husbandry approaches enabled the construction of global12 and conditional13 NaV1.7 knockout mice, the phenotype of which was analogous to the pain-free phenotype observed in patients with CIP: anatomically normal with complete insensitivity to painful mechanical, thermal and chemical stimuli. In conditional NaV1.7 knockout mice, the deletion of SCN9A in both sensory and sympathetic neurons was required for generating the same phenotype observed in humans even though NaV1.7 is mainly expressed in the PNS.13 Although both global and conditional knockout mice display anosmia,9,12 conditional knockout rats retain olfactory function with a pain-free phenotype.14 The epigenome engineering approach that utilised CRISPER-dCas and zinc finger proteins to ablate the expression of NaV1.7 in mice led to long-lasting analgesic efficacy. In some cases, the analgesic effect lasted for up to 44 weeks.15 This genetic evidence clearly and strongly demonstrates that NaV1.7 inhibition is a promising therapeutic approach for developing analgesic agents with fewer adverse effects.
The VGSC family consists of nine subtypes (NaV1.1–NaV1.9, Table 2) with a variety of reported roles.16 The VGSC family is classified by sensitivity to the neurotoxin tetrodotoxin (TTX); specifically, the functions of NaV1.1–NaV1.4, NaV1.6 and NaV1.7 are inhibited by TTX, whereas NaV1.5, NaV1.8 and NaV1.9 are TTX-resistant. NaV1.1 and NaV1.2 are mainly expressed in the CNS, and mutation studies revealed that both neuronal sodium channels are associated with epilepsy.17 Although NaV1.3 is mainly expressed in the embryonic stage, the up-regulation of SCN3A was reported in inflammatory or NP conditions.18 NaV1.5 is highly expressed in cardiac muscle, and SCN5A mutations cause primarily inherited cardiomyopathies, including congenital long QT syndrome type 3 and Brugada syndrome.19 NaV1.7 is preferentially expressed in dorsal root ganglion (DRG) and sympathetic neurons in the PNS, and its expression in olfactory sensory neurons was reported.20 Given the distribution of NaV1.7, its selective inhibition is a promising strategy for novel analgesics with superior safety profiles and fewer CNS and CV adverse effects. NaV1.8 is highly expressed in the DRG, and the ablation of SCN10A in rodents led to deficits in nociception. As gain-of-function mutations of SCN10A result in painful peripheral neuropathy in humans,21 NaV1.8 is a therapeutic target for analgesics. In fact, selective NaV1.8 inhibitors displayed efficacy in rodent models of NP.22,23 However, NaV1.8 expression in cardiac muscle is considered to be a potential risk associated with CV side effects and a drawback compared to NaV1.7.24
VGSC family.
| Subtype | Gene | TTX sensitivity | Major expression sites |
|---|---|---|---|
| NaV1.1 | SCN1A | Sensitive | PNS, CNS |
| NaV1.2 | SCN2A | Sensitive | CNS |
| NaV1.3 | SCN3A | Sensitive | PNS, CNS (embryonic) |
| NaV1.4 | SCN4A | Sensitive | Skeletal muscle |
| NaV1.5 | SCN5A | Resistant | Cardiac muscle |
| NaV1.6 | SCN8A | Sensitive | PNS, CNS |
| NaV1.7 | SCN9A | Sensitive | PNS |
| NaV1.8 | SCN10A | Resistant | PNS, cardiac muscle |
| NaV1.9 | SCN11A | Resistant | PNS |
Two fibres, specifically Aδ- and C-fibres, play predominant roles in the transaction of pain signals in the PNS (Table 3). When peripheral sensory neurons, termed nociceptors, in the DRG receive a nociceptive pain sensation, they transmit the sensation as electrical impulses to the CNS via the spinal cord, where it synapses onto neurons in the dorsal horn. In the DRG, two small-to-medium–diameter nociceptors, termed myelinated Aδ-fibres and unmyelinated C-fibres, transduce nociceptive pain sensation with a high threshold, whereas Aα- and Aβ-fibres, which are large-diameter myelinated fibres, detect innocuous stimuli without contributing to pain signalling.25 Aδ-fibres rapidly transmit signals from the PNS to the CNS via the spinal cord, known as “first pain” in response to a stimulus, whereas C-fibres transduce “second pain” that is more diffuse and dull and that is perceived with a temporal delay relative to the inciting stimulus.
Fibres in the PNS.
| Fibre | Diameter | Myelinated | Nociceptor |
|---|---|---|---|
| Aα- and Aβ-fibres | Large | Myelinated | Proprioception |
| Aδ-fibre | Medium | Lightly myelinated | Nociception |
| C-fibre | Small | Unmyelinated | Nociception |
Although genetic evidence strongly suggests that selective inhibition of NaV1.7 is a promising analgesic approach, the structural similarity of VGSC family members has hampered this strategy. A pore-forming α subunit and a stabilising β subunit comprise the backbone of VGSCs, and they exhibit high amino acid sequence homology in the extracellular and transmembrane domains.26 Conventional NaV1.7 inhibitors are less subtype-selective with inhibitory potency in the micromolar range. In 2010, Pfizer disclosed a highly potent selective NaV1.7 inhibitor in their patent.27 This fuelled the development of a new generation of highly potent selective NaV1.7 inhibitors for the potential treatment of pain disorders, and inevitably, many pharmaceuticals and biotech firms initiated research and development on aryl sulphonamide derivatives followed by the disclosure of potent selective NaV1.7 inhibitors in patents or papers. Although many highly potent selective NaV1.7 inhibitors have been disclosed and some of them have been examined for their analgesic potency in clinical trials, none has reached the market.
In this article, we review conventional NaV1.7 inhibitors, some of which have been successfully launched into the market. Then, the highly potent selective sulphonamide and acyl sulphonamide derivatives are reviewed. By demonstrating the PK/PD discrepancy of preclinical studies relative to in vivo models and clinical results, we discuss potential reasons behind the disconnect between preclinical results and clinical outcomes and strategies for developing ideal analgesic agents.
2. Structure of VGSC
After the first crystal structure of VGSC from Arcobacter butzleri was reported in 2011,28 the crystal structure of NaV1.7 bound to sulphonamide derivative29 and the cryo-electron microscopy structures of human NaV1.7 with its auxiliary β subunit bound to TTX or saxitoxin (STX)30 were disclosed. These studies contributed to clarification of the whole structure of NaV1.7 with the detailed binding mode of such VGSC inhibitors.31
VGSC consists of a pore-forming α-subunit and a β-subunit. The α-subunit plays a significant role in channel function, whereas the β-subunit is a multifunctional signalling molecule that also regulates sodium ion conductance. Although the majority of channelopathies including CIP, PEPD and IEM are caused by mutations in the α-subunit, it was also reported that mutations in genes encoding the β-subunit lead to various channelopathies.32
The α-subunit consists of four domains (DI–DIV), each of which features six α-helical transmembrane segments designated S1–S6 (Fig. 1A and B). S1–S4 helical segments form a voltage-sensing domain (VSD), which is responsible for sensing the charge in the cell membrane. In particular, positively charged S4 helices contribute to regulating the state of VGSC by their movement. The pore domain (PD) is connected to each VSD, comprising S5–S6 segments and extracellular linkers (P-loop), which enables sodium ion conductance in an ion-selective manner.
Fig. 1. The structure of VGSC. A) The topology of VGSC pore-forming α subunit. There are six α-helices (S1–S6) in each domain (DI–DIV). Two helices (S5–S6) form the channel pore, and four helices (S1–S4) form a voltage sensor, in which positively charged residues present in each S4 contribute to the conformational change of VGSC via membrane voltage. B) Top view of hNaV1.7 pore-forming α-subunit with its domains presented in a space-filling model. C) and D) Side and top views of hNaV1.7 pore-forming α-subunit with its domains and binding sites presented in ribbon representation (PDB entry: 5EK0).29 Each domain is presented by the following colours: DI VSD (blue), DI PD (light blue), DII VSD (orange), DII PD (light orange), DIII VSD (yellow), DIII PD (light yellow), DIV VSD (green) and DIV PD (light green).

At least nine binding sites of VGSC are known, as presented in Table 4 and Fig. 1C and D. This section briefly reviews each binding site and the resulting pharmacological effects because such binding sites have been extensively reviewed.31
The binding site of VGSC.
| Binding site | Ligands | Binding domains |
|---|---|---|
| Site 1 | TTX, STX | P-loops of DI, DII, DIII and DIV |
| Site 2 | BTX, VTD, ACT | DI-S6, DIV-S6 |
| Site 3 | α-Scorpion toxins (OD1, AaH II, Lqh II, LqhaIT), sea anemone toxins (ATX-II, anthopleurin), spider toxins (δ-atracotoxin) | Extracellular loops of DIV S3–S4 |
| Site 4 | β-Scorpion toxins (Tz1, Css4), spider toxins (ProTx-II, HwTx-IV, GpTx-1, δ-palutoxins), antibody (SVmab) | Extracellular loops of DII S1–S2 and DII S3–S4 |
| Site 5 | PbTx, CTX | DI-S6, DIV-S5 |
| Site 6 | δ-Conotoxin (δ-SVIE, TxVIA, GmVIA) | DIV-S4 |
| Site 7 | Pyrethroids, DDT | DIII-S6 |
| LA binding site | Local anaesthetics (lidocaine), antiarrhythmics (mexiletine, flecainide), anticonvulsants (carbamazepine, lamotrigine) | DI-S6, DIII-S6, DIV-S6 |
| SA binding site | Sulphonamides (PF-05089771, DS-1971a, GDC-0276/RG7893, GDC-0310/RG6029) | Extracellular voltage sensor of DIV-S4 |
Binding site 1
Neurotoxins including TTX and STX inhibit VGSCs through binding site 1. Binding site 1 is localized to the extracellular region in the pore loop, in proximity to the ion selectivity filter. Neurotoxins bind directly to extracellular pore to inhibit sodium ion inward flow.30
Binding site 2
VGSC activators, such as the alkaloids batrachotoxin (BTX), veratridine (VTD) and aconitine (ACT), as well as several diterpenes and macrolide hoiamides bind to the open state at site 2 to stabilise the open state for activation. Binding site 2 is located in the S6 region of DI and DIV.
Binding site 3
α-Scorpion toxins, several spider toxins and anthopleurin from sea anemones bind to site 3 of VGSCs in the resting state to impair inactivation and induce a prolonged open state. Binding site 3 is found at the extracellular S3–S4 loops of DIV.
Binding site 4
Long-chain peptide toxins, such as β-scorpion toxins, several spider toxins and recombinant SVmab (rSVmab)33 inhibit VGSCs by binding to site 4 and acting as gating modifiers that shift the activation threshold to more negative membrane potentials. Binding site 4 is located in segments S1–S2 and S3–S4 of DII.
Binding site 5
Lipophilic cyclic polyethers, such as brevetoxins (PbTx) and ciguatoxins (CTX), activate VGSCs by binding to site 5. These toxins bind to the activated state preferentially and shift the activation threshold to more negative membrane potentials. Binding site 4 is scattered in DI-S6 and DIV-S5.
Binding site 6
δ-Conotoxins impede inactivation by binding to site 6, a subsite of site 3. The location of site 6 is believed to be extracellular DIV-S4.
Binding site 7
Some insecticides, including pyrethroids and DDT, inhibit channel inactivation by binding to site 7, thereby causing persistent activation. Binding site 7 is located in DIII-S6.
Local anaesthetic (LA) binding site
Non-selective VGSC inhibitors, including local anaesthetics, class I cardiac anti-arrhythmics, anti-convulsants, and anti-depressants, bind to the LA binding site. This binding site recognises an aromatic ring and a basic moiety as a pharmacophore. Lidocaine (1), mexiletine (2), carbamazepine (3) and lacosamide (4), which are presented in Table 6, are known to bind to this site. As this site is almost conserved across VGSCs, these drugs inhibit VGSCs in a non-selective manner.31 The LA binding site is located in the inner cavity of the pore region, which comprises residues in DI-S6, DIII-S6 and DIV-S6. The LA binding site displays significant overlap with binding site 2.
Conventional VGSC inhibitors with their in vitro profiles, free plasma concentrations and NaV1.7 coveragea.
|
|
|
|
|
|---|---|---|---|---|
| Funapide (5)55 | Vixotrigine (6) | AZD3161 (7)60 | 8: Merck62 | |
| hNaV1.1 IC50 | — | 15.2 μMb,d,46 | — | |
| hNaV1.5 IC50 | 0.084 μM | 3.2 μMb,d,46 | 12.6 μMb | 2.38 μM (Ki)c,e |
| hNaV1.7 IC50 | 0.054 μM | 6.1 μMb,d,46 | 0.079 μMb,g | 0.44 μM (Ki)c,e |
| Free plasma concentration | — | 25 nM@1 mg kg−1 (CFA model)57 | 5 μM@23 mg kg−1h (rat formalin) | 0.53 μM@1 mg kg−1h,i (rat PK) |
| 6.0, 74 nM@0.5, 5 mg kg−1 (subchronic, CCI model)57 |
|
|
|
|
|
|---|---|---|---|---|
| 9: Merck61 | 10: AbbVie63 | 11: Amgen64 | 12: Amgen65 | |
| hNaV1.1 IC50 | — | — | — | — |
| hNaV1.5 IC50 | 17.9 μMe,j | >33 μMb,c | 3.9 μMb,c | 0.95 μMb,c |
| hNaV1.7 IC50 | 0.69 μMe,j | 0.34 μMc,e | 0.15 μMb,k | 0.37 μMb,k |
| Rodent NaV1.7 IC50 | — | — | 0.049 μM (rat)b,k | 0.46 μM (mouse)b,k |
| Rodent PPBf | 99% (rat) | — | 82.2% (rat) | 96.3% (mouse) |
| Free plasma concentration | 0.72 μM@in vivo EC50 (rat SNL) | 5.7 μM@in vivo EC50 (acute, rat MIA)h | 61, 280 nM@30, 100 mg kg−1 (rat formalin) | 0.82 μM@300 mg kg−1 (mouse histamine) |
| 1.8 μM@in vivo EC50 (subchronic, rat MIA)h | ||||
| NaV1.7 coverage in plasma@efficacious dosagea | — | — | >5.7-Fold@100 mg kg−1 (rat formalin) | 1.8-Fold@300 mg kg−1 (mouse histamine) |
|
|
|
|
|---|---|---|---|
| 13: Daiichi Sankyo47 | 14: AstraZeneca69 | 15: Amgen | |
| hNaV1.1 IC50 | >100 μMb,c | — | — |
| hNaV1.5 IC50 | >100 μMb,c | >30 μMb,m | 1.1 μMe,n,70 |
| hNaV1.7 IC50 | 5.2 μMb,l | 0.41 μMb,g | 0.17 μMe,n,70 |
| Rodent NaV1.7 IC50 | 11.8 μM (mouse)b,l | 1.6 μM (rat)b,g | 0.39 μM (rat)e,n,71 |
| Rodent PPBf | 98.96% (mouse) | 86.8 % (rat) | 97.3% (rat)71 |
| Free plasma concentration | 2.7 nM@30 mg kg−1i (mouse PK) | 400 nM@43 mg kg−1 (rat formalin) | 14, 38 nM@3, 10 mg kg−1 (rat formalin)70 |
| 67 nM@10 mg kg−1 (rat CFA)71 | |||
| NaV1.7 coverage in plasma@efficacious dosagea | 0.000025-Fold@3.3 mg kg−1i (PSNL mouse) | 0.25-Fold@43 mg kg−1 (rat formalin) | 0.035-, 0.097-fold@3, 10 mg kg−1 70 (rat formalin) |
| 0.17-Fold@10 mg kg−1 71 (rat CFA) |
NaV1.7 coverage = free plasma concentration/in vitro NaV1.7 IC50.
Value in the automated patch-clamp assay.
Value in an inactivated state.
Value at Vhold = −60 mV.
Value in the manual patch-clamp assay.
Plasma protein binding.
Value at Vhold = −65 mV.
Plasma concentration.
Calculated from Cmax of the PK study.
Value at Vhold = −120 mV.
Value in a slow-inactivated state induced by prolonged depolarisation to −20 mV.
Value at Vhold = −30 mV.
Value at Vhold = −90 mV.
Value when approximately 20% of the channels were inactivated.
SA binding site
Sulphonamides, which were first disclosed by Pfizer, are known to inhibit VSD4 deactivation by binding to the activated state of voltage-sensing domain IV (VSD4), thereby stabilising the inactivated state of NaV1.7.29 The profile of these molecules is discussed in the following sections.
3. In vitro screening technologies
Over several decades, the functional activity of VGSCs has been studied in multiple in vitro assay systems. Electrophysiological techniques such as the patch-clamp assay are regarded as the gold standards for the physiological and pharmacological study of ion channel function. Although the manual patch-clamp assay provides the most reliable, high temporal resolution, direct measurement of ion channel function, it is extremely low-throughput, and it requires highly skilled operators. Recently, various automated patch-clamp assay systems have been developed and launched, and they provide high-quality and high-throughput data on ion channel function.34–36 Fluorescence-based techniques, such as fluorescent imaging plate reader (FLIPR)- and fluorescence resonance energy transfer (FRET)-based membrane potential assays, are also widely used for high-throughput screening (HTS). Although these techniques have superior throughput in general, their temporal resolution and biological relevancy are inferior to those of electrophysiological techniques. Ionic currents cannot be directly measured using these techniques, and their relatively high false-positive/negative rates because of compound-induced fluorescence or compound–dye interactions represent a major disadvantage.34,37 AstraZeneca's research group reported that the Li+ ion flux assay was a robust and reliable assay for the HTS of VGSC targets rather than FLIPR- and FRET-based membrane potential assays.38 However, in ion influx assays, the application of VTD, a VGSC activator, can produce the same drawback as fluorescence-based membrane potential assays.34,39 The features of in vitro screening technologies for VGSC drug discovery are summarised in Table 5. This review focuses on the in vitro activities measured by the gold standards, patch-clamp assay.
In vitro screening technologies for VGSC drug discovery.
| Assay type | Representative example | Temporal resolution | Information content | Throughput |
|---|---|---|---|---|
| Binding | Radioligand binding assay | Low (h) | Low (not functional) | Mid–high |
| Ion flux | Radioisotope influx assay | Mid (s to min) | Mid | Mid–high |
| AAS for Li+, Tl+ | ||||
| Fluorescence dye | FLIPR membrane potential assay | Mid (s) | Mid | High |
| FRET-based membrane potential assay (e.g., VIPR) | High (ms to s) | Mid | High | |
| Electrophysiology | Automated patch-clamp assay | High (μs to ms) | High | Mid–high |
| Manual patch-clamp assay | High (μs to ms) | High | Low |
4. Perspective of state-dependent properties in electrophysiological assays
VGSCs are extremely flexible, and they can exist in three distinct voltage-dependent conformational states: resting, open and inactivated states (Fig. 2A).40 In the resting state, the pore is closed for sodium ion conductance. When the membrane is depolarized, the voltage sensor in S4 helices moves outward to enhance pore opening, which enables sodium ion conductance within 1–2 ms. After depolarizing the membrane, VGSCs shift to an inactivated state via fast inactivation, in which the pore is still open but the inactivation gate located between DIII and DIV prevents ion conductance. Then, the channel moves to a slow inactivated state in response to prolonged depolarization or rapid repetitive stimulations. Fast inactivation occurs on a millisecond time scale, whereas slow inactivation occurs on the timescale of seconds to minutes. The activation of voltage sensor S4 across DI–DIII contributes to channel activation, whereas the activation of DIV-S4 leads to the movement of the IFM motif in the inactivation gate, resulting in channel inactivation. Finally, membrane hyperpolarisation leads to the channel resting state.
