

Abstract
Steroid-resistant nephrotic syndrome (SRNS) patients with genetic mutations most commonly have histology of focal segmental glomerulosclerosis (FSGS) and do not respond to immunosuppressive drugs. We report the molecular screening results of 18 pediatric SRNS cases presented to our nephrology clinic. Three pathogenic variants have been detected, two previously reported and one novel variant. The reported pathogenic variants have been detected in NPHS1 and NPHS2 genes. A novel pathogenic variant has been detected in the inverted formin 2 gene ( INF2 ) gene. We did not detect any variant of the WT1 gene. There were 13 males. Mean age of study participants at enrollment was 69 months. There were 12 cases of primary SRNS. The mean duration from onset of symptoms to SRNS diagnosis was 13 months. FSGS and minimal change disease (MCD) were present in the same number of cases. The response rate (complete or partial) to immunosuppressive drugs was seen in only one patient in the genetic SRNS group ( n = 3), while the response rate in nongenetic cases ( n = 15) was 80%. Two nonresponders in the genetic SRNS group had FSGS for histopathology and pathogenic variants (NPHS2 and INF2). The other three nonresponders in the nongenetic SRNS group had both FSGS ( n = 1) and MCD ( n = 2) histopathology. There were two deaths in the study cohort of the nongenetic SRNS group. This study highlights the screening of the SRNS cohort by a panel of extended genes rather focussing on the three most common genes ( NPHS1 , NPHS2 , and WT1 ). This further confirms the molecular etiology of SRNS in three cases and extends the list of pathogenic variants of genetic SRNS in the North Indian population. This is the first study in the eastern part of Uttar Pradesh in India.
Keywords: steroid-resistant nephrotic syndrome, focal segmental glomerulosclerosis, genetic, pediatric
Introduction
Steroid-resistant nephrotic syndrome (SRNS) is a rare condition, accounting for ∼15% of all childhood cases of idiopathic nephrotic syndrome. 1 Focal segmental glomerulosclerosis (FSGS) is more commonly identified as histopathology and is more frequently associated with podocytopathy. Genetic mutation screening has become an important procedure in SRNS, whenever available. 2 The most common gene found to be mutated in sporadic form of SRNS is NPHS2 . 3 There are 53 genes associated with podocytopathy in SRNS patients. 4 Monogenic cause has been reported in 10 to 30% of SRNS cases by full exome testing. 4 Study from the largest international cohort found disease-causing mutation in monogenic SRNS genes in 29.5% of families (526/1,783). 5 India contributed 127 families and mutation was detected in 25 families (19.7%). 5 Mutations were present in NPHS1 , NPHS2 , and WT1 genes commonly. 5 This cohort also found Indian founder allele in NPHS1 (Arg367Cys), NPHS2 (Arg229Gln), and SMARCAL1 (Arg586Trp) in Indian patients. 5 Indian studies also reported mutations with a cumulative frequency of 3.7% in 540 SRNS patients tested in different centers. 6 7 8 9 10 Indian studies targeted mainly NPHS1 , NPHS2 , and WT1 genes. 6 7 8 9 10 There is a need to extend gene coverage in Indian SRNS patients due to population heterogeneity. So, we tested our cohort of 18 SRNS patients for 37 genes using next-generation sequencing (NGS) technology. Three pathogenic variants have been detected in three genes. Two children had pathogenic variants of the NPHS1 and NPHS2 genes. Another child had a novel pathogenic change in the inverted formin 2 ( INF2 ) gene. INF2 had been reported to cause autosomal dominant type of SRNS (FSGS5). 11 The above study led to molecular diagnosis in three cases in Indian SRNS patients.
Methodology
The study was conducted at the tertiary center in the eastern part of Uttar Pradesh for a period of 2 years from September 2017 to July 2019. Ethical clearance was obtained from the Institute Ethical Committee. Study recruited 18 cases of SRNS for molecular screening of 37 genes for hereditary podocyte disorders and Alport's syndrome. The coding sequences and adjacent intronic fragments of 37 genes ( ACTN4 , ADCK4 , ANLN , APOL1 , ARHGAP24 , ARHGDIA , C14orf142 , CD151 , CD2AP , COL4A3 , COL4A4 , COL4A5 , COQ2 , COQ6 , CRB2 , DGKE , EMP2 , GLA , INF2 , LAMB2 , LMX1B , MAGI2 , MTTL1 , MYH9 , MYO1E , NPHS1 , NPHS2 , PAX2 , PDSS2 , PLCE1 , PTPRO , SCARB2 , SMARCAL1 , TRPC6 , TTC21B , WDR73 , and WT1 ) were analyzed. Sequencing was performed on MiSeq platform (Illumina) using Multiplicom – FSGS MASTRTM version 5. Genomic DNA was extracted from patients and was subjected to high-throughput NGS.
