

Abstract
Irradiation of aryl esters of N-hydroxyphthalimides in the presence of unactivated olefins promotes a mild and regioselective hydroesterification. Optimal results are obtained with the aid of fac-Ir(dFppy)3 in CH2Cl2. Terminal and 1,1-disubstituted olefins provide primary esters, and trisubstituted olefins provide secondary esters. The anti-Markovnikov selectivity is consistent with alkyl radical intermediates, which are also indicated by the formation of cyclized products from dienes. Mono-acylated diols are formed from tri- and tetrasubstituted olefins in the presence of water.
Keywords: photocatalysis, hydroesterification, dihydroxylation, aminohydroxylation, anti-Markovnikov
Graphical Abstract
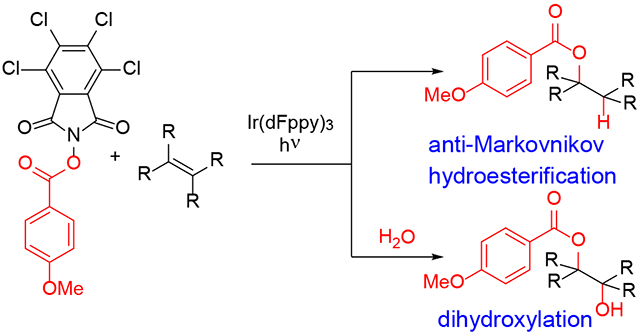
Addition of oxygen nucleophiles to unactivated olefins provides a direct route to alcohols, ethers and esters, which are basic building blocks for organic synthesis. Unfortunately, hydration, hydroetherification and hydroesterification of olefins generally fails (Scheme 1a). Indeed, dehydration of alcohol derivatives to form the corresponding olefin is more common than the reverse addition reactions. The poor performance of direct hydration of olefins has given rise to oxidation/reduction sequences to form the corresponding oxygenated products. Oxymercuration/reduction, hydroboration/oxidation and Wacker oxidation/reduction can provide alcohols and ethers with varying levels of regiocontrol and efficiency.1 We postulated that N-(acyloxy)phthalimides (1) might be coaxed into adding to olefins wherein visible light irradiation might provide the driving force to overcome thermodynamic limitations of direct hydration-type reactions.2
Scheme 1.

Background
Alkyl N-(acyloxy)phthalimides have traditionally been used as acylating reagents. Pioneering studies from Okada and Odo revealed their propensity to undergo 1-electron reduction followed by N-O bond fragmentation to release alkyl radicals following decarboxylation.3 This reactivity underpinned the development of decarboxylative additions to olefins, arenes and alkynes, as well as decarboxylative borylation, olefination and cross-coupling.4, 5, 6 Aryl N-(acyloxy)phthalimides have been exploited in decarboxylative borylation reactions,7 but the decreased stability of aryl radicals relative to alkyl radicals hinders decarboxylative processes. For example, aryl phthalimide esters provided a source of benzoyl esters in a Ni-catalyzed oxidation of aryl zinc reagents.8 A separate study generated benzoyloxy radicals from the corresponding phthalimide ester, which then underwent intramolecular C-H abstraction followed by alkylation.9
To determine if aryl N-(acyloxy)phthalimides could effect the hydroesterification of olefins, we exposed phthalimide ester 1a to cyclohexene in the presence of various photocatalysts. Key results are shown in Table 1. Several organic catalysts provided moderate yields of 4a, including acridinium-based catalysts 5 and 6 as well as the carbazole-decorated benzene 7 (entries 1-3).10 Substantial quantities of the corresponding benzoic acid 3 were recovered in these experiments, prompting us to investigate organometallic photoredox catalysts.11,12 While Ru(bpy)3Cl2 (8) proved unsuccessful, three Ir-based catalysts provided ester 4a in high yield (entries 5-8). The cleanest reaction mixtures were obtained using fac-Ir(dFppy)3 (12), which provided high yields of ester 4a and only trace quantities of benzoic acid 3. The reaction proceeds without external heating or cooling, and does not require acids, bases, oxidants or reductants. Purging the headspace with nitrogen gas provides sufficient protection from oxygen. Control reactions revealed the requirement for irradiation and photocatalyst. Heating alone in the presence of catalyst provided no desired product (entry 10). In the absence of catalyst, irradiation at 427 nM alone provided no desired product (4a), and the phthalimide ester was recovered (entry 11). Trace amounts of 4a (8%) were observed when the reaction mixture was irradiated at 365 nM without catalysts (Table S2 in the SI), but best results were obtained with radiation at 427 nM in the presence of Ir catalyst 12 (427 nM > 440 nM > 390 nM).