Fig. 2. Voltage-dependent conformational changes of VGSCs. A) Three distinct voltage-dependent conformational states: resting, open and inactivated states. B) An example of voltage protocol for an automated patch-clamp assay that permits evaluation of both resting (Vrest) and inactivated (V1/2) states of hNaV1.7.36.

Thus, inhibition of VGSC can be achieved in two distinguished manners: 1) direct pore-blocking mechanism and 2) stabilisation of a certain state, which inhibits shifting to the next state. Many VGSC inhibitors including medicinal drugs preferentially bind and interact with specific conformations or states. This state-dependent inhibition is also associated with the accumulation of inhibition, also called use-dependent inhibition or frequency-dependent inhibition.41 State-dependent inhibition is considered to impart functional selectivity to drug effects. For example, if a drug preferentially binds to a specific channel conformation and the conformation is dominant in a specific disease state or in the target organ or tissue for drug treatment, state-dependent inhibition can confer great benefits regarding both efficacy and safety. In fact, the clinical utility of state-dependent and/or use-dependent VGSC inhibitors has been demonstrated in cardiac arrhythmia,42 epilepsy43 and chronic pain.44,45 Therefore, it is extremely important to evaluate real channel function and drug effects according to individual conformational states. The patch-clamp assay is an unparalleled technique that fulfils the aforementioned demands based on its comprehensive and flexible analyses. Recently, efficient and effective pulse protocols for automated patch-clamp systems that permit the evaluation of both resting and inactivated channel states have been reported.22,36,46 In primary screening at our laboratory, the effects of compounds in both resting (Vrest) and inactivated (half-maximal voltage [V1/2]) states were determined with one protocol using an automated patch-clamp system (Fig. 2B).47,48
In chronic pain states, especially NP, ectopic discharges from primary sensory neurons represent a characteristic phenomenon. This pathological phenomenon is considered to result from the membrane potential oscillation mechanism rather than the traditional Hodgkin–Huxley model, which features a repetitive firing process.44 In rat DRG neurons, membrane potential oscillations exhibit voltage-sensitive properties. Namely, the prevalence of oscillations and consequent ectopic discharges is higher in depolarised states than in the resting state, and furthermore, those changes are enhanced after sciatic nerve injury.49 It is apparent that TTX-sensitive VGSCs contribute to the generation of membrane potential oscillations in DRG neurons. Thus, it might be useful to evaluate the effects of drugs on VGSCs under more depolarised states (i.e., pathological states) as well as physiological states. In fact, we found a series of compounds that selectively inhibit NaV1.7 currents in a more depolarised state (at the holding potential of −30 mV) but not in the resting or inactivated state, resulting in potent analgesic effects with a wide safety margin in NP model mice.47
5. Conventional VGSC inhibitors
Non-selective VGSC inhibitors have been studied for decades, and their utility has been proven in clinical settings.42–44,50 Their common characteristics are weak VGSC inhibitory activity with modest subtype selectivity. Their inhibitory activity is usually in the micromolar range.
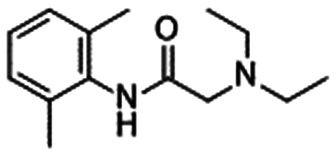
Lidocaine (1) is a classical non-selective VGSC inhibitor that has been used as local anaesthesia, whereas mexiletine (2) can be used as an oral analgesic agent. Carbamazepine (3) treats CNS disorders, including epilepsy. As presented in Table 6, these compounds weakly inhibit VGSC without high subtype selectivity.36,46 Classical non-selective VGSC inhibitors are useful; in particular, topical lidocaine (lidocaine patch) can significantly relieve various pain disorders by restricting systemic exposure.52 Thus, if a certain safety window can be obtained according to the formulation or route of administration, non-selective VGSC inhibitors could be launched for the treatment of pain disorders.
Lacosamide (4) is a broad VGSC inhibitor that was efficacious in patients with diabetes and NP in phase 2 clinical trials without an approval for NP indications.53 It was reported that lacosamide attenuated cold (from 10 mg kg−1, IP), warm (from 3 mg kg−1, IP) and mechanical allodynia (30 mg kg−1, IP) in rats with streptozotocin (STZ)-induced diabetes.54
Funapide (5: XEN402/XPF-002/TV-45070/FX301) was developed by Xenon and Teva for the treatment of several pain disorders as a topical formulation, and it is under development as FX-301, an extended-release, locally-delivered, thermosensitive hydrogel formulation, by Flexion Therapeutics. Funapide is a potent state-dependent VGSC inhibitor with IC50 values of 601, 84, 173 and 54 nM for NaV1.2, NaV1.5, NaV1.6 and NaV1.7, respectively.55
Biogen Inc. is developing vixotrigine (6: raxatrigine/BIIB074/CNV1014802/GSK1014802), which both inhibits multiple subtypes of VGSCs and exhibits MAO-B inhibitory activity.46,56–58 Vixotrigine was reported to inhibit multiple VGSCs in a state- and use-dependent manner. The consecutive administration of vixotrigine significantly reversed mechanical allodynia in a chronic constriction injury (CCI) model of NP at a dose of 0.5 or 5 mg kg−1, p.o. BID (twice daily). Free plasma concentrations on day 8 after administration for 0.5 h per day were 6.0 and 74 nM at doses of 0.5 and 5 mg kg−1, respectively. A single oral dose of vixotrigine was also efficacious in a Complete Freund's Adjuvant (CFA) rodent model with an ED50 of 0.91 mg kg−1. The free plasma concentration reached 25 nM at a dose of 1 mg kg−1, p.o.57 Thus, the in vivo efficacious free plasma concentration was more than 100-fold smaller than that reported for each human VGSC in vitro because it was noted that rodent NaV1.7 and NaV1.8 activities were comparable to those of humans. Vixotrigine is under development for the potential treatment of NP and trigeminal neuralgia.59
AZD3161 (7) was developed by AstraZeneca, and it exhibited high hNaV1.7 inhibitory activity with good selectivity over hNaV1.5. AZD3161 displayed antinociceptive effects dose-dependently in the rat phase 1 formalin test. A statistically significant antinociceptive effect was detected at 23 mg kg−1, and the plasma concentration at this dose was 5 μM.60
Merck reported the discovery of benzazepinone 8 and pyrrolo-benzo-1,4-diazine 9.61,62 Both compounds displayed comparable hNaV1.7 inhibitory activity, whereas 9 exhibited improved selectivity over hNaV1.5. Compound 8 inhibited hNaV1.7 with Ki values of 0.44 μM in the inactivated state and 24 μM in the resting state. 8 induced dose-dependent reversal in a rat spinal nerve ligation (SNL) model of NP with an ED50 of 15 mg kg−1. As Cmax was reported as 0.53 μM at a dose of 1 mg kg−1, p.o. in the PK study, Cmax is expected to approach 8 μM at ED50. No impaired motor coordination was observed at 100 mg kg−1.62 Compound 9 displayed better NaV1.5 selectivity than 8. Compound 9 inhibited hNaV1.7 with an IC50 of 0.69 μM, and it was efficacious in a rat CFA-induced inflammatory pain model at 100 mg kg−1 with statistical significance. Compound 9 was also efficacious in a rat SNL NP model with EC50 = 72 μM, which was comparable to that for NaV1.7 (IC50 = 33 μM) in the presence of 100% rat serum. Low exposure in the brain (brain Kp brain at 6 h = 0.2) was noted.61
AbbVie reported a selective NaV1.7 inhibitor 10 that inhibited hNaV1.7 with IC50 = 0.34 μM and 100-fold selectivity over hNaV1.5. Compound 10 was efficacious at 10 mg kg−1 in a monosodium iodoacetate (MIA)-induced osteoarthritis (OA) rat model (EC50 = 5.7 μM). The efficacy was enhanced by subchronic dosing twice a day for 7 days, which enhanced the efficacy at a dose of 0.3 mg kg−1 by achieving the same plasma levels as higher doses (EC50 = 1.8 μM).63
Amgen reported the discovery of compound 11. In their assay protocol, hNaV1.7 activity was measured at a slow-inactivated state induced by prolonged depolarisation to −20 mV, whereas the hNaV1.5 assay was conducted at holding partial channel inactivation. As compound 11 failed to exhibit significant efficacy in a rat formalin pain model at 100 mg kg−1 with NaV1.7 coverage of 5.7-fold, they concluded that binding to a slow-inactivated state of NaV1.7 may not significantly alter in vivo target coverage requirements.64
In the same year, Amgen disclosed the identification of the early lead compound piperazine 12, which was evaluated under the same in vitro assay protocol. Although 12 displayed comparable in vitro inhibitory activities as 11, it was efficacious in a mouse histamine-induced pruritus model with 1.8-fold target coverage. A subsequent SAR study led to the identification of the compounds with better NaV1.5 selectivity, although their in vivo efficacy was not evaluated.65 In this review article, the target coverage or NaV1.7 coverage was defined using the following formula for clear discussion on the extent of target coverage required to achieve certain in vivo efficacy: NaV1.7 coverage (fold) = free plasma concentration/in vitro NaV1.7 IC50.
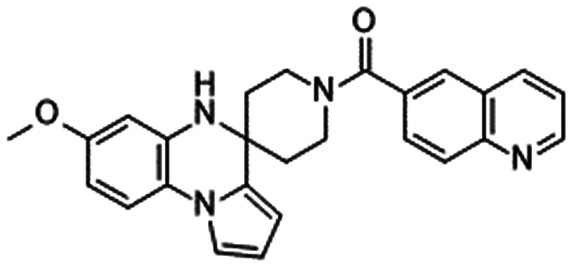
Our group disclosed compound 1347 derivatised from the report by Merck.62,66–68 Compound 13 inhibited NaV1.7 in a state-dependent manner. Although compound 13 was inactive at V1/2 (Vhold = −59 mV), it inhibited hNaV1.7 at Vhold = −30 mV (IC50 = 5.2 μM). Compound 13 was effective against thermal hyperalgesia in Seltzer (partial sciatic nerve ligation [PSNL]) model mice (ED50 = 3.3 mg kg−1). Notably, compound 13 displayed a good safety margin against CNS adverse effects with a suitable value of Kp in the brain (0.69). The target coverage of compound 13 was 0.000025-fold based on the reported PK parameters and plasma binding ability. Compound 13 exhibited modest selectivity because it did not affect other channels, receptors and transporters at 10 μM given its modest NaV1.7 IC50 values (5.2 μM for humans, 11.8 μM for mice).47
AstraZeneca disclosed an oxoisoindoline derivative 14. As compound 14 exhibited hNaV1.7 (IC50 = 0.41 μM at Vhold = −65 mV and hNaV1.7 IC50 = 3.8 μM at Vhold = −90 mV), it was considered a state-dependent NaV1.7 inhibitor. Compound 14 was inactive at several other channels but efficacious in the phase 1 reaction of a rat formalin model at 43 mg kg−1. The free plasma concentration of 14 was 400 nM at the end of the formalin assay, which corresponds to 0.25-fold of the rat NaV1.7 IC50. A site-directed mutagenesis study suggested that oxoisoindoline derivatives bound to the local anaesthetic site of NaV1.7.69
Triazine 15 is a NaV1.7 inhibitor with modest NaV1.5 selectivity that was disclosed by Amgen. Compound 15 is a state-dependent inhibitor with an IC50 of 170 nM at hNaV1.7 when 20% of the channels were inactivated and 3.6 μM in the resting state. Compound 15 was efficacious in phase 2 in a rat formalin model from 3 mg kg−1 with statistical significance, and the maximum effect at 30 mg kg−1 was equivalent to that produced by morphine. However, 15 significantly reduced movement from 20 mg kg−1. The NaV1.7 coverage was 0.035-fold at 3 mg kg−1 and 0.097-fold at 10 mg kg−1.70,7115 also reversed thermal hyperalgesia in a CFA model at 10 mg kg−1. The plasma concentration at a dose of 10 mg kg−1 reached 2.48 μM, and its target coverage was 0.17-fold. Brain exposure (0.81 μM) was observed for 15 at 10 mg kg−1. 15 was suggested not to affect the spontaneous activity or evoked responses of Aδ-fibres, but C-fibre nociceptors were indicated to regulate spontaneous discharge and cause analgesia based on the electrophysiological results of rat single nociceptive fibres in the CFA assay.71
Although some NaV1.7 inhibitors such as 11 failed to demonstrate potent in vivo efficacy, these conventional inhibitors tend to exhibit higher in vivo efficacy than predicted by PK and in vitro VGSC potency. Vixotrigine (6) displayed robust in vivo efficacy with a lower free plasma concentration (25 nM) than needed for in vitro VGSC activity (by over 100-fold at the ED50 dose), and the NaV1.7 coverage of compound 13 was extremely small at the ED50 dose. Compounds 14 and 15 were effective in a rat formalin assay with less than 1-fold NaV1.7 coverage.
One of the causes of poor PK/PD correlations is weak in vitro NaV1.7 potency. As the mode of action (MOA) of vixotrigine covers multiple targets, it is possible that this compound demonstrated potent efficacy in rodents via the synergic effect of multiple MOAs. As NaV1.6 is also involved in pain signalling pathways other than NaV1.3, NaV1.7, NaV1.8 and NaV1.9 signalling, the contribution of NaV1.6 inhibition may not be ignored.40,72 However, the potent in vivo efficacy of compound 13 can be hardly explained because it exhibited modest selectivity over other targets.47 As one possible reason is the contribution of the active metabolites, such studies are expected to resolve this issue to some extent.
Although the MOAs of potent in vivo efficacy remain controversial, some conventional VGSC inhibitors are efficacious in both animal models and patients. This indicates the possibility that these inhibitors could be future analgesics if a sufficient safety window is obtained in both preclinical animals and humans.
6. Sulphonamides
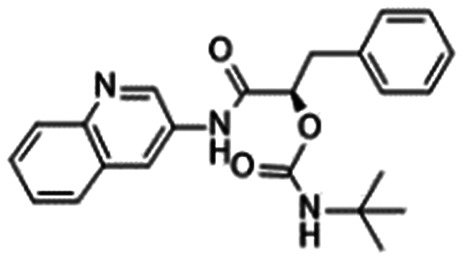
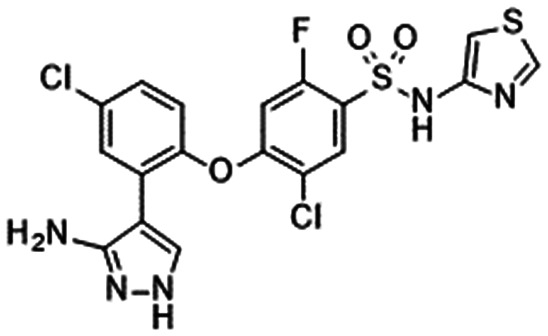
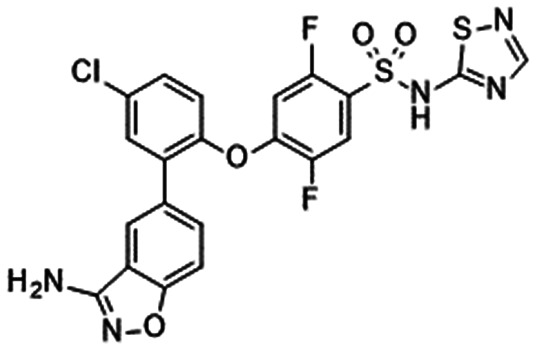
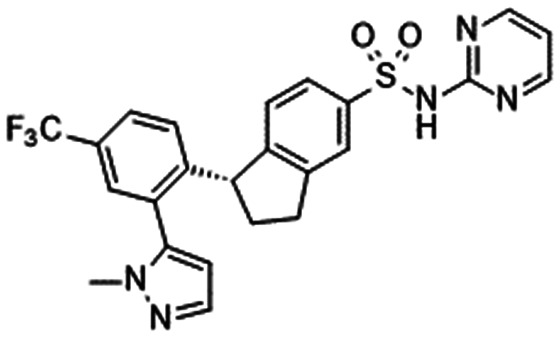
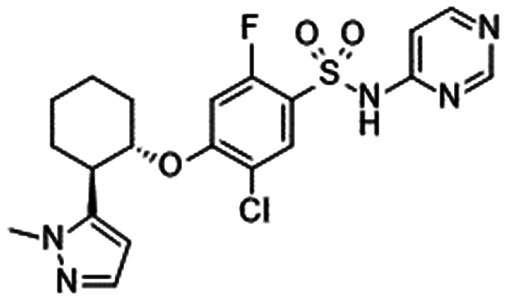
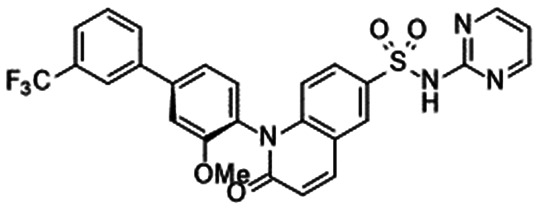
In 2010, Pfizer disclosed a highly potent selective NaV1.7 inhibitor in their patent,27 followed by the initiation of clinical trials of PF-05089771 (16).73,74 As presented in Table 7, PF-05089771 inhibited hNaV1.7 with an IC50 of 15 nM and excellent subtype selectivity over hNaV1.1 and hNaV1.5. A mutational study revealed that PF-05089771 interacted with VSD4 because the compound was not substantially affected by mutation of either the TTX or local anaesthetic binding sites, whereas its inhibitory activity was greatly affected by the hNaV1.7 M1,2,3 mutation.75 PF-05089771 inhibited hNaV1.7 in a state-dependent manner, and its IC50 for hNaV1.7 at the resting state exceeded 10 μM.75 PF-05089771 inhibited the rat orthologue with 10-fold lower potency than it inhibited the human orthologue, and the lack of potent activity against the rat orthologue was explained by the sequence divergence at VSD4, the binding site for sulphonamides.75,76 This opened a new era for the development of sulphonamide derivatives as potent selective NaV1.7 inhibitors, and many pharmaceutical and biotech firms disclosed their analogues. Some of them utilised PF-05089771 as a tool compound and reported the preclinical efficacy of their compounds. For example, Xenon and Genentech reported an evaluation of PF-05089771. In their article, they mentioned that 82-fold target coverage was required for robust efficacy in a transgenic mouse model expressing human NaV1.7 with an IEM mutation (I848T, IEM transgenic mouse).77,78 The clinical efficacy of PF-05089771 is discussed in the following section.