Results
There were 13 males. The average age of study population at the time of enrollment was 69 months. There were 12 cases of primary SRNS. The mean duration from onset of symptoms to SRNS diagnosis was 13 months. The average estimated glomerular filtration rate (eGFR) calculated was 112.62 mL/min/1.73 m 2 . FSGS and minimal change disease (MCD) were present in the same number of cases (nine each). The response rate for immunosuppressive drugs (complete and partial) in the study cohort was 72%. Despite 3 months of immunosuppressive drugs, five patients did not show any remission. Two of them had pathogenic genetic variants as a cause of SRNS, and three had sporadic SRNS. Three had FSGS for histopathology and two had MCD for histopathology. Those two, who had a genetic pathogenic variant, had an FSGS report on histopathology. The 3-month assessment showed that there were 2 deaths, 1 loss to follow-up, and 15 survivors ( Table 1 ). Patients P5 and P12 were females with sporadic SRNS with complete response (P5) and partial response (P12). The causes of death in the above-mentioned two cases were acute invasive diarrhea. Our study cohort was also broadly classified as a sporadic SRNS ( n = 15) group and a genetic SRNS ( n = 3) group. There were 15 patients in the sporadic SRNS group. Nine of the patients with sporadic SRNS had MCD histopathology and six had FSGS, while genetic SRNS ( n = 3) had FSGS for histopathology. The response rate (complete and partial) to immunosuppressive drugs at the end of 3 months in the sporadic SRNS group was 80%. Three patients in this group did not respond to immunosuppressive drugs. Two patients (P4 and P14) had histopathology MCD and one (P8) had histopathology FSGS. Genetic SRNS had a response rate of 33.33%. There were two deaths in the sporadic SRNS group. There was no death in the genetic SRNS group. Variants have been detected in four patients (P2, P9, P10, and P16) in NPHS1 , NPHS2 , INF2 , and CRB2 genes. Three variants were pathogenic ( NPHS1, NPHS2, and INF2 ) and the variant CRB2 was of unknown significance. The detection rate of the single SRNS gene was 16.66% (3/18) of the cohort. Two previously known alleles (Gly412 Cys and Thr232Ile) were detected in two patients (P2 and P10) and a novel pathogenic allele was detected in INF2 (Pro192 Thr) in patient P9 ( Fig. 1 ). All the three patients had primary SRNS. Patient P2 was male, with symptoms at 24 months of age, with FSGS on histopathology. Response to immunosuppressive drugs was shown at a follow-up period of 3 months. He had pathogenic variants in NPHS1 gene with intact renal function as shown in Table 1 .We could not comment on zygosity of this mutation as segregation analysis of parents was not done. We detected novel heterozygous pathogenic variant in INF2 gene in male patient P9 of 16 years 4 months old, who presented with primary SRNS. Child had eGFR of 101.87 mL/min/1.73 m 2 . Patient P9 had FSGS on histopathology with no response to immunosuppressive drugs at the end of 3 months ( Fig. 2A–D ). Patient P10 showed known homozygous pathogenic variant in NPHS2 gene with onset of disease at 8 years 7 months of age. Histopathology had FSGS with an eGFR of 99.1 mL/min/1.73 m 2 . No response to immunosuppressive drugs was reported at the end of 3 months of follow-up. A novel variant of the CRB2 gene was also detected in a 2-year 2-month-old male child (P16). As per the American College of Medical Genetics and Genomics (ACMG) criteria, this variant was of unknown significance. Child had an eGFR histopathology MCD of 84.60 mL/min/1.73 m 2 . Patient P16 showed complete remission at the end of 3 months of follow-up.