Table 1.
Identification of photocatalysta
|
||||||
|---|---|---|---|---|---|---|
| entry | catalysts | ET (kcal/mol) |
E1/2 (M*/M+, V) |
light source | yield 3 (%) |
yield 4a (%) |
| 1b | 9-Mes-10-Me-Acr·ClO4 (5) | 44.7c | - | Kessil H150-BLUE | 45 | 38 |
| 2b | Acridinium·BF4 (6) | Kessil H150-BLUE | 28 | 25 | ||
| 3b | 4-CzIPN (7) | 58.8d | −1.18e | Kessil PR160L (440 nm) | 22 | 62 |
| 4b | Ru(bpy)3Cl2 (8) | 46.5f,g | −0.81f,g | Kessil H150-BLUE | 0h | 0 |
| 5 | Ir(ppy)2(dtbbpy)PF6 (9) | 49.2f | −0.96f | Kessil PR160L (440 nm) | 18 | 73 |
| 6 | fac-Ir(ppy)3 (10) | 55.2f | −1.73f | Kessil PR160L (440 nm) | 11 | 81 |
| 7 | Ir(dF(CF3)ppy)2(dtbbpy)PF6 (11) | 60.1f | −0.89f | Kessil PR160L (440 nm) | 40 | 10 |
| 8 | fac-Ir(dFppy)3 (12) | 60.1f | −1.28f | Kessil PR160L (440 nm) | <5 | 87 |
| 9 | fac-Ir(dFppy)3 (12) | Kessil PR160L (427 nm) | <5 | 91 | ||
| 10 | fac-Ir(dFppy)3 (12) | None (40 °C) | 0h | 0 | ||
| 11 | None | Kessil PR160L (427 nm) | 0h | 0 | ||
|
||||||
Unless described otherwise, the reactions were conducted with substrate 1a (44.0 mg, 0.1 mmol, 1.0 equiv), CH2Cl2 (5.0 mL), catalyst (1 mol%), cyclohexene (5.0 equiv), without heating or cooling (actual reaction solution temperature: 40 - 45 °C) under a nitrogen atmosphere. 8 mL glass vials were irradiated with LED as indicated for 36 h. Yields were obtained by 1H NMR integrations by using 1,3,5-trimethoxybenzene as internal standard.
5 mol% catalyst.
Ref 10.
Ref 20.
Ref 17.
Ref 12.
Values for Ru(bpy)3(PF6)2.
Substrate 1a recovered.
Additional optimization data is provided in the supporting information, but some observations warrant discussion. The non-chlorinated version of 1a was nearly as effective as 1a (81% yield), as was the 2,4-dimethoxy congener (77% yield). However, alternative aryl esters were much less effective (see Figure S2 in the SI). For example, the parent benzoyloxy phthalimide and heteroaromatic phthalimide esters all provided complex reaction mixtures, as did the 2-methoxy and 3-methoxy analogs of 1a. Several common additives for photoredox reactions decreased yields, including amines, Hantzsch ester, silanes, thiols, and copper salts (Table S3). The reaction proceeded to varying degrees in different solvents. Dichloromethane, dichloroethane, acetone and acetonitrile were similarly effective (Table S4). Modest amounts of 4a (10-60%) were observed in chloroform, ethyl acetate, toluene, THF and methanol, while no product was observed in polar solvents such as trifluoroethanol, DMSO or DMF.
