

Abstract
Coronary heart disease (CHD) is closely related to oxidative stress and inflammatory response and is the most common cardiovascular disease (CVD). Iron is an essential mineral that participates in many physiological and biochemical reactions in the human body. Meanwhile, on the negative side, iron has an active redox capacity, which leads to the accumulation of reactive oxygen species (ROS) and lipid peroxidation. There is growing evidence that disordered iron metabolism is involved in CHD's pathological progression. And the result of disordered iron metabolism is associated with iron overload-induced programmed cell death, often called ferroptosis. That features iron-dependent lipid peroxidation. Ferroptosis may play a crucial role in the development of CHD, and targeting ferroptosis may be a promising option for treating CHD. Here, we review the mechanisms of iron metabolism in cardiomyocytes (CMs) and explain the correlation between iron metabolism and ferroptosis. Meanwhile, we highlight the specific roles of iron metabolism and ferroptosis in the main pathological progression of CHD.
1. Introduction
Coronary atherosclerotic heart disease, also known as CHD, refers to localized myocardial ischemia, hypoxia, and even necrosis due to atherosclerosis (AS) [1]. As the population ages and people's lifestyles change, the morbidity and mortality of CHD are also increasing yearly. CHD is not only the most common CVD but also one of the leading causes of death worldwide [2]. And AS is the pathological basis of CHD, which involves inflammation, lipid deposition, plaque formation, and calcification. In addition, pathological changes such as vascular endothelial damage, arterial wall plaque stability damage, CM death, myocardial fibrosis (MF), and myocardial hypertrophy (MH) are also involved in the progress of CHD [3, 4].
Iron is an essential metal for the body and is the primary raw material for manufacturing hemoglobin and myoglobin. In addition, iron is critical for cellular viability and participates in a wide range of biochemical and physiological processes, including oxygen storage and transportation, mitochondrial respiration, DNA synthesis and repair, and enzymatic reactions in cells. However, excessive iron has toxic effects on the body. Iron has an active redox capacity, making it easy for free iron to receive and contribute electrons. The most important mechanism of iron biotoxicity is the involvement of excess intracellular ferrous iron (Fe2+) in the Fenton or Haber-Weiss reaction [5]. The interaction of Fe2+ with oxygen or hydrogen peroxide catalyzes the production of large amounts of ROS, leading to lipid peroxidation and further severe organ damage [6]. At the same time, Fe2+ is oxidized to ferric iron (Fe3+). Iron is also associated with the pathological mechanisms of various diseases such as hemochromatosis, cancer, and CVD [7, 8].
Recent studies have shown that dysregulation of iron metabolism is associated with CHD [9, 10]. Severe iron overload is involved in vascular injury and CM death, promoting the development of AS, myocardial infarction (MI), and heart failure (HF) [11]. And these results are related to iron overload-induced programmed cell death, or ferroptosis, a new form of programmed cell death discovered in recent years. Ferroptosis is featured by iron-dependent lipid peroxidation, unlike autophagy, apoptosis, and pyroptosis [12]. Some studies have directly or indirectly proved that ferroptosis exists in ischemic heart disease and plays an essential role in the process of CM death [13–15]. However, the effect of ferroptosis on CHD remains unclear. Here, we review the mechanisms of iron metabolism and regulation in CMs and explain the correlation between iron metabolism and ferroptosis. Moreover, we focus on the specific role of ferroptosis in the pathological progression of CHD.
2. Iron Homeostasis in CMs
Iron-mediated injury plays an essential role in many CVD, and studies on iron metabolism in the heart have attracted many scientists (Figure 1).
Figure 1.
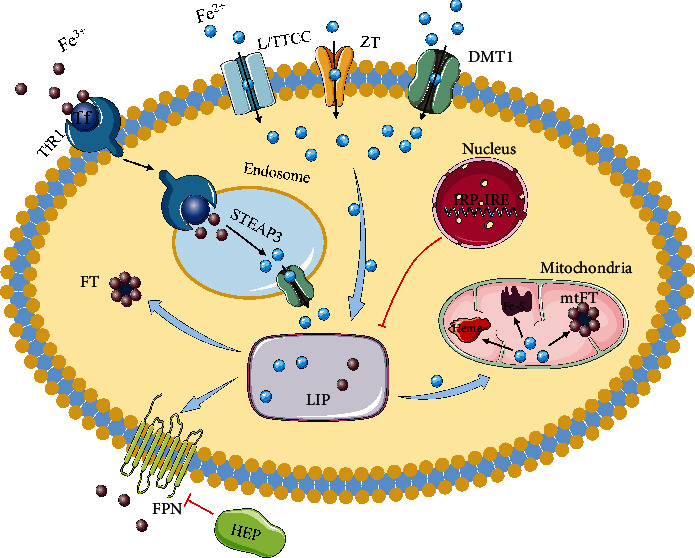
Iron homeostasis in cardiomyocytes. TBI enters cardiomyocytes via TfR1. TBI is reduced to Fe2+ by STEAP3 after release in the endosome, and Fe2+ is transferred to the cytoplasm by DMT1. NTBI enters via DMT1, LTCC, TTCC, and ZT. After entering the CMs, iron becomes part of the LIP and works through different pathways. A portion of iron is used by mitochondria to produce heme and Fe-S, and a portion is stored in FT. Furthermore, another part of iron is exported through the FPN and regulated by HEP. And cardiac iron homeostasis is regulated by IRP-IRE. TBI: Tf-bound iron; NTBI: non-Tf-bound iron; TfR1: transferrin receptor 1; STEAP3: six-transmembrane epithelial antigen of prostate 3; DMT1: divalent metal transporter 1; LTCC: L-type calcium channel; TTCC: T-type calcium channel; ZT: zinc transporters; LIP: labile iron pool; FT: ferritin; FPN: ferroportin; Fe-S: iron–sulfur cluster; mtFT: mitochondrial ferritin; HEP: hepcidin; IRE: iron-responsive elements; IRP: iron regulatory protein.
2.1. Iron Import in CMs
Systemic iron can form transferrin-bound iron (TBI) in the blood by binding to the transferrin (Tf), but binding sites in Tf have a limited high affinity for Fe3+. Studies have shown that the Tf saturation level is about 30% [16]. When high plasma iron concentration exceeds Tf iron binding capacity, iron mainly binds to serum albumin and citric acid to produce non-transferrin-bound iron (NTBI). TBI and NTBI both have access to CMs, but the pathways and regulatory mechanisms are different. CMs accumulate TBI through Tf receptors (TfRs), whereas accumulation of NTBI is via the divalent metal transporter 1 (DMT1), calcium channels, and zinc transporters.
2.1.1. TBI-Dependent Pathways
Under physiological conditions, iron mainly combines with Tf and enters CMs through TfR1 on the cell membrane [17, 18]. Fe3+ is released in endosomes and then reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate 3 (STEAP3). Subsequently, Fe2+ is transferred to the cytoplasm by DMT1. TfR1 gene has a conserved stem-loop structure in the 3′ untranslated region (UTR) called the iron response element (IRE). Iron regulatory protein (IRP) is the main protein that controls the balance of iron metabolism in CMs. IRP1 and IRP2, two forms of IRPs, have been found to act by binding to IRE [19]. The binding of IRP1 and IRP2 to IRE protected TfR1 mRNA from intranuclear degradation when CMs were iron deficient. That ensures the stability of TfR1 mRNA, increases its expression, and promotes iron absorption. Downregulation of TfR1 expression and a significant decrease in iron concentration occurred in mice with IRP gene knockout in CMs [20]. When iron overload occurs in CMs, IRP1 is converted to functional cytoplasmic aconitase via iron-sulfur clusters (Fe-S). At the same time, IRP1 loses its IRE-binding activity and the degradation of IRP2 increases. DMT mRNA with IRE in its 3′ UTRs has also been identified. IRE binding to IRP inhibits DMT mRNA degradation and facilitates iron uptake, which is consistent with the regulation of TfR1 [21].
2.1.2. NTBI-Dependent Pathways
When iron overload occurs, plasma NTBI levels are elevated. There is a consensus that NTBI is potentially toxic and can cause tissue damage by increasing oxidative stress and inducing tissue iron overload. NTBI cannot enter cells through TfR1 but DMT1, L-type calcium channel (LTCC), T-type calcium channel (TTCC), zinc transporters, and other non-transferrin receptor-dependent pathways [22]. Studies have shown that LTCC and TTCC are the main pathways for NTBI to enter CMs. Efonidipine is a dual TTCC and LTCC blocker. In thalassemic mice, efonidipine reduced cardiac iron accumulation and improved cardiac function. However, it did not affect the expression of cardiac ferroportin (FPN) [23]. In addition, ZRT/IRT-like protein 14 (ZIP14) and DMT1 may be involved in the uptake of NTBI in CMs [20]. A vivo study found that ZIP14 is consistently expressed in iron overloaded hearts but is not upregulated in response to increased iron deposition. It is speculated that ZIP14 is involved in the uptake of NTBI by CMs, but there may be other pathways involved in the uptake of NTBI [24]. And the control of NTBI import by DMT1 is regulated by the IRP/IRE system. It should be noted that the entry of NTBI into CMs through calcium channels and zinc transporters is not affected by IRP/IRE, but the specific regulatory mechanism remains unclear.