Table 1. Clinical features, molecular diagnosis, and outcome data of SRNS patients ( n = 18) .
| Patient identification | Gender | Age at enrollment in the study (mo) | SRNS (1—primary ; 2—secondary) | Onset of symptoms to steroid resistance (mo) | eGFR (mL/min/1.73 m 2 ) | Histopathology (MCD/FSGS) | Response to treatment (CR/PR/NR) | Gene involved | Genetic variant | Status of variant according to the ACMG criteria 18 | Outcome at 3 mo |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 19 | 1 | 3 | 117.5 | MCD | CR | – | Alive | ||
| P2 | M | 40 | 1 | 16 | 87.76 | FSGS | CR | NPHS1 | c.1_274del?(;)c.1234G > T; p.Gly412 Cys | Pathogenic; cannot comment on zygosity as segregation studies were not done in parents | Alive |
| P3 | M | 14 | 1 | 4 | 147.64 | FSGS | CR | – | Alive | ||
| P4 | M | 122 | 2 | 2 | 69.17 | MCD | NR | – | Alive | ||
| P5 | F | 14 | 1 | 5 | 167.26 | MCD | CR | – | Death | ||
| P6 | M | 135 | 1 | 4 | 151.7 | MCD | CR | – | Alive | ||
| P7 | M | 91 | 2 | 13 | 92.51 | FSGS | PR | – | Alive | ||
| P8 | M | 62 | 2 | 23 | 113.57 | FSGS | NR | – | Alive and developed CKD | ||
| P9 | F | 196 | 1 | 2 | 101.87 | FSGS | NR | INF2 | c.574C > A;p.Pro192Thr | Likely pathogenic(heterozygous) | Alive |
| P10 | M | 110 | 1 | 7 | 99.1 | FSGS | NR | NPHS2 | c.695C > T; p.Thr232Ile | Pathogenic (homozygous) | Alive |
| P11 | M | 128 | 1 | 12 | 130.0 | MCD | CR | – | Alive | ||
| P12 | F | 26 | 1 | 4 | 112.8 | MCD | PR | – | Death | ||
| P13 | M | 47 | 1 | 6 | 100.1 | FSGS | CR | – | Alive | ||
| P14 | M | 42 | 2 | 10 | 121.14 | MCD | NR | – | Lost to follow-up | ||
| P15 | F | 27 | 1 | 3 | 90.86 | MCD | PR | – | Alive | ||
| P16 | M | 26 | 1 | 7 | 84.60 | MCD | CR | CRB2 | c.941–3C > T(;) c.2177G > A; p.Arg727His |
Unknown significance | Alive |
| P17 | M | 120 | 2 | 96 | 140.42 | FSGS | CR | – | Alive | ||
| P18 | F | 29 | 2 | 18 | 99.12 | FSGS | CR | – | Alive |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; CR, complete response; eGFR, estimated glomerular filtration rate; FSGS, focal segmental glomerulosclerosis; MCD, minimal change disease; NR, not response; PR, partial response; SRNS, steroid-resistant nephrotic syndrome.
Fig. 1.
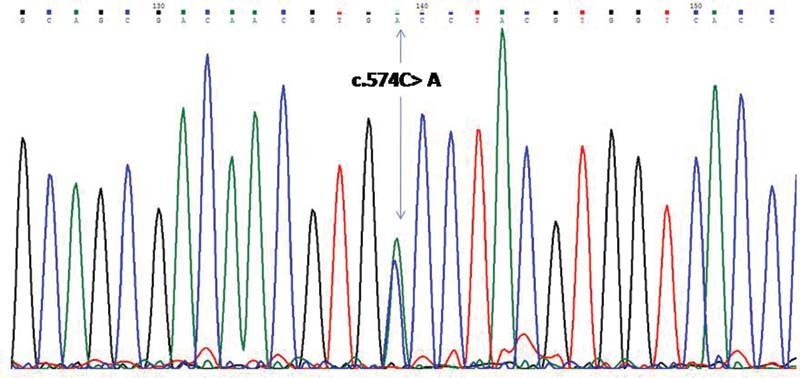
Chromatogram of patient P9 showing pathogenic variant: c.574C > A;p.Pro192 Thr.
Fig. 2.
