

Abstract
Earlier we showed by affinity cross-linking of initiating substrates to Escherichia coli primase that one or more of the residues Lys211, Lys229, and Lys241 were involved in the catalytic center of the enzyme (A. A. Mustaev and G. N. Godson, J. Biol. Chem. 270:15711–15718, 1995). We now demonstrate by mutagenesis that only Lys241 but not Lys211 and Lys229 is part of the catalytic center. Primase with a mutation of Arg to Lys at position 241 (defined as K241R-primase) is almost unable to synthesize primer RNA (pRNA) on the single-stranded DNA-binding protein (SSB)/R199G4oric template. However, it is able to synthesize a pppApG dimer plus trace amounts of 8- to 11-nucleotide (nt) pRNA transcribed from the 5′ CTG 3′ pRNA initiation site on phage G4 oric DNA. The amount of dimer synthesized by K241R-primase is similar to that synthesized by the wild-type primase, demonstrating that the K241R mutant can initiate pRNA synthesis normally but is deficient in chain elongation. In the general priming system, the K241R-primase also can synthesize only the dimer and very small amounts of 11-nt pRNA. The results of gel retardation experiments suggested that this deficiency in pRNA chain elongation of the K241R mutant primase is unlikely to be caused by impairment of the DNA binding activity. The K241R mutant primase, however, can still prime DNA synthesis in vivo and in vitro.
Synthesis of a small primer RNA (pRNA) by primase is required to provide a primed template for DNA polymerase to initiate complementary strand synthesis in most viral and chromosomal DNA replication systems (9). Primases, like all RNA polymerases, differ from DNA polymerase in that they can initiate complementary strand synthesis de novo and have sequence-specific recognition sequences on DNA where assembly of the initiation complex takes place (9). In Escherichia coli (8, 29) and lambda phage (29), primase initiates pRNA synthesis preferentially at the T residue of a 5′ CTG 3′ template sequence and extends the pRNA chain to between 10 and 30 nucleotides (nt). In the single-stranded DNA (ssDNA) coliphages G4 (5, 13), St-1, and φK (17), however, E. coli primase initiates at a single and specific 5′ CTG 3′ sequence. This specificity has allowed a detailed analysis of the interaction of primase with its template (16, 19, 21).
In a previous study (10), we took advantage of the specific initiation of pRNA synthesis by primase at the phage G4 oric (G4oric) sequence to use affinity labeling to directly identify the active center of primase. We used as the first substrate ATP with an aldehyde group attached to the 5′ phosphate. This aldehyde group could be covalently cross-linked to Lys (K) residues at or close to the substrate binding site of primase. Adding [α-32P]GTP as the second substrate resulted in the newly synthesized pppAp32G dinucleotide pRNA being cross-linked to the nearest Lys residue in the catalytic center. By using chemical mapping, we identified one or more of the residues Lys211, Lys229, and Lys241 as the cross-linking target. The chemical cleavages were not precise enough to separate the three Lys residues. However, they all fall in a region of primase that contains some amino acids that are conserved in all bacterial primases (24) and that contains sequence motifs that have been proposed by sequence comparison with other DNA and RNA polymerases to be functional (7, 26; also see Fig. 1). This region of primase between amino acids 200 and 350, therefore, most probably contains the active center of primase where the enzyme interacts with the template DNA, binds nucleoside triphosphate (NTP) substrates, and synthesizes phosphodiester bonds. The affinity labeling experiments showed that the Lys residue (one of the residues Lys211, Lys229, and Lys241) that cross-linked to the first NTP substrate (the modified ATP) is within 2 Å from the site of catalysis. Because of its charged nature, the cross-linked Lys residue will most likely be involved in some function at the active center.
FIG. 1.

Structure of primase and conservation of amino acid sequence surrounding Lys211, Lys229, and Lys241. (A) Domain structure of primase (data taken from references 22 and 25). (B) Conserved amino acid sequences and motifs at the catalytic center of primase. Amino acids that are conserved in all 16 sequenced prokaryote primases are in bold type; the data were taken from Szfranski et al. (24). Conserved motifs 3 to 6 are from Ilyana et al. (7), and the RNA polymerase (RNAP), and dnaG boxes are from Verlasalovic et al. (26). The Lys211, Lys229, and Lys241 residues which were changed to Arg residues by oligonucleotide mutagenesis are indicated with arrows.
To identify which of the three Lys residues was cross-linked to the pRNA dinucleotide and to analyze its function, we have mutagenized all three of the Lys residues to Arg (R) residues and examined the effect on pRNA synthesis. We found that K211R and K229R mutant proteins were not functionally impaired. K241R mutant protein, however, was partially impaired and was able to initiate pRNA synthesis by polymerizing the first two ribonucleotides (pppApG) on the template but was almost unable to extend the dinucleotide to a longer pRNA chains. The K241R mutant protein still possesses DNA binding activity. Lys241 is, therefore, part of the active center of primase and is involved in the mechanism of pRNA chain elongation.
MATERIALS AND METHODS
Expression plasmids and cells.
pGNG7 contains the E. coli dnaG gene cloned into the BamHI and EcoRI sites of pET-21d (18). This vector adds a six-His tag to the C terminus of the expressed protein. The dnaG gene was excised from pGNG1 (3). pGEX-47 contains the N-terminal P47 domain of primase (22, 25) cloned into pGEX-2TK (Pharmacia). pGEX-K211R, pGEX-K229R, and pGEX-K241R contain P47 with Lys-to-Arg changes at amino acid positions 211, 229, and 241. E. coli HMS174 F− recA1 hsdR (rK− mK+) RifR (18) was used for plasmid construction, mutagenesis, and expression of primase from pGNG7. E. coli BL21(DE3) F− ompT hsdB (rB− mB−) gal dcm (DE3) (18) was used to express the pGEX plasmids. Cells were normally grown in Luria broth (LB) (15) containing 50 μg of ampicillin per ml and induced for protein expression by adding 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG). When plasmid protein overexpression was induced with lambda phage CE6 (18), cells were grown in minimal M9 medium supplemented with 1% Casamino Acids (15) containing 100 μg of ampicillin per ml.