2.2. Iron Export from CMs
Once iron enters CMs, it becomes part of the labile iron pool (LIP). Iron enters the mitochondria as a feedstock for heme and Fe-S. In addition, ferritin (FT) stores some of the iron, and FPN exports the excess iron. Iron can enter CMs through multiple pathways, but only FPN is the export pathway, suggesting that CMs are particularly sensitive to iron overload. One study specifically knocked out FPN in the hearts of mice and found that their cardiac function was severely impaired, which was associated with cardiac iron overload [9]. However, there was no significant change in systemic iron status, suggesting that CMs have their unique regulation mechanism of iron metabolism. Notably, IREs are also present in the 5′ UTR of FPN mRNA and FT mRNA. The combination of IRE and IRP inhibits the translation of FPN and FT and prevents intracellular iron export and storage. Another more critical mechanism of iron export regulation is the role of the hepcidin (HEP)-FPN axis in the heart. The HEP-FPN axis in the heart is not affected by systemic HEP. Myocardial iron deficiency or hypoxia promotes local HEP expression and limits iron export by degrading FPN [10, 25]. A study prepared mouse models of HEP-resistant FPN knockout and cardiomyocyte-specific deletion of HEP. Both models showed severe cardiac dysfunction and iron deficiency in CMs rather than systemic iron deficiency [26]. A recent study found that knock-in HEP-resistant FPN in mouse pulmonary arterial smooth muscle cells (SMCs) leads to pulmonary hypertension and HF, suggesting that the HEP-FPN axis also plays a crucial role in regulating vascular homeostasis [26].
3. Effect of Iron Accumulation on Cardiac Ischemic and Hypoxic Injuries
During the development of CHD, we can observe abnormalities in multiple cell death signaling cascades. These include apoptosis, autophagy, pyroptosis, and ferroptosis (Table 1). Disordered iron metabolism runs through the whole pathological progression of CHD, and iron overload is considered an essential pathological factor of cardiovascular injury [9]. The data suggested that nearly 1/3 of the iron in CMs is distributed in mitochondria, catalyzing electron transport through the reversible oxidation state of iron and providing the energy required for normal cardiac function. Mitochondrial function is associated with disordered iron metabolism in the development of CVD [27]. When myocardial ischemia and hypoxia occur, LIP is imbalanced. Iron overload leads to the peroxidation of oxygen radicals in the cytoplasm and mitochondria. And it then will damage DNA, proteins, and lipids, resulting in cardiotoxicity [27]. In addition, AS was shown to be exacerbated significantly in FPN knockout mice, suggesting that iron overload has a promotive effect on AS. This effect is associated with iron overload-induced pathological changes, including dyslipidemia, altered vascular permeability, sustained endothelial activation, and elevated proinflammatory mediators. However, iron clearance mediated by transferrin or iron chelators and a low-iron diet may rescue NTBI-mediated toxicity [28].
Table 1.
Comparison of different forms of programmed cell death.
| Cell death mode | Morphological characteristics | Biochemical features | Characteristic molecules | References |
|---|---|---|---|---|
| Apoptosis | Chromatin condensation, nuclear fixation, cell shrinkage, membrane blistering, and formation of apoptotic bodies | DNA fragmentation, no leakage of cell contents, no inflammatory reaction | Caspase 3, caspase 7, caspase 8, BCL-2, Bax, P53, Fas | [34, 35] |
|
| ||||
| Autophagy | Accumulation of double-membraned autophagic vesicles | Increased lysosomal activity | Beclin 1, mTOR, ATG5, ATG7, LC3, TFEB, DRAM-3 | [35] |
|
| ||||
| Pyroptosis | Nuclear consolidation, plasma membrane pore formation, cell swelling and rupture | DNA fragmentation and inflammatory cascade response | NLRP3, ASC, pro-caspase 1, IL-1β, IL-18 | [4, 36] |
|
| ||||
| Ferroptosis | Mitochondrial shrinkage, increased membrane density, decreased mitochondrial cristae, and outer membrane rupture | Iron overload, lipid peroxidation, mitochondrial membrane potential changes | ACSL4, LPCAT3, xCT, GPX4, Fer-1, OxPLs, TfR1, SLC7A11, Nrf2, NCOA4 | [37, 38] |
Iron metabolism and oxidative stress in CMs are closely related to autophagy. Autophagy is a process of cellular self-feeding that relies primarily on lysosomes for intracellular degradation and recycling, promoting cellular repair or accelerating cell death. Thus, autophagy plays a dual role in maintaining cellular homeostasis and promoting cell renewal and metabolism [29]. Autophagy is activated by various environmental stressors, such as energy depletion, nutrient deficiency, and endoplasmic reticulum stress [30]. Appropriate autophagy can protect CMs by reducing oxidative stress and weakening myocardial inflammation, but excessive autophagy can lead to CM death and thus aggravate cardiac functional impairment. In the early stage of myocardial ischemia and hypoxia, appropriate autophagy facilitates the removal of mitochondria from damaged tissues, reduces the production of ROS, and maintains CM homeostasis to protect the heart from ischemic injury. However, in the late phase of myocardial ischemia and hypoxia, disordered iron metabolism induces CM autophagy to release iron stored in FT [31]. In addition, ferritinophagy is mediated by NCOA4 and exacerbates myocardial injury by increasing cellular unstable iron levels through degradation of cellular FT and induction of TfR1 expression, which subsequently sensitizes cells to ferroptosis [32]. Erastin is a classical ferroptosis inducer that acts on multiple molecular structures to induce ferroptosis. According to published reports, NCOA4 can prevent erastin-induced ferroptosis [33]. Therefore, timely regulation of autophagic by maintaining iron homeostasis during myocardial ischemia and hypoxia can reduce autophagy-induced myocardial injury.
4. Effect of Iron and ROS on Ferroptosis in the Heart
Ferroptosis is a newly discovered form of regulated cell death driven by iron-dependent lipid peroxidation. A growing number of studies have demonstrated that lipid peroxidation is a key trigger and landmark event in ferroptosis [39]. And high levels of intracellular NTBI are a prerequisite for triggering ferroptosis [40]. Extensive ROS generated by intracellular iron via the Fenton reaction and the Haber-Weiss reaction can directly produce a chain reaction with polyunsaturated fatty acids (PUFAs) in membrane phospholipids [38]. Further analysis has shown that AA and AdA are essential phospholipids to facilitate the peroxidation reaction [41]. PUFAs are highly sensitive to lipid peroxidation due to their unstable double bonds. With the help of ACSL4 and LPCAT3, PUFAs in cell membranes undergo synthesis, activation, and incorporation into phospholipids to produce PUFA-phosphatidyl ethanolamine (PUFA-PE) [42]. That makes the cell membrane easier to be attacked by ROS and produces more lipid peroxides. Also closely associated with ferroptosis is lipoxygenase (LOX). It can catalyze the peroxidation reaction of PUFA-PE [42]. Harmful lipid peroxides are scavenged by the intracellular antioxidant system when they accumulate. However, when the antioxidant system is weakened, lipid peroxides will not be scavenged in time, leading to an attack on the cytoplasmic membrane and morphological changes associated with ferroptosis [43]. Notably, ferroptosis inhibitors has been proven to inhibit the occurrence of ferroptosis [15, 44]. Research has shown that MI can lead to high levels of ROS production in the myocardium and that ferrostatin-1 (Fer-1) can significantly reduce the area of MI [45].
5. Regulatory Pathways of Ferroptosis
Under physiological conditions, the antioxidant response in the body is in a relative balance. When this balance is disrupted, it causes the accumulation of free radicals and triggers ferroptosis [46]. The System Xc- glutathione (GSH)-glutathione peroxidase 4 (GPX4) axis is the central redox mechanism inhibiting ferroptosis [47]. GSH is an essential antioxidant in the oxidative stress response, and cystine is one of the basic raw materials for GSH synthesis. GPX4 is an antioxidant enzyme that scavenges lipid peroxides and prevents the conversion of iron-dependent conversion of lipid peroxides to more reactive lipid radicals [48]. The reduction of toxic lipid peroxides to nontoxic lipid alcohols by GPX4 depends on the electrons provided by GSH [43]. System Xc- is a cystine/glutamate antiporter composed of SLC7A11 and SLC3A2, which mediates the exchange of extracellular cystine with intracellular glutamate and is responsible for the transport of cystine into the cell [49]. When selectively inhibiting System Xc-, cystine uptake will be reduced, and GSH synthesis in the organism will also be reduced [50]. Notably, erastin causes GSH depletion and GPX4 inactivation by inhibiting System Xc-, inducing ferroptosis [51]. The expression of SLC7A11 and GPX4 was significantly decreased in H/R-induced H9C2 cells. In addition, the naringin ameliorated cardiomyocyte ferroptosis via the System Xc-/GPX4 axis. However, the protective effect could be counteracted by erastin [52]. The data confirmed that the System Xc-GSH-GPX4 axis plays an essential role in the process of myocardial injury.
In addition to the System Xc-GSH-GPX4 axis, several pathways of ferroptosis regulation have been identified (Figure 2). Among them, the GCH1-BH4 pathway and the FSP1-CoQ-NADPH pathway are the other two independent mechanisms unaffected by GPX4 [47]. BH4 is a potent antioxidant. The expression of GCH1 triggers the production of BH4, thus exerting an antioxidant effect to inhibit ferroptosis [53, 54]. CoQ10 is another strong oxidant, and its fully reduced state, CoQH2, can trap lipid peroxide radicals and prevent peroxidative damage to the plasma membrane. FSP1 is a novel oxygen reductase that inhibits ferroptosis. It can catalyze CoQ10 regeneration dependent on NADPH and inhibit GPX4 deficiency-induced ferroptosis [55, 56].
