

Abstract
The Cornelia de Lange syndrome (CdLS) is a genetic disorder characterized by multisystemic malformations. CdLS is due to mutations in one of the following genes: NIPBL , SMC1A , SMC3 , RAD21 , and HDAC8 . On the other hand, 10q11.2 deletions cause a wide range of presentations in patients. Approximately 40 cases with variable deletions of 10q11.2 have been reported in literature. Some of the reported cases involve the coexistence of duplication or deletion affecting one copy of the chromosome. However, deletion of chromosome 10q11.22-q11.23 and CdLS syndrome caused by NIPBL gene mutations have not been reported previously. This report, therefore, is the first to report their coexistence together.
Keywords: the Cornelia de Lange syndrome, NIPBL gene , 10q11.2 micro deletion
Introduction
The Cornelia de Lange syndrome (CdLS; OMIM no.: 122470, 300590, 610759, 614701, and 300882) is a cohesinopathy that is characterized by dysmorphic features, prenatal and postnatal growth retardation, hearing loss, ocular problems, hirsutism, congenital malformations of gastrointestinal tract including diaphragmatic hernia, intestinal malrotation, piloric stenosis, gastroesophageal reflux, cardiac anomalies, genitourinary malformations, and upper extremity defects ranging from small hands to severe reduction defects of the forearms. 1 The syndrome was originally reported in 1933 by Cornelia de Lange, although Brachmann described a child with similar phenotype at autopsy in 1916. 2 Prevalence is between 1/10,000 and 1/100,000 with most cases being sporadic.
The typical facial features in CdLS include hirsutism, arched eyebrows, synophyrs, long eyelashes, short nose with depressed nasal bridge, anteverted nares, long and smooth philtrum, and thin lips. In addition, multiorgan abnormalities including limb anomalies, congenital heart defects, hearing loss, myopia, gastrointestinal malformation, genitourinary malformation, and neurologic disease are observed in patients with CdLS. Furthermore, short stature occurs proportionally and hypertrichosis is commonly observed. 3 4 Previous studies have shown that upper extremity defects ranging from clinodactyly to severe reduction defects of the forearms have been reported in approximately 30% of individuals with CdLS. Although, cardiac anomaly is not a major criterion of the disease, approximately 50% of CdLS patients presented with congenital heart defects. 4 5 Approximately 50% of individuals with CdLS have ocular anomalies with the most common being ptosis, myopia, nystagmus, and strabismus. Many individuals with CdLS demonstrate neuropsychiatric features including seizures, autistic behavior, and self-destructive tendencies. Most individuals with CdLS have been reported to have intellectual disabilities with IQs ranging from 30 to 86 (mean: 53). 4 5
CdLS is caused by autosomal dominant or X-linked mutations in the cohesin core subunits encoded by SMC1A, SMC3 , and RAD21 genes or cohesion components encoded by NIPBL and HDAC8 genes. 6 Mutations in the NIPBL (nipped-B-like protein) gene account for 60% of classical CdLS cases while mutations in the other four genes account 5% only of the reported cases. 4 To date, approximately 35% of clinically diagnosed CdLS patients do not carry any identifiable gene mutation. 7 CdLS is defined as cohesinopathy because the encoded proteins of these genes are components of the cohesin complex.
Partial deletion of the distal long arm of chromosome 10 (10q11.2) has several clinical characteristics and they have been described in asymptomatic cases, as well as in patient showing neurological symptoms. Deletions of 10q11.2 have been reported in patient with intellectual disability, psychomotor developmental delay, cardiac anomalies, skeletal system anomalies, and neurological symptoms such as sleep apnea, ptosis, and hypotonia. 8 9 Some people are mildly affected by a deletion of 10q11.2 but others show more marked effects. They are rarer but also present a variety of clinical features. To date, over 30 cases with variable deletions of 10q11.22-q11.23 have been reported in literature.
We report a 10q11.22–11.23 microdeletion and CdLS with novel NIPBL mutation in the same individual.
