

SUMMARY
Long-lived rodents have become an attractive model for the studies on aging. To understand evolutionary paths to long life, we prepare chromosome-level genome assemblies of the two longest-lived rodents, Canadian beaver (Castor canadensis) and naked mole rat (NMR, Heterocephalus glaber), which were scaffolded with in vitro proximity ligation and chromosome conformation capture data and complemented with long-read sequencing. Our comparative genomic analyses reveal that amino acid substitutions at “disease-causing” sites are widespread in the rodent genomes and that identical substitutions in long-lived rodents are associated with common adaptive phenotypes, e.g., enhanced resistance to DNA damage and cellular stress. By employing a newly developed substitution model and likelihood ratio test, we find that energy and fatty acid metabolism pathways are enriched for signals of positive selection in both long-lived rodents. Thus, the high-quality genome resource of long-lived rodents can assist in the discovery of genetic factors that control longevity and adaptive evolution.
In Brief
Zhou et al. generate “chromosome-level” genome assemblies for the Canadian beaver and naked mole rat. They characterize genome features and identify common substitutions in long-lived rodents that support enhanced tolerance of cells to DNA damage. The study also provides a valuable genome resource for aging research.
Graphical Abstract
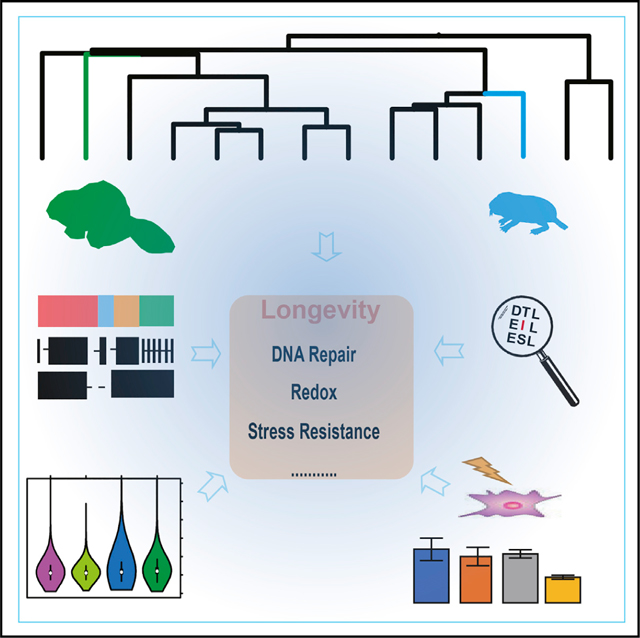
INTRODUCTION
Discerning the genetic factors that affect the aging process, in particular how Nature uses them to control lifespan, is one of the important yet unanswered questions in biology and evolution (Gladyshev, 2013). Rodent species differ more than 10-fold in maximum lifespan, and long-lived rodents have been observed to show low susceptibility to certain age-related diseases (Gorbunova et al., 2008). Therefore, analyses of their genomes could help discover genetic factors responsible for such diversity of lifespan. Motivated by this idea, an initial genome assembly of the naked mole rat (NMR), a rodent best known for its longevity (maximum lifespan >35 years; Ruby et al., 2018), was generated with a contig N50 of 19.3 kb and scaffold N50 of 1.6 Mb (Kim et al., 2011). It represented the first case of a mammalian genome being sequenced with the explicit purpose of providing insights into longevity. The initial NMR assembly was subsequently improved to scaffold N50 approaching 21 Mb (Fang et al., 2014). In addition, another NMR genome was independently sequenced and assembled, with similar characteristics (Keane et al., 2014). Analyses of these genome assemblies revealed several unique features and molecular mechanisms related to NMR phenotypes, such as cancer resistance, protein synthesis, visual function, etc. The North American beaver is an organism with the second longest lifespan (>23 years) known for rodents. This species is famous for its ability to modify the environment by building complex dams and lodges, which sets them apart from other mammals (Gorbunova et al., 2008). To date, two beaver genome assemblies have been reported (Lok et al., 2017), although extensive genome analyses have not been performed.
It should be noted that rodents have achieved long lives at least four times independently, and two contrasting combinations of longevity and body mass are recognized: i.e., species with large body mass and long lifespan (e.g., beaver and porcupine) and species with small body mass and long lifespan (e.g., the NMR; Austad, 2005). Therefore, comparative analyses of these rodents and their closely related relatives that are characterized by small body mass and short lifespan (e.g., mouse and rat) could be useful for understanding how lifespan coevolved with body mass along the radiation of rodents. It was proposed that the ability of organisms to effectively cope with both intrinsic and extrinsic stresses is linked with longevity (Kourtis and Tavernarakis, 2011). With these goals in mind, we prepared high quality chromosome-level genome assemblies of the longest-lived rodents, the beaver and NMR. By carrying out extensive comparative genomic analysis and experimental validation of cellular stress response in long-lived rodents, we revealed common paths to longevity and uncovered the role of genome stability.
RESULTS AND DISCUSSION
Comparative Assembly and Genome Synteny
We first generated a preliminary beaver genome assembly (2.76-Gb estimated genome size, 49.32-kb contig N50, and 55.69-kb scaffold N50) using short reads sequenced on the Illumina HiSeq platform. Then three Chicago HiRise libraries (Putnam et al., 2016) were prepared and sequenced (Figure S1). Approximately 500 million reads that were generated and processed through the HiRise assembler yielded a total of ~741,906 scaffolds (Figures S1 and S2). Next, we employed PBJelly (English et al., 2012) to fill the gaps of this HiRise assembly using the previously generated long PacBio reads (Lok et al., 2017). Finally, we realigned all paired-end Illumina data to polish this beaver genome assembly with the help of Pilon (Walker et al., 2014). The resulting de novo assembly had an average depth of coverage of ~137× and N50 of 24.3 Mb (Figures 1A and 1B), representing a substantial improvement both in contig and scaffold length compared to previous assemblies. We found that 95.0% of the BUSCO (Simão et al., 2015) orthologs were complete and correct in our beaver genome assembly, which was higher than in the previously reported beaver genome (83.1%). We further employed homology and de novo methods as well as RNA sequencing (RNA-seq) to predict 26,515 protein-coding genes in the beaver genome, which is comparable to what is predicted for other mammals.