Figure 2.
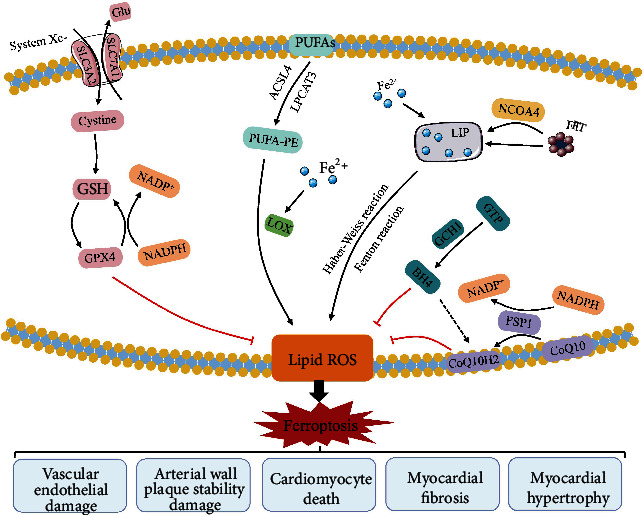
The regulatory mechanisms of ferroptosis in the pathological progression of CHD. There are three major independent regulatory pathways of ferroptosis: the System Xc-GSH-GPX4 axis, the GCH1-BH4 pathway, and the FSP1-CoQ-NADPH pathway. In addition, iron metabolism and lipid peroxidation are the main mechanisms. Abbreviations: PUFAs: polyunsaturated fatty acids; PUFA-PE: polyunsaturated fatty acid-phosphatidyl ethanolamine; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOX: lipoxygenase; SLC7A11: subunit solute carrier family 7 member 11; SLC3A2: solute carrier family 3 member 2; Glu: glutamate; GSH: glutathione; GPX4: glutathione peroxidase 4; GTP: guanosine triphosphate; BH4: tetrahydrobiopterin; GCH1: guanosine triphosphate cyclohydrolase 1; FSP1: ferroptosis suppressor protein 1; NCOA4: nuclear receptor coactivator 4; NADPH: nicotinamide adenine dinucleotide phosphate; ROS: reactive oxygen species; FT: ferritin; LIP: labile iron pool; CoQ10: coenzyme Q10.
6. Ferroptosis Involvement in the Pathological Progression of CHD
The primary pathological mechanism of CHD lies in the formation, growth, and even rupture of atherosclerotic plaques, resulting in luminal narrowing or blockage. Reduced myocardial perfusion induces MI and eventually leads to the progression of HF [3]. Several studies have shown that iron metabolism and ferroptosis are involved throughout the development of CHD and influence the key pathological changes of CHD. These include vascular endothelial damage, arterial wall plaque stability damage, CM death, MF, and MH [11, 37] (Table 2).
Table 2.
Ferroptosis involvement in the pathological progression of CHD.
| Histological type | Interventions | Features or changes | Pathways or signals | References |
|---|---|---|---|---|
| Vascular endothelial damage | Knock out FPN genes | Increase NTBI, induce chronic iron overload, increase vascular oxidative stress levels, promote AS | [28] | |
| High sugar and high lipid diet | Iron overload, elevated ROS level, downregulation of GPX4 and lipid peroxidation | HMOX1 increase | [57] | |
| PDSS2 | Inhibit VEC ferroptosis and AS progression | Nrf2 activation | [58] | |
| miR-17-92 overexpression | Reduce erastin-induced growth inhibition and ROS generation of HUVEC | A20-ACSL4 axis | [59] | |
| Fluvastatin | Reverse ox-LDL-induced decreases in GPX4 and xCT levels | Regulate GPX4 and xCT | [60] | |
| PM2.5 | Increase ROS production and iron content, decrease GSH, GSH-Px, and NADPH levels, promote lipid peroxidation | [40] | ||
|
| ||||
| Arterial wall plaque stability damage | High-iron diet | Iron overload, accelerate inflammation and the formation of macrophage-derived foam cells | [61] | |
| Macrophage-specific FPN1 deficiency | Iron overload, increase oxidative stress and systemic inflammation levels, inhibit ABC transporter protein expression, increase numbers of macrophages, decrease collagen | Downregulate LXRα expression | [62] | |
| High levels of uric acid | Induce the formation of macrophage-derived foam cells and lipid peroxidation | Nrf2/SLC7A11/GPX4 signaling pathway | [63] | |
| Cigarette smoke extract | Increase PTGS2 expression, GSH depletion, and lipid peroxidation, SMC ferroptosis | [64] | ||
|
| ||||
| CM death | Models of HF after MI | Downregulate FTH levels, increase oxidative stress and free iron levels, decrease CM viability | [14] | |
| Erastin, isoprenaline | Increase free iron levels, promote lipid peroxidation, and decrease CM viability | [65] | ||
| Fer-1, puerarin | Inhibit ferroptosis, reduce the loss of CMs | Upregulate the expression of GPX4 and FTH1 | [65] | |
| MI models | Downregulate the levels of GPX4 protein and GPX4 mRNA expression, increase CM death | Reduce GPX4 level | [66] | |
| HUCB-MSC exosomes | Inhibit H/R-induced CM ferroptosis, attenuate myocardial injury | miR-23a-3p/DMT1 axis | [13] | |
| Dexmedetomidine | Inhibit ROS production, maintain the structural integrity of mitochondria, inhibit ferroptosis, attenuate myocardial I/R injury | SLC7A11/GPX4 axis | [67] | |
| Propofol | Reduce SOD and iron accumulation, decrease lipid peroxidation levels, and increase the expression of antioxidant enzymes | AKT/P53 signaling pathway | [68] | |
| HF models | Downregulate GPX4 and FTH1 protein levels | TLR4-NOX4 pathway | [69] | |
|
| ||||
| MF | Inject iron dextran | Increase MDA levels, decrease glutathione peroxidase levels, leading to the occurrence of MF | [70] | |
| miR-375-3p | Promote MF due to CM ferroptosis | Downregulate GPX4 | [71] | |
| Dexmedetomidine | Inhibit CM ferroptosis after myocardial I/R, reduce the area of MF | SLC7A11/GPX4 signaling pathway | [67] | |
|
| ||||
| MH | Apelin-13 | Increase iron and ROS levels in mitochondria of CM, induce mitochondrial damage | Induce the expression of SFXN1 and NCOA4 | [72] |
| Knock out xCT | Increase PTGS2, MDA, and ROS levels, exacerbate Ang II-induced MH | Downregulate xCT | [50] | |
| Beclin 1 haploinsufficient | Elevate levels of SLC7A11, GPX4, and NCOA4, promote autophagy and ferroptosis, and exacerbate low ambient temperature-induced MH | [73] | ||
6.1. Vascular Endothelial Damage
Vascular endothelial cells (VECs) are border cells between blood and the vascular wall. They have highly selective permeability and are biological barriers to the interchange of material and protection of the inner surface of blood vessels [74]. Normal VECs have the effects of regulating vascular tone, procoagulation, antithrombosis, and anti-inflammatory, which are critical for maintaining vascular homeostasis [75]. VEC dysfunction and morphological damage are the beginning of AS and participate in the occurrence and development of CHD. In the early stages of AS, risk factors lead to increased adhesion molecule expression and dysregulation of antioxidant effects [76, 77]. All these alterations lead to the adhesion of leukocytes (especially monocytes) to VECs. Monocytes adhering to the vascular intima gradually migrate to the intima and differentiate into macrophages in response to inflammatory factors and the expression of receptors that facilitate lipid uptake [78]. As a result, lipid components of the blood, especially low-density lipoprotein (LDL), are absorbed and gradually deposited in the intima. LDL is oxidatively modified to oxidize LDL (ox-LDL) in the subintima. The oxidative process and toxic effects of ox-LDL can lead to or aggravate vascular endothelial damage and dysfunction, thereby significantly promoting lipid deposition [79]. Subsequently, scavenger receptors on the surface of macrophages can rapidly recognize ox-LDL and phagocytose it to form foam cells. Foam cells and lipids accumulate massively under the intima, producing an early lesion of AS known as fatty streaks [80, 81].
Although the pathological mechanism of AS is complex, current studies emphasize that oxidative stress is a key factor in the occurrence and development of AS. ROS generated during oxidative stress can oxidize lipids and proteins, induce inflammatory responses, and directly damage vascular cells, leading to endothelial dysfunction [82, 83]. Iron is considered potentially toxic because of its intense oxidative activity. When iron is overloaded, the body can produce large amounts of oxygen free radicals that injure VECs and other target cells [6, 84]. FPN is the only pathway of iron export in cells. If FPN is lost, intracellular and systemic iron overload will occur [9]. In one study, deletion of apoE and FPN genes in mice led to increased NTBI and induced chronic iron overload, subsequently increasing vascular oxidative stress levels and promoting AS [28]. These results may result from iron overload-induced endothelial activation and dysfunction. In contrast, restricting dietary iron intake or treating with iron chelators inhibited the progression of AS in mice [28].