With optimal conditions identified, we determined the generality of the reaction (Scheme 2). A wide range of terminal olefins underwent hydroesterification in good yields and moderate-good regioselectivity. For example, linear alkenes reacted with regioselectivities around 3:1 (4b, 4c). Steric bulk near the olefin markedly increased regioselectivity, with a cyclohexyl group adjacent or even one carbon away from the olefin steering addition to the terminal carbon (4d, 4e). tert-Butyl ethylene was a poor substrate (4f), and in general, substrates lacking allylic C-H’s did not perform well. Aryl groups (4g, 4h), olefins (4i, 4j) and halides (4k, 4l) were accommodated. Existing oxygenation did not compromise reactivity, as demonstrated by a protected alcohol (4m), epoxide (4n), ketone (4o) and esters 4p, 4q. 1,1-Disubstituted olefins reacted in similar yields as terminal olefins but with complete regioselectivity favoring oxygenation of the terminal carbon. Internal olefins generally provided higher yields than terminal and 1,1-disubstituted olefins. Linear (4w, 4x) and cyclic (4z – 4ee) disubstituted alkenes performed well. Trisubstituted (4ff) and even tetrasubstituted olefins (4y) were competent reaction partners. Complete regioselectivity was observed for cyclic ethers (4gg, 4hh) using the non-chlorinated phthalimide 1b. para-Methoxy benzoic acid accounted for the remaining mass balance in most cases along with tetrachlorophthalimide, and the reaction performed similarly on a 0.1 and 1 mmol scale.
Scheme 2.

Scope of the hydroesterificationa
arr: regioisomer ratio. Isolated yields. Reaction conditions: 1a (0.1 mmol), olefin (0.5 mmol), Ir(dFppy)3 (1 mol%). Irradiation with Kessil PR160L-427 nm LED. Arrow indicates position of olefin in starting material. b36 h reaction time. c24 h reaction time. d1 mmol 1a. eNon-chlorinated phthalimide 1b (1,3-dioxoisoindolin-2-yl 4-methoxybenzoate) was used.
Under the standard conditions, but in the presence of added water, we observed difunctionalization of 1,1-disubstituted, tetrasubstituted and activated disubstituted olefins (Scheme 3). Monobenzoyl-protected diols (13) were obtained in moderate yield and complete regioselectivity. Under these modified conditions, terminal olefins and internal disubstituted olefins continued to generate the hydroesterification products (4) with trace amounts of the diol being observed. This difference likely arises from the relative ease of accessing tertiary carbocations vs. secondary carbocations (see below).
Scheme 3.

Difunctionalization of olefinsa
aReaction conditions: 1a (0.1 mmol), olefin (0.5 mmol), Ir(dFppy)3 (1 mol%), H2O (10 equiv), 20 mM in CH2Cl2. Irradiation for 24 h with Kessil PR160L-427 nm LED.
Some electron-rich olefins gave unexpected oxidation products (Scheme 4). For example, three olefins provided aminohydroxylation products in which both the benzoate and phthalimide moieties of 1a were transferred to the olefin.13 2,3-Dimethoxy-1,3-butadiene reacted with complete regiocontrol (14a) while dihydrofuran and trans-stilbene generated single stereoisomers (14b, 14c).14 1,1-Diphenylethylene, styrene and a silyl enol ether were oxidized under the standard conditions, but in slightly different ways. Enol ester 15 was obtained from diphenylethylene whereas the bis-benzoate 16 was formed in low yield from styrene. The only substrate we have observed to undergo allylic oxidation was a silyl enol ether, which yielded allylic benzoate 17.15
Scheme 4.

Unexpected oxidations with 1aa
aReaction conditions: 1a (0.1 mmol), olefin (0.5 mmol), Ir(dFppy)3 (1 mol%), 20 mM in CH2Cl2. Irradiation for 24 h with Kessil PR160L-427 nm LED.
Mechanistic considerations.
Radical intermediates in the reaction were implicated by several cyclizations and fragmentations (Scheme 5). For example, dienes 18, 20 and 22 underwent addition/cyclization consistent with intramolecular radical addition to the pendent olefins. Cyclopropyl-containing substrate 24 generated naphthalene 25 in a process that likely involves cyclopropyl ring-opening followed by cyclization onto the arene and aerobic oxidation. Finally, α-pinene was converted to cyclohexene 27, suggestive of radical fragmentation of the cyclobutyl ring.