Figure 1. Beaver and NMR Genome Assemblies Support Robust Synteny Analysis in Rodents.

(A) Schematic diagram of beaver and NMR genome assemblies. (Left) North American beaver. (Right) NMR.
(B) Genome statistics of beaver and NMR genome assemblies.
(C) Circle plots of mouse genome synteny to beaver (left) and NMR (right) genomes.
(D) Comparative organization of genes in eutherian PARs. Evolutionary strata (S3–S5) of human sex chromosomes are referenced and shown at the bottom.
To better understand the molecular basis of long lifespan, we also prepared an improved version of the NMR genome, because the previously reported NMR assemblies were prepared from short sequence reads. We developed a robust procedure, which used Hi-C linking information, a sequencing-based method that measures the frequency of contact between pairs of loci (Lieberman-Aiden et al., 2009; Rao et al., 2014), to generate a genome assembly with chromosome length scaffolds. We started from the latest NMR draft assembly (Fang et al., 2014) that comprised 2.69 Gb of sequence (contig N50 length: 19.3 kb) partitioned among 175,458 scaffolds (scaffold N50: 21.31 Mb). We then generated and applied in situ Hi-C data (~153 Gb) to split, anchor, order, and orient the scaffolds in this initial assembly using a 3D-DNA pipeline (Dudchenko et al., 2017). The resulting Hi-C assembly consisted of 30 large scaffolds (lengths from 40.7 to 132.1 Mb), which covered 90.7% of the total genome assembly (3.04 Gb; Figure S1C). We next generated PacBio long reads (~60 Gb) and used them to fill gaps in the Hi-C assembly (Figure S1D), followed by its polishing using an alignment of short reads (~260 Gb). The resulting NMR genome assembly had a total length of 2.89 Gb with an ~4.5-fold improvement in scaffold and contig N50. Our new assembly also contains fewer missing base pairs (represented by undetermined bases, 48 per kb) than the initial assembly based on short reads (109 per kb). We annotated the improved NMR assembly for protein-coding genes using RNA-seq reads and coding sequences from published NMR genome assemblies with the help of Gene Model Mapper (GeMoMa; Keilwagen et al., 2016), finding 54,202 transcripts and 29,195 genes.
These long-range assemblies of the two longest-lived rodents presented an opportunity to perform broad genome analyses. First, we computed their synteny to human and mouse genomes using SyMAP (Haug-Baltzell et al., 2017). We used the published PacBio assembly of the beaver genome for comparison and found that the increased contiguity of our assemblies vastly improved the ability to compute synteny between beaver and human genomes, with the percentage of the genome covered by synteny blocks increasing from 69% to 91%. In addition, 299 of the 503 synteny blocks were longer than 10 Mb, compared to all synteny blocks being shorter than 10 Mb in the previous version of PacBio assembly. We assessed the quality of the NMR Hi-C assembly by comparing it to the human (hg38) and mouse (mm10) genome references. In particular, the NMR 30 super-scaffolds, spanning 91% and 87% of mouse and human chromosome length, respectively, and most scaffolds in the new NMR assembly corresponded to contiguous regions of mouse or human chromosomes, although often with some intra-chromosomal rearrangements (Figure 1C).
By building synteny between human and NMR (“Hystricomorpha” species) genomes, we identified several human synteny blocks (HSA 6, 15/14, X, 1, 10/1, 12/22, 13, 8, 18, 8/4/8) that were a part of the putative rodent ancestor genome architecture (Graphodatsky et al., 2008; Romanenko et al., 2012). Cytogenetic analyses of hystricomorph species suggested that the HSA 8/12 and HSA 15/20 associations may define rodents (Romanenko et al., 2012). We found that HSA 15/20 was syntenic to superscaffold 17 in the NMR genome assembly (Figure S2). However, no association of HSA 8/12 was identified. We also did not detect the occurrence of HSA 9/11, which is considered to be ancestral for Glires (the group combining Rodentia and Lagomorpha), further supporting the idea that the occurrence of this association cannot be the result of convergence in some Glires groups or the loss of an ancestral feature in certain branches (Sannier et al., 2011). Using the synteny relationship, we found a large region (>10 Mb) in human chromosomes (located in HSA 1, 9, 13,14, 15, 16, 21, 22) with the absence of synteny to both NMR and mouse. However, nearly all human genes located in these regions had orthologs in NMR and mouse genome assemblies. Interestingly, one region in human X chromosome (X: 1– 8848114) showed synteny to the NMR but lacked synteny to the mouse (Figure S2). This region overlaps with the genome segment called “pseudoautosomal region 1” (PAR1), which behaves like an autosome and recombines during meiosis (Mangs and Morris, 2007). This PAR has unique structural and functional properties not found in other parts of the genome, and genes in this region were implicated in a variety of human disorders, such as short stature, asthma, psychiatric disorders, and leukemia (Raudsepp and Chowdhary, 2015). Further analyses of gene synteny in this region across rodent phylogeny revealed that the NMR, beaver, and thirteen-lined ground squirrel harbored the most syntenic genes to human genes located in PAR1, whereas murid rodents essentially lost gene synteny (Figure 1D). Moreover, a significant positive correlation (Pearson correlation coefficient = 0.70; phylogenetic generalized least-squares test p = 0.007) was observed for the number of syntenic genes in the PAR1 region and maximum longevity in the examined 12 rodents. Thus, the new NMR genome assembly represents a valuable resource to validate putative karyotypes of ancient rodents, and the genomes of long-lived rodents have fewer rearrangements and changes in syntenic genes compared to the mouse.