The imbalance of redox reactions and iron overload as the main characteristics of ferroptosis provide indirect evidence to explore the role of ferroptosis in vascular endothelial damage in AS [85]. Direct evidence suggested that VEC ferroptosis was observed in diabetic AS mouse models [57]. The mice showed iron overload, downregulation of GPX4, and lipid peroxidation following upregulation of HMOX1. In addition, the study also found that Fer-1 alleviated the increase in ROS and endothelial dysfunction induced by a high-fat-high-sugar diet [57]. Ox-LDL is commonly used to prepare animal models of AS, and multiple studies have found that AS induced by ox-LDL is associated with ferroptosis [60, 63]. The prenyldiphosphate synthase subunit 2 (PDSS2) is a primary regulator of AS [86]. In ox-LDL-induced human coronary AS models, PDSS2 inhibited VEC ferroptosis and AS progression by promoting Nrf2 activation, thereby inducing the proliferation of human coronary endothelial cells [58]. Research has shown that miR-17-92 protected VECs from erastin-induced ferroptosis by targeting the A20-ACSL4 axis [59]. As a classical lipid-lowering drug, fluvastatin reversed the reduction of GPX4 and xCT levels induced by ox-LDL, thus achieving inhibition of VEC ferroptosis and vascular protection [60]. The discovery provides scientific evidence for the new role of statins in the prevention and treatment of AS. In terms of environment and health, studies have shown that PM2.5 accelerates the progression of CVD in numerous ways [87]. A recent study showed that PM2.5-induced vascular endothelial damage is also associated with ferroptosis [40]. The data showed that PM2.5 increases ROS production and iron content in VECs. In addition, the study provided the primary evidence for ferroptosis induced by iron uptake and storage dysfunction (disordered iron metabolism) by monitoring transferrin receptor, ferritin light chain (FTL), and ferritin heavy chain (FTH1) expression. This study further found that PM2.5 decreased the levels of GSH, glutathione peroxidase (GSH-Px), and NADPH. It was concluded here that PM2.5 induces VEC ferroptosis [40]. Although the mechanism of vascular endothelial damage is still unclear, the role of ferroptosis in triggering endothelial dysfunction has been widely proved. And Fer-1 and iron chelators can reverse the toxicity to a certain extent. Therefore, exploring the relationship between ferroptosis and endothelial damage is relevant to the mechanism research and prevention of CHD.
6.2. Arterial Wall Plaque Stability Damage
With the development of coronary AS, macrophages constantly phagocytize lipids and transform them into foam cells. At the same time, macrophages secrete inflammatory cytokines, which aggravate the local inflammatory response and further promote macrophage death [88]. That will further accelerate the formation of plaques and destabilize them. Under the stimulation of inflammation, SMCs in the middle membrane of the coronary artery migrate to the intima. Subsequently, they fuse with SMCs in the intima, proliferate, and secrete extracellular matrices (ECMs), such as elastin and interstitial collagen. SMCs and ECMs are the main components of the fibrous cap. They cover the foam cells and lipids accumulated under the intima. Then, the lipid core of the plaque gradually forms [89]. Under the stimulation of long-term hypoxia and inflammation, foam cells in the lipid core die in various ways and release lipids and cell debris. Significant amounts of cell components accumulate in the central area of plaques, forming a lipid-rich pool called the necrotic core [90]. As the pathological basis of coronary atherosclerotic plaque formation, necrotic core and fibrous cap are closely related to the unstable progress of the plaque. Angina pectoris occurs when the coronary arteries gradually narrow with plaque growth and block the blood supply to the heart muscle. When acute thrombosis obstructs large coronary vessels, blood flow is rapidly interrupted, leading to MI and unstable angina [91]. The foremost common cause of thrombosis is the rupture of vulnerable plaques. It has been established that the characteristics of vulnerable plaques include massive monocyte/macrophage infiltration, bulky lipid-rich necrotic cores, thin fibrous caps, and fewer SMCs [92]. Macrophages under the fibrous cap can reduce ECMs by their phagocytic function. The macrophages also can secrete plenty of matrix metalloproteinases to hydrolyze ECMs within the fibrous cap. As a result, the fibrous cap becomes thin and brittle, causing the plaque to become unstable and rupture to form a thrombus [80, 93]. In contrast, SMCs can secrete ECMs, which are the main ingredient of the fibrous cap, thus improving plaque stability [89]. Therefore, understanding further the role of macrophages and SMCs in plaque stability may provide new ideas for treating CHD.
Recent studies have shown that increased free iron can promote pathological processes such as oxidative stress and lipid peroxidation, accelerating inflammation and the formation of macrophage-derived foam cells, thus affecting plaque stability [61]. There are two primary sources of reactive iron in atherosclerotic plaques. One is plaque rupture and bleeding, and the other is the phagocytosis and rapid lysis of red blood cells by macrophages [94]. Considering the significant role of macrophages in systemic iron metabolism and the formation and progression of atherosclerotic plaques, Apoe−/− mouse models of macrophage-specific FPN1 deficiency were prepared in a study to investigate the effects and mechanisms of iron overload in macrophages on the progression of CHD [62]. The results indicated that iron overload in macrophages inhibited ATP-binding cassette (ABC) transporter protein expression by downregulating the liver X receptor α (LXRα) expression. This progression increased oxidative stress and systemic inflammation levels, promoted foam cell formation, and restricted lipid efflux, ultimately contributing to AS progression. In addition, iron overload also causes several changes in the composition of atherosclerotic plaques, with an increase in the number of macrophages and a decrease in collagen within the plaques, which make the plaques more prone to rupture [62]. However, iron chelation therapy increased ABC transporter protein expression, reversed lipid deposition, and reduced the surface volume of atherosclerotic plaques, ultimately slowing the progression of AS [62]. In another study, high levels of uric acid inhibited the Nrf2/PTGS2/GPX4 signaling pathway, induced the formation of macrophage-derived foam cells, and lipid peroxidation, thereby promoting the progression of AS. However, all these changes were reversed by Fer-1 [63]. There are no systematic studies to explore the mechanism between macrophage ferroptosis and CHD. Even so, several studies have shown that ROS accumulation, lipid oxidation, and iron deposition in macrophages are critical features of advanced atherosclerotic plaques [95]. Thus, macrophage ferroptosis may play an essential role in coronary AS and vulnerable plaque formation. And regulation of macrophage ferroptosis may be a promising approach to enhance plaque stability and delay the progression of CHD.
Unlike macrophages, SMCs impair the stability of atherosclerotic plaques mainly by affecting fibrous cap components [89]. A study found that iron overload stimulated SMCs to migrate, proliferate abnormally, and calcify, causing them to acquire a macrophage-like phenotype. In addition, iron overload increased ROS production to create a prooxidant microenvironment, which promoted foam cell formation and plaque instability progression. Moreover, researches demonstrated that removing excess iron and reducing the production of ROS can reverse these results mentioned above [96–98]. Notably, cigarettes are a major risk factor for AS. A study found that cigarette smoke extract (CSE) induced ferroptosis in vascular SMCs but not VECs [64]. The data showed increased PTGS2 expression, GSH depletion, and lipid peroxidation in vascular SMCs. However, GPX4 overexpression had no significant effect on CSE-induced ferroptosis [64]. The triggering mechanisms of ferroptosis in SMCs and VECs may be different. Therefore, inhibiting ferroptosis in SMCs may also be a new research direction for plaque stabilization.
6.3. Cardiomyocyte Death
The main pathological change of CHD is the formation and development of atherosclerotic plaques. The plaques gradually increase in size or even fall off, narrowing or blocking the lumen of the arterial blood vessels, resulting in insufficient blood supply to the coronary arteries. And the lack of perfusion causes local damage and mass death of CMs [99]. CMs are terminally differentiated cells with extremely limited regenerative capacity [100]. The mass death of CMs will result in structural and functional defects in the heart and cause HF [101]. The best way to prevent ischemic damage to the heart is to restore blood flow to the myocardial tissue, also known as reperfusion. However, reperfusion itself can also cause damage to the myocardium, called myocardial ischemia/reperfusion (I/R) injury. The mechanisms involved are oxidative stress, calcium overload, and mitochondrial damage, all of which can cause CM death [102]. Therefore, how to prevent CM death is key to improving and restoring cardiac function after MI and myocardial I/R injury. New insights into how cells are programmed to die have provided new ideas for salvaging myocardial injury in recent years. A study showed increased iron deposition and ROS in CMs around the MI region during ischemia and early reperfusion, suggesting that ferroptosis is a prominent form of CM death [103].