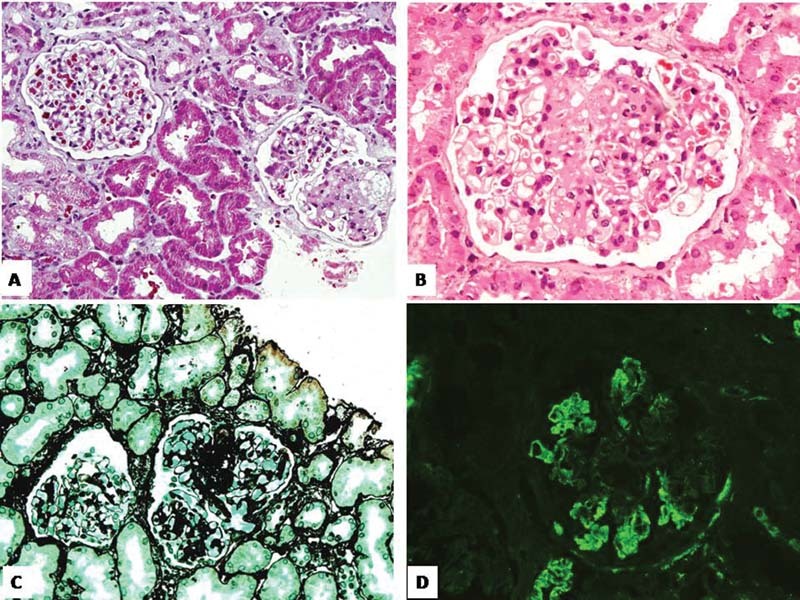
( A, B ) Glomerulus at low and high power showing segmental sclerosis in tuft (Masson's trichrome stain ×200 and hematoxylin and eosin stain ×400) ( C ) Sclerosed tuft is positive on silver stains (silver methenamine stain ×200). ( D ) C3 stain on immunofluorescence showing nonspecific deposits in the sclerosed segment (×200) in patient (P9).
Discussion
SRNS has both genetic and phenotypic heterogeneities. We performed molecular analysis in a group of 18 patients with SRNS. In three patients, we found a single gene as the cause of SRNS. The three genes in our cohort were NPHS1 , NPHS2 , and INF2 . We did not find a mutation in the WT1 gene. The rate of mutation detection was 16.66%. Patient P2 had mutations in the NPHS1 gene with exon 1 and exon 2 deletion in one allele and the other pathogenic variant was Gly412 Cys allele. Gly412Cys had previously been reported by Heeringa et al in 2008 in two brothers with congenital nephrotic syndrome. 12 Patient P10 had also previously reported pathogenic variant Thr232Ile as reported by Tonna et al. 13 There are limited data on mutation analysis in Indian pediatric SRNS cases. To make our findings more meaningful and logical, we tabulated pathogenic variant data from all Indian studies published to date ( Table 2 ). We found only five studies of South Indian children, and there was no North Indian study. 6 7 8 9 10 Data were collected from 20 patients who had pathogenic variants from five different studies in South India. 6 7 8 9 10 The cumulative pathogenic variant detection rate in the earlier studies was 3.7% (20/540) in NPHS1 , NPHS2 , WT1 , and PLCe1 genes. The most common gene involved was NPHS2 in 15 patients, followed by WT1 in 3 patients, NPHS1 and PLCe1 in 1 patient. The most common exons in the NPHS2 gene were 1 (five patients), 4 (five patients), 5 (two patients), 8 (two patients), and splice site (one patient). We also found pathogenic variant in exon 5 in our patient as previously reported by Tonna et al. 13 The most common pathogenic genotype reported in NPHS2 was Arg168His (three patients) and Arg71X (three patients). 6 8 10 Diagnosis of pathogenic variants was associated with poor outcomes in Indian studies. FSGS is the most common histopathological lesion reported from the earlier studies in SRNS. 6 7 8 9 10 Large studies have found a monogenic cause of SRNS in 26.2% (49 of 187 patients) of the United Kingdom cohort, 29.5% (526 of 1,783 families) of the international multiethnic SRNS cohort, and 28.3% (34 of 120) of the Chinese cohort. 4 5 14 The most common genes found in the majority of patients above the United Kingdom and International SRNS registry were NPHS1 , NPHS2 , and WT1 genes. 4 5 This contrasts with the Wang et al's study which found a lower percentage of mutations in NPHS1 (5.83%), NPHS2 (3.33%), and WT1 (5.83%) genes compared with the United Kingdom cohort and international cohort. 14 Indian studies focused mainly on NPHS1 , NPHS2 , and WT1 genes. It is only 3.7%. This finding is in contrast to the international cohort report, which included 127 Indian families and found mutations in 25 families. 5 Mutations were commonly found in NPHS1 , NPHS2 , and WT1 genes. 5 Patient selection, with ethnic variations present in large Indian populations, could be possible explanation of above difference (3.7% versus 19.68%). However, the findings in Table 2 are consistent with reports from Chinese (3%), Japanese (0%), Korean cohort (0%), and Vasudevan et al from India (4%) in sporadic SRNS cases. 10 15 16 17
Table 2. Comparative summary chart of all Indian pathogenic variants, histopathology type, and their outcome.