Mutagenesis.
Mutations in P47 were made by PCR mutagenesis (6) using pGEX-47. Similar protocols were used to make the Lys241-to-Arg mutation in pGNG7 containing wild-type primase with a C-terminal His tag (HT-primase). Mutant oligonucleotides were purchased from DNA Agency. The resulting P47 mutant plasmids containing the Lys-to-Arg changes at amino acid positions 211, 229, and 241 were designated pGEX-K211R, pGEX-K229R, and pGEX-K241R, respectively, and the primase plasmid containing Lys241-to-Arg mutation was designated pGNG7-K241R. All mutant plasmids were completely sequenced to establish that no additional mutations had been induced by the PCR mutagenesis procedure.
Preparation of primase and P47-primase.
Wild-type primase and K241R mutant primase were prepared from HMS174 cells containing pGNG7 and pGNG7-K241R. The proteins were overexpressed by infecting the cells with lambda phage CE6 at a multiplicity of infection of 10 by the protocol previously described (18). The His-tagged primase was purified from cell lysates with nickel resin by the protocol recommended by the manufacturer of the His tag kit (Novagen). The bound primase was eluted with 0.5 M NaCl containing 0.1% imidazole. The eluted fractions containing primase were dialyzed against 20 mM Tris-HCl containing 50 mM NaCl, 0.1 mM dithiothreitol, and 15% (vol/vol) glycerol overnight at 4°C. These fractions were divided into small aliquots and stored at −70°C. The P47 N-terminal domain of primase (22) was prepared as a glutathione S-transferase (GST) fusion protein by inducing E. coli BL21 cells containing pGEX-47 with 0.4 mM IPTG for 3 h at 37°C. The harvested cells were treated with lysozyme and lysed with Triton X-100 as previously described (3). The GST fusion proteins were affinity purified from the lysate as recommended by the supplier (Pharmacia) of the pGEX-2TK vector. P47 was cleaved from the fusion protein with thrombin. The cleaved protein has a 7-amino-acid protein kinase labeling site added to the N terminus which does not affect its functional activity (unpublished observation). The mutant P47 proteins P47-K211R, P47-K229R, and P47-K241R were similarly prepared from pGEX plasmids pGEX-K211R, pGEX-K229R, and pGEX-K241R, respectively.
Preparation of DnaB Helicase.
DnaB helicase was prepared from BL21 cells containing the wild-type dnaB gene cloned into pET-11c (14). Induction of the cells and purification of DnaB helicase were performed as previously described (14).
pRNA synthesis on an SSB/R199G4oric template.
pRNA synthesis on an SSB/R199G4oric template was done as previously described (5). The reaction mixture (25 μl) contained 10 pmol of primase or P47, 4 pmol of ssDNA-binding protein (SSB) (purchased from U.S. Biochemicals), 0.12 pmol of R199/G4oric ssDNA, 100 μM ATP, 20 μM GTP, 20 μM CTP, 20 μM UTP, and 10 μCi of [α-32P]GTP (3,000 Ci/mM; Du Pont-New England Nuclear) in a buffer containing 20 mM Tris-HCl (pH 7.5), 8 mM dithiothreitol, 8 mM MgCl2, and 4% sucrose. The reaction mixture was incubated for 20 min at 30°C, and synthesis was stopped by adding 1/10 volume of both 0.5 M EDTA and 3 M sodium acetate, and the pRNA was precipitated with 3 volumes of 95% ethanol overnight at −20°C. The RNA was recovered by centrifugation and resuspended in 25 μl of denaturing sample buffer (formamide containing 0.05% bromophenol blue). Aliquots were analyzed either on a 23% polyacrylamide–7 M urea gel (acrylamide to bisacrylamide ratio of 8:1), using a Hoeffer minigel (8 cm by 9 cm by 0.75 mm) run at 300 V at 50°C, or on an 20% polyacrylamide–7 M urea gel (acrylamide to bisacrylamide ratio of 19:1), using a standard DNA sequencing gel (20 by 40 cm) run at 30 W. The gels were frozen and autoradiographed. To quantitate the amounts of different pRNA species synthesized by wild-type and mutant primase, autoradiographs of the analytical polyacrylamide gels were scanned with a PhosphorImager. The results obtained were therefore relative to each other and not absolute values.
pRNA synthesis in the general priming system.
The reaction conditions were the same as described for pRNA synthesis in the SSB/R199G4oric system, except the R199/G4 ssDNA template was replaced with an equimolar amount of R199 ssDNA (i.e., no G4oric) and SSB was replaced with 40 pmol of DnaB helicase hexamer (primase-to-DnaB helicase ratio of 1:4).
Gel retardation.
Gel retardation was done as previously described (20). The interactants used in binding reaction mixtures (12.5 μl) were ∼10 fmol of 5′-end 32P-labeled 278-nt G4oric fragment, 10 pmol of SSB (tetramer), 15 pmol of primase or HT-primase and 36 pmol of HT-primase-K241R. MgCl2 (8 mM) was added to the reaction mixtures, polyacrylamide gel, and the electrophoresis buffer (20). Electrophoresis was performed at 4°C.
In vitro coupled RNA and DNA syntheses.