In HF mouse and rat models after MI, FTH levels were significantly downregulated, and oxidative stress and free iron levels were significantly increased. And then, desferrioxamine, an iron chelator, reversed these results and improved the viability of CMs [14]. Iron overload as a crucial mechanism for ferroptosis occurrence has attracted scientists to explore the underlying mechanisms of ferroptosis involvement in CM death. Either erastin or isoproterenol-treated H9C2 cells significantly increased free iron levels and promoted lipid peroxidation, thereby decreasing the viability of CMs. This result suggested that ferroptosis is associated with CM death. Both Fer-1 and puerarin upregulated the expression of GPX4 and FTH1 and inhibited ferroptosis [65]. In another study, the levels of GPX4 protein and GPX4 mRNA expression were downregulated in mice during early and midstage MI. To further determine the role of GPX4 in CM ferroptosis, a study transfected H9C2 cells with GPX4 siRNA. The results showed a significant increase in malondialdehyde (MDA) and superoxide dismutase (SOD) levels. And Fer-1 reduced CM death induced by GPX4 downregulation and inhibited lipid peroxide production, which suggested that ferroptosis is involved in CM death and myocardial injury after MI and is partially associated with reduced GPX4 levels [66]. In addition, studies have demonstrated that NADPH oxidase (NOX), a key enzyme for ROS production, is highly expressed in CMs [104]. In the descending aortic banding procedure-induced HF models, ferroptosis and autophagy were associated with massive CM death, and they were regulated by the TLR4-NOX4 pathway. It mainly showed downregulation of TLR4 and NOX4 expression, decreased LC3B-II and Belcin1, and upregulation of p62, GPX4, and FTH1 protein expression. It also significantly reduced CM death and improved cardiac function [69]. Drugs targeting the associated pathways of autophagy and ferroptosis in CMs may also be new therapeutic strategies for CHD, but others in both need to be studied in depth. Currently, CM ferroptosis during MI and HF has not been well studied. Are these mechanisms still applicable to clinical practice? Is blocking the process of ferroptosis in CMs effective? Addressing these questions may lead to new treatments to protect CMs from ferroptosis and delay the progression of MI to HF.
The exosome of MSCs derived from HUCB-MSC is known to alleviate myocardial injury caused by MI in mice. In the CM H/R models, investigators found that overexpression of DMT1 promoted CM ferroptosis. However, HUCB-MSC exosomes could inhibit H/R-induced CM ferroptosis through the miR-23a-3p/DMT1 axis and attenuate myocardial injury [13]. In addition, dexmedetomidine was demonstrated to inhibit myocardial I/R-induced ferroptosis via the SLC7A11/GPX4 axis [67]. Iron overload and ferroptosis have also been found in myocardial I/R. In one study, propofol inhibited ferroptosis via the AKT/P53 signaling pathway, thereby protecting CM from I/R injury. Specifically, it reduced SOD and iron accumulation, decreased lipid peroxidation levels, thereby increasing the expression of antioxidant enzymes [68]. Ferroptosis as one of the mechanisms of CM death after myocardial I/R has been widely demonstrated, and inhibiting ferroptosis may be an effective way to attenuate myocardial I/R injury.
6.4. Myocardial Fibrosis
MF after MI is a process of self-repair and inflammatory response of the myocardium [105]. The pathology is characterized by the proliferation of myofibroblasts in the myocardial tissue, secretion and excessive deposition of ECMs, and disorders in the ratio and arrangement of various types of collagen [106, 107]. After MI, local ischemia and hypoxia lead to the mass death of CMs. The body then produces a repair response that includes cardiac fibroblast activation, proliferation, and phenotypic transformation to form myofibroblasts. The above pathological changes lead to a replacement fibrotic process, in which fibroblasts and myofibroblasts produce fibrous scars to replace the damaged tissue [108]. Furthermore, the process of replacement fibrosis will reduce further dilatation of the infarcted area and maintain the structural integrity of the ventricles, thus preventing cardiac rupture. In addition, increased intraventricular mechanical pressure and inflammatory response after MI can induce expansion of connective tissue in the noninfarcted region and cause reactive fibrosis in the noninfarcted area. Reactive fibrosis can alter ventricular compliance and increase ventricular wall stiffness, thus affecting cardiac systolic and diastolic function and synchronicity, eventually leading to HF, worsening arrhythmias, and even sudden death [106, 109]. MF is a significant manifestation of cardiac remodeling and an essential factor influencing the prognosis of MI. Therefore, inhibition of the progression of reactive fibrosis in the peripheral myocardium of the infarct area would be an ideal treatment after MI.
In one study, iron overload increased oxidative stress levels and led to MF in gerbil hearts, as evidenced by increased MDA levels and decreased GPX4 levels [70]. It is suggested that iron overload may foster MF development by inducing lipid peroxidation damage. In addition, GPX4 participates in various pathological processes such as inflammation, cellular repair, oxidative stress, and ferroptosis. And GPX4 is closely associated with the development of fibrotic diseases [48]. Recent studies on ferroptosis intervention in MF have focused on GPX4. In myocardial I/R injury mouse models, miR-375-3p was found to promote MF development by downregulating GPX4 expression. However, both miR-375-3p inhibitors and Fer-1 significantly attenuated MF in these mice and enhanced the antioxidant capacity of cardiac fibroblasts in vitro [71]. In another study, dexmedetomidine activated the SLC7A11/GPX4 signaling pathway, inhibited CM ferroptosis after myocardial I/R in mice, and significantly reduced the area of MF. Predictably, the extent of myocardial injury and fibrosis area markedly increased following erastin treatment [67]. All these studies have confirmed the role of ferroptosis in MF. However, the current studies on its pathogenesis are relatively homogeneous, without distinguishing the location and type of MF occurrence. Future studies should delve into the effects of ferroptosis on reactive fibrosis and its mechanisms.
6.5. Myocardial Hypertrophy
CHD, especially MI, leads to CM damage or death due to ischemia and hypoxia, resulting in a localized cardiac function deficit. Under prolonged stimulation, peripheral CMs gradually hypertrophy to compensate for partial cardiac function [110, 111]. The early stage of MH is a beneficial compensatory response for the organism, but its compensatory capacity is limited. The late-stage shows increased myocardial oxygen consumption and reduced cardiac compliance and contractility, leading to loss of compensatory effect of pathological MH, further increasing the risk of HF and malignant arrhythmias [112, 113]. Studies have shown that MH is an independent risk factor for increased morbidity and mortality from various CVD during clinical practice [114, 115].
Apelin-13 can accelerate the progression of MH. In a study, apelin-13 induced hypertrophy and elevated free iron levels in H9C2 cells. It was further observed that apelin-13 increased iron and ROS levels in mitochondria of CM and triggered mitochondrial damage. This experiment showed that apelin-13-stimulated MH was closely related to NCOA4-mediated ferritinophagy and sideroflexin 1 (a mitochondria iron transporting protein) mediated mitochondrial iron overload [72, 116]. The initial relationship between ferroptosis and MH has also been established. In addition, one study used angiotensin II to induce MH in mice and found that oxidative stress and ferroptosis occurred [50]. Knockdown of xCT increased PTGS2, MDA, and ROS levels and exacerbated Ang II-induced MH in mice [50]. In addition, Beclin 1 is a homolog of the yeast autophagy gene Atg6/Vps30, an essential molecule in the autophagic process. A study found elevated levels of SLC7A11, GPX4, and NCOA4 in Beclin 1 haploinsufficient mice. They promoted autophagy and ferroptosis and exacerbated low-temperature-induced MH [73]. Currently, there is no direct evidence that post-MI ferroptosis is associated with MH. However, MH induced by other methods is strongly associated with iron overload and ferroptosis. These studies further imply the potential of ferroptosis as a therapeutic target for MH after MI.
7. Ferroptosis as a Novel Therapeutic Target for CHD
As discussed above, ferroptosis has been found to play a significant role in the pathological progression of CHD [11, 37]. Accordingly, targeting ferroptosis will become a new therapeutic strategy for treating CHD. With intensive studies on the mechanisms and regulatory pathways of ferroptosis, three main methods of ferroptosis inhibition have been identified.
First, iron chelators can bind to iron in the body to effectively increase iron excretion, thus blocking the redox reaction caused by iron overload. Currently, the main iron chelators used in clinical practice are deferoxamine, deferiprone, and deferasirox [117]. Several studies have shown that iron chelators have cardiovascular protective effects, such as improving vascular endothelial function, inhibiting SMC proliferation, and protecting CMs [64, 118]. Second, genetic manipulation of ferroptosis has been shown to inhibit ferroptosis and reduce myocardial injury [119]. These include upregulation of GPX4 and overexpression of SLC7A11 [66, 120]. However, this approach is currently not clinically applicable. Last, the cardioprotective effects of antioxidants have also been widely demonstrated [121]. Fer-1 is one of the most common antioxidants. It can upregulate the expression of GPX4 and FTH1, thus inhibiting lipid peroxidation [65]. And Fer-1 can slow down the progression of AS and reduce the area of MI [45, 57, 63]. Vitamin E is a common clinical antioxidant that can inhibit ferroptosis by inhibiting LOX [41]. Therefore, antioxidants may be the most promising ferroptosis inhibitors for widespread use. Excavating drugs with inhibiting ferroptosis from clinically available drugs may provide new options for treating CHD more quickly, such as vitamin E, fluvastatin, puerarin, dexmedetomidine, and propofol [41, 60, 65, 67, 68].
8. Conclusions
This article reviews the mechanisms of iron metabolism in CMs. It also focuses on the role of iron metabolism and ferroptosis in the crucial pathological changes of CHD. In contrast, the current research on the mechanisms of ferroptosis is still at an early stage, and most findings are obtained from animal and cellular experiments. We cannot conclude whether inhibition of ferroptosis is totally beneficial at different stages of human CHD. Inhibition of vascular and cardiac ferroptosis may bring new benefits to patients with CHD. Therefore, exploring the mechanisms and clinical feasibility is necessary for future studies. At this point, we have some unanswered questions that need to be concerned. (1) What are the roles and mechanisms of iron overload and ferroptosis in intraplaque angiogenesis? (2) Is ferroptosis involved in cardiomyocyte proliferation a target for cardiac regeneration? (3) How do we select methods to regulate ferroptosis in the pathological progression of CHD? With ongoing research, the mystery of ferroptosis will be further uncovered. Maintaining iron homeostasis and targeting ferroptosis will be promising strategies for the staged treatment of CHD.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant number 81774232) and QI HUANG Scholars (Junping Zhang) Special Funding (National Traditional Chinese Medicine People's Education Letter [2021] grant number 203).