Characterization of Substitutions at Disease-Causing Sites
Using human protein sequence as a reference and our established genome syntenic relationships, we screened for substitutions in orthologs across 12 rodent assemblies. We found a total of 406,924 substitutions in mouse and 327,538 and 375,829 amino acid substitutions in beaver and NMR genomes, respectively. Consistent with prior studies (Gao and Zhang 2003; Jordan et al., 2015), hundreds of them matched human disease-causing sites (1,627 substitutions associated with 601 genes, 1,347 substitutions associated with 563 genes, 1,567 substitutions associated with 613 genes in mouse, beaver, and NMR, respectively). We then examined which type of human disease is significantly associated with such amino acid substitutions in long-lived rodents (STAR Methods). Our comparative analysis of 12 rodent genomes revealed that 51 and 55 human diseases were predicted to be significantly enriched in beaver and NMR substitutions, respectively (Table S1). The top disease categories enriched in beaver substitutions were hypertension, hypophosphataemic rickets, ocular and skeletal abnormalities, atrial fibrillation and other disorders associated with clinical phenotypes such as high blood pressure, growth retardation, ocular and skeletal anomalies, and rapid heart rate (Figure 2). This is interesting because beavers are not known to have high blood pressure, and their heart beat rate (<130 beats/mins during swimming; Swain et al., 1988) is much lower than in murids (>400 beats/mins) and other rodents (~250 beats/min; Carpenter and Marion, 2013). In addition, the beaver is the second largest rodent (body length 29–35 inches), which is inconsistent with growth retardation. This, though, cannot reject the compensatory mutation hypothesis (Gao and Zhang, 2003); in another way, it may suggest that substitutions at “disease-causing” sites are either putatively benign or experience a functional shift that is favored by natural selection.
Figure 2. Amino Acid Substitutions at Disease-Causing Sites.

Histogram of top human diseases enriched based on the analysis of beaver (upper part) and NMR (lower part) substitutions. Clinical phenotypes and associated genes for each category are also shown.
The latter possibility can be examined by functional assays of disease-causing substitutions that are unique to particular species. One of the candidate genes we identified was PTGIS, which harbored a disease-causing substitution for hypertension (R26Q) that was unique to the beaver (Figure S3). This gene encodes prostacyclin, which plays roles in cardiac function, response to oxidative stress, and hypoxia (Kim et al., 2003; Nakayama et al., 2002). It may have evolved as a diving adaptation, because beavers, like other diving mammals, are known to optimize respiration and cardiac function for staying longer under water (Dolka et al., 2015; Irving and Orr, 1935). The anatomy of the cardiac muscle in beavers also suggests that the beaver heart shares more similarity with aquatic rather than land mammals (Bisaillon, 1982). In addition to this disease-causing substitution, PTGIS possessed 23 other unique substitutions (Figure S3) in the beaver based on University of California Santa Cruz (UCSC) 100-way coding sequence alignment. Some of these unique substitutions were identified as being under positive selection using a recently developed branch-site model that incorporates multinucleotide mutations (Venkat et al., 2018). It is an attractive possibility that “disease-causing” substitutions in the beaver represent adaptive changes to an aquatic environment.
The top disease categories enriched in the NMR’s substitutions were Leber congenital amaurosis, Usher syndrome, myotonia (non-dystrophic), and oral-facial-digital syndrome, with clinical features related to eye disorders, hearing loss, and various development disorders of the mouth, teeth, and digits (Table S1). This is consistent with poor visual function and phenotypic specialization observed in the NMR. The identified NMR genes involved in eye disorders included CRX, encoding a photo-receptor-specific transcription factor, and RPE65, a component of the vitamin A visual cycle of the retina. Both genes harbored unique disease-causing substitutions in the NMR (Figure S3) and have an important role in maintaining circadian rhythms. For example, CRX is expressed not only in photoreceptors of the retina, but also in pinealocytes of the pineal gland (Furukawa et al., 1999). Knockout of CRX affects the expression of several other photoreceptor- and pineal-specific genes and the circadian entrainment, e.g., the percentage of total activity at night, in mice (Furukawa et al., 1999). This agrees with the idea that light detection-related genes play a role in NMR life, most likely for circadian entrainment or setting seasonal rhythms (Crish et al., 2006).
Common Substitutions in the NMR and Beaver Support Stress Resistance
Residues that are similarly altered in species with convergent phenotypes may reveal clues about functional adaptations. We examined amino acid changes in orthologs across 100 vertebrate genomes and identified 869 proteins containing 974 common substitutions unique to both beaver and NMR, which is generally similar to those between long-lived and comparable short-lived lineages (Figure S4). This shows that the convergence of long lifespan in rodents is commonly driven by limited locus rather than genome-wide sequence convergence. To test the potential functional effects of these substitutions, we performed experimental validation of the substitutions identified in Atrip (p.Ser618Ile) and Rpa1 (p.Thr572Met; Figure 3A). Both proteins are critical components of the DNA damage checkpoint and repair pathway (Cortez et al., 2001; Haring et al., 2008), suggesting that these substitutions may contribute to the increased genome stability of the beaver and NMR. We expressed mouse Atrip and Rpa1 and their forms containing NMR/beaver substitutions in a mouse fibroblast cell model and found that Atrip p.Ser618Ile significantly increased cell viability compared to the control Atrip following irradiation of cells with 200 J/m2 UV light (Figure 3B), suggesting that p.Ser618Ile substitution contributes to increased resistance to UV radiation. No significant changes in cell viability were found in the case of Rpa1 p.Thr572Met substitution. In addition to other molecular damage forms, oxidative damage and endoplasmic reticulum (ER) stress could contribute to “hallmarks of aging” because of their links with cell senescence (López-Otín et al., 2013). We further tested the stable cells by treatment with rotenone and Brefeldin A, and both substitutions (Atrip Ser618Ile and Rpa1 Thr572Met) significantly improved cell viability (Figure 3B). Atrip Ser618Ile and Rpa1 Thr572Met variants also largely rescued cells from apoptosis following treatment with UV irradiation, rotenone, and Brefeldin A for 48 h (Figure 3B; Figure S5). Together, these data suggest that the tested substitutions can improve mouse cell viability by enhancing tolerance of cell stress.
