

Abstract
Overexpression of the anti-apoptotic BCL-2 proteins is associated with the development and progression of a range of cancers. Venetoclax, an FDA-approved BCL-2 inhibitor, is fast becoming the standard-of-care for acute myeloid leukemia and chronic lymphocytic leukemia. However, the median survival offered by venetoclax is only 18 months (as part of a combination therapy regimen), and one of the primary culprits for this is the concomitant upregulation of sister anti-apoptotic proteins, in particular MCL-1 (and BCL-xL), which provides an escape route that manifests as venetoclax resistance. Since inhibition of BCL-xL leads to thrombocytopenia, we believe that a dual MCL-1/BCL-2 inhibitor may provide an enhanced therapeutic effect relative to a selective BCL-2 inhibitor. Beginning with a carboxylic acid-containing literature compound that is a potent inhibitor of MCL-1 and a moderate inhibitor of BCL-2, we herein describe our efforts to develop dual inhibitors of MCL-1 and BCL-2 by scaffold hopping from an indole core to an indazole framework. Subsequently, further elaboration of our novel N2-substituted, indazole-3-carboxylic acid lead into a family of indazole-3-acylsulfonamides resulted in improved inhibition of both MCL-1 and BCL-2, possibly through occupation of the p4 pocket, with minimal or no inhibition of BCL-xL.
The anti-tumour efficacies of selective inhibitors of anti-apoptotic BCL-2 family proteins are often compromised by the concomitant upregulation of sister proteins. Herein, we describe our efforts to develop dual MCL-1/BCL-2 inhibitors.
Introduction
The BCL-2 family of proteins comprises pro-apoptotic members and anti-apoptotic counterparts that interact together to regulate apoptosis.1 In a plethora of cancers, the anti-apoptotic proteins, which include BCL-2, BCL-xL and MCL-1, are upregulated, and directly contribute to the development and progression of the cancer.2–5 In addition, MCL-1 overexpression has been associated with resistance to conventional chemotherapies.6 The primary interaction between the pro- and anti-apoptotic members is described by an α-helical BH3 “death” domain on the former that is sequestered by a hydrophobic BH3-binding groove on the latter. Key contacts within this protein–protein interaction are hydrophobic side chains projected from one face of the α-helix that nestle into sub-pockets labelled p1 through p4 in the hydrophobic groove, as well as a salt bridge through a conserved aspartate on the opposing face of the helix.1,7 The development of small-molecules that mimic the BH3 α-helix as a strategy to re-activate apoptosis has been an active area of research for over 20 years.7–11
Several of the earlier small-molecule BCL-2 antagonists, such as TW-37, are pan-BCL-2 inhibitors, binding BCL-2, BCL-xL and MCL-1 with similar affinities, a consequence of the highly conserved BH3-binding grooves on their surfaces.7 However, in the last decade or so, selective inhibitors have emerged, such as the BCL-2 inhibitor venetoclax (VEN).9 In fact, VEN has received FDA approval for acute myeloid leukemia (as well as chronic lymphocytic leukemia and small lymphocytic leukemia) in combination with a hypomethylating agent, such as azacytidine, or low-dose cytarabine,12,13 although resistance to these treatments soon emerges,14 commonly due to MCL-1 (or BCL-xL) upregulation3,15–18 (as well as to mutations in BCL-2 (in CLL)19).
Accordingly, this has fuelled research into the development of MCL-1 inhibitors,20,21 and, in the last few years, multiple academic laboratories and pharmaceutical companies have achieved subnanomolar, selective inhibitors of MCL-1,8,10,22,23 culminating in clinical trials for several chemotypes. Alarmingly, however, at least three of these trials have been suspended or terminated for safety reasons.24 There is a double-edged sword here because MCL-1 upregulation is the most common mechanism of resistance to VEN,3,15–18 yet several cell types depend on MCL-1 function for survival, including hematopoietic stem cells,25 cardiomyoctes26 and hepatocytes;27 in fact, MCL-1 knockout in mice is embryonic lethal.28 A potent inhibitor of MCL-1 might never achieve FDA approval due to an unacceptable safety profile. Inspired by the synergism observed in combination studies with a BCL-2 inhibitor and an MCL-1 inhibitor,29 we hypothesize that less potent inhibitors of MCL-1 that are equipped to also recognize BCL-2 but not BCL-xL (to avoid thrombocytopenia) may provide the balanced platform needed to neutralize BCL-2-inhibited apoptosis and concomitantly address the associated resistance mechanism of MCL-1 upregulation. In fact, Lee et al. reasoned that partial inhibition of MCL-1, which they observed through covalent, allosteric inhibitors, may be more desirable than complete inhibition of MCL-1, producing fewer side effects.30
Results and discussion
The discovery of novel chemotypes in drug discovery is crucial to increasing the probability of a drug successfully advancing through clinical trials.31 An expedited strategy towards this goal is “scaffold hopping” in which the scaffold of an existing inhibitor is modified to generate a new chemotype.32 Fesik's laboratory, Abbvie and AstraZeneca have all experienced much success with the indole-2-carboxylic acid scaffold as a common chemotype in their MCL-1 inhibitors, wherein a critical p2-binding moiety is grafted adjacent to the carboxylic acid that forms an essential salt bridge with Arg263.10,23,33 One of Fesik's earliest lead compounds (1) demonstrates nanomolar inhibition of MCL-1 (Ki = 55 nM), sub-micromolar inhibition of BCL-2 (Ki = 0.87 μM) and is inactive against BCL-xL (Ki > 15 μM).33 We, therefore, considered this was a promising starting point towards the discovery of dual MCL-1/BCL-2 inhibitors. The benzimidazole and indazole scaffolds provide excellent alternative scaffolds to the benchmark indole core and permit the preservation of the 1,2 relationship of the p2-binding group and the carboxylic acid (Fig. 1), although it is acknowledged that the proposed indazole core itself may bind differently given the relative displacement of the benzene ring component of the bicycle.
Fig. 1. Scaffold hopping from indoles to benzimidazoles and indazoles.