To extend pRNA chains with DNA polymerase III (Pol III) holoenzyme, a two-step reaction was used. First, pRNA was synthesized under the normal conditions, using the 278-nt G4oric fragment as template DNA (21), then deoxynucleotide triphosphate (dNTP) and Pol III holoenzyme was added, and the incubation continued. For pRNA synthesis, the reaction mixture (20 μl) contained 10 pmol of primase, 0.2 pmol of G4oric-278 ssDNA, 4.2 pmol of SSB, 500 μM ATP, 100 μM of CTP, 100 μM UTP, 30 μM GTP, 30 μCi of [α-32P]GTP (3,000 Ci/mmol), and 1.5 μg of bovine serum albumin in buffer containing 20 mM Tris-HCl (pH 7.5), 8 mM dithiothreitol, 8 mM MgCl2, and 4% sucrose. After incubation at 30°C for 10 min, 70 ng of Pol III*, 15 ng of Pol III β-subunit, 50 μM dCTP, 50 μM dGTP, 50 μM dTTP, 18 pmol of dATP, and 10 μCi of 35S-labeled dATP were added for DNA synthesis and the mixture (25 μl) was incubated at 30°C for a further 10 min. The primed extension products were precipitated with 75% ethanol overnight at −20°C, and after recovery, the products were analyzed on an 8% polyacrylamide–7 M urea gel as described above. The gel was dried before autoradiography.
RESULTS
Cloning the N-terminal domain of primase (P47) for mutant studies.
We had reported earlier that the 47-kDa N-terminal fragment of primase isolated from an AspN digestion had almost normal catalytic activity on the SSB-coated R199G4oric ssDNA template, failing only in synthesizing the largest pRNA products (22). As cloned copies of the 47-kDa fragment (i.e., the P47 N-terminal domain [Fig. 1A]) are more stable in cells than intact dnaG gene (data not shown), we have found it convenient to screen primase mutations in P47 and then transfer mutations of interest into intact primase for further study. P47 was expressed as a GST fusion protein so that the mutant proteins could be affinity purified away from wild-type chromosomal primase that would distort activity assays. For similar reasons, the active mutations were transferred to primase with a C-terminal six-His tag.
Effect of changing Lys211, Lys229, and Lys241 to Arg residues on pRNA synthesis of P47.
Lys211, Lys229, and Lys241 were changed to Arg (R) residues in wild-type P47 domain of primase (Fig. 1B), and pRNA synthesis activity of the mutant proteins was tested on the R199/G4oric template. The P47-K211R and P47-K229R mutant proteins were able to synthesize pRNA of the same size as the wild-type P47 protein, although the efficiency decreased to 56 and 81% of that synthesized by P47 (Fig. 2A). P47-K241R mutant protein, however, appeared to be unable to synthesize normal pRNA, except for a single radioactive band migrating just above the free [α-32P]GTP (Fig. 2A). This band was more easily observed in Fig. 2B (the quantities of proteins used were not equal). We deduced this band to be a pRNA dimer (i.e., pppApG) from its migration relative to free [α-32P]GTP. P47, P47-K211R, and P47-K229R proteins also synthesized the pppApG dimer, but unlike P47-K241R, they synthesized significant amounts of trimer (pppApGpU), tetramer (pppApGpUpA), and larger pRNA as well. The amount of dimer synthesized by P47-K241R was approximately 70 to 80% of the amount of dimer synthesized by wild-type P47 when quantitation was done carefully (e.g., Fig. 2A).
FIG. 2.

Synthesis of pRNA by P47-K211R, P47-K229R, and P47-K241R mutant proteins. pRNA was synthesized on an SSB-coated R199/G4oric DNA template, as described in Materials and Methods. (A) pRNA synthesized by P47 and the mutant P47 proteins. Care was taken to use exactly the same amount of protein in each reaction mixture and to load an exact aliquot onto the gel (20% polyacrylamide–7 M urea gel with an acrylamide-to-bisacrylamide ratio of 19:1). The leftmost gel is a film exposed overnight. (B) In a similar experiment, the pRNA products were separated on a 23% polyacrylamide gel (acrylamide-to-bisacrylamide ratio of 8:1) containing 7 M urea. The gels were autoradiographed frozen. The size markers were [γ-32P]ATP-labeled 12- and 18-nt oligonucleotides (oligo). The longer species of pRNA were 11 to 22 nt long, and the smaller species were deduced from their migration relative to that of free [α-32P]GTP (not shown) to be the dimer(pppApG), trimer (pppApGpU), and tetramer (pppApGpUpA) pRNA.
To unequivocally identify the size of the small pRNA band synthesized by P47-K241R, we used dNTPs to block chain extension at defined positions on the template DNA (Fig. 3A). Primase is able to use dNTPs for chain extension on DNA in all positions except the first two, which must be ribodeoxyribonucleotides (rNTPs) (13), and pRNA synthesis of primase can be selectively blocked by incorporating dideoxy-NTP derivatives (23). As can be seen in Fig. 3B, adding ddT to the pRNA synthesis reaction mixture instead of UTP arrested the pRNA chain synthesized by P47 at 3 nt (lane 3) and adding ddC instead of CTP arrested the chain at 9 nt (lane 4). Small pRNAs with a terminal dideoxyribonucleotide migrate slightly faster that their counterparts with normal terminal ribonucleotides; e.g., the pppApGpddT trimer synthesized in the presence of ddT (lane 3) runs faster in the polyacrylamide gel than the pppApGpU trimer synthesized in the normal reaction mixture (lane 2). By counting the radioactive bands back from the chain-terminated pRNA bands to the free [α-32P]GTP band, it was determined that the size of the first pRNA band was a dimer. P47-K241R mutant protein synthesized only the dimer under all conditions (lanes 5 to 7) and even after a long exposure of the polyacrylamide gel to film, no trimer, tetramer, or larger pRNA species were observed. P47-K241R protein, therefore, was able to initiate primer RNA synthesis and form the first phosphodiester bond but was unable to extend the initiating dimer further.
FIG. 3.

Synthesis of pRNA by the mutant P47 proteins with dideoxynucleotides to arrest chain extension. P47 and P47-K241R proteins were incubated under the normal pRNA synthesis conditions, except that dideoxynucleotides replaced specific rNTPs in order to terminate chain extension at different locations on the template. (A) Template sequence showing where the pRNA chain synthesized on G4oric would be terminated by ddTTP and ddCTP. (B) pRNA synthesized by P47 and P47-K241R in the presence of all four NTPs (lanes 2 and 5), and when ddTTP (ddT) (lanes 3 and 6) and ddCTP (ddC) (lanes 4 and 7) were added to the incubation mixture. pRNA products were separated on a 23% polyacrylamide gel (acrylamide-to-bisacrylamide ratio of 8:1) containing 7 M urea. The gel was run so that the free [α-32P]GTP remained in the gel. Two autoradiographic exposures (15 s and 30 min, respectively) were necessary to visualize both free [α-32P]GTP and the dimer or trimer pRNA.