Figure 3. Unique Substitutions in the Beaver and NMR and Stress Resistance.

(A) Schematic diagram of structure and sequence alignment of ATRIP and RPA1 proteins. Unique substitutions identified in the beaver and NMR sequences are highlighted in red.
(B) Cell viability of mouse fibroblast cells expressing Atrip and Rpa1 measured by an MTT assay following UV irradiation (200 J/m2), rotenone, and Brefeldin A (BFA) treatments. Atrip-mus and Rpa1-mus are mouse wild-type proteins. Atrip-mut and Rpa1-mut are mutant mouse proteins containing the identified beaver/NMR substitutions. The value from each test in the figure was taken by at least three replicates in the same assay.
Pathways Affected by Rapid Evolution
We further explored a variation of selection pressure that could be associated with longevity in the beaver and NMR. For this, we developed a hypothesis testing framework to measure the shift of selection pressure of genes (measured by dN/dS, ω; see STAR Methods). We assigned three discrete categories of dN/dS to 12 rodents (ωNMR for the NMR, ωbeaver for the beaver, and ωothers for the rest of the rodents; Figure 4A) and then compared nested models using a likelihood ratio test (LRT) under a specific hypothesis. We identified 168 (ωbeaver > ωNMR > ωothers) and 257 (ωNMR > ωbeaver > ωothers) rapidly evolved genes with significantly elevated selection pressure in the beaver and NMR lineages, respectively (Figure 4B). These candidate genes were enriched in biological processes such as oxidation reduction (p = 2.00 × 10−4, Fisher’s exact test), visual perception (p = 1.90 × 10−3, Fisher’s exact test), sodium ion transport (p = 7.00 × 10−4, Fisher’s exact test), long-chain fattyacyl-CoA biosynthetic process (p = 4.70 × 10−4, Fisher’s exact test) and spermatogenesis (p = 3.00 × 10−2, Fisher’s exact test; Tables S2 and S3). Interestingly, seven rapidly evolved genes in the beaver genome (BAK1, CALCA, CACYBP, CASP7, and SOD1) were associated with aging (p = 1.50 × 10−2, Fisher’s exact test; Figure 4C), and all of them, except SOD1, were associated with cancer incidence. For example, BAK1 encoding a member of the BCL2 family was proposed to play a central role in regulating cell death and tumorigenesis (Strasser and Vaux, 2018), and Siah-1-interacting protein, which is encoded by CACYBP is a component of the ubiquitin pathway and is well known to be a critical protein in tumorigenesis, such as colorectal and gastric cancers (Zhai et al., 2017). This implies that the anti-aging and cancer-resistant pathways have been tightened up in the beaver.
Figure 4. Characterization of Substitution Rates in the Beaver and NMR.

(A) Phylogeny of rodent species characterized by whole genome sequences that were used for hypothesis testing. (Right) Maximum lifespan and body mass of 12 rodent species used in this study. The divergence time of those species were retrieved from TimeTree (http://www.timetree.org). Maximum lifespan (years) and adult weight (g) data were retrieved from the AnAge database (http://genomics.senescence.info/species/).
(B) Vioplot of DlnL values of genes that were tested under four models, i.e., ωbeaver > ωNMR > ωothers, ωNMR > ωbeaver > ωothers, ωbeaver = ωNMR > ωothers, ωNMR > ωothers, and ωbeaver > ωothers. (B) Histogram of the number of rapidly evolved genes identified under three models, i.e., ωbeaver > ωNMR > ωothers, ωNMR > ωbeaver > ωothers, ωNMR > ωothers, and ωbeaver > ωothers. Candidate genes under each model that were also supported by the model ωbeaver = ωNMR > ωothers are shaded in red.
(C) Table showing top five biological processes that were significantly enriched by candidate genes in beaver and NMR.
(D) Pathway analysis of DlnL values under the models ωbeaver > ωNMR > ωothers (x axis) and ωNMR > ωbeaver > ωothers (y axis). Circle sizes are proportional to the number of genes assigned to a pathway. Pathways affected by rapid substitutions and unique in beaver and NMR lineages are shaded in orange and lime green, respectively.
We further searched for genes with elevated selection pressure in both species compared to other rodents (ωNMR > ωothers, ωbeaver > ωothers) and identified 345 such genes. Strikingly, the majority of them (85.2%) fit to the model of equally elevated selection pressure in the beaver and NMR (i.e., ωbeaver = ωNMR > ωothers; Figure 4B). A similar trend was observed for the genes identified only in the beaver (95.2%) or NMR (83.2%), suggesting that these genes could evolve synchronously in long-lived rodents. To characterize global biological pathways affected by changes of selection pressure in the beaver and NMR, we employed an approach that focused on whole pathways and scored them with a signal of rapid evolution, instead of focusing only on outlier “significant” genes (Daub et al., 2013). We tested over 1,200 pathways from the Biosystems database and for each pathway inferred significance of this “SUMSTAT” score against a null distribution of random gene sets of the same size. In addition, we applied a foreground permutation strategy to identify the unique pathways that were enriched by target lineages (see STAR Methods). In total, we found 70 and 48 unique significant pathways affected by natural selection in the beaver and NMR, respectively (Figure 4D; Tables S2 and S3). The unique pathways for the increase of selection pressure in the beaver lineage included pathways related to amino sugar and nucleotide sugar metabolism (p = 1.38 × 10−2), Parkinson’s disease (p = 2.46 × 10−2), biological oxidations (p = 2.95 × 10−2), and cellular response to heat stress (p = 3.02 × 10−2) which are either coherent with the aging process or involved molecules that maintain cell homeostasis through the control of cell proliferation and differentiation. In parallel, many top scoring candidates in the NMR lineage were associated with insulin secretion and degradation, e.g., glucagon-like peptide-1 (GLP1) regulates insulin secretion (p = 4.02 × 10−2), synthesis of bile acids and bile salts (p = 4.74 × 10−2), and triglyceride biosynthesis (p = 4.66 × 10−2).