Sulphonamide derivatives with their in vitro profiles and target coveragea.
|
|
|
|
|
|
|---|---|---|---|---|---|
| PF-05089771 (16) | Xenon/Genentech (17)79 | 18: Lupin80 | DS-1971 (19)48 | AM-0466 (20)81 | |
| hNaV1.1 IC50 | 677 nMc,d,73 | 3080 nMd,e | 72%@10 000 nMd,e | >10 000 nMd,e | >42 500 nMc,h |
| hNaV1.5 IC50 | >10 000 nMc,d,73 | 1380 nMd,e | 40%@10 000 nMd,e | >10 000 nMd,e | >42 500 nMc,h |
| hNaV1.7 IC50 | 15 nMc,d,73 3.9 nMe,f,73 | 0.4 nMd,e | 33 nMd,e | 22.8 nMd,e | 21 nMc,h |
| Mouse NaV1.7 IC50 | 8 nMc,d,75 | 0.2 nMd,e (rat: 26 nMd,e) | 33 nMd,e | 59.4 nMd,e | 30 nMc,h |
| Mouse PPBb | 98.7%,77 99.9%90 | (rat: 98.9%) | 92.9% | 96.7% | — |
| NaV1.7 coverage in plasma@efficacious dosagea | 82-Fold@in vivo IC50 (IEM mouse)77 | 10-Fold@100 mg kg−1 (IP) (rat formalin, rat aconitine) | 10-Fold@3 mg kg−1 | 0.058, 0.26-fold@1, 3 mg kg−1g (PSNL mouse) | 16-Fold@30 mg kg−1 (mouse histamine) |
| 39-Fold@10 mg kg−1g (mouse formalin) | 43-, 57-fold@100, 300 mg kg−1 (mouse capsaicin) |
|
|
|
|
|
|---|---|---|---|---|
| GNE-616 (21)82 | 22: Amgen83 | 23: Merck84 | 24: Merck | |
| hNaV1.1 IC50 | >1000 nM (Kd)e,f | — | — | — |
| hNaV1.5 IC50 | >1000 nM (Kd)e,f | 330 nMe,i | 33 000 nMc,d | >30 000 nMc,d,85 |
| hNaV1.7 IC50 | 0.38 nM (Kd)e,f | 140 nMe,i | 39 nMc,d | 8 nMc,d,85 3.1 nMe,f,77 2.4 nMe,j,93 |
| Mouse NaV1.7 IC50 | — | 60 nMe,i (rat: 70 nMe,i) | 11 nMc,d | 15 nMc,d,85 3.9 nMe,j,93,95 1.8 nMe,j,k,95 |
| Mouse PPBb | 98.70% | 99.4% (rat: 98.5%) | 85.5% | 59%,85 78%,77 76%93,95 |
| NaV1.7 coverage in plasma@efficacious dosagea | 25-Fold@in vivo EC50 (IEM mouse) | 1.3-Fold, 3.6-fold@30, 100 mg kg−1 (rat formalin) | 100-Fold@in vivo EC50 (mouse formalin) | 11-Fold@in vivo IC50 85 (mouse formalin) |
| 10-Fold, 16-fold@60, 100 mg kg−1 (mouse formalin) | 28-Fold@in vivo IC50 77 (IEM mouse) | |||
| 326-Fold@60 mg kg−1 92 | ||||
| 21-Fold@60 mg kg−1l,92 | ||||
| 6-Fold, 10-fold, 11-fold@30, 60, 100 mg kg−1 (mouse histamine) | 87-Fold@30 mg kg−1 94 | |||
| 189-Fold@30 mg kg−1m,94 (mouse formalin) |
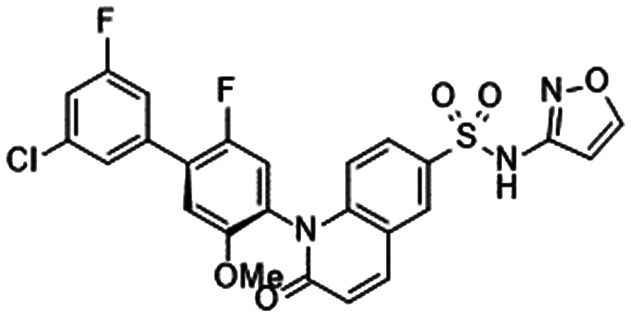
|
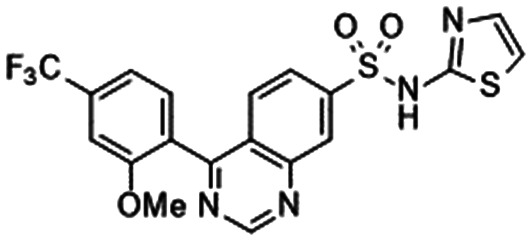
|
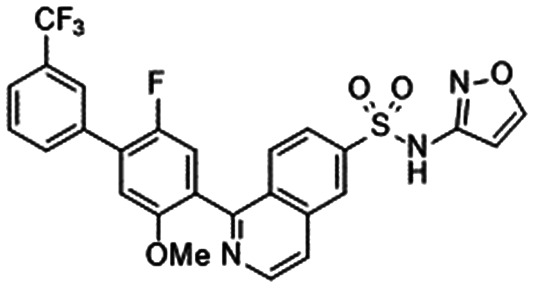
|

|
|
|---|---|---|---|---|
| AMG8379 (25)86 | 26: Amgen88 | 27: Amgen88 | 28: Pfizer89 | |
| hNaV1.1 IC50 | >14 000 nMc,n | 7300 nMe,o | 17 000 nMc | 314 nMc,d |
| hNaV1.5 IC50 | >14 000 nMc,n | 16 000 nMe,o | 12 000 nMc | 2592 nMc,d |
| hNaV1.7 IC50 | 8.5 nMc,h, 3.2 nMc,n | 140 nMe,o | 17 nMc | 0.01 nMc,d |
| Mouse NaV1.7 IC50 | 18.6 nMc,h, 16.8 nMc,n | 180 nMc,o | 36 nMc | <0.1 nMc,d |
| Mouse PPBb | 99.83% | 96.45% | 98.84% | 99.719% |
| NaV1.7 coverage in plasma@efficacious dosagea | 5.3-Fold@30 mg kg−1 (mouse histamine) | 28-Fold@60 mg kg−1 (mouse histamine) | 45-Fold@300 mg kg−1 (mouse histamine) | >62.5-Fold@5.4 mg kg−1 (i.v.) (mouse formalin) |
| 23-Fold@100 mg kg−1 (UVB-induced thermal hyperalgesia in mice) | ||||
| 21-Fold@100 mg kg−1 (mouse capsaicin) |
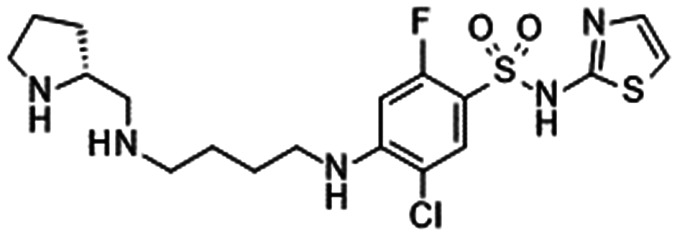
|
|
|
|
|
|---|---|---|---|---|
| 29: Merck90 | 30: Merck91 | 31: Bristol-Myers Squibb92 | 32: Bristol-Myers Squibb93 | |
| hNaV1.1 IC50 | 9000 nMc,i | — | — | — |
| hNaV1.5 IC50 | >33 000 nMc,i | >34 000 nMe,p | 1900 nMc,o | 6400 nMc,o |
| hNaV1.7 IC50 | 66 nMe,i | 87 nMe,p | 4.0 nMe,j | 78 nMe,f |
| Mouse NaV1.7 IC50 | 295 nM (rhesus)e,i | 8800 nMe,p | 7.5 nMe,j | 54 nMe,f, 34 nMq |
| 310 nM (rhesus)e,p | ||||
| Mouse PPBb | 92.63% (rhesus) | 94%, 98% (rhesus) | 92% | 97.7%, 90%r |
| NaV1.7 coverage in plasma@efficacious dosagea | 12-Fold, 42-fold@12.06, 24.12 mg kg−1 (i.v., rhesus OB) | 0.04-Fold@in vivo EC90 (mouse formalin) | >43-Fold@100 mg kg−1 | 14-Fold@100 mg kg−1 (mouse CCI) |
| 1.9-Fold@20 mg kg−1 (s.c., thermal stimulus in rhesus) | 4.8-Fold@19.2 mg kg−1 (rhesus OB) | >0.77-Fold@100 mg kg−1l (mouse formalin) | 0.5-Fold, 1-fold@30, 60 mg kg−1 (mouse CFA) | |
| 3-Fold@30 mg kg−1 | ||||
| 1-Fold@30 mg kg−1m (mouse formalin) |
NaV1.7 coverage = free plasma concentration/in vitro NaV1.7 IC50.
Plasma protein binding.
Value in the automated patch-clamp assay.
Value in an inactivated state.
Value in the manual patch-clamp assay.
Value at Vhold = −60 mV.
Calculated from Cmax of the PK study.
Value at a voltage yielding 20−50% channel inactivation.
Value at a voltage yielding approximately 20% channel inactivation.
Value at Vhold = −70 mV.
IC50 of mouse DRG NaV1.7.
The target coverage in the DRG given the plasma protein binding as an estimate of tissue binding.
NaV1.7 coverage in the DRG: free DRG concentration/in vitro IC50 of mouse DRG NaV1.7.
5-Hz use-dependent voltage protocol.
Value at Vhold = −50 mV.
Value under a hyperpolarised assay protocol.
IC50 of TTX-S currents in the mouse DRG.
Value of the mouse DRG.
The Xenon/Genentech group disclosed compound 17. They successfully obtained compound 17 with high rat NaV1.7 activity, which enabled in vivo evaluation in this animal. Because compound 17 failed to display sufficient plasma accumulation in rats following oral administration, in vivo assessments were conducted via IP administration. Compound 17 significantly reduced the pain response in the phase 2 formalin assay at 100 mg kg−1 IP. Compound 17 was also effective against acute pain evoked by aconitine as a pain stimulus at 100 mg kg−1 IP. The required NaV1.7 coverage was 10-fold for robust efficacy in both assays. Compound 17 was also efficacious against CFA-induced cold allodynia in mice.79
Lupin reported an indane derivative 18 with excellent subtype selectivity. Compound 18 induced the state-dependent blockade of hNaV1.7 and led to slow inactivation of hNaV1.7 with an IC50 of 33 nM, whereas 18 induced rapid inactivation with an IC50 of 99.7 nM. Compound 18 displayed high efficacy against veratridine-induced nociceptive behaviours from 10 mg kg−1, phase 2 formalin-induced nociceptive behaviour from 3 mg kg−1 and CCI-induced allodynia at 100 mg kg−1 with statistical significance in mice. The unbound plasma concentration of 18 at the end of the formalin study at 3 mg kg−1 was 0.343 μM, which is 10-fold higher than the mNaV1.7 IC50.80
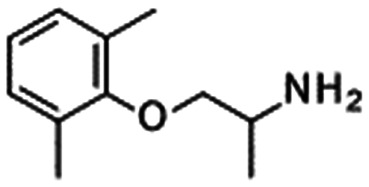
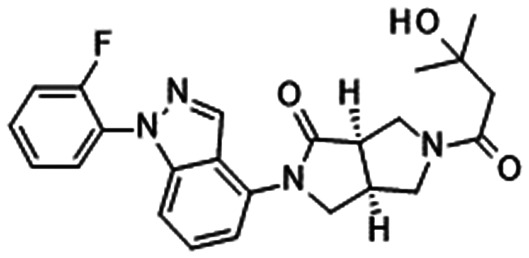
Our group disclosed the discovery of DS-1971 (19), a potent selective NaV1.7 inhibitor, which inhibited hNaV1.7 with IC50 = 22.8 nM and high subtype selectivity. DS-1971 displayed potent analgesic efficacy in PSNL mice. DS-1971 displayed efficacy against mechanical hypersensitivity from 1 mg kg−1, and it exhibited potent efficacy against thermal hyperalgesia from 0.3 mg kg−1 with statistical significance. Hence, DS-1971 displayed potent efficacy at less than 1-fold target coverage. The unique kinetics of DS-1971 was reported. DS-1971 exhibited a slower onset of inhibition than mexiletine, whereas the dissociation velocity of DS-1971a was slower than that of mexiletine. We concluded that this unique kinetics contributed to the potent efficacy of DS-1971.48
Amgen reported AM-0466 (20) with an IC50 of 21 nM for hNaV1.7. AM-0466 reduced scratching bouts in a histamine-induced pruritus mouse model in a dose-dependent manner, and a statistically significant reduction of itching behaviour was achieved at 30 mg kg−1. The unbound plasma concentration of 20 was 0.49 μM at a dose of 30 mg kg−1, which resulted in target coverage of 16-fold versus the mNaV1.7 IC50. AM-0466 was also efficacious in a mouse capsaicin-induced nociception pain model at 100 and 300 mg kg−1, which generated unbound plasma concentrations of 1.3 and 1.7 μM, respectively, resulting in target coverage values of 43- and 57-fold, respectively, over the mNaV1.7 IC50.81
Genentech reported the identification of GNE-616 (21). This compound inhibited hNaV1.7 at Kd = 0.38 nM with excellent subtype selectivity. PK/PD analysis revealed that 21 reduced nociceptive events with EC50 = 740 nM (unbound EC50,u = 9.6 nM) in an IEM aconitine mouse model, which corresponded to 25-fold hNaV1.7 coverage.82
Amgen disclosed a tool compound 22 with potent rat NaV1.7 inhibitory activity, although this compound lost high selectivity against hNaV1.5. Compound 22 was efficacious in phase 2 of a formalin rat model at 30 and 100 mg kg−1. These doses produced plasma coverage of 1.3 and 3.6-fold over the rat NaV1.7 IC50, respectively. In vivo assessments were also conducted in mice, and higher target coverage (more than 6-fold) was needed for high in vivo efficacy in the formalin test in mice with histamine-induced pruritus.83
The discovery of benzoxazolinone 23 was reported by Merck. Compound 23 was effective in phase 2 in a mouse formalin model from 20 mg kg−1, and the in vivo IC50 was calculated as 7.4 μM (unbound IC50,u = 1.1 μM), which is 100-fold higher than the in vitro potency in mice.84 The Merck group also reported the identification of compound 24, which displayed statistically significant efficacy in phase 2 in the mouse formalin model from 3 mg kg−1. The unbound plasma concentration at in vivo IC50,u was 170 nM, which was 11-fold higher than the mNaV1.7 IC50. Compound 24 is also efficacious in the histamine-induced itch assay in mice. Compound 24 induced a dose-dependent blockade of scratching events, and complete blockade was achieved at 30 mg kg−1.85 Later, the Xenon/Genentech group reported an evaluation of 24 and disclosed that 28-fold target coverage was needed to elicit significant efficacy in IEM transgenic mice.77
AMG8379 (25) was reported to block mechanically induced action potential firing in C-fibres, whereas the less active enantiomer AMG8380 had no such effects. AMG8379 was examined in a variety of mouse models. The compound displayed robust efficacy against histamine-induced scratching from 30 mg kg−1, at which the target coverage was 5.3-fold higher than the mNaV1.7 IC50. In UVB-induced thermal hyperalgesia and acute capsaicin-induced nociception, AMG8379 exhibited significant efficacy at 100 mg kg−1, and the target coverage values were 23- and 21-fold, respectively.86
Compounds 26 and 27 are selective NaV1.7 inhibitors reported by Amgen. Both compounds suppressed scratching behaviour in a histamine-induced scratching mouse model at 60 and 300 mg kg−1, which corresponded to free plasma concentrations of 5.1 and 1.59 μM, respectively, with target coverage values of 28- and 45-fold higher than the mNaV1.7 IC50, respectively. As the brain Kp of compound 26 was 0.008 in mice, the authors concluded that the efficacy was the result of NaV1.7 inhibition in the PNS.87,88
Pfizer disclosed the tool compound 28. This compound was not efficacious in a mouse formalin model with i.v. infusion even though its free plasma concentration exceeded the mNaV1.7 IC50 by more than 62.5-fold.89
Merck reported in vivo pharmacology results in rhesus monkeys. The evaluation of in vivo assays used two models: odour-induced activation of the olfactory bulb (OB) and noxious heat-evoked pain behaviours. Compound 29 was evaluated for its effects on membrane potential when approximately 20% of the channels were inactivated. Compound 29 inhibited rhesus NaV1.7 with an IC50 of 295 nM, versus 926 nM for PF-05089771 (16) in their assay protocol. Compound 29 significantly inhibited odorant-induced olfaction of OB in rhesus monkeys at two doses (12.06 and 24.12 mg kg−1, i.v.), resulting in target coverage values of 12- and 42-fold, respectively. In this assay, 16 at 9.5 mg kg−1 (i.v.) was not efficacious with target coverage of 0.12-fold based on the reported value (rhesus NaV1.7 IC50 = 926 nM, plasma concentration: 107 μM, PPB: 99.9%). Compound 29 was efficacious against thermal stimulation at 20 mg kg−1, s.c. The target coverage of 29 in this assay was 1.9-fold. Thus, Merck successfully reproduced the anosmia conditions observed in patients with NaV1.7 loss-of-function by inhibiting NaV1.7. They also revealed that heat withdrawal responses can be inhibited with lower NaV1.7 coverage than odour-induced activation of OB in rhesus monkeys because compound 29 was not efficacious in the OB assay at an NaV1.7 coverage of 3.4-fold (2.68 mg kg−1, i.v.) whereas it was efficacious in the thermal assay at 1.9-fold.90
Because NaV1.6 blockade leads to respiratory inhibition in the phrenic nerve preclinically, Merck identified compound 30 with better NaV1.6 selectivity (60-fold). They conducted an in vitro evaluation using the hyperpolarised assay protocol, which is close to the resting state. Surprisingly, compound 30 required 0.04-fold NaV1.7 coverage in plasma at in vivo EC50, whereas 4.8-fold target coverage was required in the rhesus OB assay. Based on these results, the estimated human dose varies from 40 (BID) to 4600 mg (BID), and they concluded that further optimisation was needed to acquire clinical candidates.91
Bristol-Myers Squibb reported the identification of compound 31. In their article, both compounds 24 and 31 were assessed for their ability to reduce nociceptive behaviour in a formalin test in mice, revealing that 24 was effective at 60 mg kg−1 whereas 31 was not effective at 100 mg kg−1. The target coverage values of these compounds in plasma were 326- and 43-fold, respectively. They evaluated the DRG concentrations of both compounds, and the concentrations of 24 and 31 reached 0.34 and 0.072 μM, respectively, corresponding to 21- and 0.77-fold target coverage in the DRG, respectively, given the plasma protein binding as an estimate of tissue binding. They speculated that greater target coverage in the DRG was required for high efficacy and synthesised two compounds exceeding the target coverage by more than 10-fold in the DRG. One compound successfully displayed statistically significant efficacy, whereas another failed to elicit robust efficacy even though both compounds possessed comparable properties, including similar target coverage in the DRG.92
Compound 32 achieved improved membrane permeability by avoiding the zwitterionic characteristics of 31 and better exposure in the DRG. Thus, compound 32 was efficacious in mouse CCI and CFA models at NaV1.7 coverage values of 14- and 0.5-fold, respectively. Notably, compound 32 was efficacious in the phase 2 mouse formalin model at 1-fold NaV1.7 coverage in the DRG. They also reported high exposure in the trigeminal ganglion in the mouse formalin model. They confirmed that compound 32 elevated the electrical threshold for the nociceptive flexion reflux to elicit an electromyographic response to the activation of Aδ nociceptive neurons in a highly corrected manner with plasma exposure.93
As previously described, almost all sulphonamide derivatives induced potent selective inhibition of hNaV1.7 and exhibited robust efficacy with high NaV1.7 coverage in preclinical animal studies. This high target coverage was realised by the enhancement of in vitro NaV1.7 activity. Some compounds are expected to display human efficacy at a lower dose given that efficacy is predicted by the in vitro IC50 and human PK parameters. Further, our group reported the excellent preclinical safety profile of DS-1971 (19) for the initiation of clinical trials.48 The reasons why these attractive clinical candidates did not proceed to clinical trials are discussed in the following section.
