

Summary
Despite the recognized importance of antitumor immunity, our understanding of brain tumor immunity is poor. Orthotopic injection models have been widely used for immunological analyses. However, these models have limitations in analysis of antitumor immunity because the approach involves drilling skulls and injecting tumor cells, which can induce adverse effects. We describe a protocol for the induction of spontaneous brain tumor model, isolation of single cells from brain tumor microenvironment, and analysis of the immune responses using scRNA-seq and flow cytometry.
For complete details on the use and execution of this protocol, please refer to Park et al. (2021).
Subject areas: Cell isolation, Single cell, Flow cytometry/Mass cytometry, Cancer, Sequencing, RNAseq, Immunology, Model organisms, Molecular biology
Graphical abstract
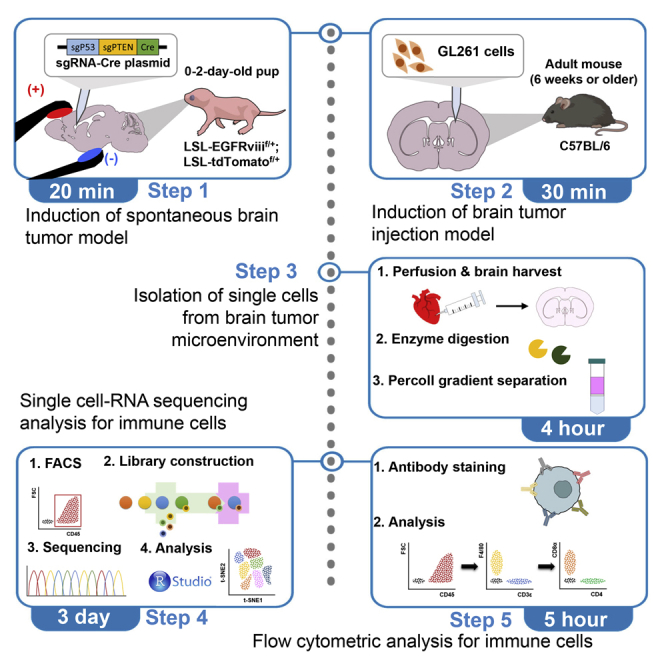
Highlights
-
•
Protocols for spontaneous brain tumor induction in mouse
-
•
Analysis of antitumor immune responses in spontaneous and injection models
-
•
Spontaneous and injection tumor models were compared in antitumor immunity
-
•
Immunological analysis of the tumor microenvironment using scRNA-seq and flow cytometry
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Despite the recognized importance of antitumor immunity, our understanding of brain tumor immunity is poor. Orthotopic injection models have been widely used for immunological analyses. However, these models have limitations in analysis of antitumor immunity because the approach involves drilling skulls and injecting tumor cells, which can induce adverse effects. We describe a protocol for the induction of spontaneous brain tumor model, isolation of single cells from brain tumor microenvironment, and analysis of the immune responses using scRNA-seq and flow cytometry.
Before you begin
The following protocol details two murine brain tumor models, a spontaneous model with EGFR, Trp53 and Pten mutations induced by the CRISPR-Cas9 system and an intracranial injection model of the orthotopic brain tumor cell line GL261 (Lee et al., 2018; Park et al., 2021). There are multiple methods capable of inducing glioblastoma multiforme in mice, including the patient-derived xenograft, orthotopic syngeneic model, and the gene-edited spontaneous model (Fomchenko and Holland, 2006). Because patient-derived tumors must be implanted into immunodeficient mice, this method is unsuitable for immunologic analysis. Thus, the orthotopic syngeneic model is more widely used. Though the orthotopic injection model is convenient, tissue damage induced by the syringe during injection is inevitable. The spontaneous model, however, has more similarities to the characteristics of tumors from glioblastoma patients than the injection model and also avoids the adverse effects of injection.
For the injection model, we used the GL261 cell line, a murine brain tumor cell line that is frequently used as a model for glioblastoma. GL261 cells possess mutations in the Kras and Trp53 genes (Szatmari et al., 2006). These cells were injected into the right hemisphere of the mouse brain for tumor development. A stereotaxic instrument was used to inject tumor cells into a certain position and control a syringe infusion pump to prevent cells from refluxing backwards.
For the spontaneous brain tumor model, we used the progeny of loxP-stop-loxP (LSL)-EGFRviii mice crossed with LSL-tdTomato mice. Tumors were induced in the brain by transfecting vector containing Cas9 and Cre recombinase with single-guide RNAs (sgRNAs) targeting Trp53 and Pten using in vivo electroporation.
Animal experiments were performed in accordance with guidelines and protocols (KA2017-41) approved by the Institutional Animal Care and Use Committee of KAIST.
Preparation of a micropipette
Timing: 10 min
Note: The micropipette produced in this procedure is utilized for injecting plasmid DNA into the brain of murine pups to induce spontaneous brain tumors.
Note: The procedure in Park, Kim et al. used a P-97 Flaming/Brown Micropipette Puller and Borosilicate glass with filament. The diameter of the filament used in this experiment was 1.50 mm on the outside and 0.86 mm on the inside, which is a relatively thick-walled glass filament. There are several glass filaments of different thicknesses that users can choose for their experiment. Furthermore, users can adjust the parameter settings to their preference. More detailed information about the parameters are provided on the manufacturer’s website (https://sutter.com/MICROPIPETTE/p-97.html).
-
1.Open the cover of the machine and load a sample glass into puller bar (Figure 1A).
-
a.Loosen the clamping knobs.
-
b.Place the glass into the V-groove of one puller bar and slide it beyond the clamp.
-
c.Tighten the clamping knob.
-
d.Pull both puller bars toward the clamp, and place the opposite side of the glass into another V-groove of the puller bar.
-
e.Tighten the clamping knob to fix the glass with both puller bars.
-
a.
-
2.
Run the RAMP TEST function to determine the adequate HEAT value.
-
3.Run a program with adjusted parameter values (Figure 1B).
-
a.HEAT: Ramp value + 5.
-
b.For thick-walled glass, P (air pressure): 500.
-
c.For injection needles, PULL: 50–75 (in this experiment: 50).
-
d.For microinjection, VELOCITY: 50–80 (in this experiment: 80).