Surprisingly, we found that the top-scoring pathways enriched in both the beaver and NMR were directly related to oxidative phosphorylation. These pathways remained significant after pruning in the analysis of each species, suggesting that their roles in the aging process of rodents may have been a major driver for selection. For example, the highest scoring candidate was the respiratory electron transport (p = 1.69 × 10−2), fatty acid degradation (p = 2.27 × 10−2), and peroxisome (p = 4.52 × 10−2). i.e., pathways that have roles in clearing circulating and cellular lipids by regulating the expression of genes involved in lipid metabolism. Genes associated with this pathway also seemed to be involved in tumorigenesis regulation via activation of different pathways (Fanale et al., 2017). Other common enriched pathways in both species were also involved in lipid metabolism, such as fatty acid metabolism, fatty acid beta oxidation, peroxisomal lipid metabolism, and ABCA transporters in lipid homeostasis (Tables S2 and S3). It is well known that lipid metabolism has an important role in the aging process and chronic diseases. For example, the NMR has lower levels of DHA-containing phospholipids compared to mice (Mitchell et al., 2007). It was hypothesized to lead to lower susceptibility to peroxidative damage and serve as an important determinant of longevity. However, even though the NMR is resistant to tumorigenesis, it may develop age-associated lesions, including lipofuscinosis and heart disease (Delaney et al., 2013). Information on lipid metabolism in the beaver is relatively scarce. An earlier study reported that dietary polyunsaturated fatty acids do not accumulate in the beaver (Käkelä and Hyvärinen, 1996), suggesting that this species may be resistant to peroxidation too. Of note, changes of membrane composition are dynamic and could be influenced by unique diets of the beaver and NMR. In addition, the selective signal of lipid metabolism may be due to other metabolic processes and pathways. We believe future experimental studies analyzing genes key to lipids and lipid metabolism in other long-lived rodents may be particularly important for understanding longevity.
Conclusions
Our analysis of the beaver and NMR genomes identified changes in genes and pathways putatively associated with adaptations to the species’ ecology and acquired longevity. Substitutions at disease-causing sites may contribute to phenotypic adaptation of particular species and long-term survival of such substitutions, making them candidates for signatures of natural selection. By testing substitutions common to both long-lived species, we found that some substitutions enhanced tolerance of cells to DNA damage, oxidative damage, and ER stress, suggesting future use of long-lived rodent genomes in discovering genetic factors of lifespan control. Indeed, no single molecular damage form can be a major constraint on lifespan, but a combination could. When examined, the whole pathways affected by natural selection in the beaver and NMR compared to short-lived rodents, we identified oxidation reduction and lipid metabolism as the most affected systems, suggesting common mechanisms may be used to achieve longevity of these species.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vadim Gladyshev (vgladyshev@rics.bwh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The naked mole rat and beaver whole-genome shotgun projects have been deposited in GenBank under accession codes RPDE00000000 and RPGA00000000, respectively. All PacBio reads and short-read data have been deposited in the Short Read Archive under accession code SRR8182754-SRR8182767 and SRR8204318, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Naked mole rat was sampled from a breeding colony housed at the University of Rochester. Beaver was trapped in New York State.- The animals were sacrificed and DNA was isolated for subsequent sequencing. All animal experiments were approved by the University of Rochester Committee on Animal Resources (UCAR).
METHOD DETAILS
Sequencing and assembly
Whole-genome shotgun sequences were generated on the Illumina HiSeq platform. The initial assembly was generated using All-Path-LG with default parameters and all sequence data. The assembled scaffolds from this initial assembly were further extended using Chicago library with Dovetail Genomics’ HiRise scaffolder as previously described (Putnam et al., 2016). Around 10X coverage PacBio reads were downloaded and used to fill gaps within scaffolds by locally mapping the reads associated with each gap using PBJelly (English et al., 2012). Finally, we corrected the sequence errors in the assembly with Pilon pipeline(Walker et al., 2014), helping align the reads with BWA-mem (Li and Durbin, 2009) and parsing with SAMtools (Li et al., 2009). To improve the NMR genome assembly, we generated in situ Hi-C data (~60 × sequence coverage)(Rao et al., 2014) to split, anchor, order, orient, and merge scaffolds. Briefly, NMR embryonic fibroblast cell lines were cultured following the manufacturer’s recommendations, and around three million cells were used for in situ Hi-C (Rao et al., 2014). The resulting libraries were sequenced using an BGISEQ-500 sequencing platform. We constructed 30 superscaffolds (or pseudo-chromosomes) that occupied 90% of the genome using 3D-dna pipeline (Dudchenko et al., 2017). Then, we generated ~10X coverage PacBio reads to fill the gaps within pseudo-chromosomes using PBJelly (English et al., 2012) and corrected the sequence errors using Pilon pipeline (Walker et al., 2014) with published short reads (~100X coverage). BUSCO (Simão et al., 2015) was used to evaluate completeness of the genome assemblies. Briefly, BUSCO assesses the genome by searching for the presence of near-universal single-copy orthologs from OthoDB v9 (Zdobnov et al., 2017). Absence of these conserved genes indicates incompleteness of the genome. In our analysis, we used the mammalian gene set consisting of 4104 single-copy genes present in more than 90% mammalian species.