Activity of primase containing the Lys241-to-Arg mutation.
To confirm the results obtained with P47, we made the same Lys241-to-Arg mutation in wild-type primase and tested its effect on pRNA synthesis. The mutation was made in HT-primase so that we could use affinity chromatography to obtain mutant primase that was free of endogenous chromosomal wild-type primase. First, we compared the activity of the HT-primase with primase without the His tag to make sure that the C-terminal His tail and method of affinity purification did not affect the pRNA synthesis activity of primase. Figure 4A shows that the synthesis activity of the HT-primase was the same as that of primase without a His tag. Then, we examined the synthesis activity of the mutant. K241R mutant primase synthesized pRNA in the same pattern when NTP concentrations were increased 10-fold (see Materials and Methods). As can be seen in Fig. 4B, the HT-primase-K241R was able to synthesize the pppApG dimer on the R199/G4oric template; however, traces of longer pRNA chains (8 to 11 nt) could be seen after much longer exposure. In this respect, the primase K241R mutant differed slightly from the Lys241 mutant in the P47 domain. Quantitation showed that HT-primase-K241R synthesized more pppApG dimer than the wild-type HT-primase (approximately two- to threefold). Thus, although mutation of Lys241 in primase does not affect initiation and synthesis of the first phosphodiester bond, it caused serious impairment on further pRNA chain extension.
FIG. 4.

pRNA synthesis by primase-K241R. A six-His tag was fused to the N terminus of primase by using the vector pET-21d, and HT-primase was prepared by affinity purification on Ni resin. In the same vector, Lys241 was changed to Arg (HT-primase-K241R) by site-directed mutagenesis. (A) Comparison of the activity of HT-primase with two independent preparations of wild-type primase that does not contain a His tag. In this experiment, [α-32P]UTP was used to label the pRNA products. Care was taken to use identical amounts of primase in each reaction mixture and to load identical aliquots of the reaction mixture on the polyacrylamide gel. (B) pRNA synthesis by HT-primase and HT-primase-K241R was assayed under the standard conditions with [α-32P]GTP (see Materials and Methods). In both experiments, the synthesis products were separated on a 20% polyacrylamide–7 M urea gel (20 by 40 cm). To visualize the dimer pRNA in panel B, most of the [α-32P]GTP was run out of the leftmost gel and the bottom region of the frozen gel was autoradiographed for a shorter time, as shown in the rightmost gel.
Activity of K241R-primase in the general priming system.
In the presence of DnaB helicase, primase can synthesize pRNA on ssDNA in the absence of SSB (general priming system [1]). In this in vitro system, pRNA chains are initiated at many sites on the template and are extended into very long species ranging in size from 10 to 60 nt. To test the effect of the Lys241-to-Arg mutation on pRNA synthesis in the general priming system, we used HT-primase (P47 cannot synthesize pRNA in this system because the C terminus of primase is required for interaction with the helicase [25]). As can be seen in Fig. 5, with the HT-primase-K241R, synthesis of the 10- to 60-nt pRNA was almost completely blocked. The K241R mutant primase synthesized less dimer than in the SSB/G4oric system and in addition synthesized a small amount of trimer and 11-nt pRNA. In the presence of DnaB helicase, therefore, primase with a Lys241-to-Arg mutation still behaved as an elongation mutant.
FIG. 5.
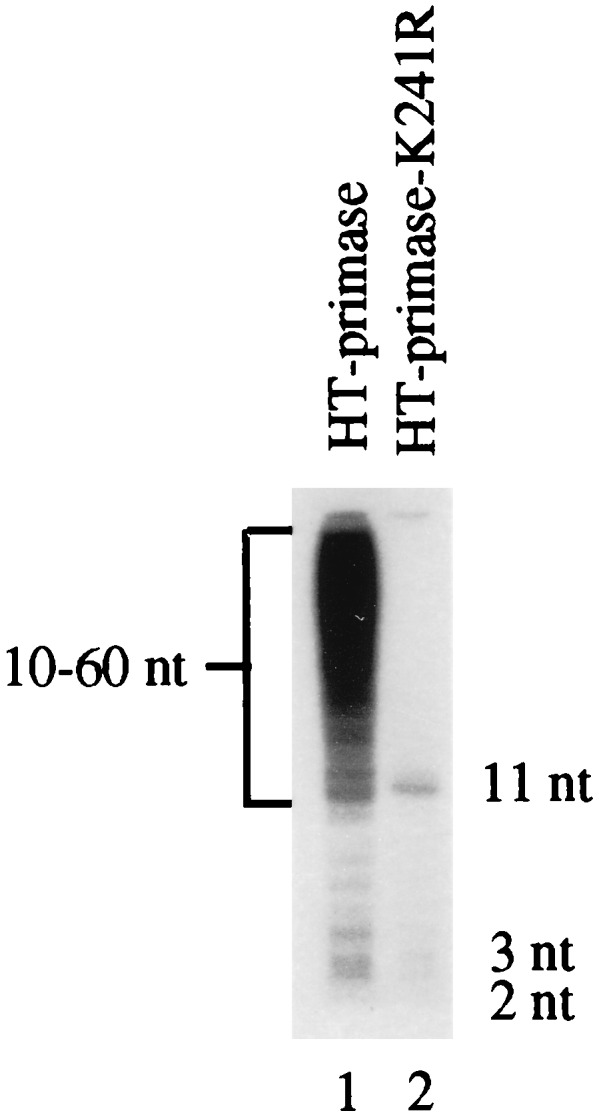
pRNA synthesis by HT-primase-K241R in the general priming system. The pRNA synthesis of HT-primase and mutant HT-primase-K241R was assayed on R199 ssDNA in the presence of DnaB helicase. The products were separated on an small 18% polyacrylamide gel containing 7 M urea.