Case Presentation
A 12-year-old Turkish girl was referred to our department of medical genetics because of short stature. Our case is the first child of a consanguineous Turkish couple. Her mother and father are 34 and 39 years old, respectively. The proband was born by cesarean delivery, after a normal pregnancy at 40 weeks of gestation. Birth weight was 4,000 g (75th centile) with a length of 48 cm (25th centile) and a head circumference of 35 cm (10th centile). Apgar's score was within normal limits. She had a history of strabismus surgery 6 years ago. Although her motor development is in normal range, there was a delay in her speech development. She had intellectual disability and her communication and social skills were impaired.
At 12 years of age, some dysmorphic features were observed ( Fig. 1 ), including high arched and thick eyebrows, hirsutism, synophrysis, long eyelashes, flattened nasal root, antevert nares, pointed jaw, long philtrum, low-set ear, 2 cm × 2 cm cafe au lait spot on abdomen, clinodactyly on fifth finger in both hands and four to five metatarsal bone shortness. The main physical findings include weight of 29 kg (10th centile), height of 125 cm (<3rd centile), and head circumference of 50 cm (<3rd centile). Further evaluation revealed that she had minimal mitral valve insufficiency in echocardiography (ECO), varus deformity on foot X-ray, and low growth hormone. She also had gastroesophageal reflux disease. Brain magnetic resonance imaging (MRI) and hearing examination were normal but she had a history of strabismus surgery 6 years ago. No additional abnormalities were detected and the patient had no family history of similar phenotype, and her brother was healthy and morphologically normal.
Fig.1.
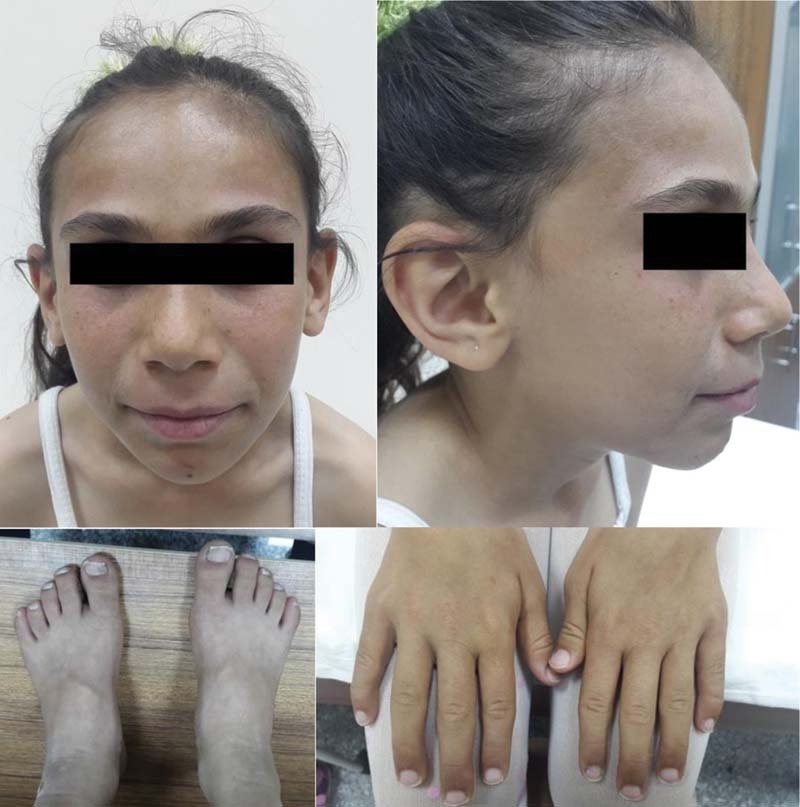
Clinical photographs of the patient at 12 years of age. Dysmorphic facial features, including a high arched and thick eyebrows, synophrys, flattened nasal root, antevert nares, pointed jaw, long philtrum, low-set ear, clinodactyly on fifth finger in both hands, and four to five metatarsal bone shortness.
