

Abstract
The emergence and global dissemination of Severe Acute Respiratory Syndrome virus 2 (SARS-CoV-2) variants of concern (VOCs) have been described as the main factor driving the Coronavirus Disease 2019 pandemic. In Brazil, the Gamma variant dominated the epidemiological scenario during the first period of 2021. Many Brazilian regions detected the Delta variant after its first description and documented its spread. To monitor the introduction and spread of VOC Delta, we performed Polymerase Chain Reaction (PCR) genotyping and genome sequencing in ten regional sentinel units from June to October 2021 in the State of Minas Gerais (MG). We documented the introduction and spread of Delta, comprising 70 per cent of the cases 8 weeks later. Comparing the viral loads of the Gamma and Delta dominance periods, we provide additional evidence that the latter is more transmissible. The spread and dominance of Delta did not culminate in the increase in cases and deaths, suggesting that the vaccination may have restrained the epidemic growth. Analysis of 224 novel Delta genomes revealed that Rio de Janeiro state was the primary source for disseminating this variant in the state of MG. We present the establishment of Delta, providing evidence of its enhanced transmissibility and showing that this variant shift did not aggravate the epidemiological scenario in a high immunity setting.
Keywords: Genomic surveillance, phylogeny, phylogeography, viral lineages, vaccines
1. Introduction
The emergence and global spread of Severe Acute Respiratory Syndrome virus 2 (SARS-CoV-2) variants of concern (VOCs) extensively shaped the Coronavirus Disease 2019 (COVID-19) pandemic (Tao et al. 2021). VOCs are defined by particular characteristics, such as enhanced transmissibility and immunological escape, and their circulation has been shown to cause detrimental changes to COVID-19 epidemiology (CDC 2021; Cherian et al. 2021; Faria et al. 2021; Garcia-Beltran et al. 2021; Volz et al. 2021; Naveca et al. 2021a; Tegally et al. 2021b; Viana et al. 2022; WHO 2022). Specifically, VOC establishment and dissemination have been repeatedly associated with upsurges in cases and deaths (Cele et al. 2021; Faria et al. 2021). Five VOCs have been identified: Alpha, initially identified in the UK (Volz et al. 2021); Beta, identified in South Africa (Tegally et al. 2021b); Gamma, initially identified in Brazil (Faria et al. 2021; Naveca et al. 2021a); Delta, initially identified in India (Cherian et al. 2021); and Omicron, also identified in South Africa (Viana et al. 2022).
Evidence supports that the distinguished epidemiological characteristics reported for VOCs and Variant of Interest (VOI) are genetically determined, mainly driven by non-synonymous mutations on the Spike gene (Greaney et al. 2021; Tao et al. 2021). Two regions in the Spike protein are considered the primary targets for neutralising monoclonal and polyclonal antibodies: the receptor-binding domain (RBD), which is responsible for the main interactions with the host cell, and the N-terminal domain (NTD), capable of interacting with auxiliary receptors (Andreano et al. 2020; Liu et al. 2020; McCallum et al. 2021). Mutations in the spike protein may diminish the efficiency of neutralising antibodies elicited by vaccines or previous infections, culminating in escape from the humoral response. Examples of RBD mutations include K417N/T (VOCs Beta and Gamma), L452R/Q (Delta and Lambda), and E484K (Alpha, Beta, and Gamma) (Deng et al. 2021; Nonaka et al. 2021; Wang et al. 2021a, 2021b). Moreover, mutations in the NTD region can confer resistance against multiple neutralising monoclonal antibodies, as in VOC Alpha (∆144), Gamma (L18F), Beta (∆242-244), and Omicron (∆69-70) (McCarthy et al. 2021; Resende et al. 2021; Xie et al. 2021). Mutations in the RBD may also lead to an increased affinity between the spike protein and angiotensin-converting enzyme 2, the human cellular receptor used by SARS-CoV-2 for docking and entry in the host cell (Lan et al. 2020; Ge et al. 2021). An example is the N501Y mutation (Leung et al. 2021; Tian et al. 2021) shown to impact virus entry and transmissibility, which is present in four out of five currently recognised VOCs (Alpha, Beta, Gamma, and Omicron). Likewise, the mutation P681H/R (close to the S1/S2 furin cleavage site; identified in VOCs Alpha and Delta) has also been reported as mediating faster viral entry (Saito et al. 2022). Noticeably, the evolution of VOCs has been marked by the parallel emergence of several functionally essential mutations (Thye et al. 2021). The combined effect of VOC mutations leads to their enhanced fitness and epidemiological success, replacing previously circulating lineages across various settings (Schmidt et al. 2021; Thye et al. 2021).
Brazil is one of the countries most severely impacted throughout the pandemic and stage for VOCs’ emergence and widespread dissemination (Faria et al. 2021; Moreira et al. 2021a, 2021b; Naveca et al. 2021a). While the epidemiological scenario was heterogeneous around the country in 2020, with the circulation of many lineages, a significant change occurred with the emergence of VOC Gamma. This variant emerged in Manaus (November 2020) (Faria et al. 2021; Naveca et al. 2021a) and caused a massive epidemic wave in the following months, despite a previous estimate of an attack rate of around 70 per cent, near the herd immunity threshold (Buss et al. 2021). During the first period of 2021, multiple reports indicated the spread of VOC Gamma to all Brazilian regions, becoming the main lineage driving the Brazilian epidemic (Tosta et al. 2021; Volpato et al. 2021; Moreira et al. 2021b). A significant consequence of the predominance of VOC Gamma throughout the country was the considerable increment in infections and case fatality rates (Brizzi et al. 2021; Faria et al. 2021).
A novel shift in the Brazilian epidemiological scenario was observed in the second period of 2021 due to the country’s introduction and dissemination of the VOC Delta (Lamarca et al. 2021). The first imported Delta cases from India were documented on boat workers in Maranhão state (Northeast region) on 20 May 2021 (Dos Santos, Ferreira, and da Silva et al. 2021). This variant was detected in Rio de Janeiro (RJ) state in the next month, evidencing an autochthonous transmission (Lamarca et al. 2021), and was later identified in different Brazilian states (Lamarca et al. 2021; Patané et al. 2021; Naveca, Souza, and Nascimento et al. 2021b). Since then, genomic monitoring initiatives have detected VOC Delta across all states and increased its frequency (Brasil 2021; last accessed on 20 October 2021). Nevertheless, the dynamics of the introduction and establishment of VOC Delta in Brazil remain to be clarified. Specifically, the mechanism involved in lineage replacement has not been thoroughly investigated.
Besides the introduction of VOCs Delta in the country, another significant change occurred in Brazil: the advancement of large-scale vaccination programs. More than 82 per cent of the Brazilian population has already completed the SARS-CoV-2 vaccine cycle (see https://conselho.saude.gov.br/vacinometro, last accessed on 10 March 2022). There are four different types of vaccines against COVID-19 in use in Brazil. The first one is CoronaVac (Sinovac), which began to be used in late January 2021 in all Brazilian states. AstraZeneca (ChAdOx1—SARS-CoV-2) started to be administered in late February 2021. Pfizer (BNT162b2) began to be used in early May 2021, and Janssen (JNJ-78436735) started to be used in late June 2021 (see https://conselho.saude.gov.br/vacinometro, last accessed on 10 March 2022). Given the high capacity of immunological escape reported for VOC Delta (Planas et al. 2021), it is unclear how establishing this variant will impact the number of cases and deaths throughout the country.
In this context, we present a genomic epidemiology survey developed in Minas Gerais (MG), the second most populous Brazilian state. MG offers continuous transit of people and transport, as it connects three different regions (Northeast, Southeast, and Central-West) of the country. To monitor the introduction and establishment of VOC Delta in the state, we conducted a population-based study covering ten regional sentinel units from epidemiological weeks (EWs) 25 to 40. Through this effort, we documented the first case of Delta variant in MG and its epidemic growth, reaching a frequency above 70 per cent 8 weeks later. Analysing a large SARS-CoV-2 Polymerase Chain Reaction (PCR)-genotyping data set from MG, we could compare viral loads between Gamma and Delta dominance periods, providing further indirect evidence that the latter variant is more transmissible. In contrast to the significant lineage displacement previously reported in the state, the Delta spread did not culminate with increases in the numbers of cases and deaths, most likely due to the advancement of vaccination coverage. Finally, we selected 224 samples classified as Delta in PCR genotyping to be sequenced. Analysis of the novel Delta genomes allowed the estimation of the dynamics of viral dissemination into the state, revealing that Rio de Janeiro state alone contributed to more than three-quarters of total Delta imports into MG. Altogether, our findings uncover the epidemiology of SARS-CoV-2 in a significant Brazilian state in the second period of 2021 and provide context to the scenarios observed throughout the country.
2. Methods
2.1. Study design
Prospection of cases, collection of samples, and generation of clinical and epidemiological data were obtained from the Observatório de Vigilância Genômica do Estado de Minas Gerais network (OviGen), which integrates the Health Secretary of Minas Gerais (SES-MG) and five different laboratories: the state reference laboratory Fundação Ezequiel Dias (FUNED); Laboratório de Biologia Integrativa (LBI), Núcleo de Ações e Pesquisas em Apoio Diagnóstico (NUPAD), and CT Vacinas from Universidade Federal de Minas Gerais (UFMG); and Instituto Hermes Pardini. To assess the epidemiological situation of different geographical regions of MG, we searched for SARS-CoV-2 variants in 2,407 positive samples for RT-qPCR in ten out of the twenty-eight Regional Health Units (RHUs) distributed all over the state. The RHUs were selected given their geographic position, as they border on other states and/or have a large population flow. The RHUs covered in this study were Belo Horizonte, Coronel Fabriciano, Juiz de Fora, Manhuaçu, Montes Claros, Pedra Azul, Pouso Alegre, Teófilo Otoni, Uberaba, and Unaí (Supplementary Fig. S1). All samples used were obtained from the public health programme of COVID-19 diagnostic in MG state. Random samples were selected according to the number of SARS-CoV-2 cases and population size of each RHU, following the specifications of ECDC (2021). Each RHU contributed with up to twenty samples per week between EWs 29 and 40. Additionally, six RHUs (Belo Horizonte, Manhuaçu, Pedra Azul, Teófilo Otoni, Uberaba, and Unaí) also provided samples from EWs 25 to 28. For some EWs, it was not possible to recover the estimated sample number (twenty samples/RHU) due to the unavailability of samples in RHUs (e.g. EWs 33, 35, and 36) (Supplementary Table S1). Our study was approved by a Research Ethics Committee under protocol Certificado de Apresentação de Apreciação Ética (CAAE) 33202820.7.1001.5348.