We prepared the indole-based MCL-1 inhibitor 1,33 as a positive control, and the corresponding benzimidazole 2 and 2,3-substituted indazole 3 (full synthetic procedures for the syntheses of 2 and 3 are provided in the ESI†). The non-chlorinated benzimidazole was used to avoid ambiguity in the position of the chlorine atom upon alkylation of the benzimidazole nucleus. As shown in Table 1, the control indole 1 performed as expected, confirming the functionality of our fluorescence polarization competition assay (FPCA).34–40 Unfortunately, the benzimidazole and the indazole elicited negligible inhibition of MCL-1. At this stage, we elected to pursue only the indazole scaffold, since the benzimidazole scaffold exhibited significant polarity (TLC) that may compromise binding in the hydrophobic cleft, although this may be further investigated in the future. First, we shortened the three-carbon linker between the indazole core and the p2-binding 3,5-dimethyl-4-chlorophenyl moiety (4), but this did not restore binding affinity. Next, we used a more rigid benzyl linker (5), which provided some recovery in inhibition of MCL-1 (Ki = 2.36 μM) relative to the parent indole 1. Since the synthesis of N2-substituted indazole 5 also yielded the corresponding N1 isomer in the same pot, we evaluated compound 6 as well, which performed comparably to 5. Lastly, shifting the chlorine from the 5- to the 6-position (7) resulted in a marginal increase in binding affinity (Ki = 1.76 μM).
Binding affinities of initial “scaffold-hopping” MCL-1 inhibitors.
| Compound number | Structure | K i, μM (MCL-1) |
|---|---|---|
| 1 |