Informed consent was obtained from the family before blood sampling and DNA analysis. Conventional chromosome, NIPBL gene mutation analysis and array comparative genomic hybridization analysis have been performed at the Medical Genetic Laboratory of Haseki Education and Research Hospital in Turkey. First, conventional chromosome analysis was performed on 72-hour lymphocyte cultures according to standard techniques. The proband had Giemsa's trypsin banding (GTG) technique. Her karyotypes were normal, 46,XX. Family members' karyotypes were normal. Then, molecular genetic testing (targeted next-generation sequencing on MiSeq) was performed by mutation analysis. Entire coding exons and their flanking regions of the genes were analyzed ( Fig. 2 ). Next-generation sequencing revealed that the proband had a novel heterozygous c.245A > G (p.Asn82Ser) variant in NIPBL gene. It has been identified as pathogenic by mutation taster, DANN, EIGEN, FATHMM-MKL, M-CAP, PolyPhen-2, and PrimateAI databases. However, according to the American College of Medical Genetics and Genomics (ACMG) sequence variant classification guideline, 10 the variant was classified as variant of uncertain significance. This variant was absent in healthy control population databases (GnomAD) and also has not been reported in the disease mutation databases Clinvar or Human Gene Mutation Database (HGMD).
Fig. 2.
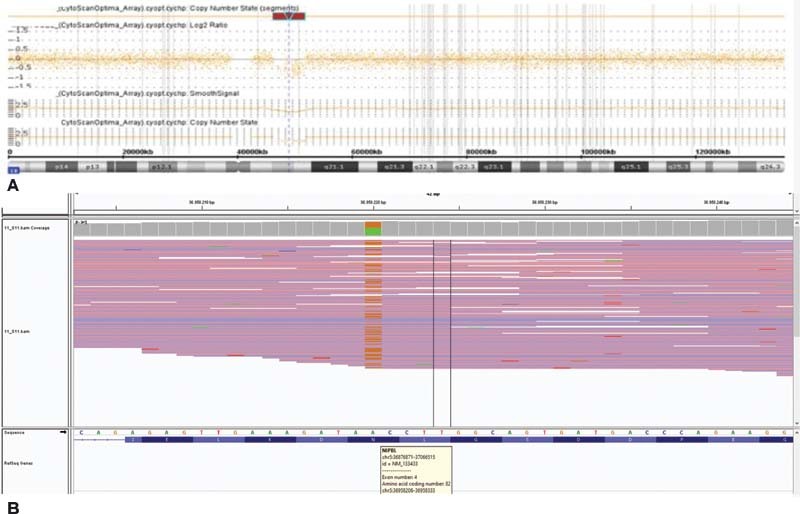
( A ) Result of CytoScan 315K array exhibiting deletion in 10q11.22 to 10q11.23. ( B ) Targeted next-generation sequencing (MiSeq)] revealed that proband had a novel heterozygous c.245A > G (p.Asn82Ser) variant in NIPBL gene.
Genomic DNA was isolated from peripheral blood leukocytes using the standard procedures. The investigation of copy number changes was performed by array comparative genomic hybridization (acgh) using the Whole Human Genome Comparative Genomic Hybridization Microarray 315K (Affymetrix Cytoscan Optima) as recommended by the manufacturer. Cytoscan 315K array analysis of the proband showed the following copy number variations (cnvs): arr[grch37] 10q11.22q11.23(46,293,590_51,857,206)x1 pat. Cnvs represent an overall of 5.564-Mb, including losses of DNA. It included 22 genes listed in OMIM. The father showed a total of 4.804-Mb of cnvs, including: arr (grch37) 10q11.22q11.23 (46,981,607–51,785,817) × 1. The mother and brother arrays were normal.