Scheme 5.

Evidence for radical intermediatesa
aReaction conditions: 4,5,6,7-tetrachloro-1,3-dioxoisoindolin-2-yl 4-methoxybenzoate (1a, 0.1 mmol), olefin (0.5 mmol), Ir(dFppy)3 (1 mol%), 20 mM in CH2Cl2. Irradiation for 24 h with Kessil PR160L-427 nm LED. bIrradiation for 36 h.
We attempted to identify the source of the hydrogen that is added to the olefin along with the benzoate group. Remarkably, the reaction was substantially suppressed in CD2Cl2 (eq 1). The recovered product showed partial deuteration at the newly introduced H/D. Control experiments with mixed solvents (CH2Cl2 + CD2Cl2) confirmed that there were no catalyst poisons in the CD2Cl2 (79% yield of 4a). Similar results were obtained with other substrates and with CD3CN. Additionally, no deuterium incorporation was observed when CD3OD was included in the reaction mixture. This evidence suggested that CH2Cl2 could be the source of the added hydrogen, and that a kinetic isotope effect could be operative in a hydrogen atom transfer step. A reciprocal experiment with cyclohexene-d10 provided consistent results (eq 2). The newly introduced hydrogen was predominantly 1H, but partial 2H incorporation suggested some C-D abstraction from the allylic position of the starting material.
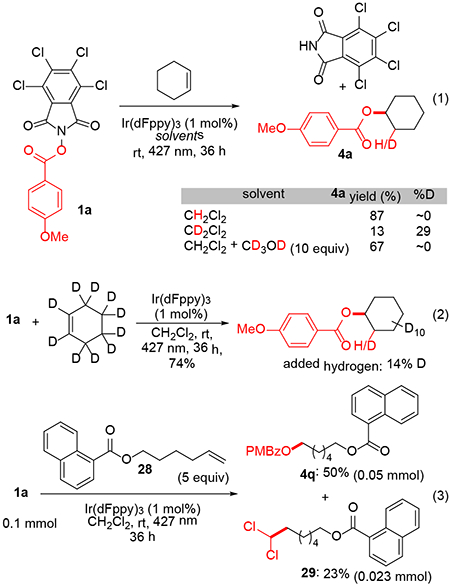 |
The overall reaction is redox neutral with regard to the olefin, but the N-hydroxyphthalimide ester undergoes net 2e− reduction. To better understand the redox profile of the reaction, we subjected non-volatile substrate 28 to the hydroesterification (eq 3) to search for any oxidized products. In addition to the expected product 4q, we observed the dichloromethyl adduct 29.16 While this observation provides evidence for formation of the dichloromethyl radical through H-atom transfer, it does not resolve the overall redox balance for the transformation because 1a + CH2Cl2 29 is also redox neutral. We did not observe any oxidized products (e.g. diene, allylic alcohol derivative, dimer) derived from 28, which was recovered from the reaction unreacted. Our current interpretation is that dichloromethane provides the H atom for the hydroesterification, but the ultimate fate of the dichloromethyl radical is not clear.
Steady-state Stern-Volmer quenching experiments demonstrated that benzoyloxy phthalimide 1a effectively quenched the fluorescence of fac-Ir(dFppy)3 (12) whereas cyclohexene had no effect (Figure S3, S4). We considered electron transfer and energy transfer as possible mechanisms to activate the phthalimide ester. The excited state of fac-Ir(dFppy)3 is less reducing (−1.28V vs SCE) than would be required to reduce benzoyloxy phthalimide (E1/2 = −1.4V).7 Moreover several catalysts that provide reasonable yields are much less reducing in the excited state (4-CzIPN, −1.18V; Ir(ppy)2(dtbbpy)PF6, −0.96V). 10,12,17 Even more striking, two acridinium catalysts (5, 6, see Table 1) catalyzed the reaction with 5-9 turnovers. These catalysts are unlikely to act as excited state reductants, as SET would form dicationic intermediates. Overall, we consider it more likely that the reaction proceeds through energy transfer from the excited state of 12 to the phthalimide ester,18,19 although there is not a clear correlation between catalyst triplet energy levels and reaction efficiency.20 In this regard, it is possible that different catalysts operate through slightly different mechanisms.