DNA binding by the K241R mutant in gel shift.
The deficiency of the K241R mutant primase in pRNA elongation could be caused by impairment of its ability to bind a DNA template. To test this possibility, we used gel retardation, as we had earlier shown that wild-type primase can induce a gel shift of SSB-coated G4oric ssDNA and this shift was associated with primase pRNA synthesis activity (19, 20). A radioactively 32P-labeled, 278-nt G4oric ssDNA fragment was used as a DNA probe. In control reactions, wild-type primase and HT-primase bound to the saturated SSB-G4oric binding complex to produce a further gel shift (Fig. 6, lanes 3 and 4), showing that the HT-primase behaved normally in this reaction. The HT-primase-K241R mutant was also capable of binding to the SSB-G4oric complex to induce the same shifted band (lane 5), although not all of the SSB-G4oric complexes were shifted. This partial shift might be caused by a low concentration of the K241R protein preparation used in the experiment. We concluded that the mutant primase was capable of binding template DNA.
FIG. 6.
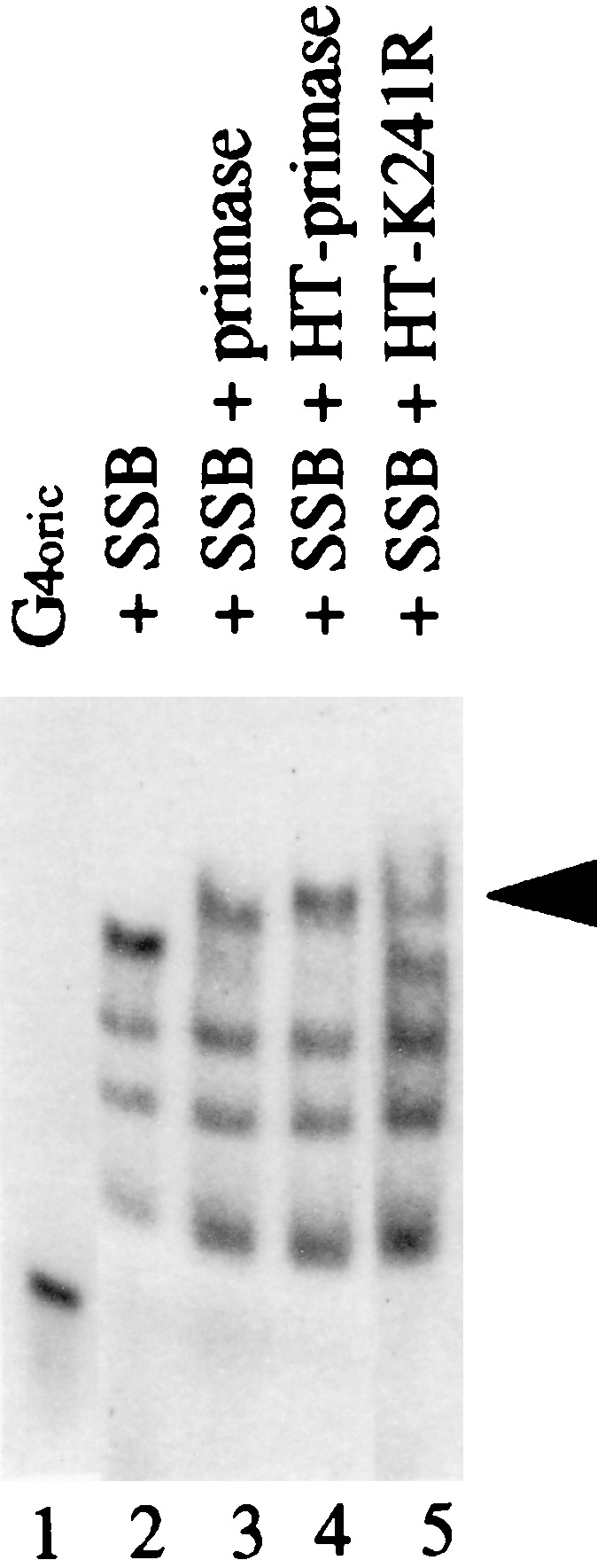
Gel retardation assay of G4oric DNA binding by HT-primase-K241R. The wild-type or mutant primase was incubated with 32P-labeled G4oric 278-nt ssDNA fragment and SSB, and binding complexes were then analyzed on a natural 4% polyacrylamide gel as described in Materials and Methods. The gel was dried before autoradiography. Lane 1, G4oric ssDNA alone; lane 2, DNA plus SSB; lane 3, DNA, SSB, and primase (no His tag); lane 4, DNA, SSB, and HT-primase; lane 5, DNA, SSB, and HT-primase-K241R. The shifted band induced by primase binding is indicated with an arrowhead.
Priming DNA synthesis by the K241R mutant in vivo and in vitro.