Discussion
CdLS is caused by dominant mutations in genes that alter the activity of the cohesin protein complex. Cohesin is highly conserved from yeast to human, and plays roles in sister chromatid cohesion, DNA repair, chromosome segregation, and the regulation of gene transcription. 11 Mutations in structural and regulatory cohesin proteins including those encoded by NIPBL, SMC1A, SMC3, RAD21 , and HDAC8 genes account for at least %65 patients with CdLS. 3 6
NIPBL is the human homolog of the Drosophila melanogaster Nipped-B gene. NIPBL, a cohesin loader, has been implicated in transcriptional control and genome organization. Localized at 5p13.2, the 47 exons NIPBL gene encodes the NIPBL, also known as SCC2 or delangin. The exact function of NIPBL is unknown but it is believed to be essential for controlling the activity of chromosomes during cellular division. 10 11
According to the HGMD, 457 mutations have been identified so far for the NIPBL gene with most of them being nonsense or missense mutations. Previous reports have shown that patients with CdLS, NIPBL mutations are more severely affected in limb anomalies, growth, and cognitive function compared with those with mutations in SMC1A, SMC3, RAD21 , and HDAC8 genes or in those with unknown molecular etiology. 13
CdLS is characterized by typical facial features, short stature, cardiac anomalies, hirsutism, hypoplastic genitalia, and upper extremity defects. Our patient had similar clinical signs. She has short stature, mitral valve insufficiently, hirsutism, high-arched eyebrows, synophrys, long eyelashes, antevert nares, clinodactyly on fifth finger in both hands, and four to five metatarsal bone shortness. Most of individuals with CdLS have the disorder as the result of a de novo variant. Some parents were found to have germline mosaicism for the causative pathogenic variant. 14 In our patient, the de novo heterozygous variant (c.245A > G) detected in the NIPBL gene was not previously described.
10q11.2 deletions cause a wide range of effects in patients. Phenotype–genotype correlation has not been reported in this region deletions. They have been described in asymptomatic individuals, as well as in patients showing intellectual disability, developmental delay, global growth retardation, epilepsy, seizures, hypotonia, gastroesophageal and vesicoureteral refluxes, constipation, sleep apnea, eczema, and skeletal system anomalies. 8 9 15 The effect in some individuals may change depending on genetic and/or nongenetic modifiers. The only clinical feature common to a majority of patients were intellectual disability and developmental delay. 9
To date, over 30 cases with variable deletions of 10q11.22-q11.23 have been reported in literature. Chen et al reported a 22-week fetus with a 10q11.21q11.23 (45,946,150–50,945,014) × 1 deletion. Pregnancy was terminated in 24 weeks, and it was reported that there was no structural anomaly in the fetus with facial dysmorphism (hypertelorism, micrognathia, and low-set ear). 16 Stankiewicz et al identified 24 patients with common clinical feature development retardation and intellectual disability with 10q11.21-q11.23 deletion. 9 It has been shown that 10q11.21q11.23 region is a recurrent deletion site, although the fracture regions vary. 8 Other cases of a 10q11.2 deletion showed a development delay patient, 17 Additionally, Langley et al reported a patient with hyperactivity disorder. Previously reported 10q11.22q11.23 region deletions are shown in ( Table 1 ). 18
Table 1. Summary of patients with overlapping 10q11.2 deletions characterized by array CGH in the same region reported in the literature and in the DECIPHER database.
| Patient | H19 start–end deletion | Size (Mb) | İnherited | Developmental delay/ID | Dysmorphism | Other features | Addition gene mutation/additional copy number changes |
|---|---|---|---|---|---|---|---|