| Patient number | Age of onset | Histopathology report | Variant change | ACMG criteria report | Zygosity | Type of mutation | Gene involved | Exons | Outcome | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | 3.5 y | FSGS | R71X; Arg71X | Pathogenic | Homozygous | Nonsense | NPHS2 | 1 | ESRD by 5 y and death by 6 y | 6 |
| Case 2 | 2.5 y | FSGS | R71X; Arg71X | Pathogenic | Homozygous | Nonsense | NPHS2 | 1 | CKD stage 3 (at 4.5 y) | 6 |
| Case 3 | 1.5 y | DMS | R752X; Arg752X | Pathogenic | Homozygous | Nonsense | PLCe1 | 7 | ESRD in 1 y of diagnosis at 2.5 y of age | 6 |
| Case 4 | 1.2 y | FSGS | g.179521737C > T (nucleotide change) | Pathogenic | Homozygous | Splice site | NPHS2 | Splice site | ESRD by the age of 3 y | 6 |
| Case 5 | 10 mo | MHC | G968V; Gly968Val | Pathogenic | Homozygous | Missense | NPHS1 | 21 | Remission in last follow-up | 6 |
| Case 6 | 4 y | FSGS | P316S; Pro316Ser | Pathogenic | Homozygous | Missense | NPHS2 | 8 | Alive | 7 |
| Case 7 | 3 y | FSGS | 42dElG; 42delGly | Pathogenic | Homozygous | Frameshift | NPHS2 | 1 | Alive | 7 |
| Case 8 | NM | FSGS | L167P;Leu167Pro | Pathogenic | Homozygous | Missense | NPHS2 | 4 | – | 8 |
| Case 9 | NM | FSGS | R168H; Arg168His | Pathogenic | Homozygous | Missense | NPHS2 | 4 | – | 8 |
| Case 10 | NM | FSGS | R168H; Arg168His | Pathogenic | Homozygous | Missense | NPHS2 | 4 | – | 8 |
| Case 11 | NM | FSGS | R168H; Arg168His | Pathogenic | Homozygous | Missense | NPHS2 | 4 | – | 8 |
| Case 12 | NM | FSGS | R196G;Arg196Gly | Pathogenic | Homozygous | Missense | NPHS2 | 5 | – | 8 |
| Case 13 | NM | FSGS | S46P;Ser46Pro | Pathogenic | Homozygous | Missense | NPHS2 | 1 | – | 8 |
| Case 14 | NM | FSGS | Q219L;Gln219Leu | Pathogenic | Homozygous | Missense | NPHS2 | 5 | – | 8 |
| Case 15 | NM | FSGS | S192F;Ser192Phe | Pathogenic | Homozygous | Missense | NPHS2 | 8 | – | 8 |
| Case 16 | NM | FSGS | P175S;Pro175Ser | Pathogenic | Homozygous | Missense | NPHS2 | 4 | – | 8 |
| Case 17 | 5 y | FSGS | IVS 9 + 4 C > T; | Pathogenic | Splice site mutation | WT1 | Intron 9 | Renal transplant and hormonal replacement therapy | 9 | |
| Case 18 | 6 y | FSGS | IVS 9 + 4 C > T | Pathogenic | Heterozygous | Splice site mutation | WT1 | Intron 9 | ESRD, death | 9 |
| Case 19 | 2 y | MCN | IVS 9 + 4G > A | Pathogenic | Heterozygous | Splice site mutation | WT1 | Intron 9 | Infrequent relapsing nephrotic syndrome | 9 |
| Case 20 | 2 y | Not mentioned | R71X;Arg71X | Pathogenic | Heterozygous | Missense | NPHS2 | 1 | Progressed to stage CKD V by 6 y | 10 |
| P2 | 3 y 4 mo | FSGS | c.1_274del?(;) c.1234G > T; p.Gly412 Cys | Pathogenic | Cannot be commented | Frameshift, missense | NPHS1 | Exon 1, 2 deletion; exon 10 | Alive and responded to immunosupressive drugs | Present study |
| P9 | 16 y 4 mo | FSGS | c.574C > A;p.Pro193 Thr | Pathogenic | Heterozygous | Missense | INF2 | 4 | Alive and nonresponsive to immunosupressive drugs | Present study |