-
e.For thick-walled glass, use the DELAY mode. DELAY: 40–200 (in this experiment: 90).
-
a.
-
4.
Loosen the clamping knobs and remove the pipettes from the puller bars.
Figure 1.

Micropipette-producing machine for plasmid DNA injection
(A) P-97 Flaming/Brown Micropipette Puller opening the cover (left). Right image shows a magnified view of the micropipette on the pullers and clamp (red rectangular part of the left image).
(B) The image for running a program to heat the glass.
Preparation of murine brain tumor cells (GL261)
Note: GL261 cells are commonly used at 2–3 passages after thawing for intracranial injection.
-
5.
Maintain GL261 cells in DMEM CM at 37°C in an atmosphere of 95% air and 5% CO2.
-
6.Harvest the cells when they reach 80%–90% confluence.
-
a.Discard the cell culture medium from the flask.
-
b.Wash cells with PBS.
-
c.Add trypsin-EDTA solution to the flask.
-
d.Incubate for 5 min at 37°C in an atmosphere of 95% air and 5% CO2.
-
e.Neutralize the trypsin-EDTA solution by adding FBS-containing media.
-
f.Transfer the cell suspension and centrifuge at 4°C and 440 rcf for 5 min.
-
g.Discard the supernatant and add DMEM CM.
-
a.
-
7.
Count the number of live cells by using trypan blue and a cell counter.
-
8.
Centrifuge at 4°C and 440 rcf for 5 min, then remove the supernatant.
-
9.
Resuspend the cell pellet with cold PBS to the optimal concentration.
Note: In this experiment, we used a concentration of 5 × 104–1 × 105 cells / 2 μL.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45.1-AF700 (clone:A20) | BioLegend | 110723 |
| CD3ε-PECy7 (clone:145-2C11) | BD Biosciences | 552774 |
| CD11b-BV510 (clone: M1/70) | BioLegend | 101263 |
| CD4-BV421 (clone: GK1.5) | BD Biosciences | 562891 |
| CD62L-FITC (clone: MEL-14) | BioLegend | 104406 |
| CD44-PerCP-Cy5.5 (clone: IM7) | BioLegend | 103032 |
| CD8α-APCCy7 (clone: 53-6.7) | BioLegend | 100714 |
| gdTCR-APC (clone: GL3) | BioLegend | 118116 |
| Anti-CD16/32 (clone: 2.4G2; Fc blocker) | Lab generated | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Propidium Iodide solution | BioLegend | 421302 |
| 7-AAD | BioLegend | 420404 |
| DMEM | Corning | 10-013-CVRC |
| RPMI1640 | Corning | 10-040-CVRC |
| Fetal Bovine Serum (FBS) | Corning | 35-015-CV |
| Bovine Serum | Gibco | 16140-078 |
| DPBS (without calcium and magnesium) | Corning | 21-031-CVC |
| Percoll | GE Healthcare | 17-0891-01 |
| Penicillin-streptomycin (100×) | GenDEPOT | CA-005-010 |
| HBSS (with phenol red and sodium bicarbonate, without calcium chloride and magnesium sulfate) | Welgene | LB003-03 |
| Trypsin-EDTA solution (0.5 M) | Welgene | ML005-01 |
| Fast Green FCF | Sigma-Aldrich | F7252 |
| Collagenase IV | Worthington | LS004189 |
| DNase I | Roche | 10104159001 |
| 10% Tween-20 | Bio-Rad | 1662404 |
| Qiagen Buffer EB | QIAGEN | 19086 |
| NH4Cl (Ammonium Chloride) | Daejung | 1060-4105 |
| KHCO3 (Potassium Bicarbonate) | Daejung | 6580-4405 |
| UltraPureTM Glycerol | Invitrogen | 92008 |
| Ethanol | Sigma-Aldrich | 64-17-5 |
| Critical commercial assays | ||
| Chromium Single Cell 3′ reagent Kits v3 | 10× Genomics | |
| Agilent High Sensitivity DNA kit | Agilent | 5067-4626 |
| HiSeq X Ten Reagent Kit v2.5 | Illumina | FC-501-2501 |
| PhiX Control v3 | Illumina | FC-110-3001 |
| SPRIselect Reagent Kit | Beckman Coulter | B23318 |
| Deposited data | ||
| scRNA-seq raw data | (Park et al., 2021) (Kim et al., unpublished) |
GSE199459; GSE159542; GSE201557 |
| Experimental models: Cell lines | ||
| Mouse: GL261 | Laboratory of Injune Kim | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: LSL-tdTomato (C57BL6J, male) | Laboratory of Jeong Ho Lee | RRID:IMSR_JAX:007914; |
| Mouse: LSL-EGFRviii (FVB, male) | Laboratory of Jeong Ho Lee | NCI Mouse repository:01X68 |
| Mouse: C57BL/6J (wild type, male, 8 weeks-old) | DBL | RRID:IMSR_JAX:000664 |
| Recombinant DNA | ||
| Plasmid: sgRNA-p53/Pten-Cas9-Cre | Laboratory of Jeong Ho Lee | Backbone:pU6-(BbsI)_CBh-Cas9-T2A-BFP plasmid (Addgene plasmid 64323) |
| Software and algorithms | ||
| Cell Ranger 3.1.0 | 10× Genomics | |
| R studio | https://rstudio.com | |
| Seurat_4.1.0 | (Hao et al., 2021) | https://satijalab.org/seurat/ |
| FlowJo | BD Biosciences | https://www.flowjo.com |
| Other | ||
| P-97 Flaming/Brown Micropipette Puller | Sutter instrument | P-97 |
| Nepa21 electroporator | Nepa Gene | |
| CellDropTM Automated Cell Counter | DeNovix | CellDrop FL-UNLTD |
| Digital mouse stereotaxic frame | World Precision Instruments | 505314 |
| Animal Anesthesia Vaporizers type 1 | RWD | R580S |
| Syringe Pump | KD Scientific | LEGATO 130 |
| Hamilton Syringe | Hamilton | 803 |
| HiSeq X Ten | Illumina | |
| MiniAmp Thermal Cycler | Thermo Fisher Scientific | A37834 |
| Chromium Controller | 10× Genomics | |
| 2100 Bioanalyzer | Agilent | G2939BA |
| LSRFortessaTM X-20 Cell Analyzer | BD Biosciences | |
| FACSAria II | BD Biosciences | |
| Micro Drill | SAESHIN | |
| Reagent | Dilution ratio | Volume |
| CD45.1-AF700 (clone:A20) | 1:200 | |
| CD3ε-PECy7 (clone:145-2C11) | 1:200 | |
| CD11b-BV510 (clone: M1/70) | 1:200 | |
| CD4-BV421 (clone: GK1.5) | 1:200 | |
| CD62L-FITC (clone: MEL-14) | 1:200 | |
| CD44-PerCP-Cy5.5 (clone: IM7) | 1:200 | |
| CD8α-APCCy7 (clone: 53-6.7) | 1:200 | |
| gdTCR-APC (clone: GL3) | 1:200 | |
| FACS buffer | N/A | |
| Total | N/A | 100 μL/sample |
Materials and equipment
DMEM CM
| Reagent | Final concentration | Volume |
|---|---|---|
| Fetal Bovine serum | 10% | 50 mL |
| Penicillin-streptomycin (100×) | 1× | 5 mL |
| DMEM | N/A | 445 mL |
| Total | N/A | 500 mL |
Can be stored at 4°C for up to one month.