Abbreviations
- DNA:
Deoxyribonucleic acid
- Bcl-2:
B cell lymphoma 2
- Bax:
Bcl-2-associated X protein
- mTOR:
Mammalian target of rapamycin
- ATG:
Autophagy-related protein
- LC3:
Light chain 3
- TFEB:
Transcription factor EB
- DRAM-3:
Damage-regulated autophagy modulator 3
- NLRP3:
NOD-like receptor protein 3
- ASC:
Apoptosis-associated speck-like protein
- IL-1β:
Interleukin-1beta
- IL-18:
Interleukin-18
- ACSL4:
Acyl-CoA synthetase long-chain family member 4
- LPCAT3:
Lysophosphatidylcholine acyltransferase 3
- xCT:
Cystine-glutamate antiporter
- OxPLs:
Oxidized phospholipids
- SLC7A11:
Subunit solute carrier family 7 member 11
- SLC3A2:
Subunit solute carrier family 3 member 2
- Nrf2:
Nuclear factor E2-related factor 2
- NCOA4:
Nuclear receptor coactivator 4
- AA:
Arachidonic acid
- AdA:
Adrenic acid
- GCH1:
Guanosine triphosphate cyclohydrolase 1
- BH4:
Tetrahydrobiopterin
- CoQ10:
Coenzyme Q10
- CoQH2:
Dihydroubiquione
- FSP1:
Ferroptosis suppressor protein 1
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- PTGS2:
Prostaglandin synthase 2
- TLR4:
Toll-like receptor 4
- AKT:
Protein kinase B
- HMOX1:
Heme oxygenase-1
- HUVEC:
Human umbilical vein endothelial cells
- H/R:
Hypoxia/reoxygenation
- PTGS2:
Prostaglandin-endoperoxide synthase 2.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Authors' Contributions
X.F. was responsible for conceptualization; X.F. and J.Z. were responsible for methodology; X.F. and A.L. were responsible for formal analysis; X.F. and H.L. were responsible for writing—original draft preparation; X.F., A.L., Z.Y., L.L., and J.Z. were responsible for writing—review and editing; H.L. and D.G. were responsible for visualization; Z.Y. and X.G. were responsible for supervision; J.Z. was responsible for funding acquisition. All authors have read and agreed to the published version of the manuscript.
References
- 1.Libby P., Theroux P. Pathophysiology of coronary artery disease. Circulation . 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 2.Leong D. P., Joseph P. G., McKee M., et al. Reducing the global burden of cardiovascular disease, part 2: prevention and treatment of cardiovascular disease. Circulation Research . 2017;121(6):695–710. doi: 10.1161/CIRCRESAHA.117.311849. [DOI] [PubMed] [Google Scholar]
- 3.Narula N., Olin J. W., Narula N. Pathologic disparities between peripheral artery disease and coronary artery disease. Arteriosclerosis, Thrombosis, and Vascular Biology . 2020;40(9):1982–1989. doi: 10.1161/ATVBAHA.119.312864. [DOI] [PubMed] [Google Scholar]
- 4.Ji N., Qi Z., Wang Y., et al. Pyroptosis: a new regulating mechanism in cardiovascular disease. Journal of Inflammation Research . 2021;14:2647–2666. doi: 10.2147/JIR.S308177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wardman P., Candeias L. P. Fenton Chemistry: An Introduction. Radiation Research . 1996;145(5):523–531. [PubMed] [Google Scholar]
- 6.Galaris D., Barbouti A., Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research . 2019;2019, article 118535 doi: 10.1016/j.bbamcr.2019.118535. [DOI] [PubMed] [Google Scholar]
- 7.Gozzelino R., Arosio P. Iron homeostasis in health and disease. International Journal of Molecular Sciences . 2016;17(1) doi: 10.3390/ijms17010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer C., Volani C., Komlódi T., et al. Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function. Antioxidants . 2021;10(11):p. 1818. doi: 10.3390/antiox10111818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakhal-Littleton S., Wolna M., Carr C. A., et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proceedings of the National Academy of Sciences of the United States of America . 2015;112(10):3164–3169. doi: 10.1073/pnas.1422373112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakhal-Littleton S. Mechanisms of cardiac iron homeostasis and their importance to heart function. Free Radical Biology & Medicine . 2019;133:234–237. doi: 10.1016/j.freeradbiomed.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S., Zhang X. Iron in cardiovascular disease: challenges and potentials. Frontiers in Cardiovascular Medicine . 2021;8, article 707138 doi: 10.3389/fcvm.2021.707138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y., Zhang W., Zhou X., et al. Roles of ferroptosis in cardiovascular diseases. Front Cardiovasc Med . 2022;9:p. 911564. doi: 10.3389/fcvm.2022.911564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song Y., Wang B., Zhu X., et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biology and Toxicology . 2021;37(1):51–64. doi: 10.1007/s10565-020-09530-8. [DOI] [PubMed] [Google Scholar]
- 14.Omiya S., Hikoso S., Imanishi Y., et al. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. Journal of Molecular and Cellular Cardiology . 2009;46(1):59–66. doi: 10.1016/j.yjmcc.2008.09.714. [DOI] [PubMed] [Google Scholar]
- 15.Zhou B., Zhang J., Chen Y., et al. Puerarin protects against sepsis-induced myocardial injury through AMPK-mediated ferroptosis signaling. Aging . 2022;14(8):3617–3632. doi: 10.18632/aging.204033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cazzola M., Huebers H. A., Sayers M. H., MacPhail A. P., Eng M., Finch C. A. Transferrin saturation, plasma iron turnover, and transferrin uptake in normal humans. Blood . 1985;66(4):935–939. doi: 10.1182/blood.V66.4.935.935. [DOI] [PubMed] [Google Scholar]
- 17.Paterek A., Mackiewicz U., Mączewski M. Iron and the heart: a paradigm shift from systemic to cardiomyocyte abnormalities. Journal of Cellular Physiology . 2019;234(12):21613–21629. doi: 10.1002/jcp.28820. [DOI] [PubMed] [Google Scholar]
- 18.Xu W., Barrientos T., Mao L., Rockman H. A., Sauve A. A., Andrews N. C. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Reports . 2015;13(3):533–545. doi: 10.1016/j.celrep.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouault T. A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nature Chemical Biology . 2006;2(8):406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 20.Haddad S., Wang Y., Galy B., et al. Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. European Heart Journal . 2016;38(5):ehw333–ehw372. doi: 10.1093/eurheartj/ehw333. [DOI] [PubMed] [Google Scholar]
- 21.Kühn L. C. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics . 2015;7(2):232–243. doi: 10.1039/c4mt00164h. [DOI] [PubMed] [Google Scholar]
- 22.Silva A. M. N., Rangel M. The (bio)chemistry of non-transferrin-bound iron. Molecules . 2022;27(6):p. 1784. doi: 10.3390/molecules27061784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumfu S., Chattipakorn S. C., Fucharoen S., Chattipakorn N. Dual T-type and L-type calcium channel blocker exerts beneficial effects in attenuating cardiovascular dysfunction in iron-overloaded thalassaemic mice. Experimental Physiology . 2016;101(4):521–539. doi: 10.1113/EP085517. [DOI] [PubMed] [Google Scholar]
- 24.Nam H., Wang C.-Y., Zhang L., et al. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica . 2013;98(7):1049–1057. doi: 10.3324/haematol.2012.072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian Z. M., Chang Y. Z., Leung G., et al. Expression of ferroportin1, hephaestin and ceruloplasmin in rat heart. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease . 2007;1772(5):527–532. doi: 10.1016/j.bbadis.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Lakhal-Littleton S., Wolna M., Chung Y. J., et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife . 2016;5, article e19804 doi: 10.7554/eLife.19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoes M. F., Grote Beverborg N., Kijlstra J. D., et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. European Journal of Heart Failure . 2018;20(5):910–919. doi: 10.1002/ejhf.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinchi F., Porto G., Simmelbauer A., et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. European Heart Journal . 2020;41(28):2681–2695. doi: 10.1093/eurheartj/ehz112. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y., Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death and Differentiation . 2015;22(3):367–376. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G., Mariño G., Levine B. Autophagy and the integrated stress response. Molecular Cell . 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito J., Omiya S., Rusu M.-C., et al. Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. eLife . 2021;10, article e62174 doi: 10.7554/eLife.62174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park E., Chung S. W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death & Disease . 2019;10(11):p. 822. doi: 10.1038/s41419-019-2064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou W., Xie Y., Song X., et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy . 2016;12(8):1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elmore S. Apoptosis: a review of programmed cell death. Toxicologic Pathology . 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong Y., Chen H., Gao J., Liu Y., Li J., Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. Journal of Molecular and Cellular Cardiology . 2019;136:27–41. doi: 10.1016/j.yjmcc.2019.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Jia C., Chen H., Zhang J., et al. Role of pyroptosis in cardiovascular diseases. International Immunopharmacology . 2019;67:311–318. doi: 10.1016/j.intimp.2018.12.028. [DOI] [PubMed] [Google Scholar]