A potential mechanism for the hydroesterification is outlined in Scheme 6. Energy transfer between the photoactivated [Ir]* species and hydroxyphthalimide ester 1a could generate the excited reagent 1a*. Homolysis of the N-O bond could yield para-methoxybenzoyl radical (PMBzO·) and the phthalimide radical. Addition of the former to the olefin followed by C-H abstraction, possibly from solvent, could generate the observed product. However, several observations are difficult to reconcile with homolysis of 1a*. First, terminal olefins react more slowly than internal olefins, even though both substrates would form similar secondary radicals. Second, in reactions involving slower-reacting substrates, such as terminal olefins, the hydroxyphthalimide ester 1a can be recovered. Homolysis of the N-O bond is unlikely to be reversible, suggesting that the olefin could be intimately involved in consumption of 1a*. Third, we have observed no evidence of allylic C-H abstraction or oxidation other than a single example (product 17). By contrast, Li and coworkers showed allylic C-H oxidation of cyclohexene with benzoyloxy radicals.15,21 Fourth, we observed no evidence of decarboxylation, which has also been reported for benzoyloxy radicals.22,23,24 Finally, the modest regioselectivity observed with terminal olefins contrasts with the high selectivity observed in reported additions of O-centered radicals to olefins.13,25 Alternatively, olefin could react directly with triplet 1a* to yield the phthalimide radical and the alkyl radical 30. In this scenario, relaxation of 1a* in the presence of less-reactive substrates could explain why we recover 1a. The absence of products associated with the benzoyl radical could likewise indicate that the benzoyl radical is not an intermediate. Difunctionalization products (see Schemes 3 and 4) could arise from oxidation of the alkyl radical to the tertiary cation 31 followed by trapping with water under non-anhydrous conditions.
Scheme 6.

Potential mechanism
In summary, we have described a mild, regioselective hydroesterification of unfunctionalized olefins. While the current protocol requires an excess of olefin, it offers a single-step route to alkyl esters.
Supplementary Material
ACKNOWLEDGMENT
Funding support from the Welch Foundation (I-1612) and the NIH (RM1 GM142002).
Footnotes
No completing financial interests
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, additional optimization experiments, characterization data (PDF).
X-ray crystal structure of 14b (cif).
REFERENCES
- (1).Baiju TV; Gravel E; Doris E; Namboothiri INN Recent developments in Tsuji-Wacker oxidation. Tetrahedron Lett. 2016, 57, 3993–4000 [Google Scholar]
- (2).While this work was under review, a related study was published: Lai S-Q; Wei B-Y; Wang J-W; Yu W; Han B Photocatalytic anti-markovnikov radical hydro- and aminooxygenation of unactivated alkenes tuned by ketoxime carbonates. Angew. Chem. Int. Ed 2021, 60, 21997–22003 [DOI] [PubMed] [Google Scholar]
- (3).Okada K; Okamoto K; Morita N; Okubo K; Oda M Photosensitized decarboxylative Michael addition through N-(acyloxy)phthalimides via an electron-transfer mechanism. J. Am. Chem. Soc 1991, 113, 9401–9402. [Google Scholar]
- (4).Parida SK; Mandal T; Das S; Hota SK; De Sarkar S; Murarka S Single electron transfer-induced redox processes involving N-(acyloxy)phthalimides. ACS Catalysis 2021, 11, 1640–1683. [Google Scholar]
- (5).Parida SK; Hota SK; Kumar R; Murarka S Late-stage alkylation of heterocycles using N-(acyloxy)phthalimides. Chem. Asian J 2021, 16, 879–889. [DOI] [PubMed] [Google Scholar]