It would be of interest to know whether the elongation mutant primase could support an initiation for DNA synthesis. We explored this question first by using a genetic complementation experiment that we developed to study the activity of mutant primases (3). We had observed earlier that in the absence of IPTG induction, the pET vector-based plasmid pGNG1 (pET-3d containing the wild-type dnaG gene) could genetically complement E. coli cells containing the temperature-sensitive dnaG chromosomal mutation and enable them to grow at nonpermissive temperatures (42°C). Evidently, a small amount of primase pRNA synthesized from a cryptic plasmid promoter was sufficient to provide enough active primase to sustain normal cell growth under the nonpermissive conditions. To test the activity of K241R-primase, E. coli KY1378 containing the temperature-sensitive allele dnaG2903 was transformed with the vector pET-21d plasmid (control with no insert), pGNG7 (pET-21d containing wild-type primase), and pGNG7-K241R (pET-21d containing mutant primase). Colonies were selected at 30°C on LB plates containing 100 μg of ampicillin per ml. Single colonies were purified and grown in liquid medium at 30°C. Comparable amounts of the different transformed cells were then spread on LB plates, and the plates were incubated at 30°C (permissive temperature) or 42°C (nonpermissive temperature). As can be seen in Fig. 7, cells containing no plasmid failed to grow at both 30°C (i.e., no antibiotic resistance) and 42°C (i.e., temperature sensitive). Adding the control vector plasmid pET-21d to these cells allowed them to grow at 30°C but not at 42°C. Cells transformed with plasmid coding for the wild-type primase grew at both temperatures, as did cells containing the K241R mutant primase. The mutant primase was able to complement the deficient dnaG2903 gene at 42°C with an efficiency similar to that of the plasmid coding for wild-type primase. This result suggested that the pRNA products synthesized by K241R mutant primase in vivo were sufficient to initiate and sustain chromosomal DNA replication.
FIG. 7.

In vivo complementation chromosomal temperature-sensitive dnaG by plasmid-expressed K241R mutant primase. E. coli KY1378 containing temperature-sensitive dnaG2903 gene was transformed with pGNG7 (wild-type primase), pGNG7-K241R (mutant primase), and the control pET-21d vector (no insert). The transformed cells were plated on LB plates containing 100 μg of ampicillin per ml and incubated at 30 or 42°C overnight.
We also tested the ability of the K241R mutant to prime DNA synthesis in vitro in the G4 priming system. In the coupled RNA and DNA synthesis reaction using the 278-nt G4oric ssDNA as a template DNA, Pol III holoenzyme could synthesize a 205-nt runoff DNA fragment from pRNA synthesized by wild-type primase and P47 (Fig. 8, lanes 2 and 4). The mutant HT-primase-K241R (lane 3) and P47-K241R (lane 5) also could synthesize full-length runoff DNA, although with much lower efficiency than that by the wild-type proteins. When the mutant or wild-type primase was omitted from the reaction mixture, Pol III could not synthesize DNA (lane 6), demonstrating that DNA synthesis by Pol III here was primase dependent. From these in vivo and in vitro results, therefore, it can be concluded that the K241R mutant primase, although deficient in pRNA chain elongation, can still prime synthesis of DNA strands.
FIG. 8.
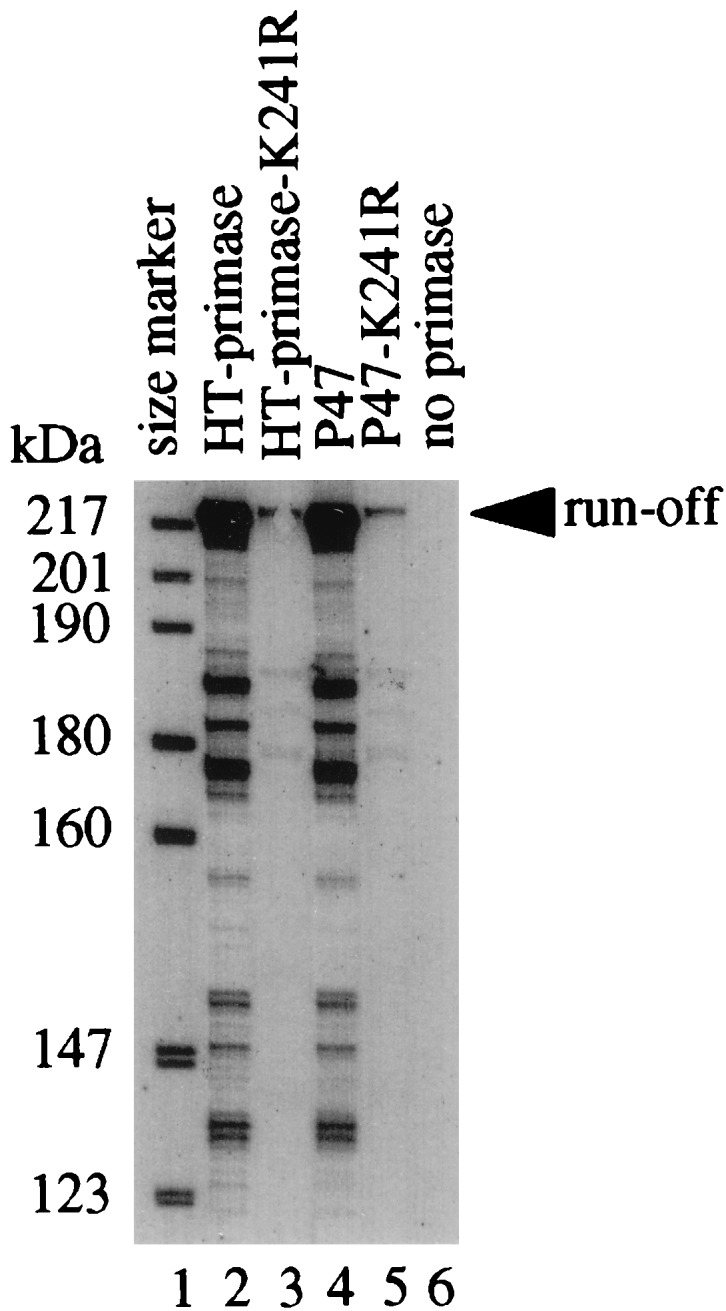
In vitro DNA synthesis by Pol III holoenzyme with pRNA synthesized by HT-primase-K241R on 278-nt G4oric template. The synthesis reaction was performed in two stages; first pRNA synthesis and then DNA synthesis. [α-32P]GTP was used to label the pRNA, and α-35S-dATP was used to label the DNA, as described in Materials and Methods. The synthesis products were analyzed on an 8% polyacrylamide–7 M urea gel, which was dried before autoradiography. Lane 1, size markers of 5′-end γ-32P-labeled fragments of pBR322 digested with MspI; lane 2, extension with wild-type HT-primase; lane 3, extension with mutant HT-primase-K241R; lane 4, extension with wild-type P47; lane 5, extension with mutant P47-K241R; lane 6, control of DNA synthesis by Pol III without adding primase in the first stage of the reaction.