|
0.247 ± 0.03 |
| 2 |

|
>10 |
| 3 |

|
>10 |
| 4 |

|
>10 |
| 5 |

|
2.36 ± 0.29 |
| 6 |
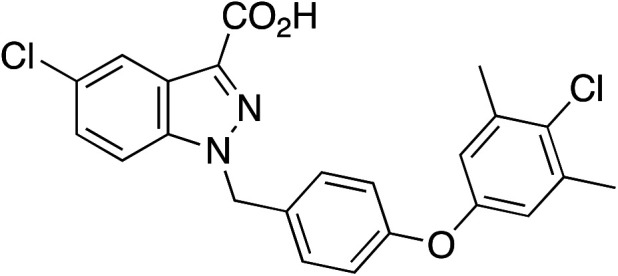
|
2.28 ± 0.34 |
| 7 |

|
1.76 ± 0.55 |
Since the synthesis of 5 was much higher yielding than that of 6 and 7, we decided to move forward with compound 5. With a new chemotype confirmed for MCL-1 inhibition, we also evaluated 5 against BCL-2 and BCL-xL. Pleasingly, 5 inhibited BCL-2 in the single-digit micromolar range (Ki = 1.48 μM) yet was inactive against BCL-xL (Ki > 10 μM), reflecting the relative BCL-2/BCL-xL affinities of the original lead 1.33 In a follow-up publication to their seminal work on the discovery of MCL-1 inhibitors, Fesik's laboratory elaborated their indole-2-carboxylic acids by converting the acid functional group into acylsulfonamides that retains the acidity of the parent group but also enables the inhibitors to occupy more of the hydrophobic BH3-binding groove by probing into the p4 pocket.41 In this way, they were able to substantially improve MCL-1 binding affinities while simultaneously preserving selectivity for MCL-1 over BCL-xL. No binding data for BCL-2 was provided. We were curious to learn if the likewise transformation of our indazole acids into indazole acylsulfonamides would also improve MCL-1 binding affinities and retain selectivity for MCL-1 over BCL-xL but also improve, or at the very least retain, binding affinities to BCL-2.
Accordingly, we prepared acylsulfonamide analogues of acid 5 (Scheme 1) in which the sulfonamide fragment was inspired by the corresponding fragment in VEN. Briefly, 5-chloroisatin (8) was transformed into 5-chloroindazole-3-carboxylic acid (9) as described previously,42 and was advanced to the next step without purification: thionyl chloride-mediated esterification of crude acid 9 yielded methyl ester 10. Alkylation of the indazole functional group can produce differing amounts of the N1- and the N2-alkylated products depending on the reaction conditions.43 We employed direct alkylation conditions of the benzylic bromide 5-(4-(bromomethyl)phenoxy)-2-chloro-1,3-dimethylbenzene with K2CO3 in DMF at RT, which produced an approximate 1 : 1 separable mixture of the N1 and N2 products. The distribution of products could be tipped in favour of N2 (in a 2 : 1) ratio by adjusting the conditions to a Mitsunobu reaction with the corresponding alcohol (see ESI† for full details). This finding is in agreement with other reports in the literature where the Mitsunobu reaction favours the formation of one isomer over another when alkylating tautomeric heterocycles.44–48
Scheme 1. (a) NaOH(aq), 45 °C, 30 min; (ii) NaNO2(aq), 10 min, 0 °C H2SO4, 30 min; (iii) SnCl2, c.HCl, 0 °C → RT, 16 h; (b) SOCl2, MeOH, reflux; (c) HOCH2C6H4O-(4-Cl-3,5-diMe)-Ph, DIAD, PPh3, THF, RT (yields a 2 : 1 ratio of 11 : 12); (d) LiOH.H2O, THF/MeOH/H2O, 3 : 1 : 1, rt; (e) R2SO2NH2 (13), EDCI, DIPEA, DMAP, CH2Cl2, rt.