2.2. VOCs and VOIs classification and geographical distribution
SARS-CoV-2-positive samples from nasopharyngeal swabs with RT-qPCR cycle threshold (Ct < 30) collected between EWs 25 and 40 (20 June 2021 and 9 October 2021) were genotyped using specific primers and probes to detect defining VOCs and Variants Being Monitored mutations to estimate the frequency of each lineage in MG state. The genotyping approach consists of an RT-qPCR using RhAmp technology (IDT Technologies, USA) to assess circulating variants. This technique uses specific primers designed to detect the following mutations of the viral spike gene: K417T (A22812C), L452R (T22917C), E484K (G23012A), N501Y (A23063T), and P681R (C23604G). All experiments were performed according to Geddes et al. (2021) and Silva et al. (2022). Samples with wild-type profiles at Positions K417, E484, and N501 and mutant at Positions L452R and P681R were classified as Delta and randomly selected for whole-genome sequencing. Further genotyping was done through partial nucleotide sequencing of the S-coding gene through Sanger sequencing (Dorlass et al. 2021). The genotyping data are provided in Supplementary Tables S1 and S2.
Spatio-temporal heatmaps were constructed from the genotyping results. Each RHU coordinate was combined with the genotyping results, and a model estimated the dispersion and interpolation of VOC Delta through the state. Spatial geostatistical modelling and prediction were carried out using the gstat (with inverse distance weighting power = 3) and predict functions from the gstat package (Gräler, Pebesma, and Heuvelink 2016). The plots were made using geobr (Pereira 2019), sf (Pebesma 2018), and ggplot2 packages (Wickham 2016). All analyses were carried out in R software (version 4.1.1) (R Core Team 2021).
2.3. Analysis of RT-qPCR Ct values in MG
RT-qPCR is the standard method for quantifying viral loads, with the Ct value inversely proportional to the amount of viral RNA available in a sample. As SARS-CoV-2 VOCs cause infections characterised by higher viral loads and enhance infectiousness, Ct values have been used to compare transmissibility among different lineages (Walker et al. 2021; Moreira et al. 2021b; King et al. 2022; Silva et al. 2022). We explored the dynamics of RT-qPCR Ct values from July (EW 26) to September (EW 39) 2021 in MG. Anonymised data collected in different units of Instituto Hermes Pardini laboratory were used to estimate differences in the distribution of Ct values in periods dominated by other VOCs. The Ct values were obtained with the TaqPath COVID-19 CE-IVD RT-PCR kit (ThermoFisher Scientific, USA), which uses three distinct viral targets to identify the presence of SARS-CoV-2 (N, ORF1ab, and S) in addition to internal process control (endogenous control) (MS2 gene). This data set contains 22,214 samples collected in 25.4 per cent (217/853) of the municipalities from MG, comprehending all twenty-eight RHUs. EWs were categorised into Delta or Gamma groups based on the frequency of the dominating VOC at the time of collection. Only weeks in which a given VOC exhibited frequency above 70 per cent were included in this analysis. That is, samples collected in weeks where Gamma showed frequency above 70 per cent had this lineage attributed to them. The same procedure was applied for VOC Delta. Differences in Ct values among periods dominated by different VOCs were then compared through linear regression, as implemented in the R software (R Core Team 2021). Ct was the predicted variable in this regression, and the lineage (Gamma or Delta, 0 or 1) was the predicting variable. Hence, the estimated slope indicates the average difference in Ct observed among periods dominated by distinct variants. Data and code used for this analysis are available on our GitHub page (https://github.com/LBI-lab/Establishment-VOC-Delta-in-Minas-Gerais-Brazil—Supplementary Material 1).
2.4. Epidemiological and clinical data
To explore the clinical outcome of SARS-CoV-2 cases classified as Delta by genotyping/sequencing, epidemiological data of approximately 56 per cent (n = 697) were obtained through SES-MG using the databases Sistema de Informações de Vigilância Epidemiológica and e-SUS, respecting patient data protection laws. We evaluated the variables: sex, age, clinical outcome, vaccine strategy, and vaccination cycle and dates (Supplementary Table S3). Sankey plots were constructed to visualise the relationships between clinical outcome, vaccine strategies, number of doses, sex, and age. The interval between vaccine doses and age and the SARS-CoV-2 RT-qPCR diagnostics were plotted in histograms. All analyses were performed in R software (R Core Team 2021) using the packages ggplot2 (Wickham 2016) and ggaluvial (Brunson 2020).
2.5. SARS-CoV-2 whole-genome sequencing
As the frequency of variants has been estimated and monitored from a more significant number of samples through RT-qPCR, we decided to sequence only Delta cases to investigate the viral spread patterns that led to this variant’s establishment in MG. Sequencing was performed using two different technologies, Illumina (Illumina, USA) and IonTorrent (ThermoFisher Scientific, USA). Only SARS-CoV-2-positive samples with Ct < 30 values for virus targets were considered. Libraries were constructed using different protocols. Illumina libraries were prepared using the QIAseq FX DNA Library Prep kit (QIAGEN, Germany) and sequenced on the Illumina MiSeq platform (Illumina, USA) with a v3 (600 cycles) cartridge, following all manufacturer’s protocols. IonTorrent libraries were prepared using the Ion AmpliSeq SARS-CoV-2 Panel (ThermoFisher Scientific, USA) and sequenced on the Ion Torrent PGM platform with a 314-chip kit (ThermoFisher Scientific, USA), according to the manufacturer’s recommendations. Three negative controls were used in all sample processing steps (cDNA synthesis, viral genome amplification, and library preparation in each batch).
2.6. Viral genome assembly and classifications
Sequencing data were processed with a custom pipeline. First, a quality control step was performed with Trimmomatic v0.39 (Bolger, Lohse, and Usadel 2014), which removed adapter and primer sequences, short reads (less than 50 nucleotides), and low-quality bases (Phred score < 30). Then, reads were mapped against the SARS-CoV-2 reference genome (GenBank accession: NC_045512) with Bowtie2 (Langmead et al. 2019). Mapping files were manipulated with samtools (Li 2011), while the bcftools (Li 2011) consensus option was used to estimate consensus genome sequences. Low-coverage sites were masked with bedtools (Quinlan and Hall 2010). The code for the described pipeline is available on GitHub (https://github.com/filiperomero2/ViralUnity). Depth thresholds differed between sequencing technologies employed. For Illumina data, sites with less than 10-fold depth were masked, while for IonTorrent, the minimum threshold was 20-fold. Sequences with less than 70 per cent genome coverage breadth were removed from downstream analysis. Consensus sequences were then classified using the Pangolin tool v.3.1.11 (O’Toole et al. 2021) or the NextClade web application v.1.7.0 (Aksamentov et al. 2021).
2.7. Phylogeographic reconstructions
2.7.1. Data sets collation
To characterise the dynamics of introductions of VOC Delta in MG, we performed a series of phylogenetic analyses. A comprehensive data set was assembled from our sequences and genomes released on the Global initiative on sharing all influenza data (GISAID). EpiCoV database (acknowledgements table is available in Supplementary Table S4). This data set encompasses all Delta (Pango lineage B.1.617.2*, NextStrain clade 21A) sequences from this variant NexStrain build (as captured on 12 October 2021, n = 5,349). In addition, sequences from all Brazilian states were randomly sampled in proportion to the number of estimated Delta cases per week in each state during the study period. All high-quality complete Brazilian sequences sampled between EWs 25 and 40 were downloaded and classified with the pangolin tool to estimate the number of Delta infections in each state. These classifications were used to estimate the frequency of VOC Delta in each state over the weeks. By multiplying this frequency by the number of weekly confirmed cases (retrieved from https://brasil.io/dataset/covid19/files/ and Supplementary Table S5), we obtained estimates of the number of Delta infections in each state over the period. These estimates were then recalculated as proportions (divided by the estimated total number of Delta infections), which were used to calculate the number of sequences to sample, considering a fixed target (n = 5,000, to approximately match the amount of NextStrain background sequences). On occasions where a given state did not harbour sufficient sequences to match the scheme, all available data were included. Detailed calculations and registered sampling gaps are available in Supplementary Table S5. This procedure led to a set of 4,149 sequences, including 358 from MG (total data set size: 9,498).
We assembled an independent secondary data set using the NextStrain background sequences and a uniform sampling scheme for Brazilian sequences to verify the influence of data set composition on the obtained results. Under this scheme, thirty sequences from each state were sampled per week throughout the study period. We set thirty as the target as this is the median number of weekly available Delta sequences for MG. As in the previous data set, they were registered whenever sampling gaps were detected (Supplementary Table S5), and all available data were included. This procedure led to a set of 5,210 sequences, including 334 from MG (total data set size: 10,559). Both sampling designs were employed based on an earlier investigation (Inward, Parag, and Faria 2022).
2.7.2. Phylogenetic analysis, analysis of temporal signal, and identification of Brazilian clades
For each data set, a maximum-likelihood phylogenetic reconstruction was performed with IQ-Tree v2.1.2 (Minh et al. 2020) under the GTR + F + I + G4 nucleotide substitution model (Tavaré 1986; Yang 1994). Shimoidara–Hasegawa-like approximate likelihood ratio test (SH-aLRT) (Guindon et al. 2010) was used to quantify phylogenetic uncertainty along tree branches. Each tree was then submitted to TempEst v1.5.3 (Rambaut et al. 2018) to (1)—find the root position that optimises the relationship between samples collection dates and genetic divergence, in accordance with the molecular clock hypothesis and (2)—identify samples with inconsistent temporal signal (outliers in the root-to-tip regression). For this study, outliers are conservatively defined as sequences that deviate more than 1.5 times the interquartile range of the distribution of the residuals. After removing outliers from the data set, rooted trees were evaluated with the TreeTime (Sagulenko, Puller, and Neher 2018) migration method. We used a two-state (Brazil/International) discrete asymmetric model to identify Brazilian VOC Delta clades.
2.7.3. Bayesian phylogeographic reconstructions
For both data sets, the previous analysis revealed that most MG data clustered within a large Brazilian clade, which comprehended thousands of genome sequences. As traditional Bayesian phylogenetic methods struggle to deal with data sets with these dimensions (Gutierrez et al. 2021), we were prompted to estimate dated phylogenies using a recently developed method from the BEASTv.1.10.5 (pre-release Thorney v0.1.1) package (Suchard et al. 2018). This method uses a Poisson model to scale genetic distances in a provided tree to time (Didelot, Siveroni, and Volz 2021), alleviating the computational burden of standard procedures. To assure the consistency of the scaling process, we used an independently estimated evolutionary rate. This rate was obtained from a full Bayesian analysis performed on a data set that comprises 10 per cent of the sequences (randomly selected, covering all temporal spans) of the major Brazilian clade identified in the proportional data set. This analysis used: (1)—the HKY + G4 model (Hasegawa, Yano, and Kishino 1984; Yang 1994); (2)—the strict clock model; and (3)—the skygrid tree prior (Gill et al. 2013). The number of grids of the latter was set to match the number of months spanned between the most recently sampled sequence and the x-intercept of the root-to-tip regression, a crude estimate of the time for the most recent common ancestor. The cut-off value was set accordingly. Except for the usage of the Hamiltonian skygrid tree, prior (Baele et al. 2020), all used options (priors and operators) were the default. Three independent Markov Chain Monte Carlo runs with 100 million generations sampling every 10,000 steps were performed for this analysis. For the BEAST Thorney runs, the number grids of the skygrid prior were set to match the number of weeks comprehended in the tree. A total of 10 independent chains of 200 million generations sampling every 20,000 steps were carried out for each data set. Logs and trees were combined with Logcombiner (Suchard et al. 2018), removing the initial 10 per cent of chains as burn-in, and convergence (effective sample sizes > 200) was verified on Tracer v1.7.1 (Rambaut et al. 2018).