- 37.Leng Y., Luo X., Yu J., Jia H., Yu B. Ferroptosis: a potential target in cardiovascular disease. Frontiers in Cell and Development Biology . 2021;9:p. 813668. doi: 10.3389/fcell.2021.813668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Cao F., Yin H.-L., et al. Ferroptosis: past, present and future. Cell Death & Disease . 2020;11(2):p. 88. doi: 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen Y., Wang X., Shen X., et al. Geniposide possesses the protective effect on myocardial injury by inhibiting oxidative stress and ferroptosis via activation of the Grsf1/GPx4 axis. Frontiers in Pharmacology . 2022;13, article 879870 doi: 10.3389/fphar.2022.879870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y., Tang PM2.5 Induces Ferroptosis in Human Endothelial Cells through Iron Overload and Redox Imbalance. Environmental Pollution . 2019;254:p. 112937. doi: 10.1016/j.envpol.2019.07.105. [DOI] [PubMed] [Google Scholar]
- 41.Kagan V. E., Mao G., Qu F., et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature Chemical Biology . 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences . 2016;113(34):E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ursini F., Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPX4. Free Radical Biology and Medicine . 2020;152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 44.Stamenkovic A., Pierce G. N., Ravandi A. Phospholipid oxidation products in ferroptotic myocardial cell death. American Journal of Physiology. Heart and Circulatory Physiology . 2019;317(1):H156–H163. doi: 10.1152/ajpheart.00076.2019. [DOI] [PubMed] [Google Scholar]
- 45.Fang X., Wang H., Han D., et al. Ferroptosis as a target for protection against cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America . 2019;116(7):2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun R., Liu M., Xu K., et al. Ferroptosis is involved in the benzene-induced hematotoxicity in mice via iron metabolism, oxidative stress and NRF2 signaling pathway. Chemico-Biological Interactions . 2022;362 doi: 10.1016/j.cbi.2022.110004. [DOI] [PubMed] [Google Scholar]
- 47.Liu M., Kong X. Y., Yao Y., et al. The critical role and molecular mechanisms of ferroptosis in antioxidant systems: a narrative review. Annals of translational medicine . 2022;10(6):p. 368. doi: 10.21037/atm-21-6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Z., Zhu Z., Liu Y., Liu Y., Zhao H. Function and regulation of GPX4 in the development and progression of fibrotic disease. Journal of Cellular Physiology . 2022;237(7):2808–2824. doi: 10.1002/jcp.30780. [DOI] [PubMed] [Google Scholar]
- 49.Sato H., Tamba M., Kuriyama-Matsumura K., Okuno S., Bannai S. Molecular cloning and expression of human XCT, the light chain of amino acid transport system Xc. Antioxidants & Redox Signaling . 2000;2(4):665–671. doi: 10.1089/ars.2000.2.4-665. [DOI] [PubMed] [Google Scholar]
- 50.Zhang X., Zheng C., Gao Z., et al. SLC7A11/XCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovascular Drugs and Therapy . 2022;36(3):437–447. doi: 10.1007/s10557-021-07220-z. [DOI] [PubMed] [Google Scholar]
- 51.Hu P., Xu Y., Jiang Y., et al. The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. European Journal of Pharmacology . 2022;928 doi: 10.1016/j.ejphar.2022.175093. [DOI] [PubMed] [Google Scholar]
- 52.Xu S., Wu B., Zhong B., et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) /system Xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered . 2021;12(2):10924–10934. doi: 10.1080/21655979.2021.1995994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraft V. A. N., Bezjian C. T., Pfeiffer S., et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Central Science . 2020;6(1):41–53. doi: 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Latremoliere A., Costigan M. GCH1, BH4 and pain. Current Pharmaceutical Biotechnology . 2011;12(10):1728–1741. doi: 10.2174/138920111798357393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bersuker K., Hendricks J. M., Li Z., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature . 2019;575(7784):688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu J., Xiong Y., Zhang Y., et al. The molecular mechanisms of regulating oxidative stress-induced ferroptosis and therapeutic strategy in tumors. Oxidative Medicine and Cellular Longevity . 2020;2020:14. doi: 10.1155/2020/8810785.8810785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meng Z., Liang H., Zhao J., et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life Sciences . 2021;284, article 119935 doi: 10.1016/j.lfs.2021.119935. [DOI] [PubMed] [Google Scholar]
- 58.Yang K., Song H., Yin D. PDSS2 inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. Journal of Cardiovascular Pharmacology . 2021;77(6):767–776. doi: 10.1097/FJC.0000000000001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao F.-J., Zhang D., Wu Y., et al. MiRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochemical and Biophysical Research Communications . 2019;515(3):448–454. doi: 10.1016/j.bbrc.2019.05.147. [DOI] [PubMed] [Google Scholar]
- 60.Li Q., Liu C., Deng L., et al. Novel function of fluvastatin in attenuating oxidized low-density lipoprotein-induced endothelial cell ferroptosis in a glutathione peroxidase4- and cystine-glutamate antiporter-dependent manner. Experimental and Therapeutic Medicine . 2021;22(5):p. 1275. doi: 10.3892/etm.2021.10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu X., Cai X., Ma R., Fu W., Zhang C., Du X. Iron-load exacerbates the severity of atherosclerosis via inducing inflammation and enhancing the glycolysis in macrophages. Journal of Cellular Physiology . 2019;234:18792–18800. doi: 10.1002/jcp.28518. [DOI] [PubMed] [Google Scholar]
- 62.Cai J., Zhang M., Liu Y., et al. Iron accumulation in macrophages promotes the formation of foam cells and development of atherosclerosis. Cell & Bioscience . 2020;10(1):p. 137. doi: 10.1186/s13578-020-00500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu W., Liu W., Xie D., et al. High level of uric acid promotes atherosclerosis by targeting NRF2-mediated autophagy dysfunction and ferroptosis. Oxidative Medicine and Cellular Longevity . 2022;2022 doi: 10.1155/2022/9304383.9304383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sampilvanjil A., Karasawa T., Yamada N., et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. American Journal of Physiology. Heart and Circulatory Physiology . 2020;318(3):H508–H518. doi: 10.1152/ajpheart.00559.2019. [DOI] [PubMed] [Google Scholar]
- 65.Liu B., Zhao C., Li H., Chen X., Ding Y., Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochemical and Biophysical Research Communications . 2018;497(1):233–240. doi: 10.1016/j.bbrc.2018.02.061. [DOI] [PubMed] [Google Scholar]
- 66.Park T.-J., Park J. H., Lee G. S., et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death & Disease . 2019;10(11):p. 835. doi: 10.1038/s41419-019-2061-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu P., Zhang J., Ding Y., et al. Dexmedetomidine post-conditioning alleviates myocardial ischemia-reperfusion injury in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation. Human Cell . 2022;35(3):836–848. doi: 10.1007/s13577-022-00682-9. [DOI] [PubMed] [Google Scholar]
- 68.Li S., Lei Z., Yang X., et al. Propofol protects myocardium from ischemia/reperfusion injury by inhibiting ferroptosis through the AKT/P53 signaling pathway. Frontiers in Pharmacology . 2022;13, article 841410 doi: 10.3389/fphar.2022.841410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X., Xu S., Zhao C., Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochemical and Biophysical Research Communications . 2019;516(1):37–43. doi: 10.1016/j.bbrc.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 70.Wang M., Liu R.-R., Wang C.-J., et al. Combined histological and hematological assessment of iron-induced organ damage in a gerbil model of iron overload. American Journal of Translational Research . 2015;7(2):385–392. [PMC free article] [PubMed] [Google Scholar]
- 71.Zhuang Y., Yang D., Shi S., et al. MiR-375-3p promotes cardiac fibrosis by regulating the ferroptosis mediated by GPX4. Computational Intelligence and Neuroscience . 2022;2022 doi: 10.1155/2022/9629158.9629158 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Tang M., Huang Z., Luo X., et al. Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radical Biology & Medicine . 2019;134:445–457. doi: 10.1016/j.freeradbiomed.2019.01.052. [DOI] [PubMed] [Google Scholar]
- 73.Yin Z., Ding G., Chen X., et al. Beclin1 haploinsufficiency rescues low ambient temperature-induced cardiac remodeling and contractile dysfunction through inhibition of ferroptosis and mitochondrial injury. Metabolism . 2020;113, article 154397 doi: 10.1016/j.metabol.2020.154397. [DOI] [PubMed] [Google Scholar]
- 74.Gutiérrez E., Flammer A. J., Lerman L. O., Elízaga J., Lerman A., Fernández-Avilés F. Endothelial dysfunction over the course of coronary artery disease. European Heart Journal . 2013;34(41):3175–3181. doi: 10.1093/eurheartj/eht351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tousoulis D., Simopoulou C., Papageorgiou N., et al. Endothelial dysfunction in conduit arteries and in microcirculation. Novel therapeutic approaches. Pharmacology & Therapeutics . 2014;144(3):253–267. doi: 10.1016/j.pharmthera.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 76.Poredos P., Poredos A. V., Gregoric I. Endothelial dysfunction and its clinical implications. Angiology . 2021;72(7):604–615. doi: 10.1177/0003319720987752. [DOI] [PubMed] [Google Scholar]
- 77.Xu S., Ilyas I., Little P. J., et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacological Reviews . 2021;73(3):924–967. doi: 10.1124/pharmrev.120.000096. [DOI] [PubMed] [Google Scholar]
- 78.Libby P., Ridker P. M., Hansson G. K. Progress and challenges in translating the biology of atherosclerosis. Nature . 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 79.Ryoo S., Bhunia A., Chang F., Shoukas A., Berkowitz D. E., Romer L. H. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis . 2011;214(2):279–287. doi: 10.1016/j.atherosclerosis.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maguire E. M., Pearce S. W. A., Xiao Q. Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vascular Pharmacology . 2019;112:54–71. doi: 10.1016/j.vph.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 81.Chistiakov D. A., Melnichenko A. A., Myasoedova V. A., Grechko A. V., Orekhov A. N. Mechanisms of foam cell formation in atherosclerosis. Journal of Molecular Medicine (Berlin, Germany) . 2017;95(11):1153–1165. doi: 10.1007/s00109-017-1575-8. [DOI] [PubMed] [Google Scholar]