| P10 | 9 y 2 mo | FSGS | c.695C > T; p.Thr232Ile | Pathogenic | Homozygous | Missense | NPHS2 | 5 | Alive and nonresponsive to immunosuppressive drugs | Present study |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; FSGS, focal segmental glomerulosclerosis; DMS, diffuse mesangial sclerosis; ESRD, end-stage renal disease; MHC, mesangial hypercellularity; MCN, minimal change nephrotic; INF2, inverted formin 2; CKD, chronic kidney disease; NM, not mentioned.
Patient P9 presented during the adolescent period with characteristics consistent with SRNS. Her work up on molecular diagnosis revealed a probable pathogenic variant P192T due to heterozygous missense mutation in exon 4 as per the criteria of the ACMG. 18 The present case did not show any hearing loss and features suggestive of peripheral neuropathy. This history was taken only to rule out the association of the clinical phenotype of Charcot–Marie–Tooth's disease with this type of FSGS. Labat-de-Hoz and Alonso recently published a summary of INF2-related mutations in the past 10 years. 19 Only 70.3% (97/138) of the cases reported FSGS histology, and 23.9% (33/138) of the cases reported FSGS + CMT. 19 Familial cases (71.7%) contributed more than sporadic cases (21.7%). 19 Variant P192T is likely to be novel as this is not reported from largest collection of database by Labat-de-Hoz and Alonso. 19 Her histopathology finding is consistent with reported FSGS histology in such patients. At present, the child is not in remission or immunosuppressive therapy. She is only on angiotensin-converting enzyme inhibitors.
Treatment response rate to immunosuppressive drugs is poor in cases of genetic SRNS compared with nongenetic SRNS cases. Largest systematic review by Malakasioti et al concluded that only 35% of cases with genetic SRNS responded fully or partially to immunosuppressive drugs and that the majority of cases with MCD responded to immunosuppressive drugs. 20 In our study, only one patient in three cases of genetic SRNS responded to treatment. Histopathological finding in the case of responsive therapy was FSGS. In our cohort, we have not done long-term follow-up. However, the long-term outcome of the genetic SRNS group is poor compared with sporadic SRNS. 21 Sixty-six per cent of the patients in genetic SRNS group developed ESRD by 44 months (median) as compared with 27% in nongenetic SRNS group (developed in 36 months). 21 In histopathology, our three genetic SRNS patients had FSGS. This finding cannot be generalized as the number of patients with genetic SRNS is very small ( n = 3). Large studies have shown both MCD and FSGS as histopathological findings in their studies. 20 21
Conclusion
In summary, this study highlights the importance of screening beyond conventional pathogenic genes ( NPHS1 , NPHS2 , and WT1 ). In our three cases, we could establish a molecular basis for SRNS by screening an extended panel of 37 genes. The study reported known variants and novel variants, thus contributed to the mutational spectrum of monogenic SRNS in Indian cases. It is time to develop an Indian panel of monogenic SRNS genes by conducting research on our heterogeneous SRNS patients.
Footnotes
Conflict of Interest None declared.