Next, we used the estimated distribution of posterior dated trees to assess the dynamics of the spread of the VOC Delta in Brazil. We conducted a Discrete Trait Analysis (DTA) (Lemey et al. 2009) with an asymmetric rates model encompassing eight states, four of them being Brazilian politically defined regions: North, Northeast, Central-West, and South; and the four federal units (states) from Southeast Brazil: RJ, São Paulo (SP), Espírito Santo (ES), and MG. We opted for this discretisation scheme as the number of sequences for states in the Southeast far exceeds the quantities available for states from other regions in the data set (Supplementary Table S6). Therefore, grouping by region sequences from less sampled federal units leads to a model with a sensible number of states and diminished bias. Additionally, it has been shown that RJ and SP are important epidemiological hubs in Brazil (Candido et al. 2020), so identifying their relative contribution to the introductions of VOC Delta in MG is a matter of interest in this study.
For each data set, the DTA was performed on a sample of 1,000 empirical dated trees, as previously reported (Candido et al. 2020). We used a robust counting method (Markov jumps) (Minin and Suchard 2007) to map all transition events involving MG and measure the relative contribution of each location to imports into the state. Using the same approach, areas measured which sites received the most exports from MG. Analyses were run sufficiently long to assure convergence and then combined, as described previously. Treeannotator (Suchard et al. 2018) was used to summarize the posterior distribution of dated geographically annotated trees into maximum clade credibility trees.
3. Results
3.1. SARS-CoV-2 Delta replacement of Gamma in MG
We genotyped SARS-CoV-2-positive samples from ten different sentinel regions (Fig. 1A) during EW 25–40 (June to October) to investigate the frequency and spread of VOC Delta in MG. Two-thousand four-hundred and seven samples were genotyped for defining lineage spike mutations (Supplementary Tables S1 and S2). Of these, 1,145 were classified as Gamma, 1,244 samples were classified as VOC Delta, and 18 were classified as other variants. In the first 3 weeks of the study (EW 25–28), VOC Gamma accounted for almost 100 per cent of cases in the state (n = 65 samples genotyped) (Fig. 1B and Supplementary Fig. S2). The first Delta case occurred on EW 29 in MG’s Manhuaçu (Northeast) and Unaí (Central-West) regions. In the following week, Delta cases were identified in the Juiz de Fora area, on the border of RJ state, Brazil’s epicentre of Delta infection. From these three regions, VOC Delta has spread across the state’s territory (Fig. 1A), and its frequency increased to values above 50 per cent on EW 34 (Fig. 1A and B). In EW 35, Unaí and Juiz de Fora were the first regions in the state with 100 per cent Delta prevalence (Fig. 1A and B), and in EW 37, the frequency of VOC Delta throughout the state was 73 per cent (8 weeks after the first case identification) (Fig. 1A). In the last week sampled (EW 40), the frequency of VOC Delta rose to 96 per cent in the whole state (Fig. 1A and B), with founder regions contributing to the community dispersion of Delta in the MG state.
Figure 1.
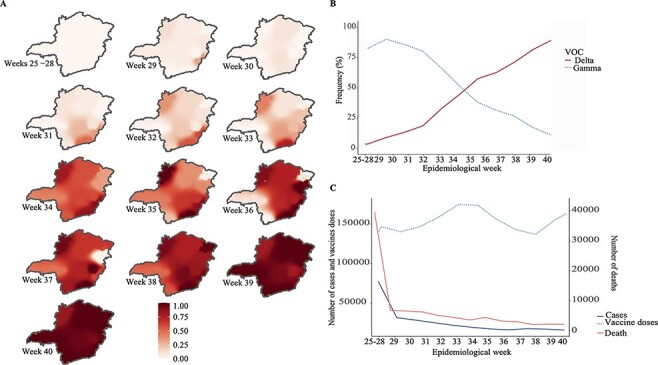
SARS-CoV-2 surveillance PCR-genotyping in Minas Gerais between June and October 2021 (EW 25–40). Spread of VOC Delta through MG. The MG maps show the frequency of VOC Delta in each epidemiological week sampled in our study (EW 25–40). (A). Replacement of VOC Gamma to Delta considering all samples genotyped in our study. Dotted line indicates the frequency information of the VOC Gamma, while solid line indicates the frequency information of the VOC Delta. For each EW, were genotyped: 92 (EW 25–28), 190, 180, 199, 201, 202, 197, 186, 161, 229, 216, 197, 153 samples respectively. Number of samples classified as Gamma during the sampled EW: 56, 179, 169, 165, 161, 156, 83, 66, 58, 53, 43, 16 and 6. Number of samples classified as Delta during the sampled EW: 0, 10, 11, 32, 36, 44, 114, 100, 98, 167, 173, 181 and 147 (B). The number of SARS-CoV-2 cases, deaths and vaccine doses applied in MG from June to October 2021 (EW 25–40). The number of cases in each EW were: 167,239 (EW 25–28), 32,882, 32,222, 29,162, 23749, 22,160, 19,635, 16,039, 16,000, 15,868, 20,549, 14,456 and 12,132. The number of death cases in each EW were: 4,225 (EW 25–28), 803, 786, 755, 633, 562, 474, 561, 438, 412, 314, 327 and 320. The number of vaccine doses applied in each EW were: 153,859 (EW 25–28), 148,598, 137,088, 135,049, 167,747, 174,475, 181,830, 165,036, 119,550, 143,218, 148,259, 174,479 and 184,341 (C).
The death numbers by COVID-19 did not follow the Delta scaling up in the state. Those numbers were compiled from SES-MG public data showing a continuous decline along EWs (Fig. 1C). For example, in June (EWs 25–28), when Gamma was dominant, 224,461 cases were diagnosed, and 5,745 deaths were confirmed (for every thirty-nine cases of COVID-19, one patient died). In October (EW 40), the prevalence of Delta was nearly 100 per cent statewide, 44,490 cases were diagnosed, and 1,035 died (one of every forty-two cases resulted in death). The period marked by a reduction in the number of cases coincides with the advancement of the vaccination programme in MG. Until October (EW 40), 64.14 per cent of the population had a complete vaccination cycle (16,057,319 and 12,263,023 with first and second doses, respectively) (Gerais 2021b) (Fig. 1C). Importantly, we report that the establishment of VOC Delta did not culminate in a novel wave of COVID-19 cases in the state.
3.2. Delta-infected individuals present higher viral loads
To assess whether VOC Delta is associated with higher viral loads in the upper respiratory tract, we considered RT-qPCR Ct values. For this, samples collected between 1 July and 21 August 2021 (EW 26-31) have been assigned to the Gamma group (n = 12,372), while samples collected from 12 to 30 September 2021 (EW 37-39) have been attributed to the Delta group (n = 2,647). The time series of the virus gene targets suggests that the rise in frequency of VOC Delta is associated with modest decays in Ct values (Fig. 2). Specifically, all weeks after this variant exceeded the frequency of 25 per cent displayed median Ct values below the median calculated for the Gamma-dominated period (median Ct values for the Gamma period: 17.31 (N), 16.99 (ORF1ab), and 17.35 (S). Median Ct values for the Delta period: 16.44, P < 0.01, β = −1.38, 95 per cent confidence interval (CI) = ±0.24 (N), 16.20, P < 0.01, β = −1.38, 95 per cent CI = ±0.24 (ORF1ab), 15.92, P < 0.01, β = −1.89, 95 per cent CI = ±0.24 (S)). Minor differences were observed for the MS2 control target (median Ct value for the Gamma period: 25.41; median Ct value for the Delta period: 24.95, P < 0.01, β = −0.41, 95 per cent CI = ±0.12). These results are consistent with the conjecture that VOC Delta induces higher viral loads in the upper respiratory tract.
Figure 2.
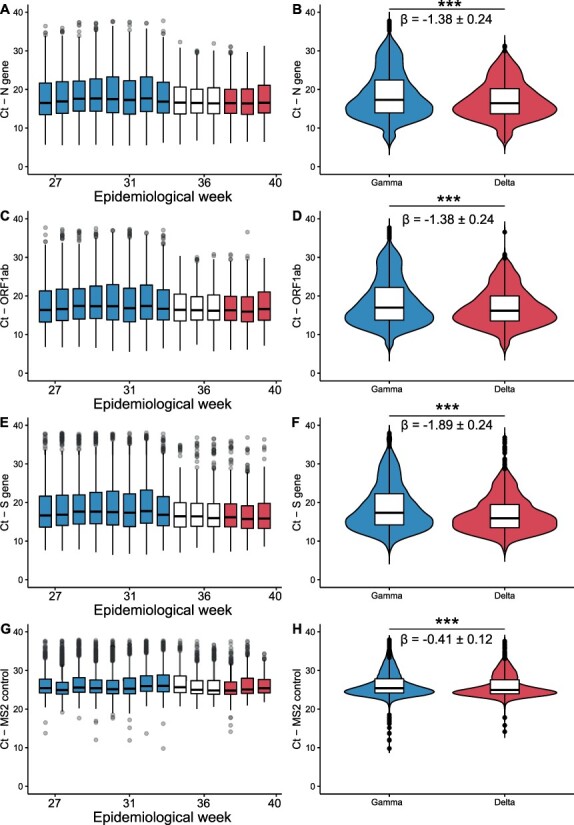
Comparative analysis of RT-qPCR Ct values between Gamma and Delta dominated periods. Boxplots depicting Ct values time series along epidemiological weeks comprehended within the study period (1 July–21 September 2021–EW 26–31) for three viral targets (N gene, A; ORF1ab, C; S gene, E) and the internal control (MS2, G). Colours mark periods where Gamma or Delta variants exhibited frequency above 75%. All weeks after Delta surpassed the frequency of 25% displayed median Ct values below the median calculated for the Gamma-dominated period (Gamma Period: 17.31 (N), 16.99 (ORF1ab), 17.35 (S); Delta period: 16.44 (N), 16.20 (ORF1ab), 15.92 (S)). Statistical comparison of Ct values distributions between periods denotes that the Delta variant might be associated with higher viral loads (N gene, B; ORF1ab, D; S gene, F). A minor decrement on Ct was also detected for the internal control (MS2), though it is 5-fold less than the smaller decrement for the viral targets. In total, 22,214 samples collected in 25.4% (217/853) of the municipalities from MG, comprehending all 28 RHUs, were used in this analysis.