RPMI CM
| Reagent | Final concentration | Volume |
|---|---|---|
| Fetal Bovine serum | 10% | 50 mL |
| Penicillin-streptomycin (100×) | 1× | 5 mL |
| RPMI1640 | N/A | 445 mL |
| Total | N/A | 500 mL |
Can be stored at 4°C for up to one month.
Isolation buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| Bovine Serum | 1% | 5 mL |
| Penicillin-streptomycin (100×) | 1× | 5 mL |
| DMEM | N/A | 490 mL |
| Total | N/A | 500 mL |
Can be stored at 4°C for up to one month.
Collagenase IV stock
| Reagent | Final concentration | Amount |
|---|---|---|
| Collagenase IV | 20 mg/mL | 5 g |
| ddH2O | N/A | 250 mL |
| Total | N/A | 250 mL |
Can be stored at −20°C for up to one month.
DNase I stock
| Reagent | Final concentration | Amount |
|---|---|---|
| DNase I | 2 mg/mL | 100 mg |
| DPBS | N/A | 50 mL |
| Total | N/A | 50 mL |
Can be stored at −20°C for up to one month.
Enzyme mixture
| Reagent | Final concentration | Volume |
|---|---|---|
| Collagenase IV stock | 2 mg/mL | 0.5 mL |
| DNase I stock | 60 μg/mL | 0.15 mL |
| Isolation buffer | N/A | 4.35 mL |
| Total | N/A | 5 mL |
Prepare the day of experiment and keep at RT.
Dissociation buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| EDTA solution (0.5 M) | 50 mM | 5 mL |
| Bovine Serum | 1% | 5 mL |
| Penicillin-streptomycin (100×) | 1× | 5 mL |
| HBSS | N/A | 485 mL |
| Total | N/A | 500 mL |
Can be stored at 4°C for up to one month.
30% Percoll solution
| Reagent | Final concentration | Volume |
|---|---|---|
| Percoll | 30% | 1.5 mL |
| RPMI1640 | N/A | 3.5 mL |
| Total | N/A | 5 mL |
Prepare the day of experiment and keep at RT.
70% Percoll solution
| Reagent | Final concentration | Volume |
|---|---|---|
| Percoll | 70% | 2.1 mL |
| DPBS | N/A | 0.9 mL |
| Total | N/A | 3 mL |
Prepare the day of experiment and keep at RT.
FACS buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| Bovine Serum | 1% | 5 mL |
| Penicillin-streptomycin (100×) | 1× | 5 mL |
| DPBS | N/A | 490 mL |
| Total | N/A | 500 mL |
Can be stored at 4°C for up to one month.
ACK lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NH4Cl (Ammonium Chloride) | 150 mM | 8.024 g |
| KHCO3 (Potassium bicarbonate) | 10 mM | 1.001 g |
| EDTA solution (0.5 M) | 10 μM | 20 μL |
| ddH2O | N/A | Fill to 1 L |
| Total | N/A | 1 L |
Autoclave the buffer and store at RT for up to one month.
Elution Solution I
| Reagent | Final concentration | Amount |
|---|---|---|
| Buffer EB | N/A | 980 μL |
| 10% Tween-20 | 1% | 10 μL |
| Reducing Agent B (in kit) | 1% | 10 μL |
| Total | N/A | 1000 μL |
Prepare the day of experiment and keep at RT.
Step-by-step method details
Section 1. Induction of spontaneous brain tumor model.
Section 2. Induction of intracranial injection model.
Section 3. Isolation of single cells from brain tumor microenvironment.
Section 4. Single cell-RNA sequencing analysis for immune cells from the brain tumor microenvironment.
Section 5. Flow cytometric analysis for immune cells from the brain tumor microenvironment.
Section 1. Induction of the spontaneous brain tumor model
This section details how to induce the spontaneous brain tumor model.
-
1.
Anesthetize pups via hypothermia by placing them on ice for at least 5 min (Figure 2A).
-
2.Prepare sgRNA-Trp53/Pten-Cas9-Cre plasmid DNA inserted into the brain of 0–2-day-old pups.
-
a.Dilute plasmid DNA stock to 2 μg/μL in ddH2O.
-
b.Add Fast green (10% final volume) to plasmid DNA solution (Figure 2B).Note: Fast Green powder is used to visually confirm injection of DNA into the brain.
-
c.Prepare a piece of parafilm in order to place several droplets (1 μL per droplet) of plasmid DNA solution.
-
a.
-
3.Prepare the micropipette for injection.
-
a.Slightly cut off the tip of a micropipette using forceps before loading the droplet (Figure 2C).
-
b.Connect the other side of a micropipette to a silicon tube for blowing out the DNA (Figure 2D).
-
c.Bring the micropipette tip to the droplet and roll it until the droplet is completely loaded (Figure 2E).
-
a.
-
4.Inject the plasmid DNA into the lateral ventricle of the brain.
-
a.Make a virtual line between the right eye and lambda.