Annotation and genome synteny
We annotated the beaver genome by employing Maker2 pipeline (Holt and Yandell, 2011), and genes were predicted through ab initio gene predictors (i.e., SNAP [Korf, 2004] and AUGUSTUS [Stanke and Waack, 2003]) and evidence-based gene calling (i.e., transcript assembly and protein sequences). Beaver transcriptomes were assembled into 16,816 transcripts, and 9,805 full-length open reading frames were retrieved.(Lok et al., 2017)These beaver transcriptomes, together with 66.7k reviewed mammalian protein sequences (from Swiss-Prot), were used to train the ab initio gene predictors, polish the predicted gene models, and evaluate each predicted gene model. Gene prediction of the NMR assembly generated in this study was conducted using GeMoMa (Keilwagen et al., 2016) with the transcriptomes from 11 NMR tissues and predicted protein sequences from published genomes. Synteny maps among beaver, NMR and mouse were created using SyMAP 4.2 (Soderlund et al., 2011), considering only scaffolds/superscaffolds of at least 100 kb. We used MCScanX (Wang et al., 2012) to identify syntenic gene blocks for human, beaver, NMR and other 15 rodents, with the gap size set to 10 genes and at least 5 syntenic genes.
Ortholog alignment and substitutions
Given that the sequences and annotation of the beaver and NMR genomes are newly generated in the current study, and the beaver is absent in the 100-species alignments made available by the UCSC genome browser, we employed a custom approach to add the correct beaver and NMR orthologous sequences based on human sequences. We first performed pairwise reciprocal nucleotide BLAST of beaver and NMR cDNA sequences with cDNA sequences of human hg19 genome and identified beaver orthologs using the proteinortho program (Lechner et al., 2011). We then performed profile alignment using the Muscle program (Edgar, 2004) for each ortholog group of these three mammals and used human sequences as a reference to add the identified beaver orthologs to the multi-species alignment. To identify the diseases enriched by substitutions in the beaver and NMR, we first conducted a pairwise comparison of protein sequences of 12 rodents, including NMR and beaver, to human sequences for each ortholog to detect amino acid changes. Then, all substitutions in each species were annotated with their disease status on the basis of HGMD (Human Gene Mutation Database) (Stenson et al., 2003), where applicable. We obtained a list of 1,283 human diseases of type “pathway” with the number of substitutions/genes of 12 rodents associated. We performed the Grubbs’ test in ‘outliers’ package (Grubbs, 1950; Komsta, 2011) to test signals of enrichment for each species and each ‘disease’. By the help of 100-species genome alignments, we also extracted the coding sequences for each ortholog to detect unique substitutions. The unique substitutions in all pairs of rodents analyzed in the present study was then counted by in house PERL script.
Experimental assays
Sequences encoding mouse Atrip and Rpa1 forms as well as their mutant variants were synthesized and cloned into the pEGFP-N2 (Addgene). All constructs were verified by sequencing. Mouse lung fibroblast cells were transfected and cultured in high-glucose DMEM supplemented with 10% FBS, and 5% penicillin-streptomycin. Stable clones were selected with 1 mg/ml puromycin (Sigma) and validated using RT-PCR. All cells were incubated in a humidified atmosphere with 5% CO2 at 37◦C. For UV irradiation assays, cells were subjected to UV irradiation at 200 J/m2 and then collected at the indicated time points. To induce oxidative damage and ER stress, cells were exposed to various concentrations of rotenone (50 mg/mL in stock) and Brefeldin A (BFA) (10 mM in stock), respectively. Cytotoxicity (MTT) assay was performed following the manufacturer’s protocol (Sigma). Results were read using a multiwell microplate reader (BioTek), and expressed as the mean ± standard error of four replicates each. Statistical analysis was performed using Mann-Whitney U test. The values depicting p < 0.05 were considered as statistically significant. As for apoptosis analysis, cells were harvested and washed once with PBS, and then resuspended in PI/Annexin-V solution (KeyGEN Biotech). At least 20000 live cells were analyzed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were analyzed by using FlowJo software.
Evolutionary models and pathway analysis
To characterize signatures of selection pressure in the beaver and NMR, we developed a model that constraints the ratio of non-synonymous to synonymous rate (dN/dS, ω) in different groups and then employs a likelihood ratio test (LRT) to compare the goodness of fit of different hypotheses. In particular, ω were estimated for three discrete categories (NMR, beaver and the rest of 10 rodents) under the Muse-Gaut (MG94) rate matrices (Muse and Gaut, 1994) combined with HKY85 model by the help of HBL language in HYPHY (Pond et al., 2005), and then compared nested models with constrained relationships among them. This allowed us to detect the shift of selection pressure associated with the beaver or NMR (relaxed model, H1), by comparing it to the null model (all rodents have the same dN/dS, H0). We ran this test five times for each gene and yielded a total of five likelihood scores for each H0 and H1. We then constructed the log-likelihood ratio score for each gene (ΔlnL) as: ΔlnL = 2(lnL H1- lnL H0) = 2(Maxi-5 (LnL H1) - Maxi-5 (LnL H0)) and employed the LRT. To profile at level of global pathways affected by natural selection in the beaver and NMR, we used a pathway enrichment approach (Daub et al., 2013) to test signals of rapid evolution. We first calculated the SUMSTAT score for each pathway, which is the sum of log-likelihood ratio score of genes under a particular pathway. A null distribution of gene sets was calculated with random sampling of the SUSMTAT score and then the significance of a target pathway was inferred. Furthermore, a foreground permutation strategy was used to remove the potential systematic biases and to identify unique pathways in target lineages during pathways enrichment analysis. Particularly, we first selected the relatives of long-lived rodents as ‘foreground’ with one of long-lived rodents to calculate DlnL of each gene as we have done in NMR and beaver. Then, we preformed the pathway enrichment for this each pair of comparable non-long-lived lineages using SUMSTAT score. The unique pathways with increase of selection pressure in NMR and beaver lineages were picked by removing the common pathways that were significantly enriched by this foreground permutation strategy.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiment validates of mutant variants of Atrip and Rpa1 were independently performed four times. Values are expressed as mean ± standard deviation (SD) and were compared using Mann-Whitney U test. The values depicting p < 0.05 were considered as statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| pEGFP-N2 | Addgene | Cat# 6081-1 |