3.3. Clinical and epidemiological aspects of the Delta establishment period in MG
Analysis of 697 confirmed positive cases for SARS-CoV-2—VOC Delta revealed that 67.86 per cent (n = 473) were in middle-aged individuals (18–60 years), 23.96 per cent (n = 167) were in older-aged individuals (>60 years), and a lower proportion (n = 57–8.18 per cent) were in younger-aged individuals (<18 years). The median age was 45 years (interquartile range = 30). Most cases were in female individuals (56.53 per cent—394). A complete summary of epidemiological data herein reported is available in Table 1. The clinical outcome was available only for 298 cases, in which most of them recovered (Recovered group) from the disease (n = 259–86.91 per cent), and thirty-nine individuals died (Death group) (13.08 per cent) (Fig. 3A). Regarding the Recovered group, 68.15 per cent (n = 169/259) were in 20–59 year individuals (median—43 years) and 56.75 per cent (n = 142/259) were female (Fig. 3A). Additionally, 81.45 per cent (n = 202/259) were vaccinated; of these, 126 received the second and fourth doses (Fig. 3B). The Death group showed 74.35 per cent (n = 29/39) individuals between 60 and 91 years of age (median—74 years). Additionally, almost 77 per cent (n = 20/39) were vaccinated with at least one shot of the CoronaVac or AstraZeneca vaccines and another 12.82 per cent (five individuals) were not vaccinated (Fig. 3B).
Table 1.
Number of Delta cases for which the clinical outcome was available, sorted by sex and age.
| Delta cases | |||||
|---|---|---|---|---|---|
| Age | Number of cases | Clinical outcome information | Sex | Number cases | Clinical outcome information |
| 18-60 | 473 | 83 | Female | 394 | 168 |
| >60 | 167 | 84 | Male | 303 | 130 |
| <18 | 57 | 24 | |||
| Total | 697 | 298 | Total | 697 | 298 |
Figure 3.
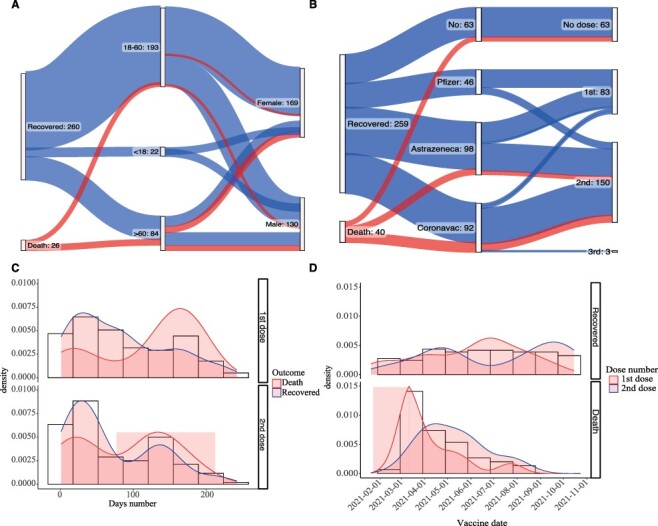
Clinical outcome and vaccination information for cases classified as Delta. Classification of clinical outcome according to age and sex (A). Classification of clinical outcomes according to the type of vaccine and the number of doses (B). The interval between vaccination time and VOC Delta infection diagnosis according to the number of doses administered in each group (C). The density of Delta cases is classified into Recovered and Death groups according to the vaccination date. The density plots indicate the number of doses (D). A total of 298 samples (298/697–43%) were used in the clinical outcome and vaccination analyses.
In general, 79.19 per cent (n = 236/298) were vaccinated with at least one dose, 50.67 per cent (n = 148/298) with the second dose, and 1.34 per cent (n = 4/298) with the third dose. The AstraZeneca vaccine was administered in almost 42 per cent (n = 98/236), followed by CoronaVac with 37.28 per cent (n = 88/236) and Pfizer with 21.18 per cent (n = 50/236). No statistical differences were found between clinical outcome and vaccine strategy, suggesting the efficacy of all vaccines available in Brazil to prevent severe disease and death, even in Delta-confirmed cases.
Particularly for vaccinated individuals, we assessed the interval between vaccination and the COVID-19 diagnosis by RT-qPCR and its clinical outcome. Most Delta-infected individuals from the Recovered group showed two peaks: one of recent vaccination (less than 100 days) and another of vaccination over 100 days (Fig. 3C). Meanwhile, in the death cases associated with Delta infection, most cases are from individuals vaccinated more than 100 days before diagnosis. We verified the months in which the individuals of the three clinical outcome groups were vaccinated. The Recovered group was the most homogeneous, with individuals vaccinated from the start of vaccination in MG until the last day of our study. In the Death group, most were vaccinated early in the year’s first period (Fig. 3D). The Delta death cases confirmed in this study are primarily cases aged over 60 years who were already vaccinated in the first period of this year with two shots (an average of 131 days after the second shot application), suggesting that vaccination time and age are the prevalent factors that may be associated with the clinical outcome despite Delta infection.
3.4. Sequencing, assembly, annotation, and classification
A total of 224 novel genome sequences have been characterised in this study, comprehending 48.44 per cent and 2.37 per cent of VOC Delta sequences previously reported for MG (n = 109) and Brazil (n = 9,480), respectively (as of 11 October 2021). This accounts for 0.036 per cent of all COVID-19 suspected cases in the state (n = 619,905; (Gerais 2021a)). Genome coverage was variable (median: 92.01 per cent, range: 70.17–100 per cent), with mean sequencing depth varying according to the sequencing methodology. Illumina median: 1,099.09×, range: 323.39–3448.06×; IonTorrent median: 1,471.8×, range: 119.20–3,313.33×. Complete sequencing statistics and sample metadata are available in Supplementary Table S7. The Nextclade tool identified all the novel genome sequences as Clade 21A (VOC Delta), in perfect agreement with the genotyping RT-qPCR results. Pangolin further classified nearly all sequences as Delta (B.1.617.2: 118; AY.4: 37, AY.6: 9, AY.25: 2, and AY.33: 1), except for seven sequences classified as B.1, likely reflecting that they missed relevant mutations for this software classification scheme due to incomplete genome coverage. These analyses support that different groups from VOC Delta co-circulate in MG. The pangolin lineage report and the complete Nextclade analysis outputs are available on our GitHub page (https://github.com/LBI-lab/Establishment-VOC-Delta-in-Minas-Gerais-Brazil—Supplementary Material 2).
3.5. Dynamics of introduction and spread of VOC Delta into Brazil and MG
We assembled two different data sets generated under distinct sampling schemes to assess the dynamics of the introduction of VOC Delta in MG. Analysis of the geographic composition of the proportional data set indicates that it effectively reflects the proportion of Delta infections in different Brazilian states and regions (P < 0.01, R2 = 0.91; Fig. 4A and B). Maximum-likelihood phylogenetic analysis performed with this data set revealed that Brazilian sequences cluster along different diverse branches of the Delta tree (Fig. 4C). A total of 113 introductions have been estimated by the TreeTime migration method, from which sixty-six were singletons (imports identified by single sequences) and forty-seven led to the emergence of local transmission clusters with variable sizes (range: 2–2,748).
Figure 4.
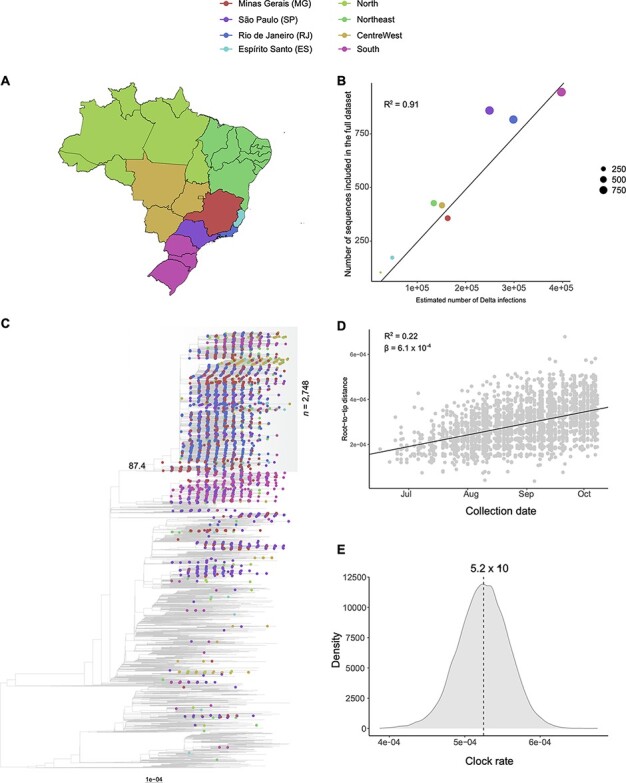
Phylogenetic analysis and investigation of the temporal signal of the proportional dataset (n = 9,498). (A) Map of Brazil with discrete locations (federal states or regions). (B) Linear regression displays a strong correlation (R2 = 0.91) between the number of estimated Delta infections and sequences in the dataset per location, reflecting the proportional sampling scheme. (C) Maximum-likelihood phylogenetic tree inferred from the dataset. Root position was defined by optimizing the correlation between sample collection dates and genetic distances under the molecular clock hypothesis. Tip shapes from Brazilian sequences are colour coded according to the map in panel A. Scale bar indicates substitutions per site. The TreeTime mugration method estimates 113 different introductions of VOC Delta in Brazil. Only three Brazilian clades with more than 100 sequences have been identified, being over 80% of all MG sequences included in the largest of them (n = 2,748; SH-aLRT = 87.4; marked in grey shading). (D) Root-to-tip distance plot inferred from the maximum-likelihood tree inferred for the major Brazilian clade. This plot suggests the dataset contains a clock-like signal (R2 = 0.22; β = 6.1 × 10–4). (E) Bayesian posterior distribution of the clock rate estimated for the major Brazilian clade (median: 5.2 × 10–4 s/s/y; 95 per cent HPD: 4.6 × 10−4–5.9 × 10–4 s/s/y). This rate was estimated on BEAST with a dataset comprehending 275 randomly selected sequences from the clade, covering all of its temporal spans.
Despite the high number of imports detected, only three Brazilian clades with more than 100 sequences have been identified, one mostly composed of sequences from the state of SP (n = 148), the other from sequences from the South region (n = 611), and a highly diverse clade with sequences from all regions (n = 2,748). As the latter clade comprehended 282 of the 345 sequences from MG (81.74 per cent) and the remaining sequences were scattered along the tree, we focused all downstream analyses on the major clade. Importantly, this clade presents a high support value (SH-aLRT = 87.4). Sequences from the major clade were assembled into a separate data set, and a maximum-likelihood tree was inferred. This tree was evaluated on TempEst and presented a substantial temporal signal (R2 = 0.22, β = 6.1 × 10–4; Fig. 4D). Bayesian analysis of a representative set of 275 sequences from this data set suggests an evolutionary rate of 5.2 × 10–4 substitutions per site per year (s/s/y; 95 per cent highest posterior density (HPD): 4.6 × 10−4–5.9 × 10–4; Fig. 4E).