-
b.Make the insertion point at about 1/3 the length of this line from the lambda (Figure 2F).
-
c.Insert the micropipette tip into this position at a depth of 2 μm from the skull.
-
d.Extrude the DNA by blowing the other side of the silicon tube.
-
e.Confirm the injection by using Fast Green to show the shape of the lateral ventricle (Figure 2G).
-
a.
Note: Fast Green staining can be visualized better by illuminating the brain with a lamp.
-
5.Apply an electric pulse to the animal after successful injection of plasmid DNA.
-
a.Prepare an electroporator and a tweezer electrode (Figure 2H).
-
i.Set the condition of pulse: 100 V, 50 ms, separated by 950 ms intervals, 5 times.
-
i.
-
b.Soak the electrode tips with PBS to make electrical pulses that pass through the brain tissue.
-
c.Hold the brain with the wet electrode and apply the pulses (Figure 2I).
-
i.Positive electrode: ahead of the eye.
-
ii.Negative electrode: the opposite position on the ventricle side.
-
i.
-
a.
-
6.
Return the pups to the cage.
-
7.
Tumor formation can be confirmed by checking for tdTomato-expressing cells (Lee et al., 2018) (Figure 2J).
Figure 2.

Induction of spontaneous brain tumors via injection of plasmid DNA into the brain
(A) Hypothermal anesthesia setup for pups.
(B) Plasmid DNA solution mixed with Fast Green.
(C) The original (a) and the tip-removed (b) micropipettes.
(D) A micropipette connected with a silicon tube.
(E) A micropipette tip loaded with the plasmid DNA solution.
(F) Injection point at approximately 1/3 the length of the virtual line between the right eye and lambda.
(G) Confirmation of plasmid DNA injection into the lateral ventricle via lightening brain with lamp.
(H) A Tweezer electrode.
(I) Transfecting plasmid DNA into the brain via electroporation.
(J) Confocal microscopy (red: tdTomato+ tumors; DAPI: nuclei) of brain tumor tissue sections from the spontaneous brain tumor model in mice with a hunched posture (20 weeks). Zoomed image of the border of the tumor tissue is shown on the right. Scale bar = 1000 μm (left) and 100 μm (right).
Section 2. Induction of the brain tumor injection model
This section explains how to induce the orthotopic syngeneic murine brain tumor model by injecting GL261 cells.
Note: The injection site could be critical, so the mouse head should be fixed during the experiments.
Note: Drilling the skull and injecting cells can cause bleeding, which could affect the immune response and tumor formation. It is important to avoid blood vessels during the experiments. Even though the injection speed could vary depending on preferences, the injection should be performed as slow as possible to avoid tissue damage.
Note: Cell type and number can vary depending on preferences and purpose, and thus should be optimized by users. Please refer to Oh et al., 2014.
-
8.Prepare the mouse for stereotaxic surgery.
-
a.Anesthetize a mouse with isoflurane.
-
b.Turn on a heating pad.
-
c.Fix the mouse on a stereotaxic instrument which connects the anesthesia machine that administers isoflurane.
-
a.
-
9.Arrange the insertion position.
-
a.Sterilize the skin of the head.
-
b.Cut the skin along the midline of the head (∼1 cm) with a surgical scissor.
-
c.Peel off the mucus on the skull with a cotton swab.
-
d.Fix a sterile pen on the syringe holder and point to the bregma to mark locations.
-
e.Set the position of the bregma as zero for the x- and y-axes.
-
f.Move to 2.0 mm anterior and 2.0 mm right, and mark this location with the pen (Figure 3).
-
g.Make a hole in the insertion position with a micro drill.
-
a.
Note: Excessive drilling can cause tissue damage and bleeding, so be careful to only drill on the skull.
-
10.Inject GL261 cells at the insertion position.
-
a.Load a Hamilton syringe with prepared GL261 cells in a volume of 2 μL.
-
b.Move the syringe to the hole and touch the tip of needle to the tissue surface, setting this location as zero for the z-axis.
-
c.Move the syringe down to 3.5 mm inferior and wait for 1 min.
-
d.Move the syringe up to 0.5 mm superior (3.0 mm inferior from the surface).
-
e.Inject the GL261 cells at a rate of 0.4 μL/min for 5 min with the syringe infusion pump (Total injection volume: 2 μL per mouse).
-
a.
-
11.
Remove the syringe carefully and slowly.
-
12.
Fill the hole with bonding material mixed with beads.
-
13.
Tighten the skin around the wound with surgical suture thread.
-
14.
Place the mouse back into its cage.
Figure 3.

Schematic image of stereotaxic surgery for the injection brain tumor model
For the injection brain tumor model, a mouse is fixed on a stereotaxic instrument and the injection point is 2.0 mm anterior and 2.0 mm right from the bregma of the brain. Several blood vessels may be present around the injection point.
Section 3. Isolation of single cells from the brain tumor microenvironment
This section explains how to isolate single cells from tissues.
Note: Single cell isolation methods can differ. Here we used enzymatic tissue dissociation.
-
15.Cardiac perfusion.
-
a.Euthanize the mouse by CO2 inhalation.
-
b.Load 10 mL of PBS into a 10 mL-syringe and then attach a 24-gauge needle.
-
c.Place and fix the mouse onto the surgery area.
-
d.Incise the ventral side of the skin and snip the thoracic cavity.Note: Be careful to avoid scratching the heart, lung, or veins.
-
e.Pin both sides of the ribs to make room for the needle injection to the heart.
-
f.Place the needle of the syringe containing PBS onto the left ventricle and make a small hole on the right atrium through which blood flows.
-
g.Push the syringe plunger with moderate force.Note: Perfusion is important to harvest cells while excluding circulating cells. Confirm whether the redness of the liver has faded after the perfusion.
-
a.
-
16.Harvest the brain tissue.
-
a.Cut the skin of the head along the midline from the neck to the eyes.
-
b.Hold the skin open on both sides.
-
c.Cut the skull along the sagittal suture beginning from the brainstem.Note: Be careful not to damage the brain tissue.
-
d.Remove both halves of the skull from the head.
-
e.Pull the brain out and place it into cold DMEM.
-
a.
-
17.Digest the brain tissue with enzymes.