Previously, N1- and N2-substituted methyl indazole-3-carboxylates were distinguished based on their polarities and NMR spectra: the N2 isomers are less polar and the protons on the carbon directly attached to the indazole nitrogen are more downfield by around 0.5 ppm, presumably due to the deshielding effect of the adjacent ester functional group. We also observed clear differences in polarity for the two isomers 11 and 12. In Fig. 2, the less polar isomer (Rf = 0.59 (hexanes/ethyl acetate, 2 : 1, versus Rf = 0.43 for the more polar product)) whose NMR spectrum is shown in panel A, also exhibited a more deshielded benzylic CH2 group by 0.39 ppm (as did the ortho-phenyl protons (see ESI†)). Thus, we assigned the less polar product as the N2-alkylated isomer 11 and the more polar product as the N1-alkylated isomer 12. Saponifications of 11 and 12 yielded carboxylic acids 5 and 6, respectively. Subsequently, acylation of sulfonamides 13a–13r with acid 5 furnished the target N2-substituted indazole-3-acylsulfonamides 14a–14r.
Fig. 2. 1H NMR spectra of the isomers (A) 11 and (B) 12. Structures assigned based on TLC polarities (11: Rf = 0.59; 12: Rf = 0.43 (hexanes/ethyl acetate, 2 : 1)) and δH values of the benzylic methylenes.

Once again, we employed the established FPCA to determine inhibitory activities of our acylsulfonamides, and the data are shown in Table 2 (ABT-737 and S63845 are dual BCL-2/BCL-xL and selective MCL-1 inhibitors, respectively). Pleasingly, we observed an improvement in MCL-1 inhibition over parent acid 5 by up to 2-fold with acylsulfonamide 14g proving the most potent MCL-1 inhibitor (Ki = 1.12 μM vs. 2.36 μM for the corresponding carboxylic acid), although with such a small improvement in affinity, it is difficult to draw any specific conclusions about the contributions of the functionalized amino group on the nitroarene ring. More interestingly, these acylsulfonamides also proved effective inhibitors of BCL-2 with 14h the best compound (Ki = 0.46 μM). Generally, the acylsulfonamides were more potent inhibitors of BCL-2 than MCL-1 by up to 3-fold. In every case, the binding affinities to BCL-xL were several-fold worse, and often >10 μM. The most potent dual MCL-1/BCL-2 inhibitor was 14a with Ki values of 1.20 μM and 0.51 μM for MCL-1 and BCL-2, respectively, and a Ki >10 μM for BCL-xL. Switching the relative positions of the nitro and amino-substituents had little impact on binding affinities.
Binding affinities of N2-substituted-indazole-3-acylsulfonamides to MCL-1, BCL-2 and BCL-xL. NA = no activity.

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound number | R2 | K i (μM) | Compound number | R2 | K i (μM) | ||||
| MCL-1 | BCL-2 | BCL-xL | MCL-1 | BCL-2 | BCL-xL | ||||
| 14a |

|
1.20 ± 0.17 | 0.51 ± 0.10 | >10 | 14k |

|
2.30 ± 0.17 | 0.90 ± 0.25 | 6.66 ± 3.14 |
| 14b |

|
1.32 ± 0.16 | 0.53 ± 0.09 | 7.99 ± 2.43 | 14l |

|
1.68 ± 0.33 | 0.96 ± 0.08 | >10 |
| 14c |

|
1.61 ± 0.10 | 0.42 ± 0.16 | NA | 14m |
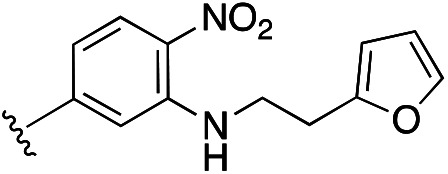
|
2.16 ± 0.61 | 0.84 ± 0.23 | >10 |
| 14d |