The rooted tree and the median estimated evolutionary rate were used to obtain a posterior distribution of dated trees of the whole clade, which was subsequently used to perform a DTA. The phylogeographic reconstruction indicates RJ as the location of origin of the major Brazilian clade (posterior probability = 1), dated to 10 April 2021 (95 per cent HPD: 7 March 2021–5 May 2021) (Fig. 5A). The model supports the virus disseminated from RJ to all other locations in the data set through multiple exportation events. The earliest introduction to MG was dated 23 May 2021 (95 per cent HPD: 12 May 2021–10 June 2021). Using the Markov jumps method, multiple imports have been inferred (median: 165, 95 per cent HPD: 158–172), with the vast majority of them from RJ (median: 141, 95 per cent HPD: 130–149), followed by ES (median: 8, 95 per cent HPD: 5–11), Central-West (median: 6, 95 per cent HPD: 3–8), South (median: 4, 95 per cent HPD: 1–7), SP (median: 3, 95 per cent HPD: 1–5), and Northeast (median: 3, 95 per cent HPD: 0–6) (Fig. 5B). Exports from MG have also been estimated (median: 18, 95 per cent HPD: 8–26), with the Central-West being the main receiving region (median: 5, 95 per cent HPD: 1–9), followed by RJ (median: 4, 95 per cent HPD: 1–8) (Fig. 5C).
Figure 5.
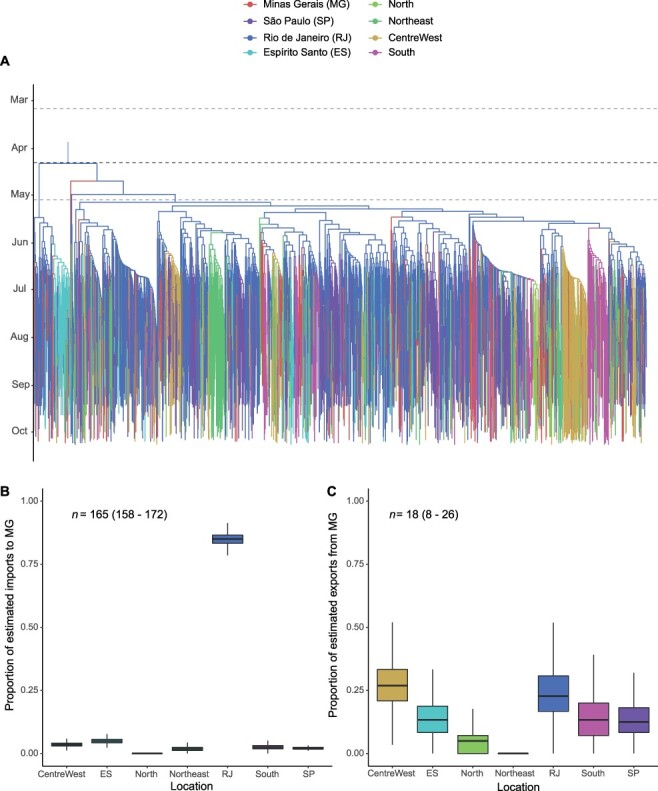
Bayesian phylogeographic reconstruction of the major Brazilian clade identified in the proportional dataset (n = 2,741). (A) Maximum clade credibility molecular clock tree annotated according to the discrete asymmetric 8-states phylogeographic model. Tree branches are colour coded according to the proposed discretisation scheme: North, Northeast, Central-West, South, Rio de Janeiro, Espírito Santo, São Paulo and Minas Gerais. The dark dashed line indicates the median age estimated for the tree root (10 April 2021), while the light dashed lines indicate its 95% HPD (7 March 2021–5 May 2021). The model suggests the ancestor of this clade occurred in Rio de Janeiro, most likely in mid-April, spreading to all other Brazilian states over the following months. (B, C) For Minas Gerais state, the Markov jumps method was used to estimate posterior distributions of total counts of imports and exports from each other. Box Plots indicate the proportion of transitions among states. (B) A total of 165 (95% HPD: 158–172) imports have been reconstructed by the model, over three-quarters of them from Rio de Janeiro. (C) Only 18 (95% HPD: 8–26) exports from MG have been reconstructed.
Similar results have been obtained for the uniform data set. A total of 126 introductions in Brazil have been estimated with the TreeTime migration model, with 69 singletons and 57 leading to the emergence of local clades. Only three clades possessed more than 100 sequences, and the major clade (n = 3,519) harboured 85.17 per cent of all MG sequences in this data set (Supplementary Fig. S3). The Bayesian timescaled phylogeographic reconstruction suggests this clade emerged in RJ (posterior probability = 1) in mid-April (95 per cent HPD: 11 March 2021–6 June 2021; Supplementary Fig. S4A), spreading to all other regions over the following months. The Markov jumps analysis suggests a total of 158 imports (95 per cent HPD: 153–164), coming mostly from RJ (median: 124, 95 per cent HPD: 113–123), followed by the Central-West (median: 12, 95 per cent HPD: 8–18), ES (median: 8, 95 per cent HPD: 5–11), Northeast (median: 7, 95 per cent HPD: 2–12), South (median: 5, 95 per cent HPD: 2–8), and SP (median: 1, 95 per cent HPD: 0–3) (Supplementary Fig. S4B). A total of eleven exports have been estimated (95 per cent HPD: 3–18), mostly to the Central-West (95 per cent HPD: 0–9) (Supplementary Fig. S4C).
These results are consistent with the analysis of PCR genotyping data, which shows a gradient of Delta frequency increase coming from east and west along with EWs 25–40 (Fig. 1). Henceforth, these phylogeographic reconstructions reinforce the importance of the epidemiological link between these locations to establish VOC Delta in MG. All annotated trees, beast XML, and log files generated in these analyses are available on our GitHub page (https://github.com/LBI-lab/Establishment-VOC-Delta-in-Minas-Gerais-Brazil and Supplementary Material 3).
4. Discussion
The epidemiological dynamics of SARS-CoV-2 in Brazil in 2021 were marked by a complex scenario of VOC co-circulation and lineage displacement events (Bittar et al. 2021; Faria et al. 2021; Sant’Anna et al. 2021; Moreira et al. 2021a). Most noticeably, throughout this year, the variant Gamma disseminated from Manaus to several country regions and led to resurgences of epidemic waves, increasing the burden on Brazilian public health (Faria et al. 2021; Freitas et al. 2021; Naveca et al. 2021a). As vaccination programmes advanced, leading to reductions in cases and deaths (Rinott, Youngster, and Lewis 2021; Roghani 2021), novel concerns emerged with reports of autochthonous circulation of VOC Delta in the country (Lamarca et al. 2021). In this context, we integrated a PCR genotyping, genomics, and epidemiological surveillance to track the lineage displacement event induced by VOC Delta in the MG state.
In this study, we genotyped 2,407 samples from MG. The identification of the first case of VOC Delta occurred in EW 29. After that, the number of cases related to the variant continued to rise, and 8 weeks later, VOC Delta was already responsible for more than 70 per cent of cases in the state. The rapid replacement of variants by Delta has already been described in other countries and states in Brazil (Dorlass et al. 2021; Kupferschmidt and Wadman 2021; Lamarca et al. 2021; Patané et al. 2021; Tegally et al. 2021a; Naveca, Souza, and Nascimento et al. 2021b). Nevertheless, despite the growth of Delta in MG, it did not lead to an increase in the number of cases and deaths. This result is the opposite of what has been described in many European and Asian countries, which experienced a third wave due to the high transmissibility rate of VOC Delta (Elliott et al. 2021; Umair et al. 2021). This difference may be related to the vaccination process in MG since approximately 88 per cent of the population have already been vaccinated with one dose and 65 per cent have been vaccinated with two doses (Gerais 2021b).
Large-scale vaccination reduces the number of cases and disease transmission in the population (Thanh Le et al. 2020). Nevertheless, previous reports indicate that the reproductive number (R0) of the VOC Delta is approximately 5–7, meaning that the transmissibility potential is higher compared with previous VOCs (Liu and Rocklöv 2021). Studies have also shown that the effectiveness of vaccines against Delta is lower, such as Pfizer or AstraZeneca, for which one dose is approximately 33 per cent effective (Lopez Bernal et al. 2021). In our study, thirty-nine individuals died from infection with the Delta variant, and 77 per cent had been vaccinated with at least one dose. The deaths were related to infections in individuals vaccinated for more than 100 days and over 60 years. This observation highlights the relevance of vaccine boosts to maintaining immunity over time. Accordingly, a third dose of Pfizer to individuals who had received two doses for at least 5 months increased the effectiveness of vaccination to 88 per cent and reduced severe cases of COVID-19 (Barda et al. 2021).
Moreover, 27,527,708 vaccine doses were administered in MG. The vaccines AstraZeneca, Pfizer, and CoronaVac were the main ones presenting 11,251,929, 8,049,386, and 7,742,744 doses, respectively, while the Janssen was applied in 483,649 individuals (Gerais 2021b). We did not find a significant difference between the clinical outcome and vaccine strategy. Nevertheless, no deaths were observed in Pfizer vaccinated persons, which may be due to the time vaccination was carried out rather than the vaccine’s effectiveness. Additionally, no infection data from individuals vaccinated with Janssen and classified as VOC Delta were recovered, which must be due to both the time of vaccination and its low frequency in the state (less than 1 per cent of the population). Our results reinforce that all vaccines helped in the reduction of cases and recovery of the individual.
While the pattern of replacement of Gamma by Delta is not surprising, given reports of the later lineage replacing VOC Alpha (Singh et al. 2021) and Beta (Tegally et al. 2021a) in India and South Africa, respectively, the mechanisms by which these events occur are not entirely established. Specifically, it is unclear whether this variant is inherently more transmissible than VOC Gamma. To assess this conjecture, we were prompted to evaluate the dynamics of viral loads measured from RT-qPCR diagnostics tests executed in the study period (Fig. 2). The analysis of the time series of distinct targets suggested a decrease in Ct values along the weeks marked by the rise in frequency of VOC Delta. The statistical comparison performed through linear models suggests that this variant induces relative increments of viral loads, i.e. reductions of Ct values (range: −1.3 to −1.8).
Nevertheless, we also report statistically significant, minor (β = −0.41) decay on Ct values measured for the MS2 internal control target. This result implies that while some random variation may be detected through our Ct comparison methodology, it incurs an effect at least 3-fold less than the one reported for the viral targets. In this sense, even discounting a random fluctuation on this same magnitude for these targets, differences of Ct around one between periods dominated by distinct VOCs would be obtained. While these differences are less than those reported between VOC Gamma and previously circulating lineages in Brazil (2.7–4 Ct values; (Faria et al. 2021; Moreira et al. 2021b; Naveca, Souza, and Nascimento et al. 2021b)), they suggest that transmissibility advantages, even if modest, shape the dynamics of viral lineages over time. Noticeably, the minor relative advantage herein reported for VOC Delta over Gamma possibly culminated in a slower substitution process (8 weeks to achieve frequency above 70 per cent) than the ones driven by the second variant across several states in the first season of 2021 (2–5 weeks; (Faria et al. 2021; Naveca et al. 2021a; Moreira et al. 2021b)). Overall, these results align with an early report from Manaus, which hypothesises that differences in transmissibility might be significant in high immunity settings (Naveca, Souza, and Nascimento et al. 2021b). Notwithstanding, the results found in our study are similar to those described previously using RT-qPCR data (Silva et al. 2022). This finding provides context to the epidemiological scenarios recently observed across other Brazilian states (Lamarca et al. 2021; Naveca, Souza, and Nascimento et al. 2021b).