DISCUSSION
Our earlier experiments with affinity labeling have mapped one or more of Lys211, Lys229, and Lys241 to be within 2 Å of the site of catalysis at the active center of primase (10). We have now shown that Lys-to-Arg mutations at positions 211 and 229 did not affect pRNA synthesis, but mutation of Lys241 to Arg did affect pRNA synthesis. Lys241 is, therefore, part of the active center of pRNA synthesis in primase. It is not, however, involved in the catalysis step of pRNA synthesis because the Arg substitution mutants of Lys241 can synthesize a template-directed pRNA pppApG dimer. Lys241 must be part of the mechanism of moving the catalytic center along the template to position it for synthesizing the next phosphodiester bond. The almost complete absence of trimer, tetramer, and larger pRNA species indicates that in the mutant primase and P47 movement relative to the template is almost completely inhibited after the dimer synthesis. The initiation process and synthesis of the first phosphodiester bond by the K241R mutant primase, however, are normal because it synthesizes approximately the same amount (or more) of dimer as the wild-type primase does.
The process of initiation of pRNA synthesis is clearly different from the extension step. Initiation involves primase binding to the template at a 5′ CTG 3′ sequence and forming a configuration that can bind the first substrate (ATP) and fix it in the correct position on the template to accept the second substrate (GTP) and catalyze the formation of a phosphodiester bond. This must be an inherently unstable reaction because the stability of binding depends upon the Km of binding of ATP and the Tm of binding of a dimer to template DNA. This is the de novo initiation step that DNA polymerases cannot perform. The extension process is dissimilar from the initiation process because the incoming NTPs are continuously added to a 3′-OH terminus of an RNA chain that is hydrogen bonded to the template rather than to single unstably bound first substrate. With increasing length of pRNA chain, the stability of the complex must increase as the Tm of binding of the product to the template DNA increases. This difference may be reflected in the absolute requirement of primase for rNTP as the first and second substrates, but an ability to accept dNTPs as well as rNTPs for primer extension (13, 28). The K241R mutation clearly separates these two steps of pRNA synthesis and defines Lys241 as a critical element of the active center. From affinity labeling studies using cross-linking arms of different lengths (10), we showed that Lys is only 2 Å from the site of catalysis and we can deduce from its function that it must be located at the 3′ terminus of the growing pRNA chain.
Similar chain extension mutants have been reported for E. coli RNA polymerase (11) and T7 RNA polymerase (4). Both, however, appear to be different from the primase mutant. Whereas K241R primase synthesized essentially on the 2-nt pRNA, the E. coli RNA polymerase mutant synthesized all of the abortive small RNA species (2, 3, 4, and 5 nt) in amounts identical to those by wild-type polymerase, and only chain extension to long runoff species was inhibited. The T7 RNA polymerase mutant was similar to the E. coli RNA polymerase mutant. Although the mutant T7 RNA polymerase protein bound to the promoter with normal efficiency, it aborted RNA synthesis at 5 nt. Experiments suggested that the T7 RNA polymerase mutant was deficient in binding of newly synthesized RNA transcript. RNA binding sites are believed to be part of the mechanism of processivity of RNA polymerases and required to keep the polymerase attached to the template (12, 27). As primases synthesize only short RNA chains under normal physiological conditions in vitro and in vivo (2, 29), they may not need an RNA binding site.
Although the K241R mutant primase is defective in the elongation of pRNA chains, it still can prime DNA synthesis. We have shown that in vivo, plasmid-encoded K241R primase was able to complement a genomic temperature-sensitive dnaG gene primase and initiate chromosomal DNA replication normally; in vitro, the K241R mutant primase could prime DNA synthesis for Pol III on a G4 DNA template but with quite low efficiency. Whether Pol III uses the dimer pRNA or the trace amount of 11-nt pRNA is not certain. However, if the dimer was used in vitro, the amount of runoff DNA synthesized by the mutant primase should be close to that of wild-type primase, which it is not. Also, to date, there is no report demonstrating that Pol III can synthesize DNA strand from a primer as small as 2 nt. The in vivo process of priming DNA replication by primase is more complicated, involving DnaB and other initiation proteins. The deficiency of the K241R mutant primase might be compensated in some way by association with other proteins at the replication fork. The mechanism of priming by the K241R mutant primase, therefore is not understood and will need further investigation.
ACKNOWLEDGMENTS
We thank Mike O’Donnell of the Rockefeller University for supplying us with Pol III holoenzyme and A. Arkady Mustaev of the New York Public Health Research Institute for helpful comments and discussion throughout this work.
This work was supported in part by NIH grant GM32898 to G.N.G.