| Dulbecco’s Modified Eagle Medium | Sigma-Aldrich | Cat# SLM-241-B |
| FBS | Thermo Fisher Scientific | Cat# 16000 |
| penicillin-streptomycin | Sigma-Aldrich | Cat# 516106 |
| rotenone | Sigma-Aldrich | Cat# 83-79-4 |
| Brefeldin A | Sigma-Aldrich | Cat# 20350-15-6 |
|
| ||
| Software and Algorithms | ||
|
| ||
| FlowJo | N/A | https://www.flowjo.com/solutions/flowjo/downloads |
| AllPath-LG | N/A | http://software.broadinstitute.org/allpaths-lg/blog/ |
| PBJelly | English et al., 2012 | https://sourceforge.net/p/pb-jelly/wiki/Home/ |
| Pilon | Walker et al., 2014 | https://github.com/broadinstitute/pilon |
| BWA-mem | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| 3D-dna | Dudchenko et al., 2017 | https://github.com/theaidenlab/3d-dna |
| BUSCO | Simão et al., 2015 | https://busco.ezlab.org/ |
| Maker2 | Holt and Yandell, 2011 | https://www.yandell-lab.org/software/maker.html |
| SNAP | Korf, 2004 | https://github.com/KorfLab/SNAP |
| AUGUSTUS | Stanke and Waack, 2003 | http://augustus.gobics.de/ |
| GeMoMa | Keilwagen et al., 2016 | http://www.jstacs.de/index.php/GeMoMa |
| SyMAP | Soderlund et al., 2011 | http://www.agcol.arizona.edu/software/symap/ |
| MCScanX | Wang et al., 2012 | http://chibba.pgml.uga.edu/mcscan2/ |
| proteinortho_v5.11 | Lechner et al., 2011 | https://www.bioinf.uni-leipzig.de/Software/proteinortho/older.html |
| Muscle | Edgar, 2004 | https://www.drive5.com/muscle/ |
| HYPHY | Pond et al., 2005 | http://www.hyphy.org/ |
|
| ||
| Deposited Data | ||
|
| ||
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182754 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182755 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182756 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182757 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182758 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182759 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182760 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182761 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182762 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182763 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182764 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182765 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182766 |
| PacBio reads of Naked mole rat | This paper | NCBI: SRR8182767 |
| Hic sequences of naked mole rat | This paper | NCBI: SRR8204318 |
| Assembled genome of American beaver | This paper | NCBI: RPDE00000000 |
| Assembled genome of naked mole rat | This paper | NCBI: RPGA00000000 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Mouse lung fibroblast cells | This paper | N/A |
Highlights.
Improved genome assemblies of the naked mole rat and Canadian beaver
Substitutions at disease-causing sites could be adaptive
Unique substitutions in long-lived rodents support stress resistance
Genome resource for studies of long-lived rodents
ACKNOWLEDGMENTS
This project was supported by National Institutes of Health (NIH) grants AG047200, AG047745, GM065204, HG008742, and AG064706.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107949.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Austad SN (2005). Diverse aging rates in metazoans: targets for functional genomics. Mech. Ageing Dev 126, 43–49. [DOI] [PubMed] [Google Scholar]
- Bisaillon A. (1982). Anatomy of the heart in the North American beaver (Castor canadensis). Anat. Anz 151, 381–391. [PubMed] [Google Scholar]
- Carpenter J, and Marion CJ (2013). Exotic Animal Formulary (Elsevier Saunders; ). [Google Scholar]
- Cortez D, Guntuku S, Qin J, and Elledge SJ (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Crish SD, Dengler-Crish CM, and Catania KC (2006). Central visual system of the naked mole-rat (Heterocephalus glaber). Anat. Rec. A Discov. Mol. Cell. Evol. Biol 288, 205–212. [DOI] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana-Murci L, Robinson-Rechavi M, and Excoffier L. (2013). Evidence for polygenic adaptation to pathogens in the human genome. Mol. Biol. Evol 30, 1544–1558. [DOI] [PubMed] [Google Scholar]
- Delaney MA, Nagy L, Kinsel MJ, and Treuting PM (2013). Spontaneous histologic lesions of the adult naked mole rat (Heterocephalus glaber): a retrospective survey of lesions in a zoo population. Vet. Pathol 50, 607–621. [DOI] [PubMed] [Google Scholar]
- Dolka I, Giżejewska A, Giżejewski Z, Kluciński W, and Kołodziejska J. (2015). Histological evaluation of selected organs of the Eurasian beavers (Castor fiber) inhabiting Poland. Anat. Histol. Embryol 44, 378–390. [DOI] [PubMed] [Google Scholar]
- Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, and Aiden EL (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, and Gibbs RA (2012). Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanale D, Amodeo V, and Caruso S. (2017). The interplay between metabolism, PPAR signaling pathway, and cancer. PPAR Res. 2017, 1830626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Seim I, Huang Z, Gerashchenko MV, Xiong Z, Turanov AA, Zhu Y, Lobanov AV, Fan D, Yim SH, et al. (2014). Adaptations to a subterranean environment and longevity revealed by the analysis of mole rat genomes. Cell Rep. 8, 1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Morrow EM, Li T, Davis FC, and Cepko CL (1999). Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet 23, 466–470. [DOI] [PubMed] [Google Scholar]
- Gao L, and Zhang J. (2003). Why are some human disease-associated mutations fixed in mice? Trends Genet. 19, 678–681. [DOI] [PubMed] [Google Scholar]