To trace the dynamics of viral dissemination that led to the introduction and establishment of VOC Delta in MG, we sequenced 224 novel genome sequences, comprehending approximately twice the amount of genetic data previously available for the state in the study period (as of 11 October 2021). These sequences have been used jointly with publicly available information on the GISAID EpiCoV database to assemble two comprehensive data sets built under different sampling schemes, proportional and uniform. The usage of two distinct data sets was intended to verify to which extent the obtained estimates were sensitive to data set composition, given that phylogeographic reconstructions are highly sensitive to sampling biases. Analysis of both data sets led to the inference of highly similar viral spread dynamics. The maximum-likelihood phylogeographic analyses recovered more than one hundred distinct imports of VOC Delta in Brazil, implying the circulation of multiple Delta subclades in the country, as previously noticed (Arantes et al. 2021; Lamarca et al. 2021; Giovanetti et al. 2022) (Fig. 4C). Nevertheless, in both data sets, a single clade harboured more than 65 per cent of all Brazilian sequences and 80 per cent of all sequences from MG. In this sense, we were prompted to investigate in detail the phylogeographic history of this clade, reasoning that it would be the most important to comprehend the process of Delta establishment in MG.
Bayesian molecular clock analysis indicates this clade originated in RJ in mid-April, approximately 1 month before the country’s first registered imported Delta case on 20 May 2021 (Dos Santos, Ferreira, and da Silva et al. 2021) (Fig. 5A; Supplementary Fig. S4A). Since then, this viral lineage has spread to all regions of the country; its earliest introduction in MG dated to late May, 2 months before the first Delta detection by the OviGen network (20 July 2021). For both data sets, the Markov jumps method has reconstructed the occurrence of more than 150 introductions into MG, with more than three-quarters of them coming from RJ (proportional: 85.3 per cent and uniform: 75.5 per cent; Fig. 5B; Supplementary Fig. S4B). Other locations such as Central-West (proportional: 3.6 per cent and uniform: 8.2 per cent), ES (proportional: 4.9 per cent and uniform: 5.1 per cent), Northeast (proportional: 1.8 per cent and uniform: 4.4 per cent), South (proportional: 2.4 per cent and uniform: 3.2 per cent), and SP (proportional: 1.8 per cent and uniform: 0.6 per cent) also contributed to the total number of imports into MG. No importation from the North region was inferred for both data sets. Less than 20 exports from MG have been estimated for both data sets, most of them to the Central-West (proportional: 27.8 per cent and uniform: 36.4 per cent), with other locations receiving slightly differing proportions among them (Fig. 5C; Supplementary Fig. S4C).
These reconstructed patterns of viral spread are consistent with the earliest report on autochthonous circulation of the VOC Delta in Brazil (RJ, late June 2021; Lamarca et al. 2021). It suggests that most viral diversity in MG came from RJ and other neighbouring locations, evidencing a clear pattern of viral dissemination. These results are coherent with the analysis of PCR genotyping data, which shows a gradient of Delta frequency increase coming from RJ, and to a lesser extent, the Central-West, along EWs 25–40 (Fig. 1A). While we could infer an intra-state dispersion dynamic through PCR data, it was impossible to assess this phenomenon through phylogenetic methods, as no clade from MG with a clear temporal signal and broad geographic span was identified. Notwithstanding, these analyses provide evidence of the epidemiological links among these locations, revealing the dynamics that led to the establishment of the VOC Delta in MG. These findings highlight the importance of interstate mobility control measures as tools to mitigate VOCs dissemination across Brazilian states.
It is relevant to emphasise that the phylogeographic reconstructions reported herein are limited since the availability of genome sequences for Brazilian states is heterogeneous. Therefore, sampling gaps—the absence of a number of sequences for a given state and week—have been recorded for both data sets (Supplementary Material 3). Nevertheless, the proportional data set displays a strong correlation between the number of genome sequences and Delta infections for each location in the proposed discretisation scheme (Fig. 4B), suggesting that this data set is adequate to explore the dynamics of viral dispersion in the country. The fact that the presented phylogeographic analyses were performed on fixed topologies, masking underlying phylogenetic uncertainty, is also a limitation. While this hinders assertions about the phylogenetic placement of some sequences, we believe that the approach used was able to reconstruct the temporal dynamics of migration events into MG. The consistency of the recovered patterns from independently assembled comprehensive data sets and the large-scale genotyping PCR analysis evidence the robustness of the presented findings.
While the molecular clock analyses suggest the existence of cryptic transmission periods at national and state levels, this likely reflects the difficulty in detecting variants at low frequency. It is conceivable that once variants start to increase in frequency, they rapidly become detectable by the many surveillance initiatives developed in the country, as was the case in MG and other states (Lamarca et al. 2021; Patané et al. 2021; Naveca, Souza, and Nascimento et al. 2021b). Nevertheless, expanding sample sizes and following a structured sampling design could enhance epidemic monitoring and lead to earlier detection of novel circulating lineages (Brito et al. 2021).
Overall, this study reports a genomic surveillance experience based on a mixed strategy, using both PCR genotyping and genome sequencing to rapidly track the epidemiological dynamics of viral lineages and serving as a model in limited-resource settings. By integrating these methods with epidemiological data from the state, we could document a fast lineage displacement event induced by VOC Delta in near real time, providing evidence that it is more transmissible than VOC Gamma. These findings explain the scenario observed in several Brazilian states, providing a rationale for epidemiological observations across various settings. In addition, through robust phylogeographic reconstructions, we established a clear epidemiological link between MG and RJ. Finally, we show that despite the fast epidemic growth displayed by VOC Delta, it did not culminate in rises in the numbers of cases and deaths in MG. This outcome is likely a consequence of the advancement of the vaccination programme at the state level, reinforcing the importance of vaccine roll-out as a central tool to control the pandemic.
Supplementary Material
Contributor Information
Paula L C Fonseca, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Filipe R R Moreira, MRC Centre for Global Infectious Disease Analysis, J-IDEA, Imperial College London, Exhibition Rd, South Kensington, London SW7 2BX, UK; Departamento de Genética, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Av. Carlos Chagas Filho 373, Cidade Universitaria, Rio de Janeiro 21941-902, Rio de Janeiro, Brazil.
Rafael M de Souza, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Natália R Guimarães, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Nara O Carvalho, Núcleo de Ações e Pesquisa em Apoio Diagnóstico-Nupad/Faculdade de Medicina/Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Av. Prof. Alfredo Balena 189, Centro, Belo Horizonte 30130-100, Minas Gerais, Brazil.
Talita E R Adelino, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Hugo J Alves, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Luige B Alvim, Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
Darlan S Candido, Department of Zoology, University of Oxford, 11a Mansfield Rd, Oxford OX13SZ, UK; Instituto de Medicina Tropical, Faculdade de Medicina da Universidade de São Paulo, Av. Dr. Enéas Carvalho de Aguiar 470, Jardim América, São Paulo 05403000, São Paulo, Brazil.
Helena P Coelho, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Alana V B Costa, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Walyson C Costa, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Alex F de Carvalho, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Bruna W F de Faria, Secretaria Municipal de Saúde de Belo Horizonte, Av. Afonso Pena 2336, Funcionários, Belo Horizonte 30130-040, Minas Gerais, Brazil.
Aline B de Lima, Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
Eneida S de Oliveira, Secretaria Municipal de Saúde de Belo Horizonte, Av. Afonso Pena 2336, Funcionários, Belo Horizonte 30130-040, Minas Gerais, Brazil.
Carolina S A de Souza, Pan American Health Organization—PAHO, Av. Das Nações SEN, Asa Norte, Brasilia 70312970, Brazil.
Fernanda G de Souza, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Rillery C Dias, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Victor E V Geddes, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Igor P Godinho, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Alessandro L Gonçalves, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Karine L Lourenço, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Rubens D M Magalhães, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Frederico S V Malta, Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
Eva L A Medeiros, Subsecretaria de Vigilância em Saúde, Secretaria de Estado de Saúde de Minas Gerais, Rodovia Papa João Paulo II 4143. Edifício Minas Gerais, Cidade Administrativa, Serra verde, Belo Horizonte 31630900, Minas Gerais, Brazil.
Fernanda S Mendes, Núcleo de Ações e Pesquisa em Apoio Diagnóstico-Nupad/Faculdade de Medicina/Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Av. Prof. Alfredo Balena 189, Centro, Belo Horizonte 30130-100, Minas Gerais, Brazil.
Pedro H B de P Mendes, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Cristiane P T B Mendonça, Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
Andre L Menezes, Secretaria Municipal de Saúde de Belo Horizonte, Av. Afonso Pena 2336, Funcionários, Belo Horizonte 30130-040, Minas Gerais, Brazil.
Diego Menezes, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Mariane T Menezes, MRC Centre for Global Infectious Disease Analysis, J-IDEA, Imperial College London, Exhibition Rd, South Kensington, London SW7 2BX, UK.
Lucyene Miguita, Departamento de Patologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Rennan G Moreira, Centro de Laboratórios Multiusuários, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Renata B Peixoto, Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Daniel C Queiroz, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Adriana A Ribeiro, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Ana Paula de B Ribeiro, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Juliana W Saliba, Pan American Health Organization—PAHO, Av. Das Nações SEN, Asa Norte, Brasilia 70312970, Brazil.
Hugo I Sato, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Joice do P Silva, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil; Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
Natiely P Silva, Núcleo de Ações e Pesquisa em Apoio Diagnóstico-Nupad/Faculdade de Medicina/Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Av. Prof. Alfredo Balena 189, Centro, Belo Horizonte 30130-100, Minas Gerais, Brazil.
Nuno R Faria, MRC Centre for Global Infectious Disease Analysis, J-IDEA, Imperial College London, Exhibition Rd, South Kensington, London SW7 2BX, UK; Department of Zoology, University of Oxford, 11a Mansfield Rd, Oxford OX13SZ, UK; Instituto de Medicina Tropical, Faculdade de Medicina da Universidade de São Paulo, Av. Dr. Enéas Carvalho de Aguiar 470, Jardim América, São Paulo 05403000, São Paulo, Brazil.
Santuza M R Teixeira, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Flávio G da Fonseca, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Ana Paula S M Fernandes, Centro de Tecnologia de Vacinas, Universidade Federal de Minas Gerais, Rua Professor José Vieira de Mendonça 770, Engenho Nogueira, Belo Horizonte 31310260, Minas Gerais, Brazil.
Danielle A G Zauli, Instituto Hermes Pardini, Av. das Nações 2448, Distrito Industrial, Vespasiano 33201003, Minas Gerais, Brazil.
José Nélio Januario, Núcleo de Ações e Pesquisa em Apoio Diagnóstico-Nupad/Faculdade de Medicina/Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Av. Prof. Alfredo Balena 189, Centro, Belo Horizonte 30130-100, Minas Gerais, Brazil.
Jaqueline S de Oliveira, Subsecretaria de Vigilância em Saúde, Secretaria de Estado de Saúde de Minas Gerais, Rodovia Papa João Paulo II 4143. Edifício Minas Gerais, Cidade Administrativa, Serra verde, Belo Horizonte 31630900, Minas Gerais, Brazil.