-
a.Prepare an enzyme mixture containing 2 mg/mL Collagenase IV and 30 μg/mL DNase I in 5 mL of isolation buffer.
-
b.Sterilize a razor with 100% ethanol.
-
c.Place the brain tissue on a 60-mm petri dish.
-
d.Chop it finely with the sterilized razor in the dish.Note: Keep samples moist with cold DPBS.
-
e.Transfer the tissue pieces into a new 15-mL tube with 5 mL of enzyme mixture.Note: Large tissues may block the pipette end. Instead, large tissues could be transferred by forceps.
-
f.Incubate at 37°C for 30 min in a shaking water bath.
-
g.After incubation, centrifuge the tube at 4°C, 440 rcf for 5 min.
-
h.Remove the supernatant and resuspend the pellet with 5 mL of the dissociation buffer.
-
i.Incubate at 37°C for 5 min in a shaking water bath.
-
a.
-
18.Dissociate cells from the tissue suspension.
-
a.Prepare a 60-mm petri dish, a syringe plunger, and 70-μm strainer.
-
b.Place the strainer on the petri dish.
-
c.Vortex the tissue suspension thoroughly and pour it into the strainer.
-
d.Grind it with the syringe plunger thoroughly.
-
e.Wash the strainer with 5 mL of the isolation buffer.
-
f.Transfer the homogenate from the strainer into a new 15-mL tube.
-
g.Repeat the wash and transfer the homogenate to a new tube.
-
h.Centrifuge the tube at 4°C, 440 rcf for 5 min.
-
a.
-
19.Separate cells from the myelin debris via Percoll density gradient centrifugation.
-
a.Prepare 30% and 70% Percoll solutions.
-
b.Remove the supernatant of the centrifugated homogenate, and resuspend the pellet with 5 mL of 30% Percoll solution.
-
c.Place a Pasteur pipette into the tube.
-
d.Add 3 mL of 70% Percoll solution below the 30% Percoll solution through the Pasteur pipette.
-
e.Centrifuge the tube at RT, 800 rcf for 20 min. Allow the centrifuge to come to a stop without applying the brakes.
-
f.Remove the myelin debris on the 70% Percoll layer.
-
g.Within the line between the 30% and 70% Percoll layers, take about 3 mL of media and transfer into a new 15-mL tube.
-
h.Add FACS buffer to a total volume of 15 mL and invert gently.
-
i.Centrifuge the tube at 4°C, 440 rcf for 5 min.
-
a.
-
20.Remove red blood cells by ACK lysis buffer.
-
a.Remove the supernatant.
-
b.Resuspend the cell pellet with 1 mL of ACK lysis buffer, and incubate at RT for 5 min.
-
c.Add 1 mL of FACS buffer, and centrifuge at 4°C, 440 rcf for 5 min.
-
d.Remove the supernatant and resuspend the cell pellet with 1 mL of RPMI CM.
-
a.
-
21.
Filter cells with a sterilized 70-μm mesh. Then count the number of cells using trypan blue and a cell counter and store at 4°C.
Section 4. Single-cell RNA sequencing (scRNAseq) analysis for immune cells from the brain tumor microenvironment
This section explains how to perform scRNAseq analysis.
Note: The platform used to perform scRNAseq analysis could vary depending on preferences, and details for analysis could be different depending on the platform. We followed the manufacturer’s protocol and there are more details for this procedure. The protocol is provided from the manufacturer’s website (https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/user-guide-chromium-single-cell-3-reagent-kits-user-guide-v3-chemistry).
-
22.Antibody staining for immune cell preparation.
-
a.Transfer an adequate amount of cells into a 5-mL round-bottom polystyrene tube.
-
b.Centrifuge the tube at 4°C, 440 rcf for 5 min.
-
c.Remove the supernatant and add 100 μL (per million cells) of Fc block diluted in FACS buffer to a 1:100 ratio.
-
d.Incubate on ice for 20 min in the dark.
-
e.Add 1 mL of FACS buffer to the sample and centrifuge at 4°C, 440 rcf for 5 min.
-
f.Remove the supernatant and add 100 μL (per million cells) of anti-CD45 antibody diluted in FACS buffer at a 1:200 ratio.
-
i.Cells from C57BL/6 mice are stained with anti-CD45.2 antibody.
-
ii.Cells from LSL-EGFRviii mice are stained with anti-CD45.1 antibody.
-
i.
-
g.Incubate the sample on ice for 30 min in the dark.
-
h.Add 1 mL of FACS buffer to the sample and centrifuge at 4°C, 440 rcf for 5 min. Repeat this step once.
-
i.Remove the supernatant and resuspend the cell pellet in 1–2 mL of FACS buffer.
-
j.To exclude dead cells, add 5 μL (per million cells) of 7-AAD (BioLegend) and incubate for 5–10 min in the dark prior to flow sorting.
-
a.
-
23.
Sort ∼1–2 × 105 7-AAD-CD45+ immune cells using the FACS Aria II (BD Biosciences) into a new 15-mL tube containing RPMI CM.
-
24.
Centrifuge at 4°C, 440 rcf for 5 min, and remove the supernatant being careful not to touch the cell pellet.
-
25.
Resuspend the cell pellet with cold PBS to a concentration of 700–1200 cells/μL.
Note: The range of optimal cell concentration is chosen based on the manufacturer’s protocol.
-
26.
Prepare a master mix for barcoding that contains RT Reagent, Template Switch Oligo, Reducing Agent B, and RT Enzyme C. Add the appropriate volume of nuclease-free water depending on the cell concentration.
-
27.
Install a Chromium Chip B in a 10× Chip Holder and add 50% glycerol solution into the unused chip wells (row #1: 75 μL, row #2: 40 μL, row #3: 280 μL).
-
28.
Prepare the Gel Bead strip by vortexing for 30 s in a 10× Vortex Adaptor.
-
29.
Mix the cell suspension with the corresponding volume of master mix for barcoding.
-
30.Load the corresponding sample onto the Chromium Chip without introducing bubbles.
-
a.row #1: 75 μL of mixture for cell suspension and master mix for barcoding.
-
b.row #2: 40 μL of Gel Beads.
-
c.row #3: 280 μL of Partitioning Oil.