|
1.30 ± 0.11 | 0.57 ± 0.09 | >10 | 14n |

|
1.95 ± 0.18 | 0.71 ± 0.11 | 7.41 ± 2.56 |
| 14e |

|
1.28 ± 0.15 | 0.62 ± 0.14 | 3.40 ± 1.19 | 14o |

|
1.00 ± 0.10 | 1.22 ± 0.50 | >10 |
| 14f |

|
1.57 ± 0.14 | 0.60 ± 0.11 | 3.45 ± 0.73 | 14p |

|
1.46 ± 0.13 | 0.46 ± 0.05 | >10 |
| 14g |
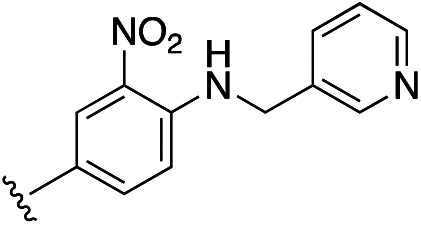
|
1.12 ± 0.07 | 1.18 ± 0.14 | 4.42 ± 0.53 | 14q |

|
1.36 ± 0.21 | 0.71 ± 0.25 | >10 |
| 14h |

|
1.46 ± 0.13 | 0.46 ± 0.05 | >10 | 14r |

|
1.42 ± 0.11 | 0.77 ± 0.18 | NA |
| 14i |

|
1.65 ± 0.18 | 1.52 ± 0.31 | >10 | 1 | — | 0.247 ± 0.03 | 1.20 ± 0.16 | >10 |
| 14j |

|
1.77 ± 0.29 | 0.66 ± 0.18 | >10 | ABT-737 | — | NA | <0.04 | <0.04 |
| S63845 | — | 0.039 ± 0.01 | NA | NA | |||||
Molecular modelling of compound 14c using SILCS methodology40 (see ESI†) provided predicted binding modes with MCL-1 and BCL-2, and reported LGFEs of −8.5 and −9.3 kcal mol−1, respectively, which is in agreement with the relative experimental binding affinities. As shown in Fig. 3A, 14c binds somewhat similarly to acylsulfonamide 49 from ref. 41, with the 3,5-dichloro-4-methylphenyl substituents probing deep into the p2 pockets, the acylsulfonamide substituents being projected into the p4 pockets, and the acidic acylsulfonamides binding Arg263. However, in stark contrast, the indazole core of 14c is forced out of the binding pocket adopted by the indole core of 49, and this may account for the reduced binding affinity of 14c to MCL-1 (Kd: 1.61 μM vs. 361 nM). Fig. 3B illustrates the binding mode of 14c in BCL-2 overlaid with that of VEN. While the larger molecule VEN clearly makes greater interactions and extends deeper into the p4 pocket, 14c appears to engage the p2 and p4 pockets, like VEN, as well as form a salt bridge between its acylsulfonamide and Arg146. In both proteins, 14c is predicted to make contacts with both the p2 and p4 pockets, but with MCL-1, the indazole core is forced away from the protein surface, while with BCL-2, the indazole core binds towards the p1 pocket. Given the predicted binding modes are significantly different, concomitant optimization of binding to both MCL-1 and BCL-2 may prove challenging, therefore It is crucial that co-crystal structures are solved to determine the specific binding modes of our lead compounds, and this information will aid in the design of next generation dual MCL-1/BCL-2 inhibitors.
Fig. 3. SILCS-MC predicted binding poses (carbons in cyan) of compound 14c with (A) MCL-1 and (B) BCL-2. Crystal orientations (carbons in pink) of compound MCL-1 inhibitor 49 from ref. 41 (PDB:5FDO) and BCL-2 inhibitor VEN (PDB:6O0K) are overlaid in (A) and (B), respectively. SILCS FragMaps for apolar, H-bond acceptor and negatively charged types are shown in green, red, and orange meshes, respectively, and rendered at a GFE level of −1.2 kcal mol−1.