Felipe C de M Iani, Fundacao Ezequiel Dias, Rua Conde Pereira Carneiro 80, Gameleira, Belo Horizonte 30510-010, Minas Gerais, Brazil.
Renato S de Aguiar, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil; Instituto D’OR de Pesquisa e Ensino, Rio de Janeiro 22281100, Rio de Janeiro, Brazil.
Renan P de Souza, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, Pampulha, Belo Horizonte 31270901, Minas Gerais, Brazil.
Data availability
All Delta consensus genome sequences characterised in this study have been deposited on GISAID and are publicly available (Supplementary Table S7).
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
We acknowledge support from the Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP 01.20.0029.000462/20 and CNPq 404096/2020-4). This project was also supported by CNPq (R.S.A.: 312688/2017-2 and 439119/2018-9; R.P.S.: 310627/2018-4), MEC/CAPES 118 (14/2020 23072.211119/2020-10), FINEP (0494/20 01.20.0026.00), UFMG-NB3, FINEP no 1139/20 (RSA), FAPEMIG (R.P.S.: APQ-00475-20), and FAPERJ (C.M.V: 26/010.002278/2019) (R.S.A: 202.922/2018) and by CADDE/FAPESP (MR/S0195/1 and FAPESP 120 18/14389-0) (N.R.F.). N.R.F. acknowledges support from the Wellcome Trust and the Royal Society (Sir Henry Dale Fellowship. 204311/Z/16/Z), Medical Research Council-São Paulo Research Foundation (FAPESP) CADDE partnership award (MR/S0195/1 and FAPESP18/143890) (https://caddecentre.org), and the Bill & Melinda Gates Foundation (INV-034540 and INV-034652). D.S.C. acknowledges support from the Clarendon Fund, the University of Oxford Department of Zoology, and Merton College.
Conflict of interest:
We declare no conflicts of interest.
CRediT authorship contribution
Paula L. C. Fonseca: Acquisition, analysis, interpretation, drafting, and revision of the manuscript. Filipe R. R. Moreira: Acquisition, analysis, interpretation, drafting, and revision of the manuscript. Rafael M. de Souza: Acquisition, analysis, interpretation, and revision of the manuscript. Natália R. Guimarães: Acquisition, analysis, interpretation, and revision of the manuscript. Nara O. Carvalho: Acquisition, analysis, interpretation, and revision of the manuscript. Talita E. R. Adelino: Acquisition, analysis, interpretation, and revision of the manuscript. Hugo J. Alves: Acquisition, analysis, interpretation, and revision of the manuscript. Luige B. Alvim: Acquisition and revision of the manuscript. Darlan S. Candido: Analysis and revision of the manuscript. Helena P. Coelho: Acquisition, analysis, and revision of the manuscript. Alana V. B. Costa: Acquisition and revision of the manuscript. Walyson C. Costa: Acquisition and revision of the manuscript. Alex F. de Carvalho: Acquisition, analysis, and revision of the manuscript. Bruna W.F. de Faria: Acquisition and revision of the manuscript. Aline B. de Lima: Acquisition and revision of the manuscript. Eneida S. de Oliveira: Acquisition and revision of the manuscript. Carolina S. A. de Souza: Acquisition, analysis, and revision of the manuscript. Fernanda G. de Souza: Acquisition and revision of the manuscript. Rillery C. Dias: Acquisition and revision of the manuscript. Victor E. V. Geddes: Acquisition and revision of the manuscript. Igor P. Godinho: Acquisition, analysis, and revision of the manuscript. Alessandro L. Gonçalves: Acquisition and revision of the manuscript. Karine L. Lourenço: Acquisition, analysis, and revision of the manuscript. Rubens D. M. Magalhães: Acquisition, analysis, and revision of the manuscript. Frederico S. V. Malta: Acquisition and revision of the manuscript. Eva L. A. Medeiros: Acquisition, analysis, and revision of the manuscript. Fernanda S. Mendes: Acquisition, analysis, and revision of the manuscript. Pedro H. B. de P. Mendes: Acquisition and revision of the manuscript. Cristiane P. T. B. Mendonça: Acquisition and revision of the manuscript. Andre L. Menezes: Acquisition and revision of the manuscript. Diego Menezes: Acquisition, analysis, and revision of the manuscript. Mariane T. Menezes: Acquisition and revision of the manuscript. Lucyene Miguita: Acquisition and revision of the manuscript. Rennan G. Moreira: Acquisition and revision of the manuscript. Renata B. Peixoto: Acquisition, analysis, and revision of the manuscript. Daniel C. Queiroz: Acquisition and revision of the manuscript. Adriana A. Ribeiro: Acquisition and revision of the manuscript. Ana Paula de B. Ribeiro: Acquisition and revision of the manuscript. Juliana W. Saliba: Acquisition, analysis, and revision of the manuscript. Hugo I. Sato: Acquisition, analysis, and revision of the manuscript. Joice do P. Silva: Acquisition and revision of the manuscript. Natiely P. Silva: Acquisition, analysis, and revision of the manuscript. Nuno R. Faria: Analysis, interpretation, and revision of the manuscript. Santuza M. R. Teixeira: Conception, acquisition, analysis, interpretation, and revision of the manuscript. Flávio G. da Fonseca: Acquisition, analysis, interpretation, and revision of the manuscript. Ana Paula S. M. Fernandes: Acquisition, analysis, interpretation, and revision of the manuscript. Danielle A. G. Zauli: Acquisition and revision of the manuscript. José Nélio Januario: Acquisition and revision of the manuscript. Jaqueline S. de Oliveira: Acquisition, analysis, and revision of the manuscript. Felipe C. de M. Iani: Conception, acquisition, analysis, interpretation, and revision of the manuscript. Renato S. de Aguiar: Conception, acquisition, analysis, interpretation, drafting, and revision of the manuscript. Renan P. de Souza: Conception, acquisition, analysis, interpretation, drafting, and revision of the manuscript.
References
- Aksamentov I. et al. (2021) ‘Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes’, The Journal of Open Source Software, 6: 3773.doi: 10.5281/ZENODO.5607694 [DOI] [Google Scholar]
- Andreano E. et al. (2020) ‘SARS-CoV-2 Escape in Vitro from a Highly Neutralizing COVID-19 Convalescent Plasma’, Immunology, 118: 36.doi: 10.1101/2020.12.28.424451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes I. et al. (2021) ‘Emergence and Spread of the SARS-CoV-2 Variant of Concern Delta across Different Brazilian Regions’, Genetic and Genomic Medicine, e2103154118.doi: 10.1101/2021.11.25.21266251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele G. et al. (2020) ‘Hamiltonian Monte Carlo Sampling to Estimate past Population Dynamics Using the Skygrid Coalescent Model in a Bayesian Phylogenetics Framework’, Wellcome Open Research, 5: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barda N. et al. (2021) ‘Effectiveness of a Third Dose of the BNT162b2 mRNA COVID-19 Vaccine for Preventing Severe Outcomes in Israel: An Observational Study’, The Lancet, S0140673621022492.doi: 10.1016/S0140-6736(21)02249-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittar C. et al. (2021) ‘The Emergence of the New P.4 Lineage of SARS-CoV-2 with Spike L452R Mutation in Brazil’, Frontiers in Public Health, 9: 745310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil . (2021), BOLETIM EPIDEMIOLÓGICO ESPECIAL - Doença Pelo Novo Coronavírus – COVID-19. <https://www.gov.br/saude/pt-br/media/pdf/2021/outubro/01/boletim_epidemiologico_covid_81_v2.pdf> accessed 20 Oct 2021.
- Brito A. F. et al. (2021) ‘Global Disparities in SARS-CoV-2 Genomic Surveillance’, Epidemiology.doi: 10.1101/2021.08.21.21262393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzi A. et al. (2021) ‘Spatial and temporal fluctuations in COVID-19 fatality rates in Brazilian hospitals’, Nat Med. 10.1038/s41591-022-01807-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson J. (2020) ‘Ggalluvial: Layered Grammar for Alluvial Plots’, Journal of Open Source Software, 5: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss L. F. et al. (2021) ‘Three-quarters Attack Rate of SARS-CoV-2 in the Brazilian Amazon during a Largely Unmitigated Epidemic’, Science, 371: 288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido D. S. et al. (2020) ‘Evolution and Epidemic Spread of SARS-CoV-2 in Brazil’, Science, 369: 1255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC . (2021), SARS-CoV-2 Variant Classifications and Definitions. <https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html> accessed 10 Nov 2021.
- Cele S. et al. (2021) ‘Escape of SARS-CoV-2 501Y.V2 From Neutralization by Convalescent Plasma’, Nature, 593: 142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian S. et al. (2021) ‘SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India’, Microorganisms, 9: 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X. et al. (2021) ‘Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant’, Cell, 184: 3426–37.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X., Siveroni I., and Volz E. M. (2021) ‘Additive Uncorrelated Relaxed Clock Models for the Dating of Genomic Epidemiology Phylogenies’, Molecular Biology and Evolution, 38: 307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlass E. G. et al. (2021) ‘Survey of SARS-CoV-2 Genetic Diversity in Two Major Brazilian Cities Using a Fast and Affordable Sanger Sequencing Strategy’, Genomics, 113: 4109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos J. et al. (2021), First Reported Cases of SARS-CoV-2 Sub-lineage B.1.617.2 In Brazil: An Outbreak in a Ship and Alert for Spread. <https://virological.org/t/first-reported-cases-of-sars-cov-2-sub-lineage-b-1-617-2-in-brazil-an-outbreak-in-a-ship-and-alert-for-spread/706> accessed 30 Nov 2021. [Google Scholar]
- ECDC . (2021), Guidance for Representative and Targeted Genomic SARS-CoV-2 Monitoring. <https://www.ecdc.europa.eu/sites/default/files/documents/Guidance-for-representative-and-targeted-genomic-SARS-CoV-2-monitoring-updated-with%20erratum-20-May-2021.pdf> accessed 10 May 2021.
- Elliott P. et al. (2021) ‘Exponential Growth, High Prevalence of SARS-CoV-2, and Vaccine Effectiveness Associated with the Delta Variant’, Science, 374: eabl9551.doi: 10.1126/science.abl9551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2021) ‘Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil’, Science, 372: 815–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas A. R. R. et al. (2021) ‘The Emergence of Novel SARS-CoV-2 Variant P.1 In Amazonas (Brazil) Was Temporally Associated with A Change in the Age and Sex Profile of COVID-19 Mortality: A Population Based Ecological Study’, The Lancet Regional Health - Americas, 1: 100021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W. F. et al. (2021) ‘Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-induced Humoral Immunity’, Cell, 184: 2372–83.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J. et al. (2021) ‘Antibody Neutralization of SARS-CoV-2 through ACE2 Receptor Mimicry’, Nature Communications, 12: 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes V. et al. (2021). A Novel RT-qPCR Assay for Detection of SARS-CoV-2 Variants Based on RhAmp Technology (IDT Technologies) V1.doi: 10.17504/protocols.io.buf2ntqe [DOI]
- Gerais M. (2021a), Informações de Casos de COVID-19 No Estado de Minas Gerais. <https://coronavirus.saude.mg.gov.br/dadosabertos>.