References
- 1.Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant. 2016;31(11):1802–1813. doi: 10.1093/ndt/gfv355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Pediatric Nephrology Association . Trautmann A, Vivarelli M, Samuel S. IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2020;35(08):1529–1561. doi: 10.1007/s00467-020-04519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbeitsgemeinschaft Für Pädiatrische Nephrologie Study Group . Ruf R G, Lichtenberger A, Karle S M. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15(03):722–732. doi: 10.1097/01.asn.0000113552.59155.72. [DOI] [PubMed] [Google Scholar]
- 4.Bierzynska A, McCarthy H J, Soderquest K. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91(04):937–947. doi: 10.1016/j.kint.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 5.SRNS Study Group . Sadowski C E, Lovric S, Ashraf S. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(06):1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siji A, Karthik K N, Pardeshi V C, Hari P S, Vasudevan A. Targeted gene panel for genetic testing of South Indian children with steroid resistant nephrotic syndrome. BMC Med Genet. 2018;19(01):200. doi: 10.1186/s12881-018-0714-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramanathan A S, Vijayan M, Rajagopal S, Rajendiran P, Senguttuvan P.WT1 and NPHS2 gene mutation analysis and clinical management of steroid-resistant nephrotic syndrome Mol Cell Biochem 2017426(1-2):177–181. [DOI] [PubMed] [Google Scholar]
- 8.Dhandapani M C, Venkatesan V, Rengaswamy N B. Report of novel genetic variation in NPHS2 gene associated with idiopathic nephrotic syndrome in South Indian children . Clin Exp Nephrol. 2017;21(01):127–133. doi: 10.1007/s10157-016-1237-0. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A S, Srilakshmi R, Karthickeyan S, Balakrishnan K, Padmaraj R, Senguttuvan P. Wilms' tumour 1 gene mutations in South Indian children with steroid-resistant nephrotic syndrome. Indian J Med Res. 2016;144(02):276–280. doi: 10.4103/0971-5916.195044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasudevan A, Siji A, Raghavendra A, Sridhar T S, Phadke K D. NPHS2 mutations in Indian children with sporadic early steroid resistant nephrotic syndrome. Indian Pediatr. 2012;49(03):231–233. doi: 10.1007/s13312-012-0057-x. [DOI] [PubMed] [Google Scholar]
- 11.Brown E J, Schlöndorff J S, Becker D J.Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis Nat Genet 2010420172–76.. Erratum in: Nat Genet 2010;42(4):361. Tonna, Stephen J [added] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Members of the APN Study Group . Heeringa S F, Vlangos C N, Chernin G. Thirteen novel NPHS1 mutations in a large cohort of children with congenital nephrotic syndrome. Nephrol Dial Transplant. 2008;23(11):3527–3533. doi: 10.1093/ndt/gfn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tonna S J, Needham A, Polu K. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol. 2008;9:13. doi: 10.1186/1471-2369-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Zhang Y, Mao J. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2017;32(07):1181–1192. doi: 10.1007/s00467-017-3590-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Ding J, Huang J. Mutations in NPHS2 in sporadic steroid-resistant nephrotic syndrome in Chinese children. Nephrol Dial Transplant. 2005;20(05):902–908. doi: 10.1093/ndt/gfh769. [DOI] [PubMed] [Google Scholar]
- 16.Cho H Y, Lee J H, Choi H J. WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23(01):63–70. doi: 10.1007/s00467-007-0620-1. [DOI] [PubMed] [Google Scholar]
- 17.Maruyama K, Iijima K, Ikeda M. NPHS2 mutations in sporadic steroid-resistant nephrotic syndrome in Japanese children. Pediatr Nephrol. 2003;18(05):412–416. doi: 10.1007/s00467-003-1120-6. [DOI] [PubMed] [Google Scholar]
- 18.ACMG Laboratory Quality Assurance Committee . Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(05):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labat-de-Hoz L, Alonso M A. The formin INF2 in disease: progress from 10 years of research. Cell Mol Life Sci. 2020;77(22):4581–4600. doi: 10.1007/s00018-020-03550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malakasioti G, Iancu D, Tullus K. Calcineurin inhibitors in nephrotic syndrome secondary to podocyte gene mutations: a systematic review. Pediatr Nephrol. 2021;36:1353–1364. doi: 10.1007/s00467-020-04695-0. [DOI] [PubMed] [Google Scholar]
- 21.German Pediatric Nephrology Association (GPN) . Büscher A K, Beck B B, Melk A. Rapid response to cyclosporin a and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2016;11(02):245–253. doi: 10.2215/CJN.07370715. [DOI] [PMC free article] [PubMed] [Google Scholar]