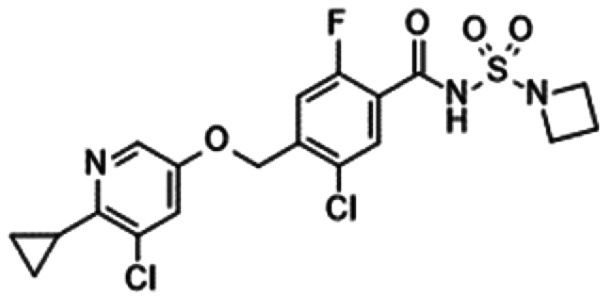
7. Acyl sulphonamides
In 2012, Pfizer described the acyl sulphonamide derivatives 33, 34 and 35 with potent NaV1.7 inhibitory activity in a patent (Table 8).95–97 These observations led to the discovery of GX-585 (36) and GX-201 (37), as reported by Xenon/Genentech. Compounds 36 and 37 inhibited hNaV1.7 with IC50 values of 15.1 and 3.2 nM, respectively, and they were efficacious in IEM transgenic mice. The in vivo IC50 values of 36 and 37 were 7.3 and 0.97 μM, respectively, and their target coverage values were 3.4- and 0.61-fold, respectively. They reported the target coverage for sulphonamide derivatives, including PF-05089771 (16) and compound 24 (Table 7), which required coverage of 82- and 28-fold, respectively, for significant efficacy in their animal model. Acyl sulphonamides 36 and 37 required less target coverage to induce robust efficacy in vivo than the sulphonamides 16 and 24. Because 36 and 37 displayed longer residence times than 16 and 24, they concluded that the improved in vivo efficacy was correlated with extremely slow dissociation from NaV1.7. They reported the efficacy of the compounds in various in vivo models of neuropathic and inflammatory pain. A radioligand study illustrated that 36 and 37 bonded competitively to VSD4, the same binding site used by sulphonamide derivatives.77
Acyl sulphonamide derivatives with their in vitro profiles and target coveragea.
|
|
|
|
|
|
|---|---|---|---|---|---|
| 33: Pfizer95 | 34: Pfizer96 | PF-05241328 (35)97 | GX-585 (36)77 | GX-201 (37)77 | |
| hNaV1.1 IC50 | — | — | — | 100 nMc,e | 192 nMc,e |
| hNaV1.5 IC50 | — | — | — | 435 nMc,f | 705 nMc,f |
| hNaV1.7 IC50 | 30 nMc,d | 90 nMc,d | 22 nMc,d | 15.1 nMc,f | 3.2 nMc,f |
| Mouse NaV1.7 IC50 | — | — | — | — | — |
| Mouse PPBb | — | — | — | 99.3% | 99.8% |
| NaV1.7 coverage in plasma@efficacious dosagea | — | — | — | 3.4-Fold@in vivo EC50 (IEM mouse) | 0.61-Fold@in vivo EC50 (IEM mouse) |
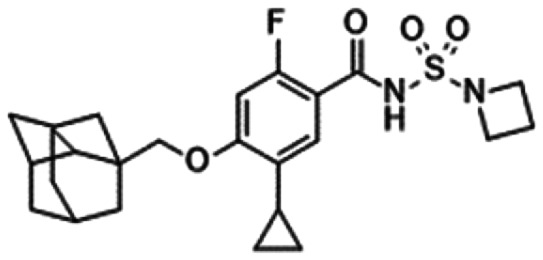
|
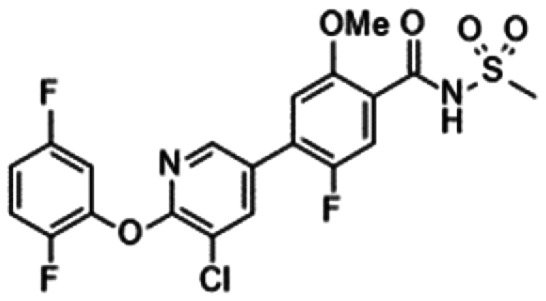
|
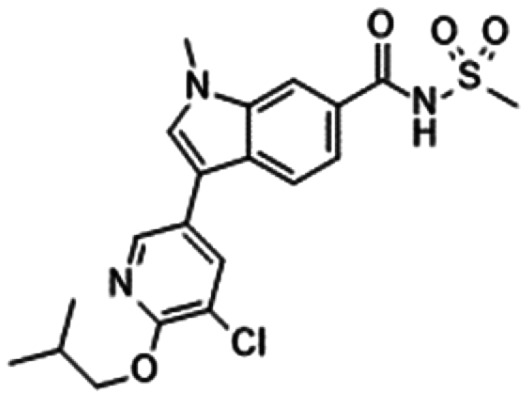
|
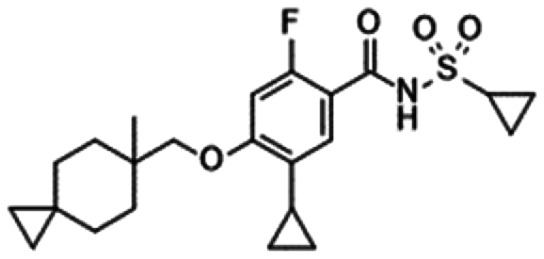
|
|
|
|---|---|---|---|---|---|
| GDC-0276 (38)78 | 39: Amgen99 | 40: Bristol-Myers Squibb94 | 41: Xenon/Genentech100 | GNE-131 (42)101 | |
| hNaV1.1 IC50 | 10.6 nMd,g | 8500 nMc,h | — | 6 nMd,g | 45 nMd,g |
| hNaV1.5 IC50 | 51 nMd,g | 11 900 nMc,h | 19 000 nMg,k | 50 nMd,g | 110 nMd,g |
| hNaV1.7 IC50 | 0.4 nMd,g | 51 nMc,h | 8 nMc,l | 0.6 nMd,g | 3 nMd,g |
| Mouse NaV1.7 IC50 | — | 115 nM, 270 nMi | 35 nMc,l, 11 nMc,l,m | 2.2 nMd,g | — |
| Mouse PPBb | 99.98% | 98.6% | 99.8%, 96.5%n | 99.9% | 99.9% |
| NaV1.7 coverage in plasma@efficacious dosagea | 1-Fold@in vivo EC50 (IEM mouse) | 36-Fold@300 mg kg−1 (15-fold@300 mg kg−1j) (mouse histamine) | 8.7-Fold@30 mg kg−1 IPo | 5-Fold@0.30 mg kg−1 (IEM mouse) | 0.17-Fold@in vivo EC50 (IEM mouse) |
| 3.4-Fold@30 mg kg−1 IP (mouse formalin) | 23-Fold@10 mg kg−1 (mouse formalin) | ||||
| 3.6-Fold@30 mg kg−1 IP (mouse CFA) | |||||
| 3.9-Fold@30 mg kg−1 IP (mouse CCI) |
NaV1.7 coverage = free plasma concentration/in vitro NaV1.7 IC50.
Plasma protein binding.
Value in the manual patch-clamp assay.
Value in an inactivated state.
Value at Vhold = −45 mV.
Value at Vhold = −60 mV.
Value in the automated patch-clamp assay.
Value at a voltage yielding 20% channel inactivation.
IC50 of native TTX-S currents in the mouse DRG.
Calculated from the mouse TTX-S IC50.
Value at Vhold = −50 mV.
Value at Vhold = −70 mV.
IC50 of mouse DRG NaV1.7.
Value in the mouse DRG.
NaV1.7 coverage in the DRG: free DRG concentration/in vitro IC50 of mouse DRG NaV1.7.
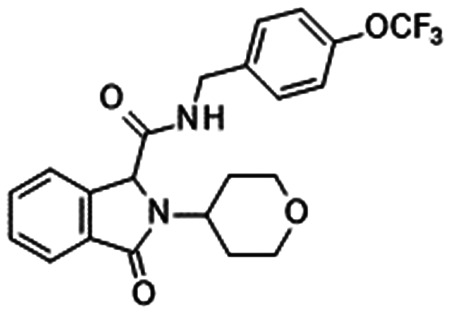
The adamantane derivative GDC-0276 (38) was disclosed by Genentech. A preclinical in vivo study of GDC-0276 was conducted using IEM transgenic mice. At the in vivo EC50 of 1.7 μM, the target coverage of 38 was 1-fold, which aligned with their conclusion that this low target coverage is sufficient for robust efficacy in acyl sulphonamides.78 Compound 16 (Table 6) displayed poor in vivo efficacy with EC50 > 18 μM in their in vivo assay. Although phase 1 clinical trials of both GDC-0276 and GDC-0310 were completed, the development of both compounds was terminated.78,98
Amgen disclosed biphenyl acyl sulphonamide derivative 39. A PK/PD study in a mouse histamine-induced scratching model demonstrated a robust reduction of scratching bouts at a dose of 300 mg kg−1, p.o. The unbound plasma concentration (Cu plasma = 4.15 μM) was more than 15-fold greater than the IC50 measured on native TTX-S currents in mouse DRG neurons (IC50 = 0.27 μM) at 300 mg kg−1.99 In comparison to another NaV1.7 inhibitor, the NaV1.7 coverage of which was calculated at the mNaV1.7 IC50, the NaV1.7 coverage of 39 was calculated as 36-fold (mNaV1.7 IC50 = 115 nM).
Bristol-Myers Squibb discovered the indole 40, which inhibited hNaV1.7 at an IC50 of 8 nM with excellent subtype selectivity over hNaV1.5. 40 was efficacious in the reversal of phase 2 formalin-induced nociceptive behaviours in mice at 30 mg kg−1 IP. At 30 mg kg−1 IP, compound 40 reversed the nociceptive behaviours to normal levels. NaV1.7 coverage in the DRG (free DRG concentration/in vitro IC50 of mouse DRG NaV1.7) was 8.7-fold, whereas NaV1.7 coverage in plasma was 3.4-fold higher than the mNaV1.7 IC50. The reference compound 24 (Table 7) reversed phase 2 formalin-induced nociceptive behaviour to normal levels at 30 mg kg−1, p.o., at which the target coverage in the DRG was 189-fold, and the coverage in plasma reached 87-fold. A close analogue of 40 that achieved a target coverage of 5.5-fold in the DRG (the target coverage in plasma: 5.4-fold) failed to display efficacy in the formalin model. Because the subtype selectivity, PK profile, off-target activity and binding kinetics of active and inactive compounds were identical, they concluded that a small difference in target coverage in the mouse DRG (8.7-fold vs. 5.5-fold) led to a significant difference in in vivo efficacy. Although a lower dose of 24 was not examined, it could be concluded that 24 needed higher target coverage than 40 for high efficacy in vivo. Compound 40 was reported to be significantly efficacious in the mouse CFA model of inflammatory pain at 30 mg kg−1 IP, whereas the effect was modest against cold allodynia in the mouse CCI model at 30 mg kg−1 IP. The target coverage values in the CFA and CCI assays were 3.6- and 3.9-fold in plasma, respectively. Sural fascicle recording studies illustrated that 40 and a close analogue were efficacious against mechanosensitivity of the mouse sural nerve at a dose of 30 mg kg−1 IP, whereas only 40 was efficacious against mechanosensitivity of the mouse sural nerve when the compound was applied directly to the nerve at 100 μM.94
Xenon/Genentech disclosed the highly potent acyl sulphonamide 41. Compound 41 inhibited both hNaV1.7 at IC50 = 0.6 nM and rat orthologues (IC50 = 3.7 nM), whereas an in vivo evaluation of 38 was performed in mice. Compound 41 was efficacious in aconitine-induced pain in IEM transgenic mice from 0.3 mg kg−1, p.o. The plasma concentration of 41 was approximately 3.1 μM at a dose of 0.3 mg kg−1 (unbound plasma concentration: 3 nM), which is approximately 5-fold higher than the hNaV1.7 IC50. Compound 41 exerted a significant effect in the phase 2 formalin assay at 10 mg kg−1, p.o. The plasma concentration of 41 was 50 μM, which corresponded to 23-fold target coverage in plasma.100
GNE-131 (42) is a less selective acyl sulphonamide that inhibited hNaV1.7 (IC50 = 3 nM). GNE-131 was efficacious in a mouse IEM model from 10 mg kg−1, and the in vivo EC50 was determined as 0.5 μM via two-point sigmoid calculation, which corresponded to hNaV1.7 coverage of 0.17-fold. Notably, they reported the discrepancy between in vivo efficacy and in vitro potency for their close analogues.101
Acyl sulphonamide derivatives possess higher plasma binding ability than sulphonamides owing to the higher acidity of the acyl sulphonamide group. As mentioned by the Xenon/Genentech group, acyl sulphonamides tend to demonstrate robust efficacy with lower target coverage than sulphonamides.77,78 As acyl sulphonamides exhibited less subtype selectivity than sulphonamides, this suggested the possible contribution of other VGSCs to the effects. It should be noticed that the less subtype-selective compound GNE-131 (42) elicited efficacy in IEM mice at low NaV1.7 coverage (0.17-fold).
Although the preclinical safety profile of acyl sulphonamides has not been disclosed to date, the acyl sulphonamide GDC-0276 (38) exhibited higher toxicity than PF-05089771, which is discussed in the following section.78,98
8. Pharmacological effects of NaV1.7 inhibitors in in vivo animal models
Animal experiments are inevitable in the research and development of analgesics, and various types of in vivo experimental animal models have been used to evaluate the pharmacological effects of NaV1.7 inhibitors. Some examples of animal models used for the pharmacological evaluation of NaV1.7 inhibitors are listed in Table 9. Although traditional nociceptive and NP models with various stimulation methods (e.g., chemical, mechanical, thermal, cold, electrical) have been widely used, it appears that each pharmaceutical company has developed a unique research strategy for the in vivo screening of NaV1.7 inhibitors.
Animal models used in the evaluation of NaV1.7 inhibitors.
| Category | Model | Endpoint | Species | Ref. |
|---|---|---|---|---|
| Pain (nociceptive model) | Hot plate | Nociceptive response (hind paw flinching/licking) induced by noxious thermal stimuli | Mouse | 77 |
| Heat thermode | Nociceptive response (forearm withdrawal) induced by noxious thermal stimuli | Rhesus monkey | 90 | |
| Acetic acid | Nociceptive response (abdominal writhing) induced by the intraperitoneal injection of acetic acid | Mouse | 111 | |
| Formalin | Nociceptive response (hind paw flinching/licking/lifting) induced by the intraplantar injection of formalin | Mouse/rat | 69, 70, 77, 80, 84, 85, 86, 90, 101 | |
| Capsaicin | Nociceptive response (hind paw licking) induced by the intraplantar injection of capsaicin | Mouse | 86 | |
| CFA | Mechanical hypersensitivity induced by the intraplantar injection of CFA | Rat | 57, 68 | |
| Thermal hypersensitivity induced by the intraplantar injection of CFA | Mouse/rat | 61, 82, 112 | ||
| Cold hypersensitivity induced by the intraplantar injection of CFA | Mouse | 77, 79 | ||
| MIA | Grip force deficit of hind limbs induced by intra-articular knee injection of MIA | Rat | 63 | |
| Ultraviolet-B (UVB) | Thermal hypersensitivity induced by UVB irradiation to the hind paw | Mouse | 86 | |
| Pain (neuropathic model) | STZ-induced diabetes | Mechanical hypersensitivity induced by diabetic neuropathy | Mouse | 77 |
| CCI | Mechanical hypersensitivity induced by CCI | Rat | 57 | |
| SNL | Mechanical hypersensitivity induced by SNL | Mouse/rat | 48, 61, 62, 67, 68 | |
| PSNL | Mechanical hypersensitivity induced by PSNL | Mouse | 48 | |
| Thermal hypersensitivity induced by PSNL | Mouse | 47, 48 | ||
| Spared nerve injury (SNI) | Cold hypersensitivity induced by SNI | Mouse | 77 | |
| Pain (target engagement model) | Scorpion toxin OD1 | Nociceptive response (hind paw licking/flinching/lifting/shaking) induced by the intraplantar injection of OD1 | Mouse | 58 |
| Aconitine | Nociceptive response (hind paw flinching) induced by the intraplantar injection of aconitine in normal mice | Rat | 79 | |
| IEM-aconitine | Nociceptive response (hind paw flinching/licking/biting) induced by the intraplantar injection of aconitine in IEM transgenic mice | Mouse | 77, 78, 82, 100, 101 | |
| Others | Itch | Scratching behaviour induced by the intradermal injection of histamine into the neck | Mouse | 83, 85, 86, 99 |
| Cough | Coughing response induced by citric acid inhalation | Guinea pig | 109 | |
| Skin blood flow | Flare response induced by the topical skin application of capsaicin | Mouse | 75 | |
| Olfactory fMRI | Olfaction response (fMRI signal) in OB induced by odour stimulation | Rhesus monkey | 90 |
Convergence/Biogen reported that their clinical compound vixotrigine (6, Table 6) displayed significant analgesic effects in the CCI and CFA models of rats at doses that did not induce sedation or ataxia.57 PF-05089771 (16, Table 7), a clinical compound developed by Pfizer/Icagen, is a breakthrough compound exhibiting potent and selective NaV1.7 inhibition, and its in vitro effects were described in several peer-reviewed papers.73–75,102,103 Although Pfizer/Icagen have not published the results of in vivo pharmacological studies of PF-05089771, their patent mentioned the synergic analgesic effects of analogues of 16 and pregabalin in the mouse formalin test.104 Pfizer/Icagen's non-clinical tool compound PF-05198007 inhibited capsaicin-induced flare response and capsaicin-induced nociceptive behaviour in mice,75,105 whereas another tool compound named PF-06456384 exerted no significant analgesic effects in the mouse formalin test.89
Generally, the evaluation of the effects of drugs on mechanical hypersensitivity (i.e., allodynia, hyperalgesia) using the von Frey test is regarded as the gold standard for in vivo screening in NP models. However, we found that NaV1.7 inhibitors are more effective against thermal hypersensitivity than mechanical hypersensitivity in NP models and screened a series of NaV1.7 inhibitors using a thermal assay (Hargreaves test) in PSNL model mice.47,48 As a result, we obtained DS-1971a (19, Table 7), a clinical compound, exerting a potent analgesic effect on thermal hypersensitivity in PSNL model mice with an ED50 of 0.32 mg kg−1, p.o., making it more potent than PF-05089771 (ED50 = 3.0 mg kg−1, p.o.). DS-1971a also significantly inhibited mechanical hypersensitivity in PSNL and SNL model mice at oral doses of at least 1 mg kg−1.48 Amgen's research group adopted histamine-induced scratching behaviour in mice (i.e., antipruritic activity) to screen their NaV1.7 inhibitors, including small molecules,81,83,86–88,100 peptides106 and antibodies,107 based on the findings from phenotype analyses of NaV1.7 knockout mice.12 Xenon/Genentech reported a target engagement assay of aconitine-induced nociceptive behaviour in IEM transgenic mice.77,82,100,101 The IEM transgenic mice displayed enhanced nociceptive responses (flinching and licking of the hind paws) elicited by the intraplantar injection of aconitine, a NaV activator, compared to the findings in wild-type mice.77 Their clinical compounds, namely GDC-0276 and GDC-0310, exerted dose-dependent analgesic effects at lower plasma concentrations than PF-05089771.78 Another target engagement assay using the scorpion toxin OD1, a NaV1.7 activator, was also reported. Vixotrigine and PF-04856264, an aryl sulphonamide, inhibited nociceptive responses evoked by the intraplantar injection of OD1.58
Concerning sulphonamides and acyl sulphonamides, large species differences in NaV1.7 blockade make the choice and interpretation of animal experiments more difficult. For instance, PF-05089771 was found to be far less potent against rat NaV1.7 than against human, mouse, dog and cynomolgus macaque NaV1.7.73,75 Namely, rats, as the most commonly used species in preclinical studies, are not appropriate for evaluating the pharmacological effects of sulphonamides and acyl sulphonamides. Therefore, many pharmaceutical companies have used mice in preclinical pharmacodynamic studies of sulphonamides and acyl sulphonamides.48,77,78,80–89,91–94,99–101 Lilly and their collaborators reported the antitussive effects of compound 801, a sulphonamide-based NaV1.7 inhibitor, on citric acid-evoked coughing in guinea pigs.108,109 Recently, Merck reported a battery of four translational assays using rhesus monkeys: 1) microneurography for the action potential propagation in unmyelinated afferents, 2) threshold tracking for the excitability of myelinated afferents, 3) heat nociception test using clinical thermode device and 4) functional magnetic resonance imaging (fMRI) for olfactory function. Their sulphonamides exerted dose-dependent and significant effects in microneurography, heat nociception and fMRI assays.90 These assays provide back-translation from clinical examination to preclinical research with non-human primates.110 It is expected that they could be used as pharmacodynamic endpoints for NaV1.7 inhibitors in clinical trials.