| Present case | 46,293,590–51,857,206 | ∼5.6 | Paternal | + | High arched and thick eyebrows, synophrysis, long eyelashes, short nose, flattened nasal root, anteverted nares, pointed jaw, long philtrum, low-set ear, 2 cm × 2 cm cafe au lait spot on abdomen, clinodactyly on fifth finger in both hands and four–five metatarsal bone shortness | Mitral valve insufficiency, varus deformity | NIPBL gene mutation |
| Proband's father | 46,981,607–51,857,817 | ∼4.9 | Unknown | – | – | Short stature | – |
| Chen et al 17 | 45,946,150–50,945,014 | 4.99 | De novo | Unknown | – | 22-weel fetus, prenatal diagnosis | – |
| Langley et al 18 | Unknown | Unknown | Unknown | – | Unknown | Hyperactivity disorder | – |
| Stankiewicz et al 8 | 45,979,000–56,907,000 | ∼10.9 | Paternal | + | Thin face, small cranium, large ears | Scoliosis, hemivertebrate, muscle strength 3/5, anal atresia, impacted teeth, jaw malocclusion, dysphagia, constipation | – |
| 45,512,000–51,585,000 | ∼6 | Paternal | + | High-arched palate, thin vermillion border, thin philtrum, narrow facies, protruding auricles | Sleep apnea, constipation | – | |
| 48,941,000–52,218,000 | ∼3.3 | Maternal | + | Large, over-folded ears, fifth finger clinodactyly | Autism spectrum disorder, decline walking skills, mild leg hyperreflexia | 13q22.3 duplication | |
| 45,480,000–51,585,000 | ∼6 | Paternal | + | – | Seizures, hypomyelination above the tentorium, apnea, constipation, abnormal rectosigmoid ratio | – | |
| 45,612,000–51,585,000 | ∼6 | Maternal | + | Telecanthus | Agenesis of corpus callosum, diffuse periventricular leukomalacia, hypotonia, scoliosis, constipation, sleep apnea | 3q13.12q13.32 deletion | |
| 45,612,000–51,585,000 | ∼6 | Maternal | + | Microcephaly | Cerebellar ataxia, intentional tremor, spastic paraparesis, increase tendon reflex, CT noted small classification within basal ganglia, seizure, stiffness, apneic episode | – | |
| 45,947,671–51,263,703 | ∼5.3 | Paternal | + | Prominent ears, tubular nose, high nasal bridge, high-arched palate, 3–4 toe cutaneous syndactyly | – | 12q23.1q23.2 duplication | |
| 46,400,346–51,237,832 | ∼4.8 | Unknown | + | – | Epilepsy, hypotonia, pigmentary retinopathy, nystagmus, swallowing dysfunction, constipation, renal reflux | – | |
| 46,400,346–51,237,832 | ∼4.8 | Maternal | + | Microcephaly, round face, high forehead, low set ears, ptosis, short palpebral fissures, beaked nose, anteverted nostrils, maxillary hypoplasia, thin lips, large uvula, hypoplastic philtrum, hypodontia, pointed chin, double hair whorl, webbed neck, cleft palate, shortened halluces, aracnodactyly of the left fifth finger | Dysgenesis of corpus callosum, choroid plexus cyst, leg length discrepancy, pulmonary stenosis, tricuspid regurgitation, hypertrophic right ventricle, hepatomegaly, lacrimal duct obstruction, sleep apnea, microphtalmia, blepharophimosis, astigmatism, hypodontia, eczema, sacral dimple, renal hypoplasia, vesicouretral reflux, urethral fistula, uretrovaginal fistula | 6p25.3p22.1 duplication, der(7)t(6;7)(p22.1;p22.3) | |
| 46,400,346–51,237,832 | ∼4.8 | Paternal | – | Epicanthal folds, micrognathia, facial asymmetry | Hypoplastic fifth digit phalanges, hypoplastic fifth nails | – | |
| 46,400,346–51,237,832 | ∼4.8 | Maternal | – | – | Ankle clonus, small umbilical hernia | – | |
| 46,400,346–51,237,832 | ∼4.8 | Unknown | + | – | Recurrent urinary tract infections, myopia, tonsillectomy, adenoidectomy | 14q31.3q32.11 deletion | |
| 46,400,346–51,237,832 | ∼4.8 | Unknown | + | Mild ptosis, short columella, short first toes | Seizures, hypotonia, clenched muscles, toe walking, optic atrophy, diarrhea, immune suppressed, chronic sinusitis and otitis media | – | |
| 46,400,346–51,237,832 | ∼4.8 | Unknown | + | – | Eczema, hypopigmentation | – | |
| 49,062,854–52,062,367 | ∼3 | Unknown | + | Downslanting eyes, ulnar deviation of hands, elbow contracture, fifth finger clinodactyly | Epilepsy, renal reflux, seizure | – | |
| 49,062,854- 52,062,367 |