Conclusions
While this work was in progress, 1-substituted 3-acylsulfonamide indazoles were reported as selective MCL-1 inhibitors (although there was no discussion of regioselectivities, we have analyzed their NMR data and we believe these are consistent with their reported identities as the N1-regioisomers).49 It will be important to ascertain if the ability of our analogous 2-substituted 3-acylsulfonamide indazoles to recognize both MCL-1 and BCL-2 is due to the different substitution pattern, the p2-binding groups or a combination of both. In this regard, we are engaged in further exploring different p2-binding groups (including from the N1 position) as well as substitutions off the benzene ring component of the indazole scaffold. Additional future work will focus on attempts to solve crystal structures of our best compounds with both proteins to help drive the design of next generation compounds. In conclusion, by scaffold hopping from an indole core to an indazole we have transformed MCL-1 selective inhibitors into dual MCL-1/BCL-2 inhibitors. This finding is significant because the BCL-2 selective inhibitor VEN provides a median survival of around only 18 months primarily due to the upregulation of MCL-1.3,5,14 A dual MCL-1/BCL-2 inhibitor may prove an essential weapon in the anti-cancer artillery.
Experimental data
All synthetic procedures, SILC-MS and the FPCA assay protocol are provided in the ESI.†
Conflicts of interest
ADM is co-founder and CSO of SilcsBio LLC.
Supplementary Material
Acknowledgments
We acknowledge the T32 training grant (NIH/NIGMS T32 GM066706) to BD and CG, the UMB CADD Center and NIH grant GM131710 to ADM, and the University of Maryland School of Pharmacy for financial support of this research to SF.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00095d
Notes and references
- Adams J. M. Cory S. Cell Death Differ. 2000;25:27–36. doi: 10.1038/cdd.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo C. D. et al. . Lancet Oncol. 2018;19:216–228. doi: 10.1016/S1470-2045(18)30010-X. [DOI] [PubMed] [Google Scholar]
- Lin K. H. et al. . Sci. Rep. 2016;6:27696. doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perini G. F. Ribeiro G. N. Pinto Neto J. V. Campos L. T. Hamerschlak N. J. Hematol. Oncol. 2018;11:65. doi: 10.1186/s13045-018-0608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. Wan J. Zhang W. Hao S. Leuk. Lymphoma. 2019;60:2170–2180. doi: 10.1080/10428194.2018.1563694. [DOI] [PubMed] [Google Scholar]
- Wei A. H. et al. . Blood Rev. 2020;44:100672. doi: 10.1016/j.blre.2020.100672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap J. L. Chen L. Lanning M. E. Fletcher S. J. Med. Chem. 2017;60:821–838. doi: 10.1021/acs.jmedchem.5b01888. [DOI] [PubMed] [Google Scholar]
- Diepstraten S. T. Anderson M. A. Czabotar P. E. Lessene G. Strasser A. Kelly G. L. Nat. Rev. Cancer. 2022;22:45–64. doi: 10.1038/s41568-021-00407-4. [DOI] [PubMed] [Google Scholar]
- Souers A. J. et al. . Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Tron A. E. et al. . Nat. Commun. 2018;9:5341. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. . ACS Med. Chem. Lett. 2020;11:1829–1836. doi: 10.1021/acsmedchemlett.9b00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juárez-Salcedo L. M. Desai V. Dalia S. Drugs Context. 2019;8:212574. doi: 10.7573/dic.212574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M. et al. . Cancer Discovery. 2016;6:1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollyea D. A. et al. . Am. J. Hematol. 2021;96:208–217. doi: 10.1002/ajh.26039. [DOI] [PubMed] [Google Scholar]
- Kaufmann S. H. et al. . Blood. 1998;91:991–1000. doi: 10.1182/blood.V91.3.991.991_991_1000. [DOI] [PubMed] [Google Scholar]