In addition to the aforementioned analgesic and/or antipruritic effects of NaV1.7 inhibitors, their side effect profiles have been published. It is notable that no sulphonamide-based NaV1.7 inhibitors exerted significant side effects on the CNS and CV system.48,77,86 This is one of the most remarkable aspects of selective NaV1.7 inhibitors and a major differentiation point versus non-selective VGSC inhibitors.
9. Clinical studies of NaV1.7 inhibitors
Many investigators including academic, biotech and mega pharma companies have been conducting research and development on selective NaV1.7 inhibitors. However, no group has successfully developed and launched a NaV1.7 inhibitor to date. Clinical trials of NaV1.7 inhibitors are listed in Table 10.
Clinical trials of NaV1.7 inhibitors.
| Compound [sponsor] | Condition/indication | Study phase | Study status | Identifiera | Ref. |
|---|---|---|---|---|---|
| Vixotrigine/raxatrigine/BIIB074/CNV1014802/GSK1014802 [Biogen/Convergence/GlaxoSmithKline] | HV (SD, resting motor threshold) | 1 | Completed 2008 | NCT00488566 | 113, 114 |
| HV (MD) | 1 | Completed 2008 | NCT00908154 | ||
| HV (SD, electrical stimulation) | 1 | Terminated 2009 | NCT00964288 | ||
| HV (MD, ambulatory blood pressure) | 1 | Completed 2009 | NCT00955396 | ||
| Lumbosacral radiculopathy (MD) | 2 | Completed 2012 | NCT01561027 | ||
| Trigeminal neuralgia (MD) | 2a | Completed 2014 | NCT01540630 | ||
| HV (MD, age, gender) | 1 | Completed 2015 | NCT02359344 | ||
| HV (MD, DDI) | 1 | Completed 2016 | NCT02551497 | ||
| HV (MD, DDI) | 1 | Completed 2016 | NCT02698267 | ||
| HV (SD, hot ADME) | 1 | Completed 2016 | NCT02751905 | ||
| IEM (MD) | 2a | Completed 2017 | NCT02917187 | ||
| HV (SD, MD, race) | 1 | Completed 2017 | NCT02831517 | ||
| HV (SD, RBA) | 1 | Completed 2017 | NCT02951221 | ||
| HV (MD, DDI) | 1 | Completed 2017 | NCT03385525 | ||
| HV (MD, DDI) | 1 | Completed 2018 | NCT03324685 | ||
| Lumbosacral radiculopathy (MD) | 2 | Completed 2018 | NCT02935608 | ||
| Lumbosacral radiculopathy (MD) | 2 | Terminated 2019 | NCT02957617 | ||
| Small fibre neuropathy (MD) | 2 | Terminated 2021 | NCT03339336 | ||
| Trigeminal neuralgia (MD) | 3 | Not yet recruiting 2021 | NCT03070132 | 59 | |
| Trigeminal neuralgia (MD) | 3 | Not yet recruiting 2021 | NCT03637387 | 59 | |
| Funapide/XEN402/XPF-002/TV-45070/FX301 [Xenon/Teva/Flexion] | IEM (MD, oral) | 2a | Completed 2010 | NCT01090622 | 115 |
| PHN (MD, ointment) | 2a | Completed 2011 | NCT01195636 | 116 | |
| IEM (MD, ointment) | 2a | Completed 2012 | NCT01486446 | ||
| HV (MD, ointment) | 1 | Completed 2015 | NCT02215941 | ||
| Knee osteoarthritis (MD, ointment) | 2 | Completed 2015 | NCT02068599 | ||
| PHN (MD, ointment) | 2 | Completed 2017 | NCT02365636 | ||
| Postoperative pain (SD, local injection) | 1 | Recruiting 2021 | NCT04826328 | ||
| PF-05089771 [Pfizer/Icagen] | HV (SD, micro-dosing, oral and intravenous) | 1 | Completed 2010 | NCT01165736 | 117 |
| HV (SD, exploratory pharmacodynamics) | 1 | Completed 2011 | NCT01259882 | ||
| HV (MD) | 1 | Completed 2011 | NCT01365637 | ||
| HV (SD, RBA) | 1 | Completed 2012 | NCT01563497 | ||
| Postoperative dental pain (SD) | 2 | Completed 2012 | NCT01529346 | ||
| HV (SD, RBA) | 1 | Completed 2012 | NCT01690351 | ||
| HV and knee OA (MD) | 1 | Completed 2012 | NCT01529671 | ||
| HV (MD, titration) | 1 | Completed 2013 | NCT01772264 | ||
| IEM (SD) | 2 | Completed 2013 | NCT01769274 | ||
| HV (SD, RBA) | 1 | Completed 2013 | NCT01854996 | ||
| HV (SD, MD, DDI) | 1 | Completed 2013 | NCT01934569 | ||
| HV (SD, pain model) | 1 | Completed 2015 | NCT02349607 | 118 | |
| Diabetic peripheral neuropathy (MD) | 2 | Completed 2015 | NCT02215252 | 119 | |
| AZD3161 [AstraZeneca] | HV (SD, UVC, intradermal injection) | 1 | Completed 2011 | NCT01240148 | |
| DSP-2230 [Sumitomo Dainippon/Sunovion] | HV (SD, MD, RBA) | 1 | Completed 2013 | ISRCTN07951717 | |
| HV (SD, capsaicin and UVB) | 1 | Completed 2013 | ISRCTN80154838 | ||
| HV (MD, renal function) | 1 | Completed 2013 | ISRCTN02543559 | ||
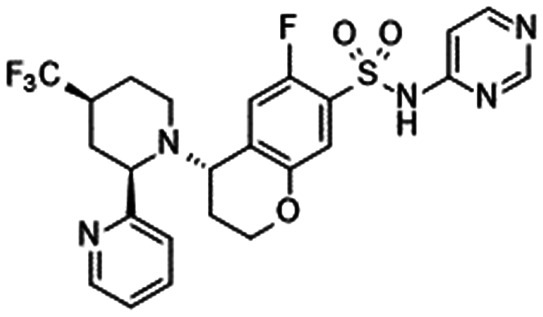
| DS-1971a [Daiichi Sankyo] | HV (SD) | 1 | Completed 2014 | NCT02107885 | |
| HV (MD) | 1 | Completed 2014 | NCT02190058 | ||
| HV (SD, age, gender, race) | 1 | Completed 2014 | NCT02261376 | ||
| HV (SD, RBA) | 1 | Completed 2015 | NCT02266940 | ||
| HV (MD, DDI) | 1 | Completed 2015 | NCT02473627 | ||
| HV (MD, gender) | 1 | Completed 2015 | NCT02564861 | ||
| Diabetic PNMD) | 2 | Withdrawn 2016 | NCT02673866 | ||
| Radiculopathy attributable to LSS (MD) | 2 | Withdrawn 2016 | JapicCTI-163 193 | ||
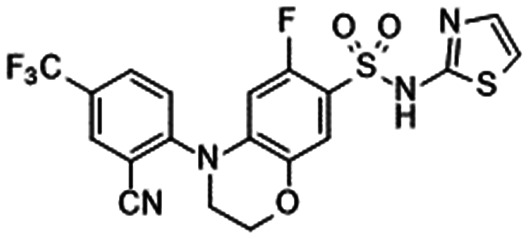
| GDC-0276/RG7893 [Xenon/Genentech/Roche] | HV (SD, RBA) | 1 | Withdrawn 2016 | NCT02856152 | 98 |
| GDC-0310/RG6029 [Xenon/Genentech/Roche] | HV (SD, MD, RBA) | 1 | Completed 2017 | NCT02742779 | |
| BIIB095 [Biogen/Convergence] | HV (SD, MD) | 1 | Completed 2019 | NCT03454126 | |
| HV and diabetic peripheral neuropathy | 1b | Withdrawn 2021 | NCT04106050 | ||
| CC8464/APS1807 [Chromocell/Astellas] | HV (SD, MD, RBA, DDI) | 1 | Completed | Not registered | |
| DSP-3905 [Sumitomo Dainippon/Sunovion] | HV | 1 | Not disclosed | Not registered |
From ClinicalTrials.gov (https://clinicaltrials.gov/ct2/home), ISRCTN registry (https://www.isrctn.com/) and Japic Clinical Trials Information (https://www.clinicaltrials.jp/cti-user/common/Top.jsp).
Vixotrigine (6, Table 6) is a current front-runner in the development of selective NaV1.7 inhibitors. Although compound 6 was initially reported as a selective NaV1.7 inhibitor, recent studies demonstrated that it is neither selective nor potent.46,56,58 Some phase 2 trials of forms of NP, such as trigeminal neuralgia, lumbosacral radiculopathy and small fibre neuropathy, were completed with mixed results. The detailed study design and outcomes of a phase 2a randomised withdrawal trial in trigeminal neuralgia have been published.113,114 Although the criterion for the primary endpoint of treatment failure was not met (33% treatment failure for vixotrigine 150 mg TID, 64% treatment failure for placebo, p = 0.0974), significant efficacy trends in the secondary endpoints (e.g., average daily pain score, number and severity of paroxysms) indicated the considerable potential for this compound in the treatment of trigeminal neuralgia.114 Two phase 3 trials in trigeminal neuralgia are planned, but recruitment has not commenced as of 2021.59
Funapide (5, Table 6) is considered a non-selective VGSC inhibitor.55 Oral administration of funapide (400 mg BID) produced analgesic effects in an exploratory clinical study of four patients with IEM, a gain-of-function mutation of NaV1.7.115 Subsequently, the compound was developed as a topical ointment to reduce systemic drug exposure and related adverse events, but phase 2 trials in postherpetic neuralgia (PHN) and knee OA failed to meet the primary endpoints (NCT02068599, NCT02365636). However, the result of subpopulation analysis in a phase 2a proof-of-concept study for PHN was worthy of notice. Patients with PHN carrying the R1150W polymorphism (arginine to tryptophan substitution at 1150), a gain-of-function NaV1.7 variant, exhibited more marked analgesic responses to funapide ointment than wild-type carriers.116 Recently, Flexion Therapeutics initiated a phase 1 trial in post-surgical pain patients undergoing bunionectomy using a thermosensitive extended-release hydrogel formulation.
Pfizer/Icagen's compound PF-05089771 (16, Table 7) is an epoch-making compound exhibiting potent and selective NaV1.7 blocking activity,73,74 and their development strategy and approach were informative and suggestive. Pfizer conducted an exploratory clinical micro-dosing study to investigate the intravenous and oral pharmacokinetic profile of four compounds (PF-05089771, PF-05150122, PF-05186462 and PF-05241328) and selected PF-05089771 as a clinical candidate (NCT01165736).117 In a phase 1 single ascending dose study of PF-05089771, exploratory pharmacodynamic parameters such as the heat pain perception threshold and odour threshold (Sniffin' Sticks) were investigated in healthy subjects, but the results have not been disclosed (NCT01259882). In another phase 1 trial for pharmacodynamic evaluation, a single oral dose of PF-05089771 (300 mg) produced no significant analgesic effects in a battery of human evoked pain models (PainCart test). Namely, PF-05089771 alone or concomitantly with pregabalin had no effects on the heat pain detection threshold under normal and UVB-exposed skin conditions and pain tolerance to electrical, pressure and cold pressor stimulation in healthy subjects (NCT02349607).118 In phase 1 multiple-dose studies, skin rash observed at oral doses of 450 and 600 mg BID was determined as the dose-limiting adverse effect (NCT01365637, NCT01529671). Therefore, an additional phase 1 trial with a 4 week titration regimen was conducted to reduce the incidence of skin rash (NCT01772264). Although the results of the additional phase 1 trial have not been disclosed, the 4 week titration regimen might not have been effective. In fact, the maximum dose was set at 150 mg BID without titration in the last phase 2 trial for patients with diabetic peripheral neuropathy because of the potential for drug–drug interactions and cholesterol elevation (NCT02215252).119 Phase 2 trials of PF-05089771 were conducted in three pathological pain states (i.e., postoperative dental pain, IEM, diabetic peripheral neuropathy). In a phase 2 trial for postoperative dental pain (NCT01529346), PF-05089771 (150, 450 and 1600 mg) significantly reduced pain, but its efficacy was far inferior to that of ibuprofen (400 mg). On the contrary, a single oral dose of PF-05089771 (1600 mg) produced analgesic effects in a phase 2 trial of five patients with IEM (NCT01769274), and the compound inhibited the hyperexcitability of iPSC-derived sensory neurons from patients with IEM in in vitro electrophysiological experiments.102 The results of a phase 2 trial in diabetic peripheral neuropathy have been published.119 PF-05089771 (150 mg BID, 4 weeks) was well tolerated, and a trend towards efficacy was noted (a decrease in the weekly average pain score). However, the effect was weaker than that of pregabalin (150 mg BID) and not statistically significant versus placebo at 4 weeks. For treatment-related adverse events, total and LDL cholesterol elevation was observed, but skin rash and CV- or CNS-related adverse events were not observed in this trial. It is interesting to note that the results of the primary analysis were somewhat different from those of the sensitivity analysis (mixed-model repeated-measures analysis). In the results of the primary analysis registered on https://ClinicalTrials.gov, the time-course changes in the weekly average pain score of the PF-05089771 treatment group were almost identical to those of the pregabalin treatment group (NCT02215252). The aforementioned outcomes of the three phase 2 trials suggest that the appropriate target indication for selective NaV1.7 inhibitors is NP rather than nociceptive pain. At present, PF-05089771 cannot be found in Pfizer's pipeline.
Daiichi Sankyo's sulphonamide derivative DS-1971a (19, Table 7) is a potent and selective NaV1.7 inhibitor with a favourable pharmacological and toxicological profile.48 A series of phase 1 trials of DS-1971a were successfully completed, and its favourable safety profile was confirmed. DS-1971a exhibited good safety and tolerability up to 1500 mg in a single ascending dose study (NCT02107885) and up to 1200 mg per day (600 mg BID or 400 mg TID) in a 14 day multiple-dose study (NCT02190058). In contrast to PF-05089771, skin rash or cholesterol elevation was not observed in any phase 1 trials. Two phase 2 trials for peripheral NP (i.e., diabetic peripheral neuropathy, radiculopathy attributable to lumbar spinal stenosis) had been planned (NCT02673866, JapicCTI-163 193), but both trials were terminated immediately before initiation because of toxicological findings in long-term toxicity studies in rats. Daiichi Sankyo has decided to discontinue the development programme of this compound.
Although several other NaV1.7 inhibitors have been found on clinical trial databases and corporate websites, their development statuses have not been updated, and some of them have already disappeared from their companies' pipelines.
AstraZeneca completed a phase 1 trial of AZD3161 (7, Table 6) in 2011 that assessed the effects of intradermal AZD3161 on quantitative sensory testing in normal and UVC-exposed skin in healthy subjects (NCT01240148). However, the study results have not been disclosed, and the compound has disappeared from the company's pipeline.
Sumitomo Dainippon/Sunovion completed three phase trials of DSP-2230, a NaV1.7/1.8 dual inhibitor, in 2013 (ISRCTN07951717, ISRCTN80154838, ISRCTN02543559). The study results have not been disclosed, and the compound has disappeared from their pipeline. Although Sumitomo Dainippon and Sunovion have also mentioned DSP-3905, a selective NaV1.7 inhibitor, on their websites, the detailed information of this compound has not been disclosed.
Genentech completed two phase 1 trials of GDC-0276/RG7893 (38, Table 8; NCT02856152) and GDC-0310/RG6029 (NCT02742779) in 2016 and 2017, respectively, and discontinued their subsequent development.78 The detailed results of phase 1 trial for GDC-0276/RG7893 were published, and various adverse events such as liver transaminase elevation, diarrhoea, dizziness and hypotension were observed in the treatment groups.98
Two phase 1 trials of BIIB095 (Biogen/Convergence) were registered on https://ClinicalTrials.gov (NCT03454126, NCT04106050), but this compound is no longer present in Biogen's pipeline.
Chromocell/Astellas announced that the FDA granted fast track designation to their candidate compound CC8464/ASP1807 for idiopathic small fibre neuropathy in October 2016.120 Although they completed a series of phase 1 trials of CC8464/ASP1807, Astellas announced the discontinuation of the research and development programme of CC8464/ASP1807 in January 2019.
10. Discussion
10.1. The relationship between in vivo efficacy and NaV1.7 coverage among three types of VGSC inhibitors
Conventional VGSC inhibitors exhibit efficacy in preclinical studies with less target coverage than sulphonamides or acyl sulphonamides. In the formalin model, compounds 14 (0.25-fold) and 15 (0.035-fold) elicited statistically significant efficacy at target coverage values of less than 1, whereas at least several folds of target coverage were required for sulphonamides 17 (10-fold), 18 (10-fold), 22 (1.3-fold in rats, 10-fold in mice), 23 (100-fold), 24 (11, 87, 326-fold) and 32 (3-fold) and acyl sulphonamides 40 (3.4-fold) and 41 (23-fold). In IEM mice, sulphonamides 16 (82-fold), 21 (25-fold) and 24 (28-fold) required higher NaV1.7 coverage than acyl sulphonamides 36 (3.4-fold), 37 (0.61-fold), 38 (1-fold), 41 (5-fold) and 42 (0.17-fold). This observation is aligned with the conclusion of Xenon/Genentech.77,78 The exception is sulphonamides 19 (0.058-fold), 30 (0.04-fold) and 32 (0.5-fold), which displayed statistically significant efficacy at target coverage values of less than 1-fold. In particular, sulphonamide 30 was effective in a mouse formalin model at 0.04-fold target coverage. As the in vitro potency of compound 30 was evaluated near the resting state, high in vitro potency in the resting state may be related to high in vivo efficacy. Different animal models were used in the evaluation of 19, 30 and 32 (19, thermal hyperalgesia in PSNL mice; 30, mouse formalin; 32, mouse CFA model). Although such exceptions are acknowledged, it is fair to conclude that sulphonamides require higher target coverage than acyl sulphonamides, whereas conventional VGSC inhibitors require the lowest target coverage. It is interesting that the subtype selectivity decreases in the same order, suggesting the possibility that other VGSC subtypes contribute to the effects or indicating synergic effects. However, the reasons for the higher required target coverage for sulphonamides or acyl sulphonamides remain unclear.