-
a.
-
31.
Attach the 10× Gasket to the Chromium Chip and place in the tray of the Controller.
-
32.
Start the Chromium Single Cell B program.
-
33.
After the run finishes, transfer 100 μL of gel based-in emulsions (GEMs) in the Recovery wells in the top row into a tube strip on ice.
-
34.
Incubate in a thermal cycler with the following steps: 53°C for 45 min, 85°C for 5 min, and hold at 4°C.
Note: After this process, the sample can be stored at 4°C for up to 72 h or at −20°C for up to week.
-
35.Purify the first-strand cDNA from the reaction mixture.
-
a.Add 125 μL of Recovery Agent to each sample at RT without pipette mixing or vortexing, and then wait 60 s.Note: This biphasic mixture consists of Recovery Agent/Partitioning Oil (pink, lower layer) and aqueous phase (clear, upper layer).
-
b.Carefully remove 125 μL of Recovery Agent/Partitioning Oil from the bottom.
-
c.Prepare Dynabeads Cleanup Mix containing Cleanup Buffer, Dynabeads MyOne SILANE, and Reducing Agent B.Note: Dynabeads MyOne SILANE should be thoroughly mixed by vortexing before using.
-
d.Add 200 μL of Dynabeads Cleanup Mix to each sample and mix by pipetting.
-
e.Incubate for 10 min at RT and mix again at ∼5 min after the start of incubation by pipetting.
-
f.During the incubation, prepare Elution Solution I containing elution buffer, 10% Tween-20, and Reducing Agent B with vortexing and spin down.
-
g.After incubation, place the sample on the High position of a 10× Magnetic Separator until the solution clears.
-
h.Remove the supernatant and wash twice with 80% ethanol.
-
i.Place the sample on the Low position of the separator and remove the remaining ethanol by air drying for 1 min.
-
j.Remove the sample from the separator, add 35.5 μL of Elution Solution I, and incubate for 2 min at RT.
-
k.Place the sample on the Low position of the separator until the solution clears.
-
l.Transfer 35 μL of the sample into a new tube strip.
-
a.
-
36.
Amplify the cDNA with Amp Mix and cDNA primers using a thermal cycler programmed with the following protocol:
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 3 min | 1 |
| Denaturation | 98°C | 15 s | 11–13 cycles |
| Annealing | 63°C | 20 s | |
| Extension | 72°C | 1 min | |
| Final extension | 72°C | 1 min | 1 |
| Hold | 4°C | forever | |
Note: The number of cycles is dependent on the number of cells inputted.
Note: After this process, the sample can be stored at 4°C for up to 72 h.
-
37.Clean up cDNA using SPRIselect.
-
a.Add SPRIselect reagent (0.6×) to each sample and incubate for 5 min at RT.
-
b.Place on the High position of the separator until the solution clears.
-
c.Remove the supernatant and wash twice with 80% ethanol.
-
d.Place the sample on the Low position of the separator and remove the remaining ethanol by air drying for 2 min.
-
e.Remove the sample from the separator, and elute with elution buffer for 2 min at RT.
-
f.Place the sample on the High position of the separator until the solution clears.
-
g.Transfer 40 μL of sample into a new tube strip.
-
a.
Note: After this process, the sample can be stored at 4°C for up to 72 h or at −20°C for up to 4 weeks.
-
38.
To perform a quality check and quantify the cDNA, dilute 1 μL of sample at 1:10 ratio using DW and use the Agilent High Sensitivity DNA Kit with Agilent 2100 Bioanalyzer.
Note: This step is followed by the manufacturer’s protocol provided in the website (https://www.agilent.com/cs/library/usermanuals/public/HighSensitivity_DNA_KG.pdf.pdf).
Note: Agilent TapeStation High Sensitivity D5000 ScreenTape can also be utilized for this process.
-
39.
Proceed with cDNA fragmentation, end repair, and A-tailing by adding the Fragmentation Buffer and Fragmentation Enzyme and then incubating in a thermal cycler with the following protocol: hold at 4°C, 5 min at 32°C, 30 min at 65°C, and hold at 4°C.
-
40.Use the SPRIselect reagent to clean up the sample.
-
a.Add 30 μL of 0.6× SPRIselect reagent to each sample and incubate for 5 min at RT.
-
b.Place on the High position of the separator until the solution clears.
-
c.Transfer 75 μL of supernatant to a new tube strip.
-
d.Add 10 μL of 0.8× SPRIselect reagent to each sample and incubate for 5 min at RT.
-
e.Place on the High position of the separator until the solution clears.
-
f.Remove the supernatant and wash twice with 80% ethanol.
-
g.Remove from the separator and elute with elution buffer for 2 min at RT.
-
h.Place on the High position of the separator until the solution clears.
-
i.Transfer sample to a new tube strip.
-
a.
-
41.
For adaptor ligation, incubate each sample with the mix containing Ligation Buffer, DNA Ligase, and Adaptor Oligos in a thermal cycler with the following protocol: 15 min at 20°C and hold at 4°C.
-
42.
Repeat the clean-up process using 0.8× SPRIselect reagent and the separator.
-
43.
For sample indexing, incubate each sample with a PCR mix containing Amp Mix, SI Primer, and an individual Chromium i7 Sample Index on a thermal cycler with the following protocol:
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 45 s | 1 |
| Denaturation | 98°C | 20 s | 6–16 cycles |
| Annealing | 54°C | 30 s | |
| Extension | 72°C | 20 s | |
| Final extension | 72°C | 1 min | 1 |
| Hold | 4°C | forever | |
Note: The number of cycles is dependent on the amount of cDNA inputted.
-
44.
Repeat the clean-up process using 0.6× SPRIselect reagent and the separator.
-
45.
To assess the quality of library construction, dilute 1 μL of sample at a 1:10 ratio using DW and use the Agilent High Sensitivity DNA Kit with Agilent 2100 Bioanalyzer.
Note: Agilent TapeStation High Sensitivity D5000 ScreenTape can also be utilized for this process.
-
46.
To sequence the library, use the Illumina HiSeq X Ten sequencing system with the HiSeq X Ten Reagent Kit v2.5 and PhiX Control v3 with the recommended cycle conditions: Read 1: 28 cycles, i7 index: 8 cycles, i5 index: cycles, and Read 2: 91 cycles.