- ——— (2021b), Vacinômetro. <https://coronavirus.saude.mg.gov.br/vacinometro> accessed 20 Oct 2021.
- Gill M. S. et al. (2013) ‘Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci’, Molecular Biology and Evolution, 30: 713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanetti M. et al. (2022) ‘Replacement of the Gamma by the Delta Variant in Brazil: Impact of Lineage Displacement on the Ongoing Pandemic’, Virus Evolution, 8: veac024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräler B., Pebesma E., and Heuvelink G. (2016) ‘Spatio-Temporal Interpolation Using Gstat’, The R Journal, 8: 204. [Google Scholar]
- Greaney A. J. et al. (2021) ‘Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition’, Cell Host & Microbe, 29: 44–57.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Gutierrez B. et al. (2021) ‘Genomic Epidemiology of SARS-CoV-2 Transmission Lineages in Ecuador’, Virus Evolution, 7: veab051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Yano T., and Kishino H. (1984) ‘A New Molecular Clock of Mitochondrial DNA and the Evolution of Hominoids’, Proceedings of the Japan Academy, Series B, 60: 95–8. [Google Scholar]
- Inward R. P. D., Parag K. V., and Faria N. R. (2022) ‘Using Multiple Sampling Strategies to Estimate SARS-CoV-2 Epidemiological Parameters from Genomic Sequencing Data’, Epidemiology.doi: 10.1101/2022.02.04.22270165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K. L. et al. (2022) ‘SARS-CoV-2 Variants of Concern Alpha and Delta Show Increased Viral Load in Saliva’, PLoS ONE, 17: e0267750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K., and Wadman M. (2021) ‘Delta Variant Triggers New Phase in the Pandemic’, Science, 372: 1375–6. [Google Scholar]
- Lamarca A. P. et al. (2021) ‘Genomic Surveillance Tracks the First Community Outbreak of the SARS-CoV-2 Delta (B.1.617.2) Variant in Brazil’, Journal of Virology, 96: JVI.01228–21.doi: 10.1128/JVI.01228-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J. et al. (2020) ‘Structure of the SARS-CoV-2 Spike Receptor-binding Domain Bound to the ACE2 Receptor’, Nature, 581: 215–20. [DOI] [PubMed] [Google Scholar]
- Langmead B. et al. (2019) ‘Scaling Read Aligners to Hundreds of Threads on General-purpose Processors’, Bioinformatics, 35: 421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K. et al. (2021) ‘Early Transmissibility Assessment of the N501Y Mutant Strains of SARS-CoV-2 in the United Kingdom, October to November 2020’, Eurosurveillance, 26: Pii=2002106.doi: 10.2807/1560-7917.ES.2020.26.1.2002106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) ‘A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data’, Bioinformatics, 27: 2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. et al. (2020) ‘Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike’, Nature, 584: 450–6. [DOI] [PubMed] [Google Scholar]
- Liu Y., and Rocklöv J. (2021) ‘The Reproductive Number of the Delta Variant of SARS-CoV-2 Is Far Higher Compared to the Ancestral SARS-CoV-2 Virus’, Journal of Travel Medicine, 28: taab124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Bernal J. et al. (2021) ‘Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant’, New England Journal of Medicine, 385: 585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M. et al. (2021) ‘N-terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2’, Cell, 184: 2332–47.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy K. R. et al. (2021) ‘Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape’, Science, 371: 1139–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., and Suchard M. A. (2007) ‘Counting Labeled Transitions in Continuous-time Markov Models of Evolution’, Journal of Mathematical Biology, 56: 391–412. [DOI] [PubMed] [Google Scholar]
- Moreira F. R. et al. (2021a) ‘Epidemic Spread of SARS-CoV-2 Lineage B.1.1.7 In Brazil’, Viruses, 13: 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Epidemiological Dynamics of SARS-CoV-2 VOC Gamma in Rio de Janeiro, Brazil’, Virus Evolution, 7: veab087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveca F. G. et al. (2021a) ‘COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence’, Nature Medicine, 27: 1230–8. [DOI] [PubMed] [Google Scholar]
- Naveca N. et al. (2021b), The SARS-CoV-2 Variant Delta Displaced the Variants Gamma and Gamma Plus in Amazonas, Brazil. <https://virological.org/t/the-sars-cov-2-variant-delta-displaced-the-variants-gamma-and-gamma-plus-in-amazonas-brazil/765> accessed 10 Nov 2021.
- Nonaka C. K. V. et al. (2021) ‘Genomic Evidence of SARS-CoV-2 Reinfection Involving E484K Spike Mutation, Brazil’, Emerging Infectious Diseases, 27: 1522–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole Á. et al. (2021) ‘Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool’, Virus Evolution, 7: veab064.doi: 10.1093/ve/veab064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patané J. et al. (2021) ‘SARS-CoV-2 Delta Variant of Concern in Brazil - Multiple Local Evolution’, Genetic and Genomic Medicine.doi: 10.1101/2021.09.15.21262846 [DOI] [Google Scholar]
- Pebesma E. (2018) ‘Simple Features for R: Standardized Support for Spatial Vector Data’, The R Journal, 10: 439. [Google Scholar]
- Pereira . (2019), Geobr: Loads Shapefiles of Official Spatial Data Sets of Brazil. <https://github.com/ipeaGIT/geobr> accessed 10 Nov 2021.
- Planas D. et al. (2021) ‘Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization’, Nature, 596: 276–80. [DOI] [PubMed] [Google Scholar]
- Quinlan A. R., and Hall I. M. (2010) ‘BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features’, Bioinformatics, 26: 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2021), R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. <https://www.r-project.org/> accessed 1 Oct 2021.
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende P. C. et al. (2021) ‘The Ongoing Evolution of Variants of Concern and Interest of SARS-CoV-2 in Brazil Revealed by Convergent Indels in the Amino (N)-terminal Domain of the Spike Protein’, Virus Evolution, 7: veab069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinott E., Youngster I., and Lewis Y. E. (2021) ‘Reduction in COVID-19 Patients Requiring Mechanical Ventilation following Implementation of a National COVID-19 Vaccination Program — Israel, December 2020–February 2021’, Morbidity and Mortality Weekly Report, 70: 326–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roghani A. (2021) ‘The Influence of COVID-19 Vaccination on Daily Cases, Hospitalization, and Death Rate in Tennessee, United States: Case Study’, Journal of Medical Internet Research X Medicine, 2: e29324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-likelihood Phylodynamic Analysis’, Virus Evolution, 4: vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A. et al. (2022) ‘Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation’, Nature, 602: 300–6.doi: s41586-021-04266-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sant’Anna F. H. et al. (2021) ‘Emergence of the Novel SARS-CoV-2 Lineage VUI-NP13L and Massive Spread of P.2 In South Brazil’, Emerging Microbes & Infections, 10: 1431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. et al. (2021) ‘The Evolving Faces of the SARS-CoV-2 Genome’, Viruses, 13: 1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. P. et al. (2022) ‘Delta Variant of SARS-CoV-2 Replacement in Brazil: A National Epidemiologic Surveillance Program’, Viruses, 14: 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J. et al. (2021) ‘SARS-CoV-2 Variants of Concern are Emerging in India’, Nature Medicine, 27: 1131–3. [DOI] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016.doi: 10.1093/ve/vey016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K. et al. (2021) ‘The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants’, Nature Reviews. Genetics, 22: 757–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré . (1986) ‘Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences’. In Providence, R.I. American Mathematical Society, c1986: Some Mathematical Questions in Biology: DNA Sequence Analysis, p. 124. Robert M. Miura. [Google Scholar]
- Tegally H. et al. (2021a) ‘Rapid Replacement of the Beta Variant by the Delta Variant in South Africa’, Epidemiology.doi: 10.1101/2021.09.23.21264018 [DOI] [Google Scholar]
- ——— et al. (2021b) ‘Detection of a SARS-CoV-2 Variant of Concern in South Africa’, Nature, 592: 438–43. [DOI] [PubMed] [Google Scholar]
- Thanh Le T. et al. (2020) ‘The COVID-19 Vaccine Development Landscape’, Nature Reviews Drug Discovery, 19: 305–6. [DOI] [PubMed] [Google Scholar]
- Thye A. Y.-K. et al. (2021) ‘Emerging SARS-CoV-2 Variants of Concern (Vocs): An Impending Global Crisis’, Biomedicines, 9: 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F. et al. (2021) ‘N501Y Mutation of Spike Protein in SARS-CoV-2 Strengthens Its Binding to Receptor ACE2’, eLife, 10: e69091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosta S. et al. (2021) ‘Early Genomic Detection of SARS-CoV-2 P.1 Variant in Northeast Brazil’, PLoS Negl Trop Dis, 15: e0009591.doi: 10.1371/journal.pntd.000959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umair M. et al. (2021) ‘Genomic Surveillance Reveals the Detection of SARS-CoV-2 Delta, Beta, and Gamma VOCs during the Third Wave in Pakistan’, Journal of Medical Virology, jmv.27429.doi: 10.1002/jmv.27429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana R. et al. (2022) ‘Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa’, Nature, 603: 679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpato F. C. Z. et al. (2021) ‘Early Detection of the SARS-CoV-2 P.1 Variant in Rio Grande Do Sul, Brazil: A Case Report’, Infection Control & Hospital Epidemiology, 1–2.doi: 10.1017/ice.2021.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz E. et al. (2021) ‘Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 In England’, Nature, 593: 266–9. [DOI] [PubMed] [Google Scholar]
- Walker A. S. et al. (2021) ‘Ct Threshold Values, a Proxy for Viral Load in Community SARS-CoV-2 Cases, Demonstrate Wide Variation across Populations and over Time’, eLife, 10: e64683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2021a) ‘Vaccine-escape and Fast-growing Mutations in the United Kingdom, the United States, Singapore, Spain, India, and Other COVID-19-devastated Countries’, Genomics, 113: 2158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Analysis of SARS-CoV-2 Variant Mutations Reveals Neutralization Escape Mechanisms and the Ability to Use ACE2 Receptors from Additional Species’, Immunity, 54: 1611–21.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . (2022), Tracking SARS-CoV-2 Variants. <https://www.who.int/activities/tracking-SARS-CoV-2-variants> accessed 28 Mar 2022.
- Wickham . (2016), Ggplot2: Elegant Graphics for Data Analysis. <https://ggplot2.tidyverse.org> accessed 1 Oct 2021.
- Xie X. et al. (2021) ‘Neutralization of SARS-CoV-2 Spike 69/70 Deletion, E484K and N501Y Variants by BNT162b2 Vaccine-elicited Sera’, Nature Medicine, 27: 620–1. [DOI] [PubMed] [Google Scholar]
- Yang Z. (1994) ‘Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods’, Journal of Molecular Evolution, 39: 306–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All Delta consensus genome sequences characterised in this study have been deposited on GISAID and are publicly available (Supplementary Table S7).