10.2. DRG concentration
Some groups disclosed the target coverage in the DRG, and the measurement of drug concentrations in the DRG may not solve the PK/PD discrepancy, as Bristol-Myers Squibb reported that compounds with similar target coverage in the DRG displayed completely different efficacy in the same in vivo model.92 It should be noted that the same company concluded that a small difference in target coverage in the mouse DRG (8.7- and 5.5-fold) may cause a significant difference in in vivo efficacy. The fact that only compound 40 was efficacious against mechanosensitivity in a mouse sural nerve when the compounds were applied directly to the nerve was a critical observation for solving the PK/PD discrepancy.94 As reported by Xenon/Genentech, the possibility of increased partitioning in DRG membranes opposed to the DRG itself should not be ignored for highly lipophilic compounds.101 Hence, although exposure in the DRG is important for in vivo efficacy, the measurement of DRG coverage does not always solve the PK/PD discrepancy.
10.3. Residence time
In addition to the IC50, the residence time for a target protein is suggested to be an important factor that determines the pharmacological effects in vivo.121 A comparative study with acyl sulphonamides and aryl sulphonamides revealed that the long residence time of NaV1.7 inhibitors likely contributes to their superior analgesic effects in vivo. Furthermore, the analgesic effects of acyl sulphonamides were dramatically enhanced by repeated dosing in a mouse chronic pain model without drug accumulation in plasma.77 These results indicate that continuous inhibition of NaV1.7 currents induced by a compound with a long residence time results in a potent analgesic effect in vivo. In fact, DS-1971, possessing a longer residence time than mexiletine, exerted a potent analgesic effect in NP model mice at a lower target coverage.48
As a long residence time in NaV1.7 and repeated dosing in vivo can contribute to potent efficacy, a longer duration in plasma or the target tissues may contribute to potent efficacy. As discussed by the Xenon/Genentech group,101 compounds with sharp PK profiles (high blood level peaks) would contribute to in vivo efficacy less than compounds without sharp PK peaks because of the lower accumulation of the latter compounds. Thus, the time above the IC50 considering the unbound fraction could be an important factor for the discussion of in vivo efficacy. Further studies are essential to clarify the relationship between the PK curve shape and in vivo efficacy by developing multiple chemical scaffolds.
10.4. In vitro assay protocol
Sulphonamides and acyl sulphonamides inhibit NaV1.7 in a state-dependent manner. They exert inhibitory activity by preferably binding to and stabilising the inactivated state of NaV1.7, whereas the inhibitory activity (IC50) is diminished in the resting state. Conversely, it is difficult to predict the certain state of NaV1.7 in vivo. A plausible solution is discovering compounds with high potency in the resting state, enabling compounds to bind and stabilise all states of NaV1.7 because inhibition in the resting state enables the retention of inhibitory activity in both inactivated and open states. In fact, compound 30 reported by Merck inhibited NaV1.7 in a conformation close to the resting state and demonstrated potent in vivo efficacy at extremely low NaV1.7 coverage (0.04-fold).91 Janssen reported a close relationship between the NaV1.7 resting state and pharmacological insensitivity to pain through the identification of the peptide JNJ63955918.122 Both cases may approximate the pathological condition observed in patients with CIP. As it is essential for humans to respond to a stimulus with a certain threshold to avoid dangerous signals, this condition is an adverse event in patients with CIP. However, it is possible to avoid such adverse events via proper dose setting. Therefore, research to develop compounds that inhibit NaV1.7 in the resting state could overcome this PK/PD discrepancy. If the current landscape is considered, less toxic sulphonamides with activity in the resting state may be the first target.
10.5. The complexity of in vivo models
Pain signals are transmitted from the PNS to the CNS, and the final behavioural decision is made by the CNS. Almost all in vivo models for evaluating analgesic agents are based on animal behaviour, and the final behavioural decision is made by the CNS. Therefore, the CNS may contribute to the PK/PD discrepancy to some extent even though the effect of NaV1.7 is restricted in the PNS.123 The PK/PD discrepancy may be caused by multiple reasons, and many MOAs targeting the CNS face the same problems. Further studies among multiple targets are needed. To connect this PK/PD discrepancy in animal models and humans, further translational research and the development of biomarkers have been awaited.
11. Conclusions
We reviewed three categories of NaV1.7 inhibitors, namely conventional VGSC inhibitors, sulphonamides and acyl sulphonamides, combined with their background, history, assay technology and assay protocol. Conventional VGSC inhibitors are generally non-selective, and their inhibitory potency is in the micromolar range. Based on these characteristics, these conventional inhibitors displayed the lowest NaV1.7 coverage at efficacious in vivo plasma concentrations among the three categories. The target coverage is usually less than 1-fold. One plausible reason for the low target coverage may be the synergic effects of inhibiting multiple ion channels. If these inhibitors exhibit a sufficient safety margin, they have the potential to be novel analgesic agents. Sulphonamide derivatives, which were first disclosed by Pfizer, induced potent selective NaV1.7 inhibition in vitro, but their in vivo efficacy in preclinical studies was generally poor given their high target coverage requirements. Conversely, acyl sulphonamide derivatives tend to require lower target coverage than sulphonamide derivatives to achieve robust in vivo efficacy. Thus, the required target coverage increases in the order of sulphonamides, acyl sulphonamides and conventional VGSC inhibitors, and the subtype selectivity decreases in the same order.
Although a clear solution for resolving the PK/PD discrepancy cannot be addressed in this review, we propose the following points for consideration to acquire clinical candidates with robust efficacy by overcoming the PK/PD disconnection: 1) longer residence time in NaV1.7 in vitro, 2) potent inhibitory activity in the resting state in vitro, 3) longer duration in plasma (longer duration above the IC50 considering the free fraction), 4) proper selection of the target indication based on the preclinical study and 5) the utilisation of sulphonamides.
We believe continuous research and development of novel NaV1.7 inhibitors are essential for launching novel analgesic agents.
Abbreviations used
- NaV
Voltage-gated sodium channel
- NSAID
Non-steroidal anti-inflammatory drug
- GI
Gastrointestinal
- CV
Cardiovascular
- NP
Neuropathic pain
- CNS
Central nervous system
- VGSC
Voltage-gated sodium channel
- PNS
Peripheral nervous system
- CIP
Congenital insensitivity to pain
- PEPD
Paroxysmal extreme pain disorder
- IEM
Inherited erythromelalgia
- TTX
Tetrodotoxin
- DRG
Dorsal root ganglion
- STX
Saxitoxin
- VSD
Voltage-sensing domain
- PD
Pore domain
- BTX
Batrachotoxin
- VTD
Veratridine
- ACT
Aconitine
- r
Recombinant
- PbTx
Brevetoxins
- CTX
Ciguatoxins
- LA
Local anaesthetics
- SA
Sulphonamides
- FLIPR
Fluorescent imaging plate reader
- FRET
Fluorescence resonance energy transfer
- HTS
High-throughput screening
- AAS
Atomic absorption spectroscopy
- VIPR
Voltage ion probe reader
- STZ
Streptozotocin
- CCI
Chronic constriction injury
- BID
Twice daily
- CFA
Complete Freund's adjuvant
- SNL
Spinal nerve ligation
- MIA
Monosodium iodoacetate
- OA
Osteoarthritis
- PSNL
Partial sciatic nerve ligation
- MOA
Mechanism of action
- OB
Olfactory bulb
- fMRI
Functional magnetic resonance imaging
- PHN
Postherpetic neuralgia
- HV
Healthy volunteer
- SD
Single dose
- MD
Multiple dose
- RBA
Relative bioavailability
- DDI
Drug–drug interaction
Conflicts of interest
Both authors are employees of Daiichi Sankyo Co., Ltd.
Supplementary Material
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00081d
Notes and references
- Fayaz A. Croft P. Langford R. M. Donaldson L. J. Jones G. T. BMJ Open. 2012;6:e010364. doi: 10.1136/bmjopen-2015-010364. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gaskin D. J. Richard P. J. Pain. 2012;13:715. doi: 10.1136/bmjopen-2015-010364. [DOI] [PubMed] [Google Scholar]
- For a review of analgesic agents, see: ; Woolf C. J. Biol. Psychiatry. 2020;87:74. doi: 10.1016/j.biopsych.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; Obeng S. Hiranita T. León F. McMahon L. R. McCurdy C. R. J. Med. Chem. 2021;64:6523. doi: 10.1016/j.biopsych.2019.06.017. [DOI] [PubMed] [Google Scholar]
- For a review of NaV1.7 inhibitors, see: ; Mulcahy J. V. Pajouhesh H. Beckley J. T. Delwig A. Bois J. D. Hunter J. C. J. Med. Chem. 2019;62:8695. doi: 10.1021/acs.jmedchem.8b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alsaloum M. Higerd G. P. Effraim P. R. Waxman S. G. Nat. Rev. Neurol. 2020;16:689. doi: 10.1021/acs.jmedchem.8b01906. [DOI] [PubMed] [Google Scholar]; Kushnarev M. Pirvulescu I. P. Candido K. D. Knezevic N. N. Expert Opin. Invest. Drugs. 2020;29:259. doi: 10.1021/acs.jmedchem.8b01906. [DOI] [PubMed] [Google Scholar]; Zhang Y. Wang K. Yu Z. J. Med. Chem. 2020;63:15258. doi: 10.1021/acs.jmedchem.8b01906. [DOI] [PubMed] [Google Scholar]
- Leon-Casasola O. A. Am. J. Med. 2013;126:S3. doi: 10.1016/j.amjmed.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Conaghan P. G. Cook A. D. Hamilton J. A. Tak P. P. Nat. Rev. Rheumatol. 2019;15:355. doi: 10.1038/s41584-019-0221-y. [DOI] [PubMed] [Google Scholar]; Osani M. C. Vaysbrot E. E. Zhou M. McAlindon T. E. Bannuru R. R. Arthritis Care Res. 2020;72:641. doi: 10.1038/s41584-019-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam U. Sloan G. Tesfaye S. Drugs. 2020;80:363. doi: 10.1007/s40265-020-01259-2. [DOI] [PubMed] [Google Scholar]; Freeman R. Durso-DeCruz E. Emir B. Diabetes Care. 2008;31:1448. doi: 10.1007/s40265-020-01259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; Goodman C. W. Brett A. S. N. Engl. J. Med. 2017;377:411. doi: 10.1007/s40265-020-01259-2. [DOI] [PubMed] [Google Scholar]
- Mulroy M. Reg. Anesth. Pain Med. 2002;27:556. doi: 10.1097/00115550-200211000-00008. [DOI] [PubMed] [Google Scholar]
- Cox J. J. Reimann F. Nicholas A. K. Thornton G. Roberts E. Springell K. Karbani G. Jafri H. Mannan J. Raashid Y. Al-Gazali L. Hamamy H. Valente E. M. Gorman S. Williams R. McHale D. P. Wood J. N. Gribble F. M. Woods G. C. Nature. 2006;444:894. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]; Goldberg Y. P. MacFarlane J. MacDonald M. L. Thompson J. Dube M. P. Mattice M. Fraser R. Young C. Hossain S. Pape T. Payne B. Radomski C. Donaldson G. Ives E. Cox J. Younghusband H. B. Green R. Duff A. Boltshauser E. Grinspan G. A. Dimon J. H. Sibley B. G. Andria G. Toscano E. Kerdraon J. Bowsher D. Pimstone S. N. Samuels M. E. Sherrington R. Hayden M. R. Clin. Genet. 2007;71:311. doi: 10.1038/nature05413. [DOI] [PubMed] [Google Scholar]; Ahmad S. Dahllund L. Eriksson A. B. Hellgren D. Karlsson U. Lund P.-E. Meijer I. A. Meury L. Mills T. Moody A. Morinville A. Morten J. O'Donnell D. Raynoschek C. Salter H. Rouleau G. A. Krupp J. J. Hum. Mol. Genet. 2007;16:2114. doi: 10.1038/nature05413. [DOI] [PubMed] [Google Scholar]
- Weiss J. Pyrski M. Jacobi E. Bufe B. Willnecker V. Schick B. Zizzari P. Gossage S. J. Greer C. A. Leinders-Zufall T. Woods C. G. Wood J. N. Zufall F. Nature. 2011;472:186. doi: 10.1038/nature09975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman C. R. Baker M. D. Parker K. A. Moffatt S. Elmslie F. V. Abrahamsen B. Ostman J. Klugbauer N. Wood J. N. Gardiner R. M. Rees M. Neuron. 2006;52:767. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]; Drenth J. P. te Morsche R. H. M. Guillet G. Taieb A. Kirby R. L. Jansen J. B. J. Invest. Dermatol. 2005;124:1333. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]; Yang Y. Wang Y. Li S. Xu Z. Li H. Ma L. Fan J. Bu D. Fan Z. Wu G. Jin J. Ding B. Zhu X. Shen Y. J. Med. Genet. 2004;41:171. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dib-Hajj S. D. Rush A. M. Cummins T. R. Hisama F. M. Novella S. Tyrrell L. Brain. 2005;128:1847. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Marshall L. Waxman S. G. Devigili G. Eleopra R. Pierro T. Lombardi R. Rinaldo S. Lettieri C. Faber C. G. Merkies I. S. Waxman S. G. Lauria G. Pain. 2014;155:1702. doi: 10.1016/j.pain.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Gingras J. Smith S. Matson D. J. Johnson D. Nye K. Couture L. Feric E. Yin R. Moyer B. D. Peterson M. L. Rottman J. B. Beiler R. J. Malmberg A. B. McDonough S. I. PLoS One. 2014;9:e105895. doi: 10.1371/journal.pone.0105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett M. S. Nassar M. A. Clark A. K. Passmore G. Dickenson A. H. Wang F. Malcangio M. Wood J. N. Nat. Commun. 2012;3:791. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]; Minett M. S. Falk S. Santana-Varela S. Bogdanov Y. D. Nassar M. A. Heegaard A.-M. Wood J. N. Cell Rep. 2014;6:301. doi: 10.1038/ncomms1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubinska B. Chen L. Alsaloum M. Rampal N. Matson D. J. Yang C. Taborn K. Zhang M. Youngblood B. Liu D. Galbreath E. Allred S. Lepherd M. Ferrando R. Kornecook T. J. Lehto S. G. Waxman S. G. Moyer B. D. Dib-Hajj S. Gingras J. Mol. Pain. 2019;15:1. doi: 10.1177/1744806919881846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A. M. Alemán F. Catroli G. F. Hunt M. Hu M. Dailamy A. Pla A. Woller S. A. Palmer N. Parekh U. McDonald D. Roberts A. J. Goodwill V. Dryden I. Hevner R. F. Delay L. dos Santos G. G. Yaksh T. L. Mali P. Sci. Transl. Med. 2021;13:eaay9056. doi: 10.1126/scitranslmed.aay9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N. Linley J. E. Baker M. D. Minett M. S. Cregg R. Werdehausen R. Rugiero F. Wood J. N. Brain. 2012;135:2585. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]; Catterall W. A. Goldin A. L. Waxman S. G. Pharmacol. Rev. 2005;57:397. doi: 10.1093/brain/aws225. [DOI] [PubMed] [Google Scholar]
- Meisler M. H. Kearney J. A. J. Clin. Invest. 2005;115:2010. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]; Schmunk G. Gargus J. J. Front. Genet. 2013;4:222. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. A. Liu S. Tanaka M. Cummins T. R. Waxman S. G. Pain. 2004;108:237. doi: 10.1016/j.pain.2003.12.035. [DOI] [PubMed] [Google Scholar]; Hains B. C. Klein J. P. Saab C. Y. Craner M. J. Black J. A. Waxman S. G. J. Neurosci. 2003;23:8881. doi: 10.1016/j.pain.2003.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme C. A. Bezzina C. R. Cardiovasc. Ther. 2010;28:287. doi: 10.1111/j.1755-5922.2010.00210.x. [DOI] [PubMed] [Google Scholar]; Zaklyazminskaya E. Dzemeshkevich S. Biochim. Biophys. Acta. 2016;1863:1799. doi: 10.1111/j.1755-5922.2010.00210.x. [DOI] [PubMed] [Google Scholar]