∼3 | Paternal | + | Frontal bossing, rounded cheeks, rounded nasal tip, mildly deficient alae nasi, micrognathia | Hypotonia, micropenis, severe eczema | – | |
| 45,927,753- 51,581,847 |
∼5.7 | De novo | + | Deep set eyes, short palpebral fissures, flat midface, prominent maxilla, high-palate, micrognathia, sparse hair, brachydactyly | Dysarthria, hypomyelination, hypoplasia of inferior cerebellar vermis, persistent cavum vergae, ataxia, spasticity, dysmetria, clonus, hypotonia, congenital nystagmus, cataracts, microphthalmia, hypermetropia, dysphagia | – | |
| 48,871,525- 50,765,047 |
∼1.9 | De novo | Unknown | – | 30-week fetus, hypospadias, intrauterine growth retardation | Confined placental mosaicism: arr cgh 2(x3) | |
| 46,384,979- 52,086,077 |
∼5.7 | Unknown | – | Round face, prominent eyes, epicanthal folds, depressed nasal bridge, arched palate | Total anomalous venous return, tachycardia | 15q21.2q21.3 duplication | |
| 46,384,979- 51,265.056 |
∼4.9 | Maternal | – | – | Hypotonia, right hand anomaly | 9q34.3 duplication | |
| 49,121,974- 50,641,724 |
∼1.5 | Unknown | + | Unknown | Unknown | – | |
| 48,102,606- 50,641,752 |
∼2.5 | Unknown | Unknown | Unknown | Unknown | 9q34.2 duplication | |
| 46,384,979- 51,672,034 |
∼5.3 | Unknown | + | Unknown | Unknown | – | |
| 46,400,346- 51,237,832 |
∼4.8 | Paternal | Unknown | Cleft palate | Unknown | – | |
| Schwartz et al 9 | 46,283,686- 51,832,220 |
5.55 | Maternal | + | Ptosis, reducible equinus, claw toes, pes cavus, camptodactyly | Decreased active fetal movement, respirator distress episodes | CHAT gene mutation |
| Liehr et al 16 | Unknown | Unknown | De novo | + | Unknown | Unknown | – |
| DECIPHER ID: 282374 | 47,011,584- 51,664,079 |
∼4.6 | Paternal | + | Unknown | Unknown | – |
| DECIPHER ID: 285824 | 48,381,471- 51,593,795 |
∼3.2 | Maternal | – | Unknown | Coloboma, growth delay, hypoplasia of the corpus callosum | 4p15.3 deletion 10q26.3 duplication |
| DECIPHER ID: 337184 | 47,011,548- 51,805,020 |
∼4.8 | unknown | + | Unknown | Autistic behavior | 15q11.2 duplication |
10q11.22q11.23 region involves two LCRs (Low Copy Repeats) (LCR1 and LCR2 are located at 10q11.22q11.23) 11 12 are located on this region, lead to chromosomal rearrangements through nonallelic homologous recombination. 14 Several recurrent deletion involving these LCRs have been reported. 9 14 10q11.2 deletions can be seen in a wide degree of expressivity, yet this severity depends on the extension of the 10q deletion region.
Characterization by mapping array performed on the proband demonstrated the presence of deletion 10q, located in 10q11.22q11.23. The deletion interval contains at least 22 OMIM listed genes, of which six have OMIM morbid entries (CHAT, ERCC6, GDF2, MSMB, RBP3, and SLC18A3). The affected child deletion size is larger than her father's deletion region and we, therefore, suggest that this deletion may have increased the severity of intellectual disability in the proband. The variable expressivity resulting from haploinsufficiency of genes in the deletion region and additional modifiers, genetic and nongenetic, may modulate the pathogenicity of the 10q11.22q11.23 microdeletions. 8
Conclusion
Here we report a CdLS case with a deletion of chromosome 10q11.22-q11.23 combined with a novel mutation in NIPBL gene. The results of the study may help broaden the mutation spectrum of the disease and contribute to a further understanding of the relationship with phenotype and genotype in CdLS syndrome and 10q11.2 deletions.
Acknowledgments
We kindly thank the all clinicians for their participation in this study.
Funding Statement
Funding None.
Conflict of interest None delcared.
Availability of Data and Materials
All data used in this study are available from the corresponding author on reasonable request.
Authors' Contributions
H.B. and Ö.Ö. designed the study; S.B. and Ö.Ö. made the examination of the patient and family; H.B. and Ö.Ö. wrote manuscript. B.T. performed the genetic studies. All authors read and approved the final manuscript.