- 82.Ardiana M., Susetyo Pikir B., Santoso A., Oky Hermawan H., Jibril Al-Farabi M. The effect of subchronic cigarette smoke exposure on oxidative stress parameters and endothelial nitric oxide synthase in a rat aorta. ARYA Atherosclerosis . 2021;17:1–7. doi: 10.22122/arya.v17i0.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Incalza M. A., D’Oria R., Natalicchio A., Perrini S., Laviola L., Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascular Pharmacology . 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 84.Marques V. B., Leal M. A. S., Mageski J. G. A., et al. Chronic iron overload intensifies atherosclerosis in apolipoprotein E deficient mice: role of oxidative stress and endothelial dysfunction. Life Sciences . 2019;233:p. 116702. doi: 10.1016/j.lfs.2019.116702. [DOI] [PubMed] [Google Scholar]
- 85.Zhang S., Xin W., Anderson G. J., et al. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death & Disease . 2022;13(1):p. 40. doi: 10.1038/s41419-021-04490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bain C. R., Ziemann M., Kaspi A., et al. DNA methylation patterns from peripheral blood separate coronary artery disease patients with and without heart failure. ESC Heart Failure . 2020;7(5):2468–2478. doi: 10.1002/ehf2.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cho E., Kang Y., Cho Y. Effects of fine particulate matter on cardiovascular disease morbidity: a study on seven metropolitan cities in South Korea. International Journal of Public Health . 2022;67 doi: 10.3389/ijph.2022.1604389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chawla A., Nguyen K. D., Goh Y. P. S. Macrophage-mediated inflammation in metabolic disease. Nature Reviews. Immunology . 2011;11(11):738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bennett M. R., Sinha S., Owens G. K. Vascular smooth muscle cells in atherosclerosis. Circulation Research . 2016;118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews. Immunology . 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bergmark B. A., Mathenge N., Merlini P. A., Lawrence-Wright M. B., Giugliano R. P. Acute coronary syndromes. Lancet . 2022;399(10332):1347–1358. doi: 10.1016/S0140-6736(21)02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cimmino G., Di Serafino L., Cirillo P. Pathophysiology and mechanisms of acute coronary syndromes: atherothrombosis, immune-inflammation, and beyond. Expert Review of Cardiovascular Therapy . 2022;20(5):351–362. doi: 10.1080/14779072.2022.2074836. [DOI] [PubMed] [Google Scholar]
- 93.Shah P. K., Falk E., Badimon J. J., et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation . 1995;92(6):1565–1569. [PubMed] [Google Scholar]
- 94.Chistiakov D. A., Orekhov A. N., Bobryshev Y. V. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiologica (Oxford, England) . 2015;213(3):539–553. doi: 10.1111/apha.12438. [DOI] [PubMed] [Google Scholar]
- 95.Martinet W., Coornaert I., Puylaert P., De Meyer G. R. Y. Macrophage death as a pharmacological target in atherosclerosis. Frontiers in Pharmacology . 2019;10:p. 306. doi: 10.3389/fphar.2019.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang Z., Song T., Truong L., et al. Important role of sarcoplasmic reticulum Ca2+releaseviaryanodine receptor-2 channel in hypoxia-induced rieske iron–sulfur protein-mediated mitochondrial reactive oxygen species generation in pulmonary artery smooth muscle cells. Antioxidants & Redox Signaling . 2020;32(7):447–462. doi: 10.1089/ars.2018.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramakrishnan L., Pedersen S. L., Toe Q. K., et al. The hepcidin/ferroportin axis modulates proliferation of pulmonary artery smooth muscle cells. Scientific Reports . 2018;8(1):p. 12972. doi: 10.1038/s41598-018-31095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malhotra R., Wunderer F., Barnes H. J., et al. Hepcidin deficiency protects against atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology . 2019;39(2):178–187. doi: 10.1161/ATVBAHA.118.312215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arbab-Zadeh A. Does “vulnerable” atherosclerotic plaque modify coronary blood flow?: how myths perpetuate. JACC: Cardiovascular Imaging . 2020;13(3):757–759. doi: 10.1016/j.jcmg.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 100.Castellan R. F. P., Meloni M. Mechanisms and therapeutic targets of cardiac regeneration: closing the age gap. Front Cardiovasc Med . 2018;5:p. 7. doi: 10.3389/fcvm.2018.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marunouchi T., Tanonaka K. Cell death in the cardiac myocyte. Biological & Pharmaceutical Bulletin . 2015;38(8):1094–1097. doi: 10.1248/bpb.b15-00288. [DOI] [PubMed] [Google Scholar]
- 102.Hausenloy D. J., Yellon D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. The Journal of Clinical Investigation . 2013;123(1):92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang W. H., Wu S., Wong T. M., Chung S. K., Chung S. S. M. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radical Biology & Medicine . 2008;45(5):602–610. doi: 10.1016/j.freeradbiomed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 104.Zhang M., Perino A., Ghigo A., Hirsch E., Shah A. M. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxidants & Redox Signaling . 2013;18(9):1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zuo P., Zhu Y., Li Y., et al. Protease-activated receptor 2 deficiency in hematopoietic lineage protects against myocardial infarction through attenuated inflammatory response and fibrosis. Biochemical and Biophysical Research Communications . 2020;526(1):253–260. doi: 10.1016/j.bbrc.2020.03.077. [DOI] [PubMed] [Google Scholar]
- 106.Gyöngyösi M., Winkler J., Ramos I., et al. Myocardial fibrosis: biomedical research from bench to bedside. European Journal of Heart Failure . 2017;19(2):177–191. doi: 10.1002/ejhf.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Venugopal H., Hanna A., Humeres C., Frangogiannis N. G. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cell . 2022;11(9):p. 1386. doi: 10.3390/cells11091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Nieuwenhoven F. A., Turner N. A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascular Pharmacology . 2013;58(3):182–188. doi: 10.1016/j.vph.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 109.Talman V., Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell and Tissue Research . 2016;365(3):563–581. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Toepfer C. N., Sikkel M. B., Caorsi V., et al. A post-MI power struggle: adaptations in cardiac power occur at the sarcomere level alongside MyBP-C and RLC phosphorylation. American Journal of Physiology. Heart and Circulatory Physiology . 2016;311(2):H465–H475. doi: 10.1152/ajpheart.00899.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khalid K., Padda J., Ismail D., et al. Correlation of coronary artery disease and left ventricular hypertrophy. Cureus . 2021;13(8, article e17550) doi: 10.7759/cureus.17550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pawlinski R., Tencati M., Hampton C. R., et al. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation . 2007;116(20):2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernardo B. C., Weeks K. L., Pretorius L., McMullen J. R. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacology & Therapeutics . 2010;128(1):191–227. doi: 10.1016/j.pharmthera.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 114.Kabir R., Sinha P., Mishra S., et al. Inorganic arsenic induces sex-dependent pathological hypertrophy in the heart. American Journal of Physiology. Heart and Circulatory Physiology . 2021;320(4):H1321–H1336. doi: 10.1152/ajpheart.00435.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frey N., Olson E. N. Cardiac hypertrophy: the good, the bad, and the ugly. Annual Review of Physiology . 2003;65(1):45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 116.Bellelli R., Federico G., Matte’ A., et al. NCOA4 deficiency impairs systemic iron homeostasis. Cell Reports . 2016;14(3):411–421. doi: 10.1016/j.celrep.2015.12.065. [DOI] [PubMed] [Google Scholar]
- 117.Entezari S., Haghi S. M., Norouzkhani N., et al. Iron chelators in treatment of iron overload. Journal of Toxicology . 2022;2022 doi: 10.1155/2022/4911205.4911205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jansová H., Macháček M., Wang Q., et al. Comparison of various iron chelators and prochelators as protective agents against cardiomyocyte oxidative injury. Free Radical Biology & Medicine . 2014;74:210–221. doi: 10.1016/j.freeradbiomed.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang F., Yang R., Xiao Z., et al. Targeting ferroptosis to treat cardiovascular diseases: a new continent to be explored. Frontiers in Cell and Development Biology . 2021;9, article 737971 doi: 10.3389/fcell.2021.737971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fang X., Cai Z., Wang H., et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circulation Research . 2020;127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- 121.Sun C., Peng F., Li J., Cui X., Qiao X., Zhu W. Ferroptosis-specific inhibitor ferrostatin-1 relieves H2O2-induced redox imbalance in primary cardiomyocytes through the Nrf2/ARE pathway. Disease Markers . 2022;2022 doi: 10.1155/2022/4539932.4539932 [DOI] [PMC free article] [PubMed] [Google Scholar]