- Gladyshev VN (2013). The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends Genet. 29, 506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Bozzella MJ, and Seluanov A. (2008). Rodents for comparative aging studies: from mice to beavers. Age (Dordr.) 30, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graphodatsky AS, Yang F, Dobigny G, Romanenko SA, Biltueva LS, Perelman PL, Beklemisheva VR, Alkalaeva EZ, Serdukova NA, Ferguson-Smith MA, et al. (2008). Tracking genome organization in rodents by Zoo-FISH 16, 261–274. [DOI] [PubMed] [Google Scholar]
- Grubbs FE (1950). Sample criteria for testing outlying observations. Ann. Math. Stat 21, 27–58. [Google Scholar]
- Haring SJ, Mason AC, Binz SK, and Wold MS (2008). Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and check-points. J. Biol. Chem 283, 19095–19111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, and Lyons E. (2017). SynMap2 and SynMap3D: web-based whole-genome synteny browsers. Bioinformatics 33, 2197–2198. [DOI] [PubMed] [Google Scholar]
- Mangs AH, and Morris BJ (2007). The human pseudoautosomal region (PAR): origin, function and future. Curr. Genomics 8, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, and Yandell M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving L, and Orr MD (1935). The diving habits of the beaver Laurence Irving. Science 82, 569. [DOI] [PubMed] [Google Scholar]
- Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Davis EE, Sunyaev SR, and Katsanis N; Task Force for Neonatal Genomics (2015). Identification of cis-suppression of human disease mutations by comparative genomics. Nature 524, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käkelä R, and Hyvärinen H. (1996). Fatty acids in extremity tissues of Finnish beavers (Castor canadensis and Castor fiber) and muskrats (Ondatra zibethicus). Comp. Biochem. Physiol. B Biochem. Mol. Biol 113, 113–124. [DOI] [PubMed] [Google Scholar]
- Keane M, Craig T, Alföldi J, Berlin AM, Johnson J, Seluanov A, Gorbunova V, Di Palma F, Lindblad-Toh K, Church GM, and de Magalhães JP (2014). The naked mole rat genome resource: facilitating analyses of cancer and longevity-related adaptations. Bioinformatics 30, 3558–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, and Hartung F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee DK, Choi JW, Kim JS, Park SC, and Youn HD (2003). Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech. Ageing Dev 124, 941–949. [DOI] [PubMed] [Google Scholar]
- Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P, et al. (2011). Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komsta L. (2011). Package ‘outliers’. CRAN. https://cran.r-project.org/web/packages/outliers/outliers.pdf. [Google Scholar]
- Korf I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, and Tavernarakis N. (2011). Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 30, 2520–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, and Prohaska SJ (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok S, Paton TA, Wang Z, Kaur G, Walker S, Yuen RK, Sung WW, Whitney J, Buchanan JA, Trost B, et al. (2017). De novo genome and transcriptome assembly of the Canadian beaver (Castor canadensis). G3 (Bethesda) 7, 755–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G. (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TW, Buffenstein R, and Hulbert AJ (2007). Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): a comparative study using shotgun lipidomics. Exp. Gerontol 42, 1053–1062. [DOI] [PubMed] [Google Scholar]
- Muse SV, and Gaut BS (1994). A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol 11, 715–724. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Soma M, Watanabe Y, Hasimu B, Sato M, Aoi N, Kosuge K, Kanmatsuse K, Kokubun S, Marrow JD, and Oates JA (2002). Splicing mutation of the prostacyclin synthase gene in a family associated with hypertension. Biochem. Biophys. Res. Commun 297, 1135–1139. [DOI] [PubMed] [Google Scholar]
- Pond SL, Frost SD, and Muse SV (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. [DOI] [PubMed] [Google Scholar]
- Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. (2016). Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudsepp T, and Chowdhary BP (2015). The Eutherian pseudoautosomal region. Cytogenet. Genome Res 147, 81–94. [DOI] [PubMed] [Google Scholar]
- Romanenko SA, Perelman PL, Trifonov VA, and Graphodatsky AS (2012). Chromosomal evolution in Rodentia. Heredity 108, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Smith M, and Buffenstein R. (2018). Naked mole-rat mortality rates defy Gompertzian laws by not increasing with age. eLife 7, e31157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannier J, Gerbault-Seureau M, Dutrillaux B, and Richard FA (2011). Conserved although very different karyotypes in Gliridae and Sciuridae and their contribution to chromosomal signatures in Glires. Cytogenet. Genome Res 134, 51–63. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, and Zdobnov EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Bomhoff M, and Nelson WM (2011). SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, and Waack S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (Suppl 2), ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, and Cooper DN (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat 21, 577–581. [DOI] [PubMed] [Google Scholar]
- Strasser A, and Vaux DL (2018). Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 25, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain UG, Gilbert FF, and Robinette JD (1988). Heart rates in the captive, free-ranging beaver. Comp. Biochem. Physiol. A Comp. Physiol 91, 431–435. [DOI] [PubMed] [Google Scholar]
- Venkat A, Hahn MW, and Thornton JW (2018). Multinucleotide mutations cause false inferences of lineage-specific positive selection. Nat. Ecol. Evol 2, 1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, and Earl AM (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simã FA, Ioannidis P, Seppey M, Loetscher A, and Kriventseva EV (2017). OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45 (D1), D744–D749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai H, Shi Y, Chen X, Wang J, Lu Y, Zhang F, Liu Z, Lei T, and Fan D. (2017). CacyBP/SIP promotes the proliferation of colon cancer cells. PLoS ONE 12, e0169959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The naked mole rat and beaver whole-genome shotgun projects have been deposited in GenBank under accession codes RPDE00000000 and RPGA00000000, respectively. All PacBio reads and short-read data have been deposited in the Short Read Archive under accession code SRR8182754-SRR8182767 and SRR8204318, respectively.