-
47.
A 10× Chromium Cell Ranger can be used according to the manufacturer’s instructions to generate three files for sequencing data: bacordes.tsv, features.tsv, and matrix.mtx.
-
48.Create Seurat objects and verify the quality of samples.
-
a.Read sequencing data using the Read10X function.
-
b.Create Seurat objects using the CreateSeuratObject function.
-
c.Check the level of nFeature_RNA, nCount_RNA, and mitochondrial gene expression.
-
d.Isolate quality-controlled objects using the Subset function.
-
e.Normalize data using the NormalizeData function.
-
f.Identify features of objects using the FindVariableFeatures function.
-
a.
-
49.Combine objects.
-
a.Find anchorsets using the FindIntegrationAnchors function.
-
b.Combine anchorsets using the IntegrateData function.
-
a.
-
50.Cluster and visualize the data.
-
a.Scale features in the dataset using the ScaleData function.
-
b.Set “integrated” as a default assay.
-
c.Reduce dataset dimension using the RunPCA function.
-
d.Reduce dataset using the RunUMAP function.
-
e.Construct a shared nearest neighbor graph from the dataset using the FindNeighbors function.
-
f.Identify clusters of cells by shared nearest neighbors using the FindClusters function.
-
g.Visualize clusters using the DimPlot function (Figure 4A).
-
a.
-
51.
Compare the distribution of clusters among different batches (Figures 4B and 4C).
-
52.
Determine the identity of clusters using cellular markers (Figure 4D).
-
53.
Compare the distribution of immune cells in the tumor tissues using a heat map (Figure 4E).
Figure 4.

scRNAseq analysis of spontaneous and injection brain tumor model
(A–E) EGFRvIII-LSL mice pups were injected with sgRNA-containing vectors. When mice displayed a hunched posture, tissues were obtained. We harvested tissues at 20 days after intracranial injection of GL261 cells into the C57BL/6J mice. Single cell suspensions were isolated and single cell-RNA sequencing (scRNAseq) was performed. (A) UMAP projection of whole samples. (B) UMAP projections split by batch. (C) UMAP projection of different samples. (D) Dot plots of marker genes for identifying cell type. (E) Heat map of relative proportions of immune cell types among samples.
Section 5. Flow cytometric analysis for immune cells from the brain tumor microenvironment
This section explains how to analyze immune cells using flow cytometry (continued from Section 3. Isolation of single cells from the brain tumor microenvironment). We used the FACS Fortessa X-20 to acquire cells and FlowJo software for analysis. The flow cytometers and software used for this analysis can vary depending on the user’s preferences.
-
54.
Transfer 1 × 106 cells from each sample to a new 5-mL polystyrene tube.
-
55.
Centrifuge the tube at 4°C, 440 rcf for 5 min.
-
56.
Remove the supernatant and add 100 μL (per million cells) of Fc block diluted in FACS buffer at a 1:100 ratio.
-
57.
Incubate on ice for 20 min in the dark.
-
58.
Add 1 mL of FACS buffer to the sample and centrifuge at 4°C, 440 rcf for 5 min.
-
59.Remove the supernatant and add 100 μL (per million cells) of antibody mixture.
-
a.Cells from C57BL/6 mice are stained with anti-CD45.2 antibody.
-
b.Cells from LSL-EGFRviii mice are stained with anti-CD45.1 antibody.
-
a.
-
60.
Incubate on ice for 30 min in the dark.
-
61.
Add 1 mL of FACS buffer to the sample and centrifuge at 4°C, 440 rcf for 5 min. Repeat this step once.
-
62.
Remove the supernatant and resuspend the cell pellet in 300 μL of FACS buffer.
-
63.
Before analysis, add 5 μL of propidium iodide solution to the sample and incubate for 5 min.
-
64.
Acquire cells using the FACS Fortessa X-20 machine (or other flow cytometer).
-
65.
Export .fcs data and load to FlowJo (or other software) for analysis.
-
66.Identify cell types using cell lineage markers.
-
a.Example data are shown in Figure 5.
-
a.
Figure 5.

Flow cytometric analysis of the spontaneous brain tumor model
(A–E) At 16 weeks after spontaneous tumor induction, the mice were sacrificed and single cells were isolated from tumor tissues. Cells were analyzed by flow cytometry. (A) Cell number of microglia (CD11b+CD45lo), CD11b+ tumor-inflitrating leukocytes (TILs; CD11b+CD45hi), and lymphoid TILs (CD11b-CD45hi). (B) The number of γδ T cells (CD45hiCD11b-CD3ε+γδTCR+). (C) The number of CD8 T cells (CD45hiCD11b-CD8a+) (left) and CD4 T cells (CD45hiCD11b-CD4+) (right). (D) The percentage of naïve (CD62L+CD44-), central memory (CM; CD62L+CD44+), double negative (DN; CD62L-CD44-), and CD8 and (E) CD4 effector T cells (CD62L-CD44+). Data were analyzed by the two-tailed unpaired Student’s t test. ∗P<0.05, ∗∗P<0.01, ∗∗∗P<0.001. Error bars represent the mean ± standard error of the mean.
Expected outcomes
If spontaneous tumor induction is successful, tumors can be observed by detecting tdTomato signals (Figure 2I). After 15–20 weeks post-injection, a large tumor mass can be detected. Antitumor immune responses could be promoted by tumor formation. scRNAseq analysis revealed that different types of immune cells are present in the tumor microenvironment (Figure 4A). Although microglia are the dominant type of immune cell in the normal brain (Figure 4B), cancerous tissues contain different immune cells that exhibit a distinct distribution (Figures 4B and 4C). By using multiple markers, the identity of clusters could be determined (Figure 4D). The orthotopic and spontaneous models contained different a composition of immune cells (Figures 4B and 4C). Spontaneous tumor tissues exhibited more T cell and macrophage infiltration. On the other hand, orthotopic tumors showed more infiltration by B cells and memory T cells (Figure 4E). These results suggest that cell injection can induce adverse immune responses. Immune cell infiltration into the tumor tissues was also confirmed by flow cytometry. More CD11b+ myeloid cells had preferentially infiltrated into the tumor area than lymphoid cells (Figure 5A). Previous research showed that γδ T cells are important antitumor cells in brain tumors (Park et al., 2021). Spontaneous brain tumor formation also promotes γδ T cell infiltration (Figure 5B). However, infiltration of CD4 and CD8 T cells was marginal and not significant (Figure 5C). Among T cell populations, tumor formation promoted the effector subsets (Figures 5D and 5E).