- Morinville A. Fundin B. Meury L. Jureus A. Sandberg K. Krupp J. Ahmad S. O'Donnell D. J. Comp. Neurol. 2007;504:680. doi: 10.1002/cne.21484. [DOI] [PubMed] [Google Scholar]; Djouhri L. Newton R. Levinson S. R. Berry C. M. Carruthers B. Lawson S. N. J. Physiol. 2003;546.2:565. doi: 10.1002/cne.21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr B. J. Souslova V. McMahon S. B. Wood J. N. NeuroReport. 2001;12:3077. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]; Faber C. G. Lauria G. Merkies I. S. J. Cheng X. Han C. Ahn H.-S. Persson A.-K. Hoeijmakers J. G. J. Gerrits M. M. Pierro T. Lombardi R. Kapetis D. Dib-Hajj S. D. Waxman S. G. Proc. Natl. Acad. Sci. U. S. A. 2012;109:19444. doi: 10.1097/00001756-200110080-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis M. F. Honore P. Shieh C.-C. Chapman M. Joshi S. Zhang X.-F. Kort M. Carroll W. Marron B. Atkinson R. Thomas J. Liu D. Krambis M. Liu Y. McGaraughty S. Chu K. Roeloffs R. Zhong C. Mikusa J. P. Hernandez G. Gauvin D. Wade C. Zhu C. Pai M. Scanio M. Shi L. Drizin I. Gregg R. Matulenko M. Hakeem A. Gross M. Johnson M. Marsh K. Wagoner P. K. Sullivan J. P. Faltynek C. R. Krafte D. S. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8520. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kort M. E. Drizin I. Gregg R. J. Scanio M. J. Shi L. Gross M. F. Atkinson R. N. Johnson M. S. Pacofsky G. J. Thomas J. B. Carroll W. A. Krambis M. J. Liu D. Shieh C. C. Zhang X. Hernandez G. Mikusa J. P. Zhong C. Joshi S. Honore P. Roeloffs R. Marsh K. C. Murray B. P. Liu J. Werness S. Faltynek C. R. Krafte D. S. Jarvis M. F. Chapman M. L. Marron B. E. J. Med. Chem. 2008;51:407. doi: 10.1021/jm070637u. [DOI] [PubMed] [Google Scholar]; Bagal S. K. Bungay P. J. Denton S. M. Gibson K. R. Glossop M. S. Hay T. L. Kemp M. I. Lane C. A. L. Lewis M. L. Maw G. N. Million W. A. Payne C. E. Poinsard C. Rawson D. J. Stammen B. L. Stevens E. B. Thompson L. R. ACS Med. Chem. Lett. 2015;6:650. doi: 10.1021/jm070637u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabel S. Ahmad S. Tirilomis P. Stehle T. Mustroph J. Knierim M. Dybkova N. Bengel P. Holzamer A. Hilker M. Streckfuss-Bömeke K. Hasenfuss G. Maier L. S. Sossalla S. Basic Res. Cardiol. 2020;115:20. doi: 10.1007/s00395-020-0780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D. Basbaum A. I. Nature. 2001;413:203. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Vetter I. Deuis J. R. Mueller A. Israel M. R. Starobova H. Zhang A. Rash L. D. Mobli M. Pharmacol. Ther. 2017;172:73. doi: 10.1016/j.pharmthera.2016.11.015. [DOI] [PubMed] [Google Scholar]; Noreng S. Li T. Payandeh J. J. Mol. Biol. 2021;433:166967. doi: 10.1016/j.pharmthera.2016.11.015. [DOI] [PubMed] [Google Scholar]
- Beaudoin S., Laufer-Sweiler M. C., Markworth C. J., Marron B. E., Millan D. S., Rawson D. J., Reister S. M., Sasaki K., Storer R. I., Stupple P. A., Swain N. A., West C. W. and Zhou S., PCT Int. Appl., WO2010079443, Jul 15, 2010
- Payandeh J. Scheuer T. Zheng N. Catterall W. A. Nature. 2011;475:353. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S. Mukund S. Deng L. Khakh K. Chang E. Ho H. Shriver S. Young C. Lin S. Johnson Jr. J. P. Wu P. Li J. Coons M. Tam C. Brillantes B. Sampang H. Mortara K. Bowman K. K. Clark K. R. Estevez A. Xie Z. Verschoof H. Grimwood M. Dehnhardt C. Andrez J.-C. Focken T. Sutherlin D. P. Safina B. S. Starovasnik M. A. Ortwine D. F. Franke Y. Cohen C. J. Hackos D. H. Koth C. M. Payandeh J. Science. 2015;350:aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Shen H. Liu D. Wu K. Lei J. Yan N. Science. 2019;363:1303. doi: 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- For a review of structural biology of VGSC, see: ; Cestele S. Catterall W. A. Biochimie. 2000;82:883. doi: 10.1016/S0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]; England S. de Groot M. J. Br. J. Pharmacol. 2009;158:1413. doi: 10.1016/S0300-9084(00)01174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stevens M. Peigneur S. Tytgat J. Front. Pharmacol. 2011;2:1. doi: 10.1016/S0300-9084(00)01174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xu L. Ding X. Wang T. Mou S. Sun H. Hou T. Drug Discovery Today. 2019;24:1389. doi: 10.1016/S0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- O'Malley H. A. Isom L. L. Annu. Rev. Physiol. 2015;77:481. doi: 10.1146/annurev-physiol-021014-071846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang S. Yoo J. Gong X. Liu D. Han Q. Luo X. Chang W. Chen G. Im S.-T. Kim Y. H. Strong J. A. Zhang M.-Z. Zhang J.-M. Lee S.-Y. Ji R.-R. Neurosci. Bull. 2018;34:22. doi: 10.1007/s12264-018-0203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Wang X. Ensign B. Li M. Wu L. Guia A. Xu J. Drug Discovery Today. 2001;6:1278. doi: 10.1016/S1359-6446(01)02095-5. [DOI] [PubMed] [Google Scholar]
- Priest B. T. Swensen A. M. McManus O. B. Curr. Pharm. Des. 2007;13:2325. doi: 10.2174/138161207781368701. [DOI] [PubMed] [Google Scholar]
- Castle N. Printzenhoff D. Zellmer S. Antonio B. Wickenden A. Silvia C. Comb. Chem. High Throughput Screening. 2009;12:107. doi: 10.2174/138620709787047993. [DOI] [PubMed] [Google Scholar]
- Felix J. P. Williams B. S. Priest B. T. Brochu R. M. Dick I. E. Warren V. A. Yan L. Slaughter R. S. Kaczorowski G. J. Smith M. M. Garcia M. L. Assay Drug Dev. Technol. 2004;2:260. doi: 10.1089/1540658041410696. [DOI] [PubMed] [Google Scholar]; Benjamin E. R. Pruthi F. Olanrewaju S. Ilyin V. I. Crumley G. Kutlina E. Valenzano K. J. Woodward R. M. J. Biomol. Screening. 2006;11:29. doi: 10.1089/1540658041410696. [DOI] [PubMed] [Google Scholar]; Laroche Y. Storme V. Meutter J. D. Messens J. Lauwereys M. Nat. Biotechnol. 1994;12:1119. doi: 10.1089/1540658041410696. [DOI] [PubMed] [Google Scholar]
- Trivedi S. Dekermendjian K. Julien R. Huang J. Lund P.-E. Krupp J. Kronqvist R. Larsson O. Bostwick R. Assay Drug Dev. Technol. 2008;6:167. doi: 10.1089/adt.2007.090. [DOI] [PubMed] [Google Scholar]
- Du Y. Days E. Romaine I. Abney K. K. Kaufmann K. Sulikowski G. Stauffer S. Lindsley C. W. Weaver C. D. ACS Chem. Neurosci. 2015;6:871. doi: 10.1021/acschemneuro.5b00004. [DOI] [PubMed] [Google Scholar]
- Bennett D. L. Clark A. J. Huang J. Waxman S. G. Dib-Hajj S. D. Physiol. Rev. 2019;99:1079. doi: 10.1152/physrev.00052.2017. [DOI] [PubMed] [Google Scholar]
- Yanagidate F. and Strichartz G. R., Handb. Exp. Pharmacol., 2007, vol. 177, p. 95 [DOI] [PubMed] [Google Scholar]
- Glaaser I. W. and Clancy C. E., Handb. Exp. Pharmacol., 2006, vol. 171, p. 99 [DOI] [PubMed] [Google Scholar]
- Yogeeswari P. Ragavendran J. V. Thirumurugan R. Saxena A. Sriram D. Curr. Drug Targets. 2004;5:589. doi: 10.2174/1389450043345227. [DOI] [PubMed] [Google Scholar]; Errington A. C. Stöhr T. Lees G. Curr. Top. Med. Chem. 2005;5:15. doi: 10.2174/1389450043345227. [DOI] [PubMed] [Google Scholar]
- Amir R. Argoff C. E. Bennett G. J. Cummins T. R. Durieux M. E. Gerner P. Gold M. S. Porreca F. Strichartz G. R. J. Pain. 2006;7:S1. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- Cummins T. R. Sheets P. L. Waxman S. G. Pain. 2007;131:243. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; Krafte D. S. Bannon A. W. Curr. Opin. Pharmacol. 2008;8:50. doi: 10.1016/j.pain.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Hinckley C. A. Kuryshev Y. Sers A. Barre A. Buisson B. Naik H. Hajos M. Mol. Pharmacol. 2021;99:49. doi: 10.1124/molpharm.120.000079. [DOI] [PubMed] [Google Scholar]
- Tanaka K. Suzuki S. Kobayashi H. Shibuya S. Naito S. Kimoto H. Domon Y. Kubota K. Kitano Y. Yokoyama T. Shimizugawa A. Koishi R. Asano D. Takasuna K. Shinozuka T. Chem. Pharm. Bull. 2020;68:653. doi: 10.1248/cpb.c20-00126. [DOI] [PubMed] [Google Scholar]
- Shinozuka T. Kobayashi H. Suzuki S. Tanaka K. Karanjule N. Hayashi N. Tsuda T. Tokumaru E. Inoue M. Ueda K. Kimoto H. Domon Y. Takahashi S. Kubota K. Yokoyama T. Shimizugawa A. Koishi R. Asano D. Sakakura T. Takasuna K. Abe Y. Watanabe T. Kitano Y. J. Med. Chem. 2020;63:10204. doi: 10.1021/acs.jmedchem.0c00259. [DOI] [PubMed] [Google Scholar]
- Amir R. Michaelis M. Devor M. J. Neurosci. 1999;19:8589. doi: 10.1523/JNEUROSCI.19-19-08589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pedroarena C. M. Pose I. E. Yamuy J. Chase M. H. Morales F. R. J. Neurophysiol. 1999;82:1465. doi: 10.1523/JNEUROSCI.19-19-08589.1999. [DOI] [PubMed] [Google Scholar]; Liu C. N. Michaelis M. Amir R. Devor M. J. Neurophysiol. 2000;84:205. doi: 10.1523/JNEUROSCI.19-19-08589.1999. [DOI] [PubMed] [Google Scholar]
- Pal R. Kumar B. Akhtar J. Chawla P. A. Bioorg. Chem. 2021;115:105230. doi: 10.1016/j.bioorg.2021.105230. [DOI] [PubMed] [Google Scholar]; Bagheri S. Haddadi R. Saki S. Kourosh-Arami M. Komaki A. Int. J. Dev. Neurosci. 2021;81:669. doi: 10.1016/j.bioorg.2021.105230. [DOI] [PubMed] [Google Scholar]
- Sheets P. L. Heers C. Stoehr T. Cummins T. R. J. Pharmacol. Exp. Ther. 2008;326:89. doi: 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- Comer A. M. Lamb H. M. Drugs. 2000;59:245. doi: 10.2165/00003495-200059020-00006. [DOI] [PubMed] [Google Scholar]; Mick G. Correa-Illanes G. Curr. Med. Res. Opin. 2012;28:937. doi: 10.2165/00003495-200059020-00006. [DOI] [PubMed] [Google Scholar]
- Perucca E. Yasothan U. Clincke G. Kirkpatrick P. Nat. Rev. Drug Discovery. 2008;7:973. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]; Rauck R. L. Shaibani A. Biton V. Simpson J. Koch B. Clin. J. Pain. 2007;23:150. doi: 10.1038/nrd2764. [DOI] [PubMed] [Google Scholar]
- Beyreuther B. Callizot N. Stöhr T. Eur. J. Pharmacol. 2006;539:64. doi: 10.1016/j.ejphar.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Goldberg Y. P. Cohen C. J. Namdari R. Price N. Cadieux J. A. Young C. Sherrington R. Pimstone S. N. Pain. 2014;155:837. doi: 10.1016/j.pain.2014.01.003. [DOI] [PubMed] [Google Scholar]
- McDermott L. A. Weir G. A. Themistocleous A. C. Segerdahl A. R. Blesneac I. Baskozos G. Clark A. J. Millar V. Peck L. J. Ebner D. Tracey I. Serra J. Bennett D. L. Neuron. 2019;101:905. doi: 10.1016/j.neuron.2019.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty D. R. Alvaro G. Derjean D. Giblin G. M. P. Gunn K. Large C. Macpherson D. T. Morisset V. Owen D. Palmer J. Rugiero F. Tate S. Hinckley C. A. Naik H. ACS Med. Chem. Lett. 2020;11:1678. doi: 10.1021/acsmedchemlett.0c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis J. R. Wingerd J. S. Winter Z. Durek T. Dekan Z. Sousa S. R. Zimmermann K. Hoffmann T. Weidner C. Nassar M. A. Alewood P. F. Lewis R. J. Vetter I. Toxins. 2016;8:78. doi: 10.3390/toxins8030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha M. Cheshire W. P. Finnigan H. Giblin K. Naik H. Palmer J. Tate S. Zakrzewska J. M. J. Pain Res. 2020;13:1601. doi: 10.2147/JPR.S247182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kers I. Csjernyik G. Macsari I. Nylöf M. Sandberg L. Skogholm K. Bueters T. Eriksson A. B. Oerther S. Lund P.-E. Venyike E. Nyström J.-E. Besidski Y. Bioorg. Med. Chem. Lett. 2012;22:5618. doi: 10.1016/j.bmcl.2012.06.105. [DOI] [PubMed] [Google Scholar]
- Yang S.-W. Ho G. D. Tulshian D. Bercovici A. Tan Z. Hanisak J. Brumfield S. Matasi J. Sun X. Sakwa S. A. Herr R. J. Zhou X. Bridal T. Urban M. Vivian J. Rindgen D. Sorota S. Bioorg. Med. Chem. Lett. 2014;24:4958. doi: 10.1016/j.bmcl.2014.09.038. [DOI] [PubMed] [Google Scholar]
- Hoyt S. B. London C. Abbadie C. Felix J. P. Garcia M. L. Jochnowitz N. Karanam B. V. Li X. Lyons K. A. McGowan E. Priest B. T. Smith M. M. Warren V. A. Thomas-Fowlkes B. S. Kaczorowski G. J. Duffy J. L. Bioorg. Med. Chem. Lett. 2013;23:3640. doi: 10.1016/j.bmcl.2013.03.121. [DOI] [PubMed] [Google Scholar]
- Frost J. M. DeGoey D. A. Shi L. Gum R. J. Fricano M. M. Lundgaard G. L. El-Kouhen O. F. Hsieh G. C. Neelands T. Matulenko M. A. Daanen J. F. Pai M. Ghoreishi-Haack N. Zhan C. Zhang X.-F. Kort M. E. J. Med. Chem. 2016;59:3373. doi: 10.1021/acs.jmedchem.6b00063. [DOI] [PubMed] [Google Scholar]
- Schenkel L. B. DiMauro E. F. Nguyen H. N. Chakka N. Du B. Foti R. S. Guzman-Perez A. Jarosh M. La D. S. Ligutti J. Milgram B. C. Moyer B. D. Peterson E. A. Roberts J. Yu V. L. Weisset M. M. Bioorg. Med. Chem. Lett. 2017;27:3817. doi: 10.1016/j.bmcl.2017.06.054. [DOI] [PubMed] [Google Scholar]
- Sparling B. A. Yi S. Able J. Bregman H. DiMauro E. F. Foti R. S. Gao H. Guzman-Perez A. Huang H. Jarosh M. Kornecook T. Ligutti J. Milgram B. C. Moyer B. D. Youngblood B. Yub V. L. Weissa M. M. Med. Chem. Commun. 2017;8:744. doi: 10.1039/C6MD00578K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. S. Felix J. P. Priest B. T. Brochu R. M. Dai K. Hoyt S. B. London C. Tang Y. S. Duffy J. L. Parsons W. H. Kaczorowski G. J. Garcia M. L. Biochemistry. 2007;46:14693. doi: 10.1021/bi7018207. [DOI] [PubMed] [Google Scholar]
- Hoyt S. B. London C. Gorin D. Wyvratt M. J. Fisher M. H. Abbadie C. Felix J. P. Garcia M. L. Li X. Lyons K. A. McGowan E. MacIntyre D. E. Martin W. J. Priest B. T. Ritter A. Smith M. M. Warren V. A. Williams B. S. Kaczorowski G. J. Parsons W. H. Bioorg. Med. Chem. Lett. 2007;17:4630. doi: 10.1016/j.bmcl.2007.05.076. [DOI] [PubMed] [Google Scholar]
- McGowan E. Hoyt S. B. Li X. Lyons K. A. Abbadie C. Anesth. Analg. 2009;109:951. doi: 10.1213/ane.0b013e3181b01b02. [DOI] [PubMed] [Google Scholar]
- Macsari I. Besidski Y. Csjernyik G. Nilsson L. I. Sandberg L. Yngve U. Åhlin K. Bueters T. Eriksson A. B. Lund P.-E. Venyike E. Oerther S. Blakeman K. H. Luo L. Arvidsson P. I. J. Med. Chem. 2012;55:6866. doi: 10.1021/jm300623u. [DOI] [PubMed] [Google Scholar]
- Bregman H. Berry L. Buchanan J. L. Chen A. Bingfan Du B. Feric E. Hierl M. Huang L. Immke D. Janosky B. Johnson D. Li X. Ligutti J. Liu D. Malmberg A. Matson D. McDermott J. Miu P. Nguyen H. N. Patel V. F. Waldon D. Wilenkin B. Zheng X. Zou A. McDonough S. I. DiMauro E. F. J. Med. Chem. 2011;54:4427. doi: 10.1021/jm200018k. [DOI] [PubMed] [Google Scholar]
- Matson D. J. Hamamoto D. T. Bregman H. Cooke M. DiMauro D. F. Huang L. Johnson D. Li X. McDermott J. Morgan C. Wilenkin B. Malmberg A. B. McDonough S. I. Simone D. A. PLoS One. 2015;10:e0138140. doi: 10.1371/journal.pone.0138140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrashdi B. Dawod B. Schampel A. Tacke S. Kuerten S. Marshall J. S. Côté P. D. J. Neuroinflammation. 2019;16:215. doi: 10.1186/s12974-019-1622-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ramachandra R. Elmslie K. S. Mol. Pain. 2016;12:1. doi: 10.1186/s12974-019-1622-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain N. A. Batchelor D. Beaudoin S. Bechle B. M. Bradley P. A. Brown A. D. Brown B. Butcher K. J. Butt R. P. Chapman M. L. Denton S. Ellis D. Galan S. Gaulier S. M. Greener B. S. de Groot M. J. Glossop M. S. Gurrell I. K. Hannam J. Johnson M. S. Lin Z. Markworth C. J. Marron B. E. Millan D. S. Nakagawa S. Pike A. Printzenhoff D. Rawson D. J. Ransley S. J. Reister S. M. Sasaki K. Storer R. I. Stupple P. A. West C. W. J. Med. Chem. 2017;60:7029. doi: 10.1021/acs.jmedchem.7b00598. [DOI] [PubMed] [Google Scholar]
- Theile J. W. Fuller M. D. Chapman M. L. Mol. Pharmacol. 2016;90:540. doi: 10.1124/mol.116.105437. [DOI] [PubMed] [Google Scholar]
- Alexandrou A. J. Brown A. R. Chapman M. L. Estacion M. Turner J. Mis M. A. Wilbrey A. Payne E. C. Gutteridge A. Cox P. J. Doyle R. Printzenhoff D. Lin Z. Marron B. E. West C. Swain N. A. Storer R. I. Stupple P. A. Castle N. A. Hounshell J. A. Rivara M. Randall A. Dib-Hajj S. D. Krafte D. Waxman S. G. Patel M. K. Butt R. P. Stevens E. B. PLoS One. 2016;11:e0152405. doi: 10.1371/journal.pone.0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K. Santos S. Chapman M. L. Krafte D. S. Marron B. E. West C. W. Krambis M. J. Antonio B. M. Zellmer S. G. Printzenhoff D. Padilla K. M. Lin Z. Wagoner P. K. Swain N. A. Stupple P. A. de Groot M. Butt R. P. Castle N. A. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2724. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankar G. Goodchild S. J. Howard S. Nelkenbrecher K. Waldbrook M. Dourado M. Shuart N. G. Lin S. Young C. Xie Z. Khakh K. Chang E. Sojo L. E. Lindgren A. Chowdhury S. Decker S. Grimwood M. Andrez J. Dehnhardt C. M. Pang J. Chang J. H. Safina B. S. Sutherlin D. P. Johnson, Jr. J. P. Hackos D. H. Robinette C. L. Cohen C. J. Cell Rep. 2018;24:3133. doi: 10.1016/j.celrep.2018.08.063. [DOI] [PubMed] [Google Scholar]