- Leverson J. D. Cojocari D. Front. Oncol. 2018;8:458. doi: 10.3389/fonc.2018.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedtke D. A. et al. . Signal Transduction Targeted Ther. 2017;2:17012. doi: 10.1038/sigtrans.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir S. K. et al. . BMC Cancer. 2017;17:399. doi: 10.1186/s12885-017-3383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tausch E. et al. . Haematologica. 2019;104(9):e434. doi: 10.3324/haematol.2019.222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Guo M. Wei H. Chen Y. J. Hematol. Oncol. 2021;14:67. doi: 10.1186/s13045-021-01079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S. Expert Opin. Ther. Pat. 2019;29:909–919. doi: 10.1080/13543776.2019.1672661. [DOI] [PubMed] [Google Scholar]
- Kotschy A. et al. . Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- Leverson J. D. et al. . Cell Death Dis. 2015;6(1):e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M. Leiva D. Lucendo E. Orzáez M. FEBS J. 2022 doi: 10.1111/febs.16136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman J. T. et al. . Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Wang X. et al. . Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vick B. et al. . Hepatology. 2009;49:627–636. doi: 10.1002/hep.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Rinkenberger J. L. Horning S. Klocke B. Roth K. Korsmeyer S. J. Genes Dev. 2000;14:23–27. doi: 10.1101/gad.14.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thomas R. L. et al. . Genes Dev. 2013;27:1365–1377. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moujalled D. M. et al. . Leukemia. 2019;33:905–917. doi: 10.1038/s41375-018-0261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. et al. . Nat. Struct. Mol. Biol. 2016;23:600–607. doi: 10.1038/nsmb.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon S. R. Ertl P. Brown N. Mol. Inf. 2010;29:366–385. doi: 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- Sun H. Tawa G. Wallqvist A. Drug Discovery Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg A. et al. . J. Med. Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X. et al. . Mol. Cancer. 2013;12:42. doi: 10.1186/1476-4598-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. et al. . Org. Biomol. Chem. 2016;14:5505–5510. doi: 10.1039/C5OB02063H. [DOI] [PubMed] [Google Scholar]
- Conlon I. L. et al. . ChemMedChem. 2020;15:1691–1698. doi: 10.1002/cmdc.202000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennen B. et al. . ChemMedChem. 2016;11:827–833. doi: 10.1002/cmdc.201500461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K.-Y. et al. . Org. Lett. 2013;15:3234–3237. doi: 10.1021/ol401197n. [DOI] [PubMed] [Google Scholar]
- Lanning M. E. et al. . Org. Biomol. Chem. 2015;13:8642–8646. doi: 10.1039/C5OB00478K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning M. E. et al. . Eur. J. Med. Chem. 2016;113:273–292. doi: 10.1016/j.ejmech.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelz N. F. et al. . J. Med. Chem. 2016;59:2054–2066. doi: 10.1021/acs.jmedchem.5b01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. L. Rodgers J. D. Synth. Commun. 2005;35:2681–2684. doi: 10.1080/00397910500214318. [DOI] [Google Scholar]
- Longworth M. Banister S. D. Mack J. B. C. Glass M. Connor M. Kassiou M. Forensic Toxicol. 2016;34:286–303. doi: 10.1007/s11419-016-0316-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. Fletcher S. Tetrahedron Lett. 2014;55:1693–1696. doi: 10.1016/j.tetlet.2014.01.065. [DOI] [Google Scholar]
- Fletcher S. Org. Chem. Front. 2015;2:739–752. doi: 10.1039/C5QO00016E. [DOI] [Google Scholar]
- Fletcher S. Tetrahedron Lett. 2010;51:2948–2950. doi: 10.1016/j.tetlet.2010.03.103. [DOI] [Google Scholar]
- Fletcher S. Shahani V. M. Lough A. J. Gunning P. T. Tetrahedron. 2010;66:4621–4632. doi: 10.1016/j.tet.2010.03.118. [DOI] [Google Scholar]
- Fletcher S. Shahani V. M. Gunning P. T. Tetrahedron Lett. 2009;50:4258–4261. doi: 10.1016/j.tetlet.2009.04.137. [DOI] [Google Scholar]
- Wan Y. Li Y. Yan C. Wen J. Tang Z. Bioorg. Chem. 2020;104:104217. doi: 10.1016/j.bioorg.2020.104217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.