- (6).Murarka S N-(acyloxy)phthalimides as redox-active esters in cross-coupling reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- (7).Candish L; Teders M; Glorius F Transition-metal-free, visible-light-enabled decarboxylative borylation of aryl N-hydroxyphthalimide esters. J. Am. Chem. Soc 2017, 139, 7440–7443. [DOI] [PubMed] [Google Scholar]
- (8).Shih B-H; Basha RS; Lee CF Nickel-catalyzed cross-coupling of aryl redoxactive esters with aryl zinc reagents. ACS Catalysis 2019, 9, 8862–8866. [Google Scholar]
- (9).Li Y; Zhang J; Li D; Chen Y Metal-free C(sp3)─H allylation via aryl carboxyl radicals enabled by donor–acceptor complex. Org. Lett 2018, 20, 3296–3299. [DOI] [PubMed] [Google Scholar]
- (10).Romero NA; Nicewicz DA Organic photoredox catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (11).Prier CK; Rankic DA; MacMillan DWC Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Teegardin K; Day JI; Chan J; Weaver J Advances in photocatalysis: A microreview of visible light mediated ruthenium and iridium catalyzed organic transformations. Org. Process Res. Dev 2016, 20, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Patra T; Das M; Daniliuc CG; Glorius F Metal-free photosensitized oxyimination of unactivated alkenes with bifunctional oxime carbonates. Nature Catalysis 2021, 4, 54–61. [Google Scholar]
- (14).Qian B; Xiong H; Zhu N; Ye C; Jian W; Bao H Copper-catalyzed diesterification of 1,3-diene for the synthesis of allylic diester compounds. Tetrahedron Lett. 2016, 57, 3400–3403. [Google Scholar]
- (15).Zhu N; Qian B; Xiong H; Bao H Copper-catalyzed regioselective allylic oxidation of olefins via C─H activation. Tetrahedron Lett. 2017, 58, 4125–4128. [Google Scholar]
- (16).An analogous product was formed alongside product 4h in approximately 20% yield. Similar dichloromethane adducts were likely formed in other cases but were difficult to isolate due to high volatility and low polarity.
- (17).Speckmeier E; Fischer TG; Zeitler K A toolbox approach to construct broadly applicable metal-free catalysts for photoredox chemistry: Deliberate tuning of redox potentials and importance of halogens in donor–acceptor cyanoarenes. J. Am. Chem. Soc 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- (18).Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy transfer catalysis mediated by visible light: Principles, applications, directions. Chem. Soc. Rev 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- (19).Strieth-Kalthoff F; Glorius F Triplet energy transfer photocatalysis: Unlocking the next level. Chem 2020, 6, 1888–1903. [Google Scholar]
- (20).Nishimoto T; Yasuda T; Lee SY; Kondo R; Adachi C A six-carbazole-decorated cyclophosphazene as a host with high triplet energy to realize efficient delayed-fluorescence OLEDs. Mater. Horiz 2014, 1, 264–269. [Google Scholar]
- (21).Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-light-promoted activation of unactivated C(sp3)─H bonds and their selective trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200–16203. [DOI] [PubMed] [Google Scholar]
- (22).Yamauchi S; Hirota N; Takahara S; Misawa H; Sawabe K; Sakuragi H; Tokumaru K A time-resolved EPR study on photodecomposition of dibenzoyl peroxides in carbon tetrachloride. J. Am. Chem. Soc 1989, 111, 4402–4407. [Google Scholar]
- (23).Oishi S; Tsubaki H; Matsuzawa H Transient spectra and photoreaction of 4-methoxybenzoyloxyl radical. Chem. Lett 1999, 28, 805–806. [Google Scholar]
- (24).Yu W-Y; Sit WN; Zhou Z; Chan ASC Palladium-catalyzed decarboxylative arylation of C─H bonds by aryl acylperoxides. Org. Lett 2009, 11, 3174–3177. [DOI] [PubMed] [Google Scholar]
- (25).Zheng M; Gao K; Qin H; Li G; Lu H Metal-to-ligand ratio-dependent chemodivergent asymmetric synthesis. Angew. Chem. Int. Ed 2021, 60, 22892–22899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.