REFERENCES
- 1.Arai K, Kornberg A. A general priming system employing only DnaB protein and primase for DNA replication. Proc Natl Acad Sci USA. 1979;76:4308–4312. doi: 10.1073/pnas.76.9.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouche J P, Rowen L, Kornberg A. The RNA primer synthesized by primase to initiate phage G4 DNA replication. J Biol Chem. 1978;253:765–769. [PubMed] [Google Scholar]
- 3.Godson G N. An over-expression plasmid for Escherichia coliprimase. Gene. 1991;100:59–64. doi: 10.1016/0378-1119(91)90350-k. [DOI] [PubMed] [Google Scholar]
- 4.He B, Rong M, Durbin R K, McAllister W T. A mutant T7 RNA polymerase that is defective in RNA binding and blocking in the early stages of transcription. J Mol Biol. 1997;265:275–288. doi: 10.1006/jmbi.1996.0741. [DOI] [PubMed] [Google Scholar]
- 5.Hiasa H, Sakai H, Tanaka K, Honda Y, Komano T, Godson G N. Mutational analysis of the primer RNA template region in the replication origin (oric) of bacteriophage G4: priming signal recognition by Escherichia coliprimase. Gene. 1989;84:9–16. doi: 10.1016/0378-1119(89)90133-9. [DOI] [PubMed] [Google Scholar]
- 6.Higuchi R. Recombinant PCR. In: Innis M A, Gelfand D H, Snisky J J, White T J, editors. PCR protocols. A guide to methods and applications. New York, N.Y: Academic Press; 1990. pp. 177–183. [Google Scholar]
- 7.Ilyana T V, Gorbalenya A E, Koonin E V. Organization and evolution of bacterial and bacteriophage primase-helicase systems. J Mol Evol. 1992;34:351–357. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 8.Kitani T, Yoda K, Ogawa Y, Okazaki T. Evidence that discontinuous DNA replication in Escherichia coliis primed by approximately 10 to 12 residues of RNA starting with a purine. J Mol Biol. 1985;184:45–52. doi: 10.1016/0022-2836(85)90042-7. [DOI] [PubMed] [Google Scholar]
- 9.Kornberg A, Baker J. DNA replication. 2nd ed. New York, N.Y: W. H. Freeman and Co.; 1992. [Google Scholar]
- 10.Mustaev A A, Godson G N. Studies of the functional topography of the catalytic center of Escherichia coliprimase. J Biol Chem. 1995;270:15711–15718. doi: 10.1074/jbc.270.26.15711. [DOI] [PubMed] [Google Scholar]
- 11.Mustaev A A, Kashlev M, Lee J, Polyakov A, Lebedev A, Zalenskaya K, Grachev M, Goldfarb A, Nikiforov V. Mapping of the priming substrate contacts in the active center of Escherichia coliRNA polymerase. J Biol Chem. 1991;266:23927–23931. [PubMed] [Google Scholar]
- 12.Nudler E, Avetissova E, Markovtsov V, Goldfarb A. Transcription processivity: protein-DNA interactions holding together the elongation complex. Science. 1996;273:211–217. doi: 10.1126/science.273.5272.211. [DOI] [PubMed] [Google Scholar]
- 13.Rowen L, Kornberg A. A ribo-deoxyribonucleotide primer synthesized by primase. J Biol Chem. 1978;253:770–774. [PubMed] [Google Scholar]
- 14.Saluja D, Godson G N. Biochemical characterization of Escherichia coli temperature-sensitive dnaB mutants dnaB8, dnaB252, dnaB70, dnaB43, and dnaB454. J Bacteriol. 1995;177:1104–1111. doi: 10.1128/jb.177.4.1104-1111.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 16.Sims J, Benz E W. Initiation of DNA replication by the Escherichia coli dnaG protein: evidence that tertiary structure is involved. Proc Natl Acad Sci USA. 1980;77:900–904. doi: 10.1073/pnas.77.2.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sims J, Capon D, Dressler D. DnaG (primase)-dependent origins of DNA replication: nucleotide sequences of the negative strand initiation sites of bacteriophages St-1, φK, and alpha 3. J Biol Chem. 1979;254:12615–12628. [PubMed] [Google Scholar]
- 18.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 19.Sun W, Godson G N. Binding and phasing of Escherichia coli single-stranded DNA-binding protein by the secondary structure of phage G4 origin of complementary DNA strand synthesis (G4oric) J Biol Chem. 1993;268:8026–8039. [PubMed] [Google Scholar]
- 20.Sun W, Godson G N. Interaction of Escherichia coli primase with a phage G4oric-E. coliSSB complex. J Bacteriol. 1996;178:6701–6705. doi: 10.1128/jb.178.23.6701-6705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun W, Godson G N. Structure of the Escherichia coli primase/single-strand DNA-binding protein/phage G4oriccomplex required for primer RNA synthesis. J Mol Biol. 1998;276:689–703. doi: 10.1006/jmbi.1997.1471. [DOI] [PubMed] [Google Scholar]
- 22.Sun W, Tormo J, Steitz T A, Godson G N. Domains of Escherichia coliprimase: functional activity of a 47-kDa N-terminal proteolytic fragment. Proc Natl Acad Sci USA. 1994;91:11462–11466. doi: 10.1073/pnas.91.24.11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swart J R, Griep M A. Primase from Escherichia coliprimes single-stranded templates in the absence of single-stranded DNA-binding protein or other auxiliary proteins. Template sequence requirements based on the bacteriophage G4 complementary strand origin and Okazaki fragment initiation sites. J Biol Chem. 1993;268:12970–12976. [PubMed] [Google Scholar]
- 24.Szafranski P, Smith C L, Cantor C R. Cloning and analysis of the dnaG gene encoding Pseudomonas putida DNA primase. Biochem Biophy Acta. 1997;1352:243–248. doi: 10.1016/s0167-4781(97)00059-6. [DOI] [PubMed] [Google Scholar]
- 25.Tougu K, Peng H, Marians K J. Identification of a domain of Escherichia coli primase required for functional interaction with the DnaBhelicase at the replication fork. J Biol Chem. 1994;269:4675–4682. [PubMed] [Google Scholar]
- 26.Verlasalovic J, Lupski J R. The Haemophilus influenzae dnaGsequence and conserved bacterial primase motifs. Gene. 1993;136:281–286. doi: 10.1016/0378-1119(93)90480-q. [DOI] [PubMed] [Google Scholar]
- 27.von Hippel P H. An integrated model of the transcription complex in elongation, termination, and editing. Science. 1998;281:660–665. doi: 10.1126/science.281.5377.660. [DOI] [PubMed] [Google Scholar]
- 28.Wickner S. DNA or RNA priming of bacteriophage G4 DNA synthesis by Escherichia coliDnaG protein. Proc Natl Acad Sci USA. 1977;74:2815–2819. doi: 10.1073/pnas.74.7.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoda K, Okazaki T. Specificity of recognition sequence for Escherichia coliprimase. Mol Gen Genet. 1991;227:1–8. doi: 10.1007/BF00260698. [DOI] [PubMed] [Google Scholar]