References
- 1.Liu J, Baynam G. Cornelia de Lange syndrome. Adv Exp Med Biol. 2010;685:111–123. [PubMed] [Google Scholar]
- 2.Ireland M, Donnai D, Burn J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am J Med Genet. 1993;47(07):959–964. doi: 10.1002/ajmg.1320470705. [DOI] [PubMed] [Google Scholar]
- 3.Kline A D, Barr M, Jackson L G. Growth manifestations in the Brachmann-de Lange syndrome. Am J Med Genet. 1993;47(07):1042–1049. doi: 10.1002/ajmg.1320470722. [DOI] [PubMed] [Google Scholar]
- 4.Kline A D, Krantz I D, Deardorff M A. Cornelia de Lange syndrome and molecular implications of the cohesin complex: abstracts from the 7th biennial scientific and educational symposium 2016. Am J Med Genet A. 2017;173(05):1172–1185. doi: 10.1002/ajmg.a.38161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayerza Casas A, Puisac Uriol B, Teresa Rodrigo M E, Hernández Marcos M, Ramos Fuentes F J, Pie Juste J.[Cornelia de Lange syndrome: congenital heart disease in 149 patients] (in Spanish) Med Clin (Barc) 201714907300–302. [DOI] [PubMed] [Google Scholar]
- 6.Avagliano L, Grazioli P, Mariani M, Bulfamante G P, Selicorni A, Massa V. Integrating molecular and structural findings: Wnt as a possible actor in shaping cognitive impairment in Cornelia de Lange syndrome. Orphanet J Rare Dis. 2017;12(01):174. doi: 10.1186/s13023-017-0723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao R, Pu T, Fang S. Patients carrying 9q31.1-q32 deletion share common features with Cornelia de Lange Syndrome. Cell Physiol Biochem. 2015;35(01):270–280. doi: 10.1159/000369694. [DOI] [PubMed] [Google Scholar]
- 8.Stankiewicz P, Kulkarni S, Dharmadhikari A V. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum Mutat. 2012;33(01):165–179. doi: 10.1002/humu.21614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz M, Sternberg D, Whalen S. How chromosomal deletions can unmask recessive mutations? Deletions in 10q11.2 associated with CHAT or SLC18A3 mutations lead to congenital myasthenic syndrome. Am J Med Genet A. 2018;176(01):151–155. doi: 10.1002/ajmg.a.38515. [DOI] [PubMed] [Google Scholar]
- 10.Sue Richards, Nazneen Aziz, Sherri Bale, David Bick, Soma Das, Julie Gastier-FosterStandards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med 20151705405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tonkin E T, Wang T J, Lisgo S, Bamshad M J, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36(06):636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 12.Dorsett D. The Drosophila melanogaster model for Cornelia de Lange syndrome: Implications for etiology and therapeutics. Am J Med Genet C Semin Med Genet. 2016;172(02):129–137. doi: 10.1002/ajmg.c.31490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deardorff M A, Kaur M, Yaeger D. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80(03):485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krajewska-Walasek M, Chrzanowska K, Tylki-Szymańska A, Białecka M. A further report of Brachmann-de Lange syndrome in two sibs with normal parents. Clin Genet. 1995;47(06):324–327. doi: 10.1111/j.1399-0004.1995.tb03974.x. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, Song J, Li H, Yuan H. Two de novo overlapping interstitial duplications at 10q22 associated with speech impairments, behavior problems, genital anomalies, developmental delay and İntellectual disability. Hereditary Genet. 2017;6:2. [Google Scholar]
- 16.Chen C P, Su Y N, Chern S R. Prenatal diagnosis of a 4.9-Mb deletion of 10q11.21 --> q11.23 by array comparative genomic hybridization. Taiwan J Obstet Gynecol. 2010;49(01):117–119. doi: 10.1016/S1028-4559(10)60025-3. [DOI] [PubMed] [Google Scholar]
- 17.Liehr T, Schreyer I, Kuechler A. Parental origin of deletions and duplications - about the necessity to check for cryptic inversions. Mol Cytogenet. 2018;11:20. doi: 10.1186/s13039-018-0369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langley K, Martin J, Agha S S. Clinical and cognitive characteristics of children with attention-deficit hyperactivity disorder, with and without copy number variants. Br J Psychiatry. 2011;199(05):398–403. doi: 10.1192/bjp.bp.111.092130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available from the corresponding author on reasonable request.