- Safina B. S. McKerrall S. J. Sun S. Chen C.-A. Chowdhury S. Jia Q. Li J. Zenova A. Y. Andrez J.-C. Bankar G. Bergeron P. Chang J. H. Chang E. Chen J. Dean R. Decker S. M. DiPasquale A. Focken T. Hemeon I. Khakh K. Kim A. Kwan R. Lindgren A. Lin S. Maher J. Mezeyova J. Misner D. Nelkenbrecher K. Pang J. Reese R. Shields S. D. Sojo L. Sheng T. Verschoof H. Waldbrook M. Wilson M. S. Xie Z. Young C. Zabka T. S. Hackos D. H. Ortwine D. F. White A. D. Johnson, Jr. J. P. Robinette C. L. Dehnhardt C. M. Cohen C. J. Sutherlin D. P. J. Med. Chem. 2021;64:2953. doi: 10.1021/acs.jmedchem.1c00049. [DOI] [PubMed] [Google Scholar]
- Focken T. Liu S. Chahal N. Dauphinais M. Grimwood M. E. Chowdhury S. Hemeon I. Bichler P. Bogucki D. Waldbrook M. Bankar G. Sojo L. E. Young C. Lin S. Shuart N. Kwan R. Pang J. Chang J. H. Safina B. S. Sutherlin D. P. Johnson, Jr. J. P. Dehnhardt C. M. Mansour T. S. Oballa R. M. Cohen C. J. Robinette C. L. ACS Med. Chem. Lett. 2016;7:277. doi: 10.1021/acsmedchemlett.5b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdas V. Talwar R. Kanoje V. Loriya R. M. Banerjee M. Patil P. Joshi A. A. Datrange L. Das A. K. Walke D. S. Kalhapure V. Khan T. Gote G. Dhayagude U. Deshpande S. Shaikh J. Chaure G. Pal R. R. Parkale S. Suravase S. Bhoskar S. Gupta R. V. Kalia A. Yeshodharan R. Azhar M. Daler J. Mali V. Sharma G. Kishore A. Vyawahare R. Agarwal G. Pareek H. Budhe S. Nayak A. Warude D. Gupta P. K. Joshi P. Joshi S. Darekar S. Pandey D. Wagh A. Nigade P. B. Mehta M. Patil V. Modi D. Pawar S. Verma M. Singh M. Das S. Gundu J. Nemmani K. Bock M. G. Sharma S. Bakhle D. Kamboj R. K. Palle V. P. J. Med. Chem. 2020;63:6107. doi: 10.1021/acs.jmedchem.0c00361. [DOI] [PubMed] [Google Scholar]
- Graceffa R. F. Boezio A. A. Able J. Altmann S. Berry L. M. Boezio C. Butler J. R. Chu-Moyer M. Cooke M. DiMauro E. F. Dineen T. A. Feric Bojic F. Foti R. S. Fremeau, Jr. R. T. Guzman-Perez A. Gao H. Gunaydin H. Huang H. Huang L. Ilch C. Jarosh M. Kornecook T. Kreiman C. R. La D. S. Ligutti J. Milgram B. C. Lin M.-H. J. Marx I. E. Nguyen H. N. Peterson E. A. Rescourio G. Roberts J. Schenkel L. Shimanovich R. Sparling B. A. Stellwagen J. Taborn K. Vaida K. R. Wang J. Yeoman J. Yu V. Zhu D. Moyer B. D. Weiss M. M. J. Med. Chem. 2017;60:5990. doi: 10.1021/acs.jmedchem.6b01850. [DOI] [PubMed] [Google Scholar]
- McKerrall S. J. Nguyen T. Lai K. W. Bergeron P. Deng L. DiPasquale A. Chang J. H. Chen J. Chernov-Rogan T. Hackos D. H. Maher J. Ortwine D. F. Pang J. Payandeh J. Proctor W. R. Shields S. D. Vogt J. Ji P. Liu W. Ballini E. Schumann L. Tarozzo G. Bankar G. Chowdhury S. Hasan A. Johnson, Jr. J. P. Khakh K. Lin S. Cohen C. J. Dehnhardt C. M. Safina B. S. Sutherlin D. P. J. Med. Chem. 2019;62:4091. doi: 10.1021/acs.jmedchem.9b00141. [DOI] [PubMed] [Google Scholar]
- La D. S. Peterson E. A. Bode C. Boezio A. A. Bregman H. Chu-Moyer M. Y. Coats J. DiMauro E. F. Dineen T. A. Du B. Gao H. Graceffa R. Gunaydin H. Guzman-Perez A. Fremeau, Jr. R. Huang X. Ilch C. Kornecook T. J. Kreiman C. Ligutti J. Jasmine Lin M. H. McDermott J. S. Marx I. Matson D. J. McDonough S. I. Moyer B. D. Nguyen H. N. Taborn K. Yu V. Weiss M. M. Bioorg. Med. Chem. Lett. 2017;27:3477. doi: 10.1016/j.bmcl.2017.05.070. [DOI] [PubMed] [Google Scholar]
- Pero J. E. Rossi M. A. Lehman H. D. G. F. Kelly III M. J. Mulhearn J. J. Wolkenberg S. E. Cato M. J. Clements M. K. Daley C. J. Filzen T. Finger E. N. Gregan Y. Henze D. A. Jovanovska A. Klein R. Kraus R. L. Li Y. Liang A. Majercak J. M. Panigel J. Urban M. O. Wang J. Wang Y. Houghton A. K. Layton M. E. Bioorg. Med. Chem. Lett. 2017;27:2683. doi: 10.1016/j.bmcl.2017.04.040. [DOI] [PubMed] [Google Scholar]
- Roecker A. J. Egbertson M. Jones K. L. G. Gomez R. Kraus R. L. Li Y. Koser A. J. Urban M. O. Klein R. Clements M. Panigel J. Daley C. Wang J. Finger E. N. Majercak J. Santarelli V. Gregan I. Cato M. Filzen T. Jovanovska A. Wang Y. Wang D. Joyce L. A. Sherer E. C. Peng X. Wang X. Sun H. Coleman P. J. Houghton A. K. Layton M. E. Bioorg. Med. Chem. Lett. 2017;27:2087. doi: 10.1016/j.bmcl.2017.03.085. [DOI] [PubMed] [Google Scholar]
- Kornecook T. J. Yin R. Altmann S. Be X. Berry V. Ilch C. P. Jarosh M. Johnson D. Lee J. H. Lehto S. G. Ligutti J. Liu D. Luther J. Matson D. Ortuno D. Roberts J. Taborn K. Wang J. Weiss M. M. Yu V. Zhu D. X. D. Fremeau, Jr. R. T. Moyer B. D. J. Pharmacol. Exp. Ther. 2017;362:146. doi: 10.1124/jpet.116.239590. [DOI] [PubMed] [Google Scholar]
- Marx I. E. Dineen T. A. Able J. Bode C. Bregman H. Chu-Moyer M. DiMauro E. F. Du B. Foti R. S. Fremeau, Jr. R. T. Gao H. Gunaydin H. Hall B. E. Huang L. Kornecook T. Kreiman C. R. La D. S. Ligutti J. Lin M.-H. Liu S. McDermott J. S. Moyer B. D. Peterson E. A. Roberts J. T. Rose P. E. Wang J. Youngblood B. D. Yu V. L. Weiss M. M. ACS Med. Chem. Lett. 2016;7:1062. doi: 10.1021/acsmedchemlett.6b00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M. M. Dineen T. A. Marx I. E. Altmann S. Boezio A. Bregman H. Chu-Moyer M. DiMauro E. F. Bojic E. F. Foti R. S. Gao H. Graceffa R. Gunaydin H. Guzman-Perez A. Huang H. Huang L. Jarosh M. Kornecook T. Kreiman C. R. Ligutti J. La D. S. Lin M.-H. J. Liu D. Moyer B. D. Nguyen H. N. Peterson E. A. Rose P. E. Taborn K. Youngblood B. D. Yu V. Fremeau, Jr. R. T. J. Med. Chem. 2017;60:5969. doi: 10.1021/acs.jmedchem.6b01851. [DOI] [PubMed] [Google Scholar]
- Storer R. I. Pike A. Swain N. A. Alexandrou A. J. Bechle B. M. Blakemore D. C. Brown A. D. Castle N. A. Corbett M. S. Flanagan N. J. Fengas D. Johnson M. S. Jones L. H. Marron B. E. Payne C. E. Printzenhoff D. Rawson D. J. Rose C. R. Ryckmans T. Sun J. Theile J. W. Torella R. Tseng E. Warmus J. S. Bioorg. Med. Chem. Lett. 2017;27:4805. doi: 10.1016/j.bmcl.2017.09.056. [DOI] [PubMed] [Google Scholar]
- Kraus R. L. Zhao F. Pall P. S. Zhou D. Vardigan J. D. Danziger A. Li Y. Daley C. Ballard J. E. Clements M. K. Klein R. M. Holahan M. A. Greshock T. J. Kim R. M. Layton M. E. Burgey C. S. Serra J. Henze D. A. Houghton A. K. Sci. Transl. Med. 2021;13:eaay1050. doi: 10.1126/scitranslmed.aay1050. [DOI] [PubMed] [Google Scholar]
- Roecker A. J. Layton M. E. Pero J. E. Kelly III M. J. Greshock T. J. Kraus R. L. Li Y. Klein R. Clements M. Daley C. Jovanovska A. Ballard J. E. Wang D. Zhao F. Brunskill A. P. J. Peng X. Wang X. Sun H. Houghton A. K. Burgey C. S. ACS Med. Chem. Lett. 2021;12:1038. doi: 10.1021/acsmedchemlett.1c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.-J. Guernon J. Shi J. Ditta J. Robbins K. J. Rajamani R. Easton A. Newton A. Bourin C. Mosure K. Soars M. G. Knox R. J. Matchett M. Pieschl R. L. Post-Munson D. J. Wang S. Herrington J. Graef J. Newberry K. Bristow L. J. Meanwell N. A. Olson R. Thompson L. A. Dzierba C. J. Med. Chem. 2017;60:2513. doi: 10.1021/acs.jmedchem.6b01918. [DOI] [PubMed] [Google Scholar]
- Wua Y.-J. Guernon J. McClure A. Luo G. Rajamani R. Ng A. Easton A. Newton A. Bourin C. Parker D. Mosure K. Barnaby O. Soars M. G. Knox R. J. Matchett M. Pieschl R. Herrington J. Chen P. Sivarao D. V. Bristow L. J. Meanwell N. A. Bronson J. Olson R. Thompson L. A. Dzierba C. Bioorg. Med. Chem. 2017;25:5490. doi: 10.1016/j.bmc.2017.08.012. [DOI] [PubMed] [Google Scholar]
- Luo G. Chen L. Easton A. Newton A. Bourin C. Shields E. Mosure K. Soars M. G. Knox R. J. Matchett M. Pieschl R. L. Post-Munson D. Wang S. Herrington J. Graef J. Newberry K. Sivarao D. V. Senapati A. Bristow L. Meanwell N. A. Thompson L. A. Dzierba C. D. J. Med. Chem. 2019;62:831. doi: 10.1021/acs.jmedchem.8b01550. [DOI] [PubMed] [Google Scholar]
- Bell A. S., Brown A. D., De Groot M. J., Gaulier S. M., Lewthwaite R. A., Marsh I. R., Millan D. S., Perez Pacheco M., Rawson D. J., Sciammetea N., Storer R. I. and Swain N. A., PCT Int. Appl., WO2012007861, Jan 19, 2012
- Rawson D. J., Storer R. I. and Swain N. A., PCT Int. Appl., WO2013088315, Jun 20, 2013
- Bell A. S., De Groot M. J., Lewthwaite R. A., Marsh I. R., Sciammetta N., Storer R. I. and Swain N. A., PCT Int. Appl., WO 2012095781, July 19, 2012
- Rothenberg M. E. Tagen M. Chang J. H. Boyce-Rustay J. Friesenhahn M. Hackos D. H. Hains A. Sutherlin D. Ward M. Cho W. Clin. Drug Invest. 2019;39:873. doi: 10.1007/s40261-019-00807-3. [DOI] [PubMed] [Google Scholar]
- DiMauro E. F. Altmann S. Berry L. M. Bregman H. Chakka N. Chu-Moyer M. Bojic E. F. Foti R. S. Fremeau R. Gao H. Gunaydin H. Guzman-Perez A. Hall B. E. Huang H. Jarosh M. Kornecook T. Lee J. Ligutti J. Liu D. Moyer B. D. Ortuno D. Rose P. E. Schenkel L. B. Taborn K. Wang J. Wang Y. Yu V. Weiss M. M. J. Med. Chem. 2016;59:7818. doi: 10.1021/acs.jmedchem.6b00425. [DOI] [PubMed] [Google Scholar]
- Sun S. Jia Q. Zenova A. Y. Wilson M. S. Chowdhury S. Focken T. Li J. Decker S. Grimwood M. E. Andrez J.-C. Hemeon I. Sheng T. Chen C.-A. White A. Hackos D. H. Deng L. Bankar G. Khakh K. Chang E. Kwan R. Lin S. Nelkenbrecher K. Sellers B. D. DiPasquale A. G. Chang J. Pang J. Sojo L. Lindgren A. Waldbrook M. Xie Z. Young C. Johnson J. P. Robinette C. L. Cohen C. J. Safina B. S. Sutherlin D. P. Ortwine D. F. Dehnhardt C. M. J. Med. Chem. 2019;62:908. doi: 10.1021/acs.jmedchem.8b01621. [DOI] [PubMed] [Google Scholar]
- Focken T. Chowdhury S. Zenova A. Grimwood M. E. Chabot C. Sheng T. Hemeon I. Decker S. M. Wilson M. Bichler P. Jia Q. Sun S. Young C. Lin S. Goodchild S. J. Shuart N. G. Chang E. Xie Z. Li B. Khakh K. Bankar G. Waldbrook M. Kwan R. Nelkenbrecher K. Karimi Tari P. Chahal N. Sojo L. Robinette C. L. White A. D. Chen C.-A. Zhang Y. Pang J. Chang J. H. Hackos D. H. Johnson J. P. Cohen C. J. Ortwine D. F. Sutherlin D. P. Dehnhardt C. M. Safina B. S. J. Med. Chem. 2018;61:4810. doi: 10.1021/acs.jmedchem.7b01826. [DOI] [PubMed] [Google Scholar]
- Cao L. McDonnell A. Nitzsche A. Alexandrou A. Saintot P.-P. Loucif A. J. C. Brown A. R. Young G. Mis M. Randall A. Waxman S. G. Stanley P. Kirby S. Tarabar S. Gutteridge A. Butt R. McKernan R. M. Whiting P. Ali Z. Bilsland J. Stevens E. B. Sci. Transl. Med. 2016;8:335ra56. doi: 10.1126/scitranslmed.aad7653. [DOI] [PubMed] [Google Scholar]
- Jo S. Bean B. P. Mol. Pharmacol. 2020;97:377. doi: 10.1124/mol.119.118380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafte D. S. and Roeloffs R., PCT Int. Appl., WO2016009303, January 21, 2016
- Zhang Y. Wang L. Peng D. Zhang Q. Yang Q. Li J. Li D. Tang D. Chen M. Liang S. Liu Y. Wang S. Liu Z. J. Biol. Chem. 2021;296:100326. doi: 10.1016/j.jbc.2021.100326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B. Murray J. K. Andrews K. L. Sham K. Long J. Aral J. Ligutti J. Amagasu S. Liu D. Zou A. Min X. Wang Z. Ilch C. P. Kornecook T. J. Lin M.-H. J. Be X. Miranda L. P. Moyer B. D. Biswas K. J. Med. Chem. 2018;61:9500. doi: 10.1021/acs.jmedchem.8b00736. [DOI] [PubMed] [Google Scholar]
- Biswas K. Nixey T. E. Murray J. K. Falsey J. R. Yin L. Liu H. Gingras J. Hall B. E. Herberich B. Holder J. R. Li H. Ligutti J. Lin M.-H. J. Liu D. Soriano B. D. Soto M. Tran L. Tegley C. M. Zou A. Gunasekaran K. Moyer B. D. Doherty L. Miranda L. P. ACS Chem. Biol. 2017;12:2427. doi: 10.1021/acschembio.7b00542. [DOI] [PubMed] [Google Scholar]
- Kocmalova M. Kollarik M. Canning B. J. Ru F. Herbstsomer R. A. Meeker S. Fonquerna S. Aparici M. Miralpeix M. Chi X. X. Li B. Wilenkin B. McDermott J. Nisenbaum E. Krajewski J. L. Undem B. J. J. Pharmacol. Exp. Ther. 2017;361:172. doi: 10.1124/jpet.116.238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L. Tsuji K. Ujihara I. Liu Q. Pavelkova N. Tsujimura T. Inoue M. Meeker S. Nisenbaum E. McDermott J. S. Krajewski J. Undem B. J. Kollarik M. Canning B. J. Eur. J. Pharmacol. 2021;907:174192. doi: 10.1016/j.ejphar.2021.174192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H. Cikurel K. Burke D. Muscle Nerve. 1998;21:137. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; Zhao F. Holahan M. A. Houghton A. K. Hargreaves R. Evelhoch J. L. Winkelmann C. T. Williams D. S. NeuroImage. 2015;106:364. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; Vallbo Å. B. J. Neurophysiol. 2018;120:1415. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; Vardigan J. D. Houghton A. K. Lange H. S. Adarayan E. D. Pall P. S. Ballard J. E. Henze D. A. Uslane J. M. J. Pain Res. 2018;11:735. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ballard J. E. Pall P. Vardigan J. Zhao F. Holahan M. A. Kraus R. Li Y. Henze D. Houghton A. Burgey C. S. Gibson C. Pharm. Res. 2020;37:181. doi: 10.1002/(SICI)1097-4598(199802)21:2<137::AID-MUS1>3.0.CO;2-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. Kuroda T. Kimoto H. Domon Y. Kubota K. Kitano Y. Yokoyama T. Shimizugawa A. Sugita R. Koishi R. Asano D. Tamaki K. Shinozuka T. Kobayashi H. Bioorg. Med. Chem. Lett. 2015;25:5419. doi: 10.1016/j.bmcl.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Ho G. D. Tulshian D. Bercovici A. Tan Z. Hanisak J. Brumfield S. Matasi J. Heap C. R. Earley W. G. Courneya B. Herr R. J. Zhou X. Bridal T. Rindgen D. Sorota S. Yang S.-W. Bioorg. Med. Chem. Lett. 2014;24:4110. doi: 10.1016/j.bmcl.2014.07.060. [DOI] [PubMed] [Google Scholar]
- Zakrzewska J. M. Palmer J. Ettlin D. A. Obermann M. Giblin G. M. P. Morisset V. Tate S. Gunn K. Trials. 2013;14:402. doi: 10.1186/1745-6215-14-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska J. M. Palmer J. Morisset V. Giblin G. M. P. Obermann M. Ettlin D. A. Cruccu G. Bendtsen L. Estacion M. Derjean D. Waxman S. G. Layton G. Gunn K. Tate S. Lancet Neurol. 2017;16:291. doi: 10.1016/S1474-4422(17)30005-4. [DOI] [PubMed] [Google Scholar]
- Goldberg Y. P. Price N. Namdari R. Cohen C. J. Lamers M. H. Winters C. Price J. Young C. E. Verschoof H. Sherrington R. Pimstone S. N. Hayden M. R. Pain. 2012;153:80. doi: 10.1016/j.pain.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Price N. Namdari R. Neville J. Proctor K. J. W. Kaber S. Vest J. Fetell M. Malamut R. Sherrington R. P. Pimstone S. N. Goldberg Y. P. Clin. J. Pain. 2017;33:310. doi: 10.1097/AJP.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones H. M. Butt R. P. Webster R. W. Gurrell I. Dzygiel P. Flanagan N. Fraier D. Hay T. Iavarone L. E. Luckwell J. Pearce H. Phipps A. Segelbacher J. Speed B. Beaumont K. Clin. Pharmacokinet. 2016;55:875. doi: 10.1007/s40262-015-0365-0. [DOI] [PubMed] [Google Scholar]
- Siebenga P. van Amerongen G. Hay J. L. McDonnell A. Gorman D. Butt R. Groeneveld G. J. Clin. Transl. Sci. 2020;13:318. doi: 10.1111/cts.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell A. Collins S. Ali Z. Iavarone L. Surujbally R. Kirby S. Butt R. P. Pain. 2018;159:1465. doi: 10.1097/j.pain.0000000000001227. [DOI] [PubMed] [Google Scholar]
- Astellas Corporate Web site, https://www.astellas.com/en/news/7921, accessed January 2022
- Copeland R. A. Pompliano D. L. Meek T. D. Nat. Rev. Drug Discovery. 2007;6:252. doi: 10.1038/nrd2281. [DOI] [PubMed] [Google Scholar]
- Flinspach M. Xu Q. Piekarz A. D. Fellows R. Hagan R. Gibbs A. Liu Y. Neff R. A. Freedman J. Eckert W. A. Zhou M. Bonesteel R. Pennington M. W. Eddinger K. A. Yaksh T. L. Hunter M. Swanson R. V. Wickenden A. D. Sci. Rep. 2017;7:39662. doi: 10.1038/srep39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagles D. A. Chow C. Y. King G. F. Br. J. Pharmacol. 2020:1. doi: 10.1111/bph.15327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.