Quantification and statistical analysis
scRNAseq data were analyzed using R software. Flow cytometry results were processed by FlowJo and analyzed by Prism 9 (GraphPad). Differential gene expression of scRNAseq data was analyzed by the false-discovery rates (FDRs) method. Immune cell infiltration was analyzed by the unpaired two-tailed Student’s t test.
Limitations
This protocol explains how to induce and analyze two different murine brain tumor models. We induced spontaneous tumor formation by Egfr, Pten, Tp53 mutation. Tumors are heterogenous and mutations can vary across patients. By using this platform technology, other mutations can be induced. On the other hand, brain tumor cell lines also vary. In this protocol, we used the GL261 cell line. However, other cell lines such as CT2A can be used for tumor formation, and immune responses can vary depending on the mutations and cell line types.
Troubleshooting
Problem 1
Spontaneous tumors did not form or the tumor formation time was too long (greater than 20 weeks) (Section 1. Induction of the spontaneous brain tumor model).
Potential solution
In some circumstances, spontaneous tumors do not form despite successful transfection, possibly because the vectors were drained rapidly if the injection site was wrong. Users should be cautious when injecting the vectors. Using a dye when injecting vectors could be helpful for monitoring the injection. The DNA amount may also affect the speed of tumorigenesis.
On the other hand, tumor formation may take too long to perform an effective analysis. Although it is difficult to control the speed of tumor formation, the mouse background may have an effect. Here we used the FVB strain for tumor induction. However, other backgrounds such as C57BL/6 could be used as an alternative.
Problem 2
Spontaneous tumor formation varied among mice (Section 1. Induction of the spontaneous brain tumor model).
Potential solution
Because spontaneous tumor induction can be affected by multiple environmental factors, tumor size, immune responses, and formation period could vary depending on samples and injection model. If luciferase-containing vectors are used, tumor formation could be monitored without sacrifice. It may be useful to match the tumor stage when users analyze antitumor immune responses.
Problem 3
In some circumstances using the injection model, tumors did not form or naturally regressed (Section 2. Induction of the brain tumor injection model). On the other hand, tumor-bearing mice die at an early time point.
Potential solution
Brain tumors can vary depending on the site. Thus, matching the tumor injection site among mice is very important. In addition, tumor injection can induce adverse effects such as bleeding or tissue damage. To avoid these issues, users should inject tumors very slowly and should not penetrate blood vessels (Figure 3). Bleeding may also affect immune responses. If tumor cells are drained rapidly just after injection, they may become immunogenic.
On the other hand, mice could die at an early time point. Penetrating skulls and intracranial injections are very stressful for mice; therefore, users should handle the animals with great caution and care.
Problem 4
Gating cell populations in flow cytometric analysis can be difficult with brain tumor samples (Section 5. Flow cytometric analysis for immune cells from the brain tumor microenvironment).
Potential solution
Flow cytometric analysis of cell populations from brain tumor samples can be difficult due to autofluorescence. Because myelin and other brain debris can be autofluorescent, we recommend removing this debris with Percoll. In addition, because a high proportion of myeloid cells may be present in the samples, autofluorescence may also be high and antibodies may bind to the FcγRs on these cells. Thus, to avoid these issues, laser intensity and antibody concentration to be used should be optimized. In addition, using a reagent to block FcγR is recommended.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Heung Kyu Lee (heungkyu.lee@kaist.ac.kr).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
The authors thank the members of the Host Defenses lab for helpful discussions and Ji Ye Kim at the BioMedical Research Center for technical service. This work was supported by the National Research Foundation of Korea (NRF-2021M3A9D3026428 and NRF-2021M3A9H3015688) funded by the Ministry of Science and ICT of Korea. This work was also supported by Samsung Science and Technology Foundation (SSTF-BA1902-05), Republic of Korea.
Author contributions
J.H.P., C.W.K., H.-J.K., H.J.K., J.H.L., and H.K.L. designed and conducted the experiments. J.H.P., C.W.K., H.-J.K., and H.K.L. conceived of the study, analyzed the data, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Jang Hyun Park, Email: janghyun.park@kaist.ac.kr.
Chae Won Kim, Email: chaewon.kim@kaist.ac.kr.
Heung Kyu Lee, Email: heungkyu.lee@kaist.ac.kr.
Data and code availability
scRNAseq data are deposited in the Gene Expression Omnibus database (GSE199459, GSE159542, GSE201557).
References
- Fomchenko E.I., Holland E.C. Mouse models of brain tumors and their applications in preclinical trials. Clin. Cancer Res. 2006;12:5288–5297. doi: 10.1158/1078-0432.CCR-06-0438. [DOI] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Lee J.E., Kahng J.Y., Kim S.H., Park J.S., Yoon S.J., Um J.Y., Kim W.K., Lee J.K., Park J., et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560:243–247. doi: 10.1038/s41586-018-0389-3. [DOI] [PubMed] [Google Scholar]
- Oh T., Fakurnejad S., Sayegh E.T., Clark A.J., Ivan M.E., Sun M.Z., Safaee M., Bloch O., James C.D., Parsa A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107. doi: 10.1186/1479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.H., Kim H.J., Kim C.W., Kim H.C., Jung Y., Lee H.S., Lee Y., Ju Y.S., Oh J.E., Park S.H., et al. Tumor hypoxia represses gammadelta T cell-mediated antitumor immunity against brain tumors. Nat. Immunol. 2021;22:336–346. doi: 10.1038/s41590-020-00860-7. [DOI] [PubMed] [Google Scholar]
- Szatmari T., Lumniczky K., Désaknai S., Trajcevski S., Hídvégi E.J., Hamada H., Sáfrány G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97:546–553. doi: 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
scRNAseq data are deposited in the Gene Expression Omnibus database (GSE199459, GSE159542, GSE201557).