

Abstract
The treatment of glioblastoma multiforme (GBM) is challenging owing to its localization in the brain, the limited capacity of brain cells to repair, resistance to conventional therapy, and its aggressiveness. Curcumin has anticancer activity against aggressive cancers, such as leukemia, and GBM; however, its application is limited by its low solubility and bioavailability. Chemoprevention curcumin analog 1.1 (CCA-1.1), a curcumin analog, has better solubility and stability than those of curcumin. In this study, we explored potential targets of CCA-1.1 in GBM (PTCGs) by an integrated computational analysis and in vitro study. Predicted targets of CCA-1.1 obtained using various databases were subjected to comprehensive downstream analyses, including functional annotation, disease and drug association analyses, protein–protein interaction network analyses, analyses of genetic alterations, expression, and associations with survival and immune cell infiltration. Our integrative bioinformatics analysis revealed four candidate targets of CCA-1.1 in GBM: TP53, EGFR, AKT1, and CASP3. In addition to targeting specific proteins with regulatory effects in GBM, CCA-1.1 has the capacity to modulate the immunological milieu. Cytotoxicity of CCA-1.1 was lower than TMZ with an IC50 value of 9.8 μM compared to TMZ with an IC50 of 40 μM. mRNA sequencing revealed EGFR transcript variant 8 was upregulated, whereas EGFRvIII was downregulated in U87 cells after treatment with CCA-1.1. Furthermore, a molecular docking analysis suggested that CCA-1.1 inhibits EGFR with various mutations in GBM, which was confirmed using molecular dynamics simulation, wherein the binding between CCA-1.1 with the mutant EGFR L861Q was stable. For successful clinical translation, the effects of CCA-1.1 need to be confirmed in laboratory studies and clinical trials.
Subject terms: Computational biology and bioinformatics, Data mining, CNS cancer, Drug screening, Target identification
Introduction
Glioblastoma multiforme (GBM) is one of the most prevalent and aggressive brain tumors1. GBM arises from astrocytes that support nerve cells and invade nearby brain cells2. It can affect children; however, it is more common in adults aged 40–75 years3. Standard therapies for GBM currently include surgery followed by radiotherapy and chemotherapy. The standard chemotherapy is temozolomide, which is administered during radiation therapy4. Targeted chemotherapy drugs, such as lomustine (chemotherapy) and bevacizumab (anti-angiogenesis), are also administered in advanced GBM5. The treatment of GBM is challenging owing to its localization in the brain, the limited capacity of brain cells to repair, resistance to conventional therapy, and its aggressiveness3.
Immune cells participate in the disease progression of liver fibrosis6 and GBM7. The importance of interactions between tumors and their microenvironment in disease progression, including GBM progression, is now well-established8. The tumor microenvironment involves chronic inflammation, involving fibroblasts, pericytes, and immune cells9. However, the immune microenvironment of GBM is extremely immunosuppressive because of the lack of immune effector cell types and tumor-infiltrating lymphocytes7, making it challenging to target immune cells. The measurement of immune cell infiltration10 in GBM is an important tool for predicting clinical outcomes11,12, as a prognostic marker and a predictor of therapeutic outcomes.
To address the hurdles limiting effective GBM treatment, we explored new therapeutic compounds related to curcumin (Fig. 1), which has anticancer activity against various aggressive cancers, such as colon cancer, leukemia, and GBM13. Curcumin has been shown to increase the sensitivity of GBM cells to cisplatin, etoposide, camptothecin, and doxorubicin14. Curcumin exerts therapeutic effects in GBM via multiple pathways, including the suppression of AKT/mTOR and activation of ERK1/2 pathways in human malignant glioma U87-MG and U373-MG with PTEN mutations15. Furthermore, the effect of curcumin on the ERK pathway promotes the activation of p21, as observed by Choi et al. 16 The curcumin-induced inhibition of GBM cell proliferation and chemoresistance is mediated by AP-1 and NF-κB14. An in vivo study by Perry et al. (2010) revealed that curcumin affects glioblastoma growth and angiogenesis in mice with U87 glioma xenografts17. In addition, Facina et al. (2021) demonstrated the anticarcinogenic effect of curcumin alone, and in combination with piperin, in bisphenol A-induced carcinogenesis in gerbil prostates18. Moreover, in vitro and in silico studies by Liang et al. (2021) successfully synthesized curcumin and its analog and suggested their potential as EGFR inhibitors, in which curcumin and its analog regulate the expression of EGFR19. A recent study has demonstrated that the curcumin analog dimethoxycurcumin promotes apoptosis, autophagy, and ROS production and suppresses cell viability in human gliomas20.
Figure 1.
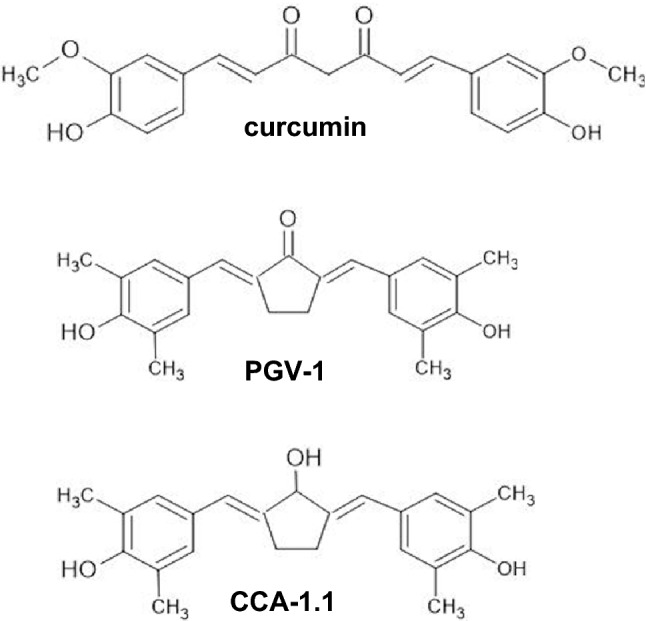
Chemical structures of curcumin, PGV-1, and CCA-1.1.
Natural products such as Tripterygium wilfordii21 and Ganoderma22 have immunomodulatory effects by inhibiting the expression of pro-inflammatory cytokines, and the production of other cytokines and antibodies. Curcumin exerts anticancer activity, in part, by modulating the immune system. A previous study has shown that curcumin increases the efficacy of immunotherapy in melanoma cells23. Additionally, curcumin is a promising immunotherapy for GBM24. A previous study showed that curcumin can be used in immunotherapy by decreasing the expression of immune checkpoint ligands and restoring the CD8 + cell function in head and neck cancer cells25. As discussed in a recent review, curcumin promotes immune function to eliminate cancer cells via several mechanisms26, however, its application is limited by its low solubility and bioavailability27.
Chemoprevention curcumin analog 1.1 (CCA-1.1), shown in Fig. 1, is a curcumin analog, with a substitution of the ketone group in the cyclopentane structure of PGV-1 (Fig. 1), a former analog, with a hydroxyl group; it has better solubility and stability than those of curcumin and PGV-128. CCA-1.1 also exhibits better anticancer activity than that of PGV-1 in several cancer cells, including luminal A MCF-7, HER2-positive HCC1954, triple-negative 4T1 breast cancer cells, K562 leukemic cells, Caco2, and WiDr colon cancer cells28. CCA-1.1 is able to induce cell cycle arrest and senescence29, increase the cytotoxicity of doxorubicin30, and hamper migration in T47D, estrogen-positive breast cancer cells31 and in WiDr colon cancer cells32. CCA-1.1 also inhibits the migration of triple-negative and HER2-positive breast cancer cells33 and induces mitotic arrest in triple-negative breast cancer34. Bioinformatics studies have explored the target genes of CCA-1.1 in colon cancer35 and triple-negative breast cancer cells36; however, similar analyses have not been performed for GBM.
In this study, we explored potential targets of CCA-1.1 in GBM (PTCG) by an integrated computational analysis and in vitro study (Fig. 2). Targets of CCA-1.1 were predicted from public databases and further analyzed for the selection of candidates. Our results indicate that CCA-1.1 not only targets certain regulatory genes in GBM but also modulates the immune environment.
Figure 2.

Work flow of the study.
Methods
Data mining
Protein targets of CCA-1.1 were predicted from several databases, including SwissTargetPrediction (http://www.swisstargetprediction.ch)37, SEA Search (https://sea.bkslab.org)38, MolTarPred (https://moltarpred.marseille.inserm.fr)39, TargetNet (http://targetnet.scbdd.com)40, BindingDB (https://www.bindingdb.org)41, DINIES (https://www.genome.jp/tools/dinies/)42, and HitPick (http://mips.helmholtz-muenchen.de/proj/hitpick)43 using the default settings for each database, as described previously35. Regulatory genes associated with GBM were obtained by searching against DISGENET https://www.disgenet.org44 with the keyword human glioblastoma and default settings for the database.
Functional annotation
Functional annotation of the PTCGs, including Gene Ontology and KEGG pathway enrichment analyses, was conducted using WebGestalt (http://www.webgestalt.org/)45 and DAVID version 6.8 (https://david.ncifcrf.gov)46, respectively. Briefly, the PTCGs were submitted to WebGestalt or DAVID as gene symbols and analyzed under default settings. FDR < 0.05 was selected as the cut-off value for the Gene Ontology analysis, and p < 0.05 was the threshold for KEGG pathway enrichment.
Disease- and drug-gene association analyses
Disease–gene and drug–gene associations were analyzed using WebGestalt (http://www.webgestalt.org/)45. Briefly, for disease–gene associations, PTCGs were submitted for an Over-Representation Analysis (ORA) using WebGestalt, selecting OMIM as the disease and functional database. For drug–gene associations, PTCGs were submitted for an ORA using WebGestalt with the DrugBank database. FDR < 0.05 was selected as the cut-off value.
Protein–protein interaction network construction and hub gene selection
A protein–protein interaction network for PTCG was constructed using STRING version 11.5 (https://string-db.org)47, with several parameters, including a confidence score of 0.4, Homo sapiens, and interaction between submitted protein symbols only. Hub genes, the genes with the top 10 degree scores were retained using the CytoHubba plugin of Cytoscape48 based on the degree score, as described previously49.
Analysis of genetic alterations in hub genes
Genetic alterations in hub genes were analyzed using cBioportal (https://www.cbioportal.org)50,51. Briefly, hub genes were submitted as gene symbols to search for alterations reported in studies of GBM. Further analyses were performed, including visualization using OncoPrint and mutation plots as well as analyses of copy number alterations and related pathways.
Expression of hub genes across glioblastoma samples
The expression levels of hub genes in GBM and normal tissues were compared using Gene Expression Profiling Interactive Analysis (GEPIA), (http://gepia.cancer-pku.cn/index.html)52. Briefly, gene symbols were submitted to GEPIA with the following parameter settings: GBM datasets, |Log2FC| cutoff = 2, p < 0.01, Jitter size of 0.4, and match TCGA normal and GTEx data.
Survival analysis for hub genes
To evaluate the prognostic value of the hub genes, Kaplan–Meier survival curves were generated using TIMER 2.0 (http://timer.cistrome.org)53,54, applying the median cutoff and the glioblastoma multiforme (GBM) dataset.
Correlations between hub genes and immune cell infiltration
Correlations between the expression levels of selected hub genes and immune cell infiltration were analyzed using TIMER 2.0 (http://timer.cistrome.org)53,54, using default settings, as described previously.
Cells
U87 cells were kindly given by Dr. Muhammad Hasan Bashari, MD., Faculty of Medicine, Universitas Padjajaran, Bandung. The U87 cells were cultured in RPMI medium, containing 10% of fetal bovine serum (FBS, Gibco), 1% of penicillin–streptomycin (Gibco), and maintained in 5% of CO2 incubator. For the cytotoxicity assay, the U87 cells (3,000 cells/ well) were seeded in a 96-well-plate and incubated for 24 h prior to treatment of CCA-1.1, temozolomide (TMZ, purchased from Sigma), or DMSO for the following 72 h. TMZ was used as a control as TMZ is the first choice for GBM treatment55. DMSO was used as a co-solvent of CCA-1.1, and TMZ, and as a control at a maximum concentration of 1% (v/v). At the end of incubation, an MTT solution was added and incubated for 3 h prior to addition of 10% of SDS solution. Cell viability was calculated as previously described56. The IC50 value was calculated with GraphPad Prism 5.0 using non-linear regression (curve fit): log (agonist) vs. normalized response-variable slope.
RNA sequencing
U87 cells were seeded, incubated, and treated with CCA-1.1 for 72 h. RNA isolation was performed using Bioline—Isolate II RNA Mini Kit, as per manufacturer’s instruction. Total RNA was then processed for mRNA enrichment, double-stranded cDNA synthesis, repair ends and addition of A overhang and A adaptor, fragment selection and PCR amplification, library quality testing, and next generation sequencing using Illumina HiSeq4000 from HiSeq-X sequencing technology. The qualities of the cleaned reads were assessed using FastQC version 0.11.9 (https://github.com/s-andrews/FastQC), and the reports were compiled using MultiQC version 1.1 (https://multiqc.info). The transcripts were quantified using the pseudo-alignment method employing Kallisto version 0.46157 with the human genome as a reference (GRCh38.p14). Differential expressed genes (DEGs) analysis was performed using EdgeR version 3.34.058 using parameters such as ILogFCI > 1 and a p value < 0.05.
Functional predictions for mutants
The effects of mutations on EGFR protein function were evaluated using several databases (with default parameters settings), including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/dokuwiki/start)59,60, Provean (http://provean.jcvi.org/index.php)61, SIFT (https://sift.bii.a-star.edu.sg)62, FATHMM (http://fathmm.biocompute.org.uk)60, and PANTHER (http://www.pantherdb.org)63. PolyPhen-2 settings were as follows: batch query input; code: 3NJP; HumDiv & HumVar classifier; canonical transcripts; missense annotations; GRCh37/hg19 genome assembly. A higher score indicated a more deleterious effect on protein function. The Provean analysis was conducted using default settings of for Human Batch Protein Prediction, in which prediction scores of less than −2.5 indicate that a mutation is deleterious. SIFT was conducted using the following parameters: database UniProt-SwissProt + TrEMBL 2010_09; median conservation of sequences: 3.00; identical query threshold: 90%. A prediction score of five indicates “tolerated.” The FATHMM analysis was performed using the following parameters: cancer-relativity inherited disease; weighted prediction; phenotype association, disease ontology. A prediction score of less than −1.5 indicates a “damaging” mutation. The coding SNP that impacted protein function was predicted using PANTHER with the following interpretations of the probability of deleterious effect (Pdel): "probably damaging" (time > 450 my, corresponding to a false positive rate of ~ 0.2 as tested using HumVar), "possibly damaging" (450 my > time > 200 my, corresponding to a false positive rate of ~ 0.4), and "probably benign" (time < 200 my). Predictions were performed by comparing the mutant to wild-type EGFR (PDB ID: 3NJP).
Molecular docking
The binding properties of curcumin and its analogs (PGV-1 and CCA-1.1) against EGFR and its mutant forms were predicted by a molecular docking analysis. Before performing the simulations, a template of the EGFR structure (UniProt code P00533) was retrieved from AlphaFold (https://alphafold.ebi.ac.uk/)64. The structures of mutant EGFR (E709K, T263P, V774M, and L861Q) were manually predicted using the MOE 2010 software, using the default step preparation. Due to the unknown binding site of each compound, the sitefinder in MOE was used to create a dummy site as the possible cavity for docking simulation. MOE 2010 (licensed from the Faculty of Pharmacy UGM) was also used for docking simulations, and the visualization of interactions. PGV-1 and CCA-1.1 structures were drawn using Marvin Sketch, and the curcumin structure was downloaded from PubChem. These structures were then subjected to conformational searches and energy minimization by MOE using the Energy Minimize Menu. For the docking simulation settings, London dG was used for both Rescoring 1 and Rescoring 2. Triangle Matcher was used for the score function and placement setting, and Forcefield was used to refine the docking results from 30 retained poses, as described previously49. The conformation with the lowest binding interaction between the ligand and receptor was determined.
Molecular dynamics simulation
The results of molecular docking were validated using molecular dynamics (MD) simulation. As the representative, we chose the binding pocket of EGFR L861Q in complex with curcumin, PGV-1, and CCA-1.1. The MD simulation was completed in NAMD 2.1465 and visualized using VMD 1.9.466. Parameterization of the proteins and ligands was prepared using CHARMM36 and CGenFF, available in the CHARMM-GUI web server67. For the solvation and neutralization steps, a cubic water box with 20-Å padding was added followed by K + and Cl− ion addition. For equilibration, the complex was minimized for 70 ps and simulated for 1 ns. Further, a 1-ns simulation (NPT ensemble, pressure 1 atm, and temperature 303 K) was conducted to finalize the MD simulation process. The visualization and trajectories of the MD results were analyzed using root-mean-square deviation (RMSD).
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Results
Data mining
We obtained 100, 4, 9, 494, 134, 71, and 1 target genes of CCA-1.1 using SwissTargetPRediction, SEA Search, MoltarPRed, TargetNet, BindingDB, DINIES, and HitPick, respectively, for a total of 618 predicted targets (Supplementary Table 1). From DISGENET, we collected 3177 GBM-related regulatory genes (Supplementary Table 2). As visualized using a Venn diagram, 268 genes (Fig. 3A, Supplementary Table 3) were targets of CCA-1.1 and involved in the regulation of GBM. These genes were identified as potential targets of CCA-1.1 in glioblastoma (PTCG) and were included in further analyses.
Figure 3.

(A) Venn diagram showing 268 potential targets of CCA-1.1 against GBM (PTCG). (B) Gene Ontology enrichment analysis of the PTCGs. (C) Disease–gene association analysis of the PTCGs. (D) Drug–gene association analysis of the PTCGs. (E) Protein–protein interaction network of the PTCGs. (F) Top 10 protein in the network, ranked by degree, as analyzed by CytoScape.
Functional annotation
A Gene Ontology analysis revealed that PTCGs were involved in various biological processes, including the response to stimulus, metabolic process, and cell communication (Fig. 3B). PTCGs were also enriched for cellular components, including the membrane, nucleus, and cytosol. In addition, PTCGs were associated with terms in the molecular functions category, including protein, ion, and nucleotide binding. A KEGG pathway enrichment analysis revealed that PTCGs were involved in several pathways, such as glioma, pathways in cancer, and p53 signaling pathways (Supplementary Table 4).
Disease–gene and drug–gene associations
A disease–gene association analysis revealed several diseases related to PTCGs, including Alzheimer’s disease, hepatocellular carcinoma, colorectal cancer, and breast cancer (Fig. 3C). A drug–gene association analysis showed that PTCGs are associated with several drugs, including ABT-869, tyrosine kinase inhibitors (sunitinib, regorafenib, ponatinib, sorafenib, imatinib, and fostamatinib), panobinostat, resveratrol, and tamoxifen (Fig. 3D).
Protein–protein interaction network and hub gene identification
Using STRING, we constructed a PPI network including 268 nodes and 4597 edges, with an average node degree of 34.3, an average local clustering coefficient of 0.523, and a PPI enrichment p value of < 1.0e − 16 (Fig. 3E). Hub genes were selected using the cytoHubba plugin of Cytoscape as the top ten target genes with respect to degree scores, including AKT1, TP53, ALB, EGFR, SRC, TNF, CASP3, MAPK1, HSP90AA1, and MAPK8 (Fig. 3F, Table 1).
Table 1 .
Top 10 proteins in the protein–protein interaction network, ranked by degree, as analyzed by CytoScape.
| Rank | Gene symbol | Gene name | Score |
|---|---|---|---|
| 1 | AKT1 | AKT serine/threonine kinase 1 | 165.0 |
| 2 | TP53 | Tumor protein p53 | 156.0 |
| 3 | ALB | Albumin | 143.0 |
| 4 | EGFR | Epidermal growth factor receptor | 135.0 |
| 5 | SRC | SRC proto-oncogene, non-receptor tyrosine kinase | 131.0 |
| 6 | TNF | Tumor necrosis factor | 128.0 |
| 7 | CASP3 | Caspase 3 | 125.0 |
| 7 | MAPK1 | Mitogen-activated protein kinase 1 | 125.0 |
| 9 | HSP90AA1 | Heat shock protein 90 alpha family class A member 1 | 112.0 |
| 10 | MAPK8 | Mitogen-activated protein kinase 8 | 111.0 |
Analysis of genetic alterations in hub genes
Genetic alterations in the ten hub genes were evaluated based on six studies of GBM using cBioportal (Fig. 4A). TCGA PanCancer Atlas68 which showed the second highest genetic alterations and the largest number of patients among the GBM studies and was selected for further analysis. We found mutation rates of 0.3–53% in hub genes in the study population, including CASP3 (0.3%), MAPK8 (0.3%), TNF (0.3%), ALB (1.1%), SRC (1.1%), HSP90AA1 (1.1%), AKT1 (1.6%), MAPK1 (1.6%), TP53 (33%), and EGFR (53%) (Fig. 4B). In a mutual exclusivity analysis, three gene pairs were significant, namely TP53–EGFR, ALB–SRC, and TNF–CASP3 (Table 2). A pathway enrichment analysis revealed that several pathways are affected by the observed genetic alterations, including RTK-RAS, TP53, PI3K, and cell cycle pathways (Supplementary Table 5). The RTK-RAS pathway was detected in two queries, EGFR and MAPK1, as well as neighboring genes, including members of the ERBB family, RAS family, and RAF family, which are involved in cellular processes including proliferation, cell survival, and translation (Fig. 4C).
Figure 4.

(A) Summary of alterations in 10 PTCG reported in GBM studies using cBioportal. (B) OncoPrint analysis of 10 PTCGs in patients with GBM from TCGA PanCancer Atlas study, as analyzed using cBioportal. (C) Pathway enrichment analysis related to genetic alterations in 10 PTCGs in patients with GBM from TCGA PanCancer Atlas, as analyzed using cBioportal. (D) Copy number alterations of 10 PTCGs in patients with GBM from TCGA PanCancer Atlas, as analyzed using cBioportal. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparison test. Mutation profiles of (E) EGFR and (F) TP53 in patients with GBM from TCGA PanCancer Atlas, as analyzed using cBioportal.
Table 2.
Mutual exclusivity analysis, as performed using cBioportal.
| A | B | p value | Tendency |
|---|---|---|---|
| TP53 | EGFR | < 0.001 | Mutual exclusivity |
| ALB | SRC | < 0.001 | Co-occurrence |
| TNF | CASP3 | 0.003 | Co-occurrence |
Copy number alterations in ALB, SRC, and TNF were not obvious (Fig. 4D). In AKT1, significant differences in mRNA levels were found between alteration types (i.e., shallow deletion, diploid, and gain); in particular, the expression of AKT1 was highest in cases with copy number gain, followed by diploid, and shallow deletion. The mRNA levels of TP53 in the shallow deletion group were significantly lower than those in the diploid and gain groups. In EGFR, we found that mRNA expression levels in the case of amplification were significantly higher than that those in the diploid and gain groups. mRNA levels of CASP3 and MAPK8 in diploids were significantly higher than those in the shallow deletion group. In addition, mRNA levels of MAPK1 and HSP90AA1 were significantly higher in the case of gain than in the diploid and shallow deletion groups. We then evaluated TP53 and EGFR mutations across patient samples in Liu et al. (2018). We found several mutations in TP53 in the p53 tetramerization domain, p53-DNA binding domain, and p53 transactivation domain (Fig. 4E). EGFR mutations occurred in many domains, such as the receptor-ligand domain, furin-like cysteine-rich region, growth factor receptor domain IV, and protein tyrosine kinase domain (Fig. 4F).
Expression of hub genes in glioblastoma samples
The mRNA levels of the hub genes AKT1, TP53, EGFR, and CASP3 were significantly higher in patients with GBM than in normal brain tissues (Fig. 5A). In addition, mRNA levels of ALB, SRC, TNF, MAPK1, HSP90AA1, and MAPK8 were not statistically significant between GBM and normal brain tissues.
Figure 5.

(A) Gene expression analysis of 10 PTCGs in patients with GBM and adjacent normal tissues from TCGA, as analyzed using GEPIA. (B) Relationships between the overall survival of patients with GBM and the expression of 10 PTCGs, as analyzed using TIMER 2.0.
Survival analysis of hub genes
The prognostic value of each hub gene was analyzed using a Kaplan–Meier plot. Among the hub genes, only ALB and MAPK8 levels were significantly associated with the survival of patients with GBM (Fig. 5B). Patients with low levels of ALB had a better overall survival than that of patients in group with high expression (p = 0.0223), whereas patients with high levels of MAPK8 had a better overall survival than that of patients with low expression levels (p = 0.0416).
Correlation between immune cell infiltration and hub genes
We explored correlations between the expression of hub genes and levels of immune cell infiltration in GBM using the TIMER 2.0 database (Table 3, Supplementary Fig. 1). We selected only four hub genes, AKT1, TP53, EGFR, and CASP3, based on their high expression levels in GBM, as analyzed by GEPIA. The expression levels of AKT1 (r = 0.311; p = 2.06 × 10−4), TP53 (r = 0.311; p = 7.36 × 10−5), EGFR (r = 0.288; p = 6.15 × 10−4), and CASP3 (r = 0.232; p = 6.24 × 10−4) were significantly and positively correlated with purity. Only CASP3 was significantly negatively correlated with B cells (r = −0.181; p = 3.44 × 10−2). The expression of CD8 + was significantly negatively correlated with the levels of AKT1 (r = −0.318; p = 1.53 × 10−4) and EGFR (r = −0.142; p = 9.75 × 10−2). CD4 + levels were significantly positively correlated with the levels of AKT1 (r = 0.187; p = 2.87 × 10−2), TP53 (r = 0.192; p = 2.49 × 10−2), and EGFR (r = 0.195; p = 2.27 × 10−2). Macrophage cells were significantly positively correlated with AKT1 expression (r = 0.219; p = 1.01 × 10−2), TP53 (r = 0.176; p = 3.92 × 10−2), and EGFR (r = 0.227; p = 7.69 × 10−3). Neutrophils were significantly positively correlated with levels of AKT1 (r = 0.266; p = 7.85 × 10−3) and TP53 (r = 0.248; p = 3.42 × 10−3). Dendritic cells showed significant positive correlations with levels of AKT1 (r = 0.439; p = 8.24 × 10−8), TP53 (r = 0.255; p = 2.63 × 10−3), EGFR (r = 0.251; p = 3.15 × 10−3), and CASP3 (r = 0.198; p = 2.01 × 10−2). Cancer-associated fibroblasts (CAFs) showed significant positive correlations with levels of AKT1 (r = 0.211; p = 1.34 × 10−2), TP53 (r = 0.211; p = 1.35 × 10−2), and CASP3 (r = 0.226; p = 7.88 × 10−3).
Table 3.
Immune cell infiltration related to the expression levels of AKT1, TP53, EGFR, and CASP3.
| Gene Name | Parameters | Purity | B cell | CD8 + | CD4 + | Macrophage | Neutrophil | Dendritic cell | Cancer Associated Fibroblast |
|---|---|---|---|---|---|---|---|---|---|
| AKT1 | R | 0.311 | 0.013 | −0.318 | 0.187 | 0.219 | 0.266 | 0.439 | 0.211 |
| P value | 2.06e-04 | 8.78e-01 | 1.53e-04 | 2.87e-02 | 1.01e-02 | 7.85e-03 | 8.24e-08 | 1.34e-02 | |
| TP53 | R | 0.311 | 0.135 | −0.008 | 0.192 | 0.176 | 0.248 | 0.255 | 0.211 |
| P value | 7.36e-05 | 1.15e-01 | 9.25e-01 | 2.49e-02 | 3.96e-02 | 3.42e-03 | 2.63e-03 | 1.35e-02 | |
| EGFR | R | 0.288 | 0.07 | −0.142 | 0.195 | 0.227 | 0.053 | 0.251 | 0.089 |
| P value | 6.15e-04 | 4.17e-01 | 9.75e-02 | 2.27e-02 | 7.69e-03 | 5.35e-01 | 3.15e-03 | 3.00e-01 | |
| CASP3 | R | 0.232 | −0.181 | 0.033 | −0.103 | −0.019 | 0.088 | 0.198 | 0.226 |
| P value | 6.24e-03 | 3.44e-02 | 7.05e-01 | 2.31e-01 | 8.29e-01 | 3.04e-01 | 2.01e-02 | 7.88e-03 |
Significant values are in bold.
CCA-1.1 performed cytotoxicity and induces the modulation of EGFR on U87 glioblastoma cells
We performed an MTT assay to measure the cytotoxicity of CCA-1.1 and TMZ, and both compounds showed cytotoxicity against U87 cells with an IC50 value of 9.8 and 40 μM, respectively (Fig. 6A,B). To check the molecular mechanism of CCA-1.1 in U87 cells, we performed next generation sequencing between untreated and CCA-1.1 treated U87 cells, and then analyzed the results for DEGs (Fig. 6C, Supplementary Table 6). The raw data of gene expression can be accessed at the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), using accession number GSE206241. Among the potential target genes, only EGFR showed significant results based on differential expression analysis, in which EGFR transcript variant 8 was upregulated in CCA-1.1 treated U87 cells, whereas EGFRvIII was downregulated in U87 cells after treatment with CCA-1.1 (Table 4). These findings confirm the bioinformatic approach which highlights the importance of EGFR as targets of CCA-1.1 in inhibition of GBM.
Figure 6.

(A) Cytotoxicity of CCA-1.1 and (B) TMZ in U87 glioblastoma cells. Cytotoxicity was determined using an MTT assay and presented as cell viability as explained in the methods section. Results are shown as the average of the three independent experiments (mean ± SD). (C) Heat map of top 100 DEGs between the U87 cells treated with CCA-1.1 and the DMSO.
Table 4.
Differentially expressed genes of hub genes in U87 glioblastoma cells upon treatment of CCA-1.1.
| No | Gene symbol | Ref seq | Gene name | Log FC | p value |
|---|---|---|---|---|---|
| 1 | EGFR | NM_001346900.2 | Epidermal growth factor receptor (EGFR), transcript variant 8, mRNA | 8.83688311 | 1.03E-05 |
| NM_001346941.2 | Epidermal growth factor receptor (EGFR), transcript variant EGFRvIII, mRNA | −6.111141843 | 0.00063995 | ||
| NM_001346898.2 | Epidermal growth factor receptor (EGFR), transcript variant 6, mRNA | −2.651710224 | 0.10586144 | ||
| NR_047551.1 | EGFR antisense RNA 1 (EGFR-AS1), long non-coding RNA | 2.325569699 | 0.30560263 | ||
| NM_201283.2 | Epidermal growth factor receptor (EGFR), transcript variant 3, mRNA | −0.969886184 | 0.5118575 | ||
| NM_001346899.2 | Epidermal growth factor receptor (EGFR), transcript variant 7, mRNA | −0.816340926 | 0.56093641 | ||
| XM_047419953.1 | Epidermal growth factor receptor (EGFR), transcript variant X2, mRNA | −0.777241106 | 0.59354503 | ||
| NM_001346897.2 | Epidermal growth factor receptor (EGFR), transcript variant 5, mRNA | 0.155644698 | 1 | ||
| 2 | TP53 | not found | Not found | Not found | Not found |
| 3 | AKT1 | NM_001014431.2 | AKT serine/threonine kinase 1 (AKT1), transcript variant 3, mRNA | −0.173979481 | 0.8970585 |
| 4 | ALB | not found | Not found | Not found | Not found |
| 5 | SRC | not found | Not found | Not found | Not found |
| 6 | TNF | NM_016292.3 | TNF receptor associated protein 1 (TRAP1), transcript variant 1, mRNA; nuclear gene for mitochondrial product | 0.095069258 | 0.94426958 |
| 7 | CASP3 | not found | Not found | Not found | Not found |
| 8 | MAPK1 | NM_138957.3 | Mitogen-activated protein kinase 1 (MAPK1), transcript variant 2, mRNA | 0.095103156 | 0.94434009 |
| 9 | HSP90AA1 | NM_005348.4 | Heat shock protein 90 alpha family class A member 1 (HSP90AA1), transcript variant 2, mRNA | −0.088807883 | 0.94722009 |
| 10 | MAPK8 | NM_001323330.2 | Mitogen-activated protein kinase 8 (MAPK8), transcript variant 16, mRNA | −0.03700038 | 1 |
Significant values are in bold.
Prediction of effects of mutations on protein function
We identified EGFR as a promising target of CCA-1.1 for GBM treatment. We further predicted the functional effects of EGFR alterations using several databases, including PolyPhen-2, Provean, SIFT, FATHMM, and PANTHER. We selected 22 EGFR mutations detected in GBM samples by Liu et al. (2018) (TCGA PanCancer); these mutations were located in the growth factor receptor domain, protein kinase-like (PK-like), receptor L domain, growth factor receptor domain IV, furin-like cysteine-rich region, protein kinase-like (PK-like), and protein tyrosine kinase (Table 5, Supplementary Table 7). The EGFR mutations in the protein kinase-like domain, namely E709K, V774M, and L861Q, were predicted to be damaging, deleterious, and cancer-related (Table 5). Another mutation, T263P, located in a furin-like cysteine-rich region, was also predicted to be associated with cancer. The V774M mutation, which occurs in the protein kinase-like domain, was predicted to be damaging and associated with cancer. In addition, L861Q, in the protein tyrosine kinase domain, was predicted to be damaging and related to cancer.
Table 5.
Prediction of EGFR mutations and activity.
| No | Mutant | PolyPhen-2 HumDiv | PolyPhen HumVar | Provean | SIFT | FATHMM | PANTHER | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | Score | Prediction | Domain Involved | Cancer | Inherited disease | |||||||
| Score | Prediction | Score | Prediction | Preservation time | Message | Pdel | |||||||||||
| 1 | A289D | 0.996 | PD | 0.993 | PD | −5.24 | Del | 0.001 | D | Growth factor receptor domain | −1.01 | Cancer | −0.01 | T | 797 | PD | 0.74 |
| 2 | A289T | 0.987 | PD | 0.974 | PD | −3.54 | Del | 0.001 | D | Growth factor receptor domain | −1.02 | Cancer | −0.02 | T | 797 | PD | 0.74 |
| 3 | A289V | 0.997 | PD | 0.989 | PD | −3.56 | Del | 0.001 | D | Growth factor receptor domain | −1.04 | Cancer | −0.04 | T | 797 | PD | 0.74 |
| 4 | C620W | 1 | PD | 0.998 | PD | −10.32 | Del | 0 | D | Growth factor receptor domain | −3.32 | Cancer | 0.00 | T | 911 | PD | 0.85 |
| 5 | C620Y | 1 | PD | 0.989 | PD | −10.32 | Del | 0 | D | Growth factor receptor domain | −3.3 | Cancer | 0.02 | T | 911 | PD | 0.85 |
| 6 | E709K | 0.974 | PD | 0.721 | Pos D | −3.38 | Del | 0.003 | D | Protein kinase−like (PK−like) | −1.93 | Cancer | 0.07 | T | 842 | PD | 0.78 |
| 7 | G598V | 0.997 | PD | 0.849 | Pos D | −8.43 | Del | 0.004 | D | Growth factor receptor domain | −2.26 | Cancer | 1.06 | T | 797 | PD | 0.74 |
| 8 | H304Y | 0 | benign | 0.005 | benign | −2.01 | Neut | 1 | T | Growth factor receptor domain | −1.02 | Cancer | −0.02 | T | 456 | PD | 0.57 |
| 9 | L62R | 0.795 | Pos D | 0.553 | Pos D | −2.02 | Neut | 0.006 | D | Receptor L domain | −0.6 | Passenger/Other | −1.27 | T | 455 | PD | 0.57 |
| 10 | P596R | 1 | PD | 0.999 | PD | −8.44 | Del | 0 | D | Growth factor receptor domain IV | −3.96 | Cancer | −0.38 | T | 911 | PD | 0.85 |
| 11 | P596S | 1 | PD | 0.968 | PD | −7.5 | Del | 0.005 | D | Growth factor receptor domain IV | −3.93 | Cancer | −0.35 | T | 911 | PD | 0.85 |
| 12 | R108G | 1 | PD | 1 | PD | −5.87 | Del | 0.01 | D | Receptor L domain | −0.81 | Cancer | −1.48 | T | 842 | PD | 0.78 |
| 13 | R108K | 1 | PD | 1 | PD | −2.59 | Del | 0.001 | D | Receptor L domain | −0.78 | Cancer | −1.44 | T | 842 | PD | 0.78 |
| 14 | R222C | 1 | PD | 1 | PD | −6.52 | Del | 0 | D | Growth factor receptor domain | −1.05 | Cancer | −0.05 | T | 797 | PD | 0.74 |
| 15 | R252C | 1 | PD | 0.993 | PD | −3.25 | Del | 0.025 | D | Furin−like cysteine rich region | −2.74 | Cancer | −2.08 | D | 456 | PD | 0.57 |
| 16 | R252P | 0.998 | PD | 0.991 | PD | −3.17 | Del | 0.058 | T | Furin−like cysteine rich region | −2.73 | Cancer | −2.06 | D | 456 | PD | 0.57 |
| 17 | S645C | 0.999 | PD | 0.982 | PD | −1.96 | Neut | 0.187 | T | − | −1.07 | Cancer | −0.94 | T | 361 | Pos D | 0.5 |
| 18 | T263P | 0.952 | Pos D | 0.913 | PD | −1.38 | Neut | 0.087 | T | Furin−like cysteine rich region | −2.51 | Cancer | −1.85 | D | 176 | Prob Benign | 0.27 |
| 19 | T363I | 1 | PD | 0.994 | PD | −5.07 | Del | 0.001 | D | Receptor L domain | −0.96 | Cancer | −1.62 | D | 842 | PD | 0.78 |
| 20 | V774M | 1 | PD | 0.994 | PD | −1.61 | Neutral | 0.001 | D | Protein kinase−like (PK−like) | −2.3 | Cancer | −0.3 | T | 456 | PD | 0.57 |
| 21 | L861Q | 1 | PD | 0.993 | PD | −5.29 | Del | 0.008 | D | Protein tyrosine kinase | −1.88 | Cancer | −1.59 | D | 797 | PD | 0.74 |
| 22 | H773_V774dup | NA | NA | NA | NA | −10.34 | Del | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
PD probably damaging, Pos D possibly damaging, Del Deleterious, Neut Neutral, D Damaging, T Tolerated.
Molecular docking and MD
We successfully predicted the structures of mutant EGFR using a template from AlphaFold (Supplementary Fig. 2). Four mutants were selected from previous experiments. Each complex protein (Fig. 7A) was docked against curcumin and its analogues, PGV-1 and CCA-1.1. The molecular docking results showed that in wild-type EGFR, PGV-1 had the lowest docking score of -13.87 kcal/mol and formed one hydrogen bond with Arg686 (Fig. 7B, Table 6). For the E709K and V774M mutant forms of EGFR, curcumin had the lowest binding energy of −11.74 kcal/Mol with three hydrogen bonds (Gly696, Pro699, and Asn700) and −11.94 kcal/Mol with two hydrogen bonds (Asn298 and Arg831), respectively. CCA-1.1 showed the lowest docking scores of −11.29 and −12.62 kcal/Mol in the T263P and L861Q mutant forms of EGFR, respectively. Interestingly, for all mutant forms, CCA-1.1 showed better binding affinity than PGV-1 (Table 6). CCA-1.1 also had much stronger binding activity (ΔG = -12.62 kcal/Mol) for the L861Q mutant than wild-type EGFR, while PGV-1 did no show a difference between mutant and wild-type EGFR. These results show that CCA-1.1 performs better than PGV-1 in the inhibition of mutant EGFR (E709K, T263P, V774, and L861Q). Taken together, these results indicate that CCA-1.1 can inhibit many EGFR variants.
Figure 7.

(A) 3D visualization of EGFR mutations, E709K (Glu709 Lys709), T263P (Thr263 Pro263), V774M (Val774 Met774), and L861Q (Leu861 Gln861). (B) Visualization of molecular docking results for wild-type EGFR and mutant EGFR (E709K, T263P, V774M, and L861Q) against Curcumin, CCA-1.1, and PGV-1. (C) Visualization of the binding interaction of compounds (Curcumin, CCA-1.1, and PGV-1) against mutant EGFR L861Q at the initial time and after 1 ns MD simulation. (D) Root mean squared deviation (RMSD) of compounds (Curcumin, CCA-1.1, and PGV-1) in complex with mutant EGFR L861Q after 1 ns MD, shown in 100 frames.
Table 6.
Molecular docking scores for curcumin, CCA-1.1, and PGV-1 against wild-type and mutant EGFR.
| Drug | Protein | Binding Energy (kcal/Mol) | No of H bonds | Interacting amino acids (Distance) |
|---|---|---|---|---|
| Curcumin | EGFR (wild-type) | −11.05 | 1 | Gln684 (2.97) |
| EGFR (E709K) | −11.74 | 3 | Gly696 (2.21), Pro699 (3.08), Asn700 (3.44) | |
| EGFR (T263P) | −10.12 | 2 | Arg686 (2.98), Glu736 (3.19) | |
| EGFR (V774M) | −11.94 | 2 | Asn298 (1.84), Arg831 (2.13) | |
| EGFR (L861Q) | −10.51 | 2 | Arg297 (1.92), Leu707 (3.00) | |
| CCA-1.1 | EGFR (wild-type) | −12.51 | 2 | Arg53 (1.80), Glu687 (1.79) |
| EGFR (E709K) | −11.02 | 3 | Val30 (2.04), Met54 (3.76), Arg297 (1.93) | |
| EGFR (T263P) | −11.29 | 1 | Tyr299 (2.02) | |
| EGFR (V774M) | −10.91 | 1 | Val30 (2.09) | |
| EGFR (L861Q) | −12.62 | 1 | Val689 (3.13) | |
| PGV-1 | EGFR (wild-type) | −13.87 | 1 | Arg686 (1.84) |
| EGFR (E709K) | −10.73 | 1 | Leu777 (2.29) | |
| EGFR (T263P) | −10.37 | – | – | |
| EGFR (V774M) | −10.58 | 1 | Leu861 (3.09) | |
| EGFR (L861Q) | −11.82 | 2 | Phe723 (2.12), Lys875 (2.85) |
The results of molecular docking were validated using MD simulation. As the representative, we chose the binding pocket of EGFR L861Q in complex with curcumin, PGV-1, and CCA-1.1. After a 1-ns simulation, CCA-1.1 displayed a minor change in the position and binding trajectory with mutant EGFR L861Q, which indicates the most stable interaction (Fig. 7C). In the presence of PGV-1, the binding pocket of mutant EGFR L861Q showed more change in position than in CCA-1.1. Further, a more dynamic change was observed with curcumin, which clarified the less stable interaction of curcumin and PGV-1 than that of CCA-1.1 (Fig. 7C). Higher-binding stability of CCA-1.1 compared with that of PGV-1 and curcumin was also demonstrated by the RMSD value of each compound after the 1-ns MD simulation. CCA-1.1 demonstrated a stable RMSD value around 1.8 nm (Fig. 7D). An increase in the RMSD value up to 2.4 and 4.6 nm was shown by PGV-1 and CCA-1.1, respectively, which demonstrates a less stable binding interaction (Fig. 7D). The results of the MD simulation confirmed the validity of the molecular docking study, indicating CCA-1.1 as the most effective EGFR inhibitor.
Discussion
We identified four targets of CCA-1.1 in GBM (i.e., TP53, EGFR, AKT1, and CASP3) by an integrative bioinformatics analysis. TP53 encodes the P53 protein, a tumor suppressor that inhibits cancer cell proliferation and promotes apoptosis69. TP53 is frequently mutated in GBM, and these mutations are mainly deletions, affecting P53 function and thereby triggering cancer progression. We also detected copy number gains, suggesting an increase in p53 expression. Both curcumin and PGV-1 compounds have been shown to increase p53 expression in breast cancer cells70. Further studies of changes in p53 expression in response to CCA-1.1 treatment in GBM are needed to support the findings of this study.
Mutations in p53 are found in almost half of human cancers71, a loss of p53 function promotes invasion, metastasis, and chemoresistance72. Mutations in p53, particularly gain-of-function mutations, increase the inflammatory response in patients with GBM73. AKT1 is a protein serine/threonine kinase that plays a role in the PI3K/AKT pathway, which regulates cell proliferation and survival74. The dysregulation of AKT is common in cancer, with reports of epigenetic modifications, mutations, and overexpression75,76. PI3K/Akt is a highly targeted pathway for glioblastoma therapy77. Several previous studies have explored the AKT-targeted anticancer effects of curcumin and its analogs. Curcumin may be effective in combination with TMZ in GBM78. Yin reported that curcumin increases the effectiveness of temozolomide against U87 glioblastoma cells by increasing ROS levels, inhibiting AKT/mTOR signaling, and promoting apoptosis79. Curcumin inhibits GBM via the pRb, p53, JAK/STST, MAPK, PI3K/Akt, and NF-κB pathways80. Another analog of curcumin, C-150, inhibits GBM progression by targeting the NF-κB, Notch, and Akt pathways81. Previous research on curcumin and PGV-1 has shown that these compounds inhibit PI3K/AKT signaling in breast cancer cells and colon cancer cells. PGV-1 inhibits NF-κB activation82 which is related to the PI3K/Akt pathway. Elucidating the mechanism by which CCA-1.1 influences the PI3K/AKT pathway will provide a scientific basis for its utilization as an anti-GBM agent.
CASP3 encodes caspase 3, which contributes to the final steps in apoptosis, and is also called an executioner caspase83. Increased caspase-3 expression in triggers GBM cell death84. The inhibition of caspase-3 in brain-resident immune cells promotes GBM progression85. Previous studies have shown that both curcumin and PGV-1 trigger apoptosis by increasing caspase expression. The curcumin analogs PGV-0 and PGV-1 stimulate the apoptosis of T47D breast cancer cells by the activation of Caspase-386. Further studies of the effect of CCA-1.1 on caspase 3 expression and activity are needed.
EGFR encodes the human epidermal growth factor receptor, a member of the tyrosine kinase receptor family87. Mutations in EGFR activate EGFR signaling, which triggers proliferation and survival in GBM88. EGFR mutations have been found in 53% of patients with GBM68, including gains or amplifications, suggesting an increase in EGFR expression. Several compounds successfully inhibit EGFR signaling, for example, Higenamine89, 20(R, S)-protopanaxatriol, a metabolite from protopanaxatriol ginsenosides90, and Tubeimoside-I, which increases the sensitivity of glioblastoma cells towards temozolomide91.
Extensive research has focused on the effects of curcumin and its analogs targeting EGFR in cancer cells. Curcumin inhibits EGFR signaling and reduces EGFR expression in cancer cells. Curcumin increases sensitivity to gefitinib by inhibiting EGFR signaling in non-small cell lung cancer92. In addition, curcumin enhances the anticancer activity of gefitinib in vitro and in vivo in lung cancer by inducing EGFR degradation93. Curcumin downregulates EGFR in colon cancer cells by reducing the transcription factor EGR194. Another study has shown that curcumin inhibits the autophosphorylation of EGFR95. Starok et al. showed that curcumin has dual effects on EGFR by inhibiting enzymatic activity of the EGFR tyrosine kinase domain and by entering the lipid bilayer, thus affecting EGFR dimerization96. A recent study by Ali et al. has shown that curcumin analog 3c has a greater inhibitory effect on leukemic cells than those of curcumin and gefitinib, and this analog inhibits EGFR activity97.
Mutations in the EGFR kinase domain have been shown to cause constitutively active ligand-independent signaling98 and to affect the sensitivity of glioma cells to temozolomide99. E709K is a mutation in EGFR exon 18 responsible for lung cancer cell resistance to gefitinib, erlotinib, AZD9291, and CO1686100. It is a rare type of EGFR mutation in lung cancer101. The T263P mutation is located in the extracellular domain of EGFR, which leads to ligand-independent signaling activation102 and tumor progression in GBM103. Moreover, the T263P EGFR mutant form has a furin-like cysteine-rich (FU-CR) domain involved in signal transduction, including an important role in promoting Wnt/β-catenin signaling104–107. L861Q is a missense mutation in the EGFR kinase domain of GBM108. The L861Q mutation increases kinase activity and tumor progression but does not increase the sensitivity of tumor cells to EGFR tyrosine kinase inhibitors109. The EGFR V774M mutation is associated with non-small-cell lung cancer progression110 and resistance to tyrosine kinase inhibitors111. A missense mutation in the EGFR kinase domain, V774M, which leads to amplification, has also been found in Japanese patients with GBM112. V774M is considered a functional mutation in lung cancer113.
In a molecular docking analysis, CCA-1.1 showed a lower docking score than that of PGV-1 in wild-type and mutant EGFR E709K, T263P, and L861Q and slightly higher docking scores for V774M. The molecular docking results for wild-type EGFR are supported by previous studies. PGV-1 exhibits the weakest interaction with EGFR and HER2 in silico114. Interestingly, CCA-1.1 showed a similar or better interaction with EGFR than PGV-128. Further, MD simulation demonstrated a more stable binding interaction of CCA-1.1 during the 1-ns simulation compared to the binding of PGV-1 and curcumin. Thus, clarifying the results of the molecular docking study. Therefore, further research on CCA-1.1 targeting EGFR is very important for its development as an anti-GBM agent.
GBM gene profiling has revealed three GBM subtypes: proneural (TCGA-PN), classical (TCGA-CL), and mesenchymal (TCGA-MES)115. GBM subtypes are characterized by abnormalities in platelet-derived growth factor alpha (PDGFRA), isocitrate dehydrogenase1 (IDH1), epidermal growth factor receptor (EGFR), and neurofibromin1 (NF1)116. Different subtypes may respond differently to therapies and show differences in the immune microenvironment117. Several studies have suggested that mesenchymal GBM is the most immunogenic, proinflammatory subtype, characterized by significant M2 macrophage and neutrophil gene expression118,119. Therefore, we expected to observe correlations between the expression of the four hub genes and the level of immune cell infiltration in GBM. In general, immune cell infiltration can be classified into two types: (1) activation of the immune response by pro-inflammatory cells and CD8 + cytotoxic T lymphocytes (CTL) and (2) suppression of the immune response to cancer cells, e.g., by regulatory T cells (Tregs). Considering the complexity of GBM and the presence of the blood–brain barrier, it is plausible that the immune response is strictly regulated, resulting in extensive immune cell infiltration120–122. Both adaptive and innate tumor-infiltrating immune cells are involved, i.e., B cells, CD8 + , and CD4 + cells as well as macrophages, neutrophils, and dendritic cells (DCs), respectively123. AKT1 and EGFR negatively affected CD8 + , while B cells were negatively correlated with CASP3 expression levels (with correlation coefficients of < 0.5). Positive correlations were observed between the expression levels of AKT1, TP53, and EGFR and the frequencies of CD4 + cells and all of the above-mentioned innate immune cells. CASP3 expression was positively correlated with DCs. Despite the low frequency of fibroblasts in the healthy brain, CAFs are found in GBM124,125. Here, we found that CAFs are positively related to AKT1, TP53, and CASP3 expression. Mu et al. reported that CD4 + plays a role in angiogenesis and the progression of GBM126. We propose that targeting the four newly identified gene candidates may be an effective approach to alter the immune response to cancer.
The cytotoxicity assay of CCA-1.1 and TMZ showed that CCA-1.1 has a better cytotoxicity than TMZ based on the IC50 values, in which the cytotoxicity against U87 cells with an IC50 value are 9.8 uM for CCA-1.1 and 40 uM for TMZ, indicating high potency of CCA-1.1 for GBM therapy. DEGs showed that among the potential target genes, only EGFR showed significant results, in which the EGFR transcript variant 8 was upregulated in CCA-1.1 treated U87 cells, whereas EGFRvIII was downregulated in U87 cells after treatment with CCA-1.1., indicating the important role of EGFR in the cytotoxicity of CCA-1.1. A previous study showed the heterogeneity of EGFR in glioblastoma cells, also referred to as EGFR truncation variants127. Moreover, genetic amplification and mutations in EGFR are the most common oncogenic events in GBM128. EGFR is encoded by the EGFR gene, producing mRNA transcript EGFR variant 1, which produces isoform a. In addition to isoform a, EGFR produces several alternatively spliced transcript variants129. Several mRNA variants encode EGFR isoforms, such as variants 1 and 8. EGFR transcript variant 1 encodes the full-length protein of EGFR, while variant 8 encodes a shorter protein. A previous study stated that all isoforms encoded by all EGFR variants could interact with their ligand, namely epidermal growth factor (EGF)130. Furthermore, Weinholdt explained that only the EGFR1 isoform had been widely studied for its biological function131. EGFRvII is an oncogenic EGFR that is responsible for sensitivity to tyrosine kinase inhibitors127.
EGFRvIII is an interesting therapeutic target in GBM therapy because EGFRvIII is present in 25–30% of the glioblastoma cell population132. EGFRvIII undergoes a 6–273 amino acid deletion at exon 2–7, encoding the extracellular domain of EGFR133, and EGFRvIII can undergo dimerization via a ligand-independent activation pathway132. EGFRvIII differs from mutant EGFR on the extracellular domain, namely due to the deletion of certain amino acids causing slow receptor internalization, as well as a slower constitutively phosphorylation level compared to EGFR isoform 1134. The results of NGS showed the downregulation of EGFRvIII due to CCA-1.1 treatment. This indicates the potential of CCA-1.1 to constitutively inhibit mutant EGFR and its potential as an inhibitor of EGFR in GBM.
According to the NCBI gene bank, EGFR transcript variant 1 has a length of 9950 bp, while EGFRvIII and EGFR transcript variant 8 have a length of 9104 and 9676 bp, respectively. Transcript variant 8 produces an EGFR isoform that is shorter than transcript variant 1 and a distinct N-terminus compared to isoform a. Research on the biological functions of other EGFR transcript variants, including transcript variant 8, are limited. In this present study, the results of NGS showed an increase in mRNA expression of EGFR variant 8 due to CCA-1.1 treatment; however, the biological role of EGFR variant 8 and the mechanism of CCA-1.1 in regulating the expression of this variant requires further study.
This study had several limitations. First, the protein targets of CCA-1.1 were curated or predicted using public databases based on a particular algorithm. Second, the results of the bioinformatics analyses need to be validated by in vitro and in vivo assays as well as clinical trials. Nevertheless, the results of this study are expected to accelerate the development of drugs for GBM.
Conclusion
Using an integrative bioinformatics approach, four CCA-1.1 targets in GBM were obtained: TP53, EGFR, AKT1, and CASP3. In addition to the potential therapeutic effects of CCA-1.1 mediated by these four proteins and the inhibition of signaling pathways, it also has the potential to modulate the immune environment. A cytotoxicity assay showed that CCA-1.1 has a better cytotoxicity than TMZ with an IC50 value of 9.8 μM compared 40 μM for TMZ. DEGs showed that among the potential target genes, only EGFR showed significant results, in which the EGFR transcript variant 8 was upregulated, whereas EGFRvIII was downregulated in U87 cells after treatment with CCA-1.1. Molecular docking results revealed that CCA-1.1 can inhibit many EGFR mutants in GBM. Further, MD simulation revealed that the binding of CCA-1.1 with the mutant EGFR L861Q is the most stable compared to those of curcumin and PGV-1. These findings require further confirmation with laboratory experiments and clinical trials for the development of GBM therapies.
Supplementary Information
Acknowledgements
The authors thank Mrs. Herwandhani Putri for writing assistance, and PT. Genetika Science for their assistance in performing mRNA Sequencing.
Author contributions
A.H. contributed to the conception and design of the study, acquisition, analysis, and interpretation of data, drafting and revising the article, and final approval of the version to be published. F.W. contributed to the acquisition and interpretation of data and drafting of the article. N.H. contributed to the acquisition and interpretation of data and drafted the manuscript. R.Y.U. synthesized CCA-1.1, performed molecular dynamics simulation, and drafted the manuscript. R.I.J. contributed to the interpretation of data and drafted the manuscript. M.I. contributed to the interpretation of data and drafted the manuscript. A.S.T. contributed to the acquisition and interpretation of the data.
Funding
This research was funded by the World Class Research (WCR) Program by the Ministry of Research and Technology and by the National Agency for Research and Innovation, Republic of Indonesia, 2021 (Contract Number 4518/UN1/DITLIT/DIT-LIT/PT/2021).
Data availability
All data produced by this study are disclosed in the manuscript and additional files. The raw data of gene expression can be accessed at the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), using accession number GSE206241.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-18348-9.
References
- 1.Taylor OG, Brzozowski JS, Skelding KA. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019 doi: 10.3389/fonc.2019.00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.So J-S, Kim H, Han K-S. Mechanisms of invasion in glioblastoma: Extracellular matrix, Ca2+ signaling, and glutamate. Front. Cell. Neurosci. 2021 doi: 10.3389/fncel.2021.663092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iacob G, Dinca EB. Current data and strategy in glioblastoma multiforme. J. Med. Life. 2009;2:386. [PMC free article] [PubMed] [Google Scholar]
- 4.Joo J-D, et al. Temozolomide during and after Radiotherapy for newly diagnosed glioblastomas: A prospective multicenter study of Korean patients. J. Korean Neurosurg. Soc. 2012;52:92–97. doi: 10.3340/jkns.2012.52.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren X, Ai D, Li T, Xia L, Sun L. Effectiveness of lomustine combined with bevacizumab in glioblastoma: A meta-analysis. Front. Neurol. 2021 doi: 10.3389/fneur.2020.603947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda M, Seki E. The liver fibrosis niche: Novel insights into the interplay between fibrosis-composing mesenchymal cells, immune cells, endothelial cells, and extracellular matrix. Food Chem. Toxicol. 2020;143:111556. doi: 10.1016/j.fct.2020.111556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pombo Antunes AR, et al. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife. 2020;9:e52176. doi: 10.7554/eLife.52176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broekman ML, et al. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018;14:482–495. doi: 10.1038/s41582-018-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson S, et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021 doi: 10.1038/s41577-021-00540-z. [DOI] [PubMed] [Google Scholar]
- 10.Barnes TA, Amir E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer. 2017;117:451–460. doi: 10.1038/bjc.2017.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kmiecik J, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013;264:71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Sokratous G, Polyzoidis S, Ashkan K. Immune infiltration of tumor microenvironment following immunotherapy for glioblastoma multiforme. Hum. Vaccin. Immunother. 2017;13:2575–2582. doi: 10.1080/21645515.2017.1303582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosieniak G, et al. Curcumin-treated cancer cells show mitotic disturbances leading to growth arrest and induction of senescence phenotype. Int. J. Biochem. Cell Biol. 2016;74:33–43. doi: 10.1016/j.biocel.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Dhandapani KM, Mahesh VB, Brann DW. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFkappaB transcription factors. J. Neurochem. 2007;102:522–538. doi: 10.1111/j.1471-4159.2007.04633.x. [DOI] [PubMed] [Google Scholar]
- 15.Aoki H, et al. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 16.Choi BH, et al. p21<sup>Waf1/Cip1</sup> expression by curcumin in U-87MG human glioma cells: Role of early growth response-1 expression. Can. Res. 2008;68:1369. doi: 10.1158/0008-5472.CAN-07-5222. [DOI] [PubMed] [Google Scholar]
- 17.Perry MC, Demeule M, Regina A, Moumdjian R, Beliveau R. Curcumin inhibits tumor growth and angiogenesis in glioblastoma xenografts. Mol. Nutr. Food Res. 2010;54:1192–1201. doi: 10.1002/mnfr.200900277. [DOI] [PubMed] [Google Scholar]
- 18.Facina CH, et al. Protective effect of the association of curcumin with piperine on prostatic lesions: New perspectives on BPA-induced carcinogenesis. Food Chem. Toxicol. 2021;158:112700. doi: 10.1016/j.fct.2021.112700. [DOI] [PubMed] [Google Scholar]
- 19.Liang Y, Zhao J, Zou H, Zhang J, Zhang T. In vitro and in silico evaluation of EGFR targeting activities of curcumin and its derivatives. Food Funct. 2021;12:10667–10675. doi: 10.1039/D1FO02002A. [DOI] [PubMed] [Google Scholar]
- 20.Luo S-M, Wu Y-P, Huang L-C, Huang S-M, Hueng D-Y. The anti-cancer effect of four curcumin analogues on human glioma cells. Onco Targets Ther. 2021;14:4345–4359. doi: 10.2147/OTT.S313961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ru Y, et al. Role of keratinocytes and immune cells in the anti-inflammatory effects of Tripterygium wilfordii Hook. F. in a murine model of psoriasis. Phytomedicine. 2020;77:153299. doi: 10.1016/j.phymed.2020.153299. [DOI] [PubMed] [Google Scholar]
- 22.Ren L, Zhang J, Zhang T. Immunomodulatory activities of polysaccharides from Ganoderma on immune effector cells. Food Chem. 2021;340:127933. doi: 10.1016/j.foodchem.2020.127933. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, et al. Curcumin micelles remodel tumor microenvironment and enhance vaccine activity in an advanced melanoma model. Mol. Ther. 2016;24:364–374. doi: 10.1038/mt.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukherjee S, et al. Liposomal TriCurin, a synergistic combination of curcumin, epicatechin gallate and resveratrol, repolarizes tumor-associated microglia/macrophages, and eliminates glioblastoma (GBM) and GBM stem cells. Molecules. 2018;23:201. doi: 10.3390/molecules23010201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, et al. The effect of Curcumin on multi-level immune checkpoint blockade and T cell dysfunction in head and neck cancer. Phytomedicine. 2021;92:153758. doi: 10.1016/j.phymed.2021.153758. [DOI] [PubMed] [Google Scholar]
- 26.Paul S, Sa G. Curcumin as an adjuvant to cancer immunotherapy. Front. Oncol. 2021 doi: 10.3389/fonc.2021.675923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suresh K, Nangia A. Curcumin: Pharmaceutical solids as a platform to improve solubility and bioavailability. CrystEngComm. 2018;20:3277–3296. doi: 10.1039/C8CE00469B. [DOI] [Google Scholar]
- 28.Utomo RY, et al. Preparation and cytotoxic evaluation of PGV-1 derivative, CCA-1.1, as a new curcumin analog with improved-physicochemical and pharmacological properties. Adv. Pharm. Bull. 2021;12:603–612. doi: 10.34172/apb.2022.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novitasari D, et al. A new curcumin analog, CCA-1.1, induces cell cycle arrest and senescence toward ER-positive breast cancer cells. Int. J. Pharm. Sci. Res. 2021;13:9. [Google Scholar]
- 30.Wulandari F, Novitasari D, Kirihata M, Kato J-Y, Meiyanto E. New curcumin analog, CCA-1.1, synergistically improves the antiproliferative effect of doxorubicin against T47D breast cancer cells. Indones. J. Pharm. 2020;31:244–256. doi: 10.22146/ijp.681. [DOI] [Google Scholar]
- 31.Wulandari F, et al. The anti-migratory activity of a new curcumin analog, CCA-1.1, against T47D breast cancer cells. Int. J. Pharm. Sci. Res. 2021;13:2877–2887. [Google Scholar]
- 32.Wulandari F, Kirihata M, Kato J-Y, Meiyanto E. Curcumin analogs, PGV-1 and CCA-1.1 exhibit anti-migratory effects and suppress MMP9 expression on WiDr cells. The Indones. Biomed. J. 2021;13:271–280. doi: 10.18585/inabj.v13i3.1583. [DOI] [Google Scholar]
- 33.Novitasari D, Jenie RI, Utomo RY, Kato JY, Meiyanto E. CCA-1.1, a novel curcumin analog, exerts cytotoxic anti- migratory activity toward TNBC and HER2-enriched breast cancer cells. Asian Pac. J. Cancer Prevent.: APJCP. 2021;22:1827–1836. doi: 10.31557/apjcp.2021.22.6.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novitasari D, et al. Curcumin-like structure (CCA-1.1) induces permanent mitotic arrest (Senescence) on Triple-negative breast cancer (TNBC) cells, 4T1. Res. J. Pharm. Technol. 2021;14:4375–4382. doi: 10.52711/0974-360X.2021.00760. [DOI] [Google Scholar]
- 35.Wulandari F, Meiyanto E, Kirihata M, Hermawan A. Bioinformatic analysis of CCA-1.1, a novel curcumin analog, uncovers furthermost noticeable target genes in colon cancer. Gene Rep. 2020;21:100917. doi: 10.1016/j.genrep.2020.100917. [DOI] [Google Scholar]
- 36.Novitasari D, Jenie RI, Kato J-Y, Meiyanto E. The integrative bioinformatic analysis deciphers the predicted molecular target gene and pathway from curcumin derivative CCA-1.1 against triple-negative breast cancer (TNBC) J. Egypt. Natl. Cancer Inst. 2021;33:1–10. doi: 10.1186/s43046-020-00056-y. [DOI] [PubMed] [Google Scholar]
- 37.Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucl. Acids Res. 2019;47:W357–W364. doi: 10.1093/nar/gkz382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 39.Peón A, et al. MolTarPred: A web tool for comprehensive target prediction with reliability estimation. Chem. Biol. Drug Des. 2019;94:1390–1401. doi: 10.1111/cbdd.13516. [DOI] [PubMed] [Google Scholar]
- 40.Yao ZJ, et al. TargetNet: A web service for predicting potential drug-target interaction profiling via multi-target SAR models. J. Comput. Aided Mol. Des. 2016;30:413–424. doi: 10.1007/s10822-016-9915-2. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Lin Y, Liu M, Gilson MK. The binding database: Data management and interface design. Bioinformatics (Oxford, England) 2002;18:130–139. doi: 10.1093/bioinformatics/18.1.130. [DOI] [PubMed] [Google Scholar]
- 42.Yamanishi Y, et al. DINIES: Drug-target interaction network inference engine based on supervised analysis. Nucl. Acids Res. 2014;42:W39–45. doi: 10.1093/nar/gku337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Vogt I, Haque T, Campillos M. HitPick: A web server for hit identification and target prediction of chemical screenings. Bioinformatics (Oxford, England) 2013;29:1910–1912. doi: 10.1093/bioinformatics/btt303. [DOI] [PubMed] [Google Scholar]
- 44.Piñero J, et al. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucl. Acids Res. 2017;45:D833–d839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucl. Acids Res. 2017;45:W130–w137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 47.Szklarczyk D, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 2019;47:D607–d613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermawan A, Putri H, Utomo RY. Comprehensive bioinformatics study reveals targets and molecular mechanism of hesperetin in overcoming breast cancer chemoresistance. Mol. Div. 2019;24:1–15. doi: 10.1007/s11030-019-10003-2. [DOI] [PubMed] [Google Scholar]
- 50.Cerami E, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.cd-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013 doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Z, et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucl. Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li T, et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Can. Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.can-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li T, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucl. Acids Res. 2020;48:W509–w514. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh N, Miner A, Hennis L, Mittal S. Mechanisms of temozolomide resistance in glioblastoma - a comprehensive review. Cancer Drug Resist. 2021;4:17–43. doi: 10.20517/cdr.2020.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Schirnding C, et al. Synergistic combination of calcium and citrate in mesoporous nanoparticles targets pleural tumors. Chem. 2021;7:480–494. doi: 10.1016/j.chempr.2020.11.021. [DOI] [Google Scholar]
- 57.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 58.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human geneticsChapter 7, Unit7.20, doi:10.1002/0471142905.hg0720s76 (2013). [DOI] [PMC free article] [PubMed]
- 60.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi Y, Chan AP. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics (Oxford, England) 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat. Protoc. 2016;11:1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 63.Thomas PD, et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Phillips JC, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020;153:044130. doi: 10.1063/5.0014475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 67.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 68.Liu J, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e411. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ozaki T, Nakagawara A. Role of p53 in cell death and human cancers. Cancers (Basel) 2011;3:994–1013. doi: 10.3390/cancers3010994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Da’i M, Jenie UA, Supardjan A, Kawaichi M, Meiyanto E. T47D cells arrested at G2M and hyperploidy formation induced by a curcumin’s analogue PGV-1. Indones. J. Biotechnol. 2007;12:1005–1012. [Google Scholar]
- 71.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212. doi: 10.1038/s41418-018-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ham SW, et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26:409–425. doi: 10.1038/s41418-018-0126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Revathidevi S, Munirajan AK. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019;59:80–91. doi: 10.1016/j.semcancer.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 75.Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs. 2019;28:977–988. doi: 10.1080/13543784.2019.1676726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song M, Bode AM, Dong Z, Lee MH. AKT as a therapeutic target for cancer. Cancer Res. 2019;79:1019–1031. doi: 10.1158/0008-5472.can-18-2738. [DOI] [PubMed] [Google Scholar]
- 77.Shahcheraghi SH, et al. Wnt/beta-catenin and PI3K/Akt/mTOR signaling pathways in glioblastoma: Two main targets for drug design: A review. Curr. Pharm. Des. 2020;26:1729–1741. doi: 10.2174/1381612826666200131100630. [DOI] [PubMed] [Google Scholar]
- 78.Sordillo LA, Sordillo PP, Helson L. Curcumin for the treatment of glioblastoma. Anticancer Res. 2015;35:6373–6378. [PubMed] [Google Scholar]
- 79.Yin H, et al. Curcumin sensitizes glioblastoma to temozolomide by simultaneously generating ROS and disrupting AKT/mTOR signaling. Oncol. Rep. 2014;32:1610–1616. doi: 10.3892/or.2014.3342. [DOI] [PubMed] [Google Scholar]
- 80.Walker BC, Mittal S. Antitumor activity of curcumin in glioblastoma. Int. J. Mol. Sci. 2020 doi: 10.3390/ijms21249435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hackler L, Jr, et al. The curcumin Analog C-150, influencing NF-κB, UPR and Akt/Notch pathways has potent anticancer activity in vitro and in vivo. PLoS ONE. 2016;11:e0149832. doi: 10.1371/journal.pone.0149832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meiyanto E, Septisetyani EP, Larasati YA, Kawaichi M. Curcumin analog pentagamavunon-1 (PGV-1) sensitizes Widr cells to 5-fluorouracil through inhibition of NF-κB activation. Asian Pac. J. Cancer Prevent.: APJCP. 2018;19:49–56. doi: 10.22034/APJCP.2018.19.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 84.Soares JM, et al. Diosmin induces caspase-dependent apoptosis in human glioblastoma cells. Anais da Academia Brasileira de Ciencias. 2019;91:e20191031. doi: 10.1590/0001-3765201920191031. [DOI] [PubMed] [Google Scholar]
- 85.Shen X, et al. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 2016;17:1282–1290. doi: 10.1038/ni.3545. [DOI] [PubMed] [Google Scholar]
- 86.Dai M, Jenie UA, Margono S, Meiyanto E, Kawaichi M. The effect of PGV-1, PGV-0 and curcumin on protein involve in G2-M phase of cell cycle and apoptosis on T47D breast cancer cell line. Jurnal Ilmu Kefarmasian Indonesia. 2012;10:99–110. [Google Scholar]
- 87.Saadeh FS, Mahfouz R, Assi HI. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers. 2018;33:22–32. doi: 10.5301/ijbm.5000301. [DOI] [PubMed] [Google Scholar]
- 88.Eskilsson E, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018;20:743–752. doi: 10.1093/neuonc/nox191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wei X, et al. Higenamine alleviates allergic rhinitis by activating AKT1 and suppressing the EGFR/JAK2/c-JUN signaling. Phytomedicine. 2021;86:153565. doi: 10.1016/j.phymed.2021.153565. [DOI] [PubMed] [Google Scholar]
- 90.Zhao J, Zhang T, Liang Y, Zou H, Zhang J. Inhibitory activities of 20(R, S)-protopanaxatriol against epidermal growth factor receptor tyrosine kinase. Food Chem. Toxicol. 2021;155:112411. doi: 10.1016/j.fct.2021.112411. [DOI] [PubMed] [Google Scholar]
- 91.Tang Q, et al. Tubeimoside-I sensitizes temozolomide-resistant glioblastoma cells to chemotherapy by reducing MGMT expression and suppressing EGFR induced PI3K/Akt/mTOR/NF-κB-mediated signaling pathway. Phytomedicine. 2022;99:154016. doi: 10.1016/j.phymed.2022.154016. [DOI] [PubMed] [Google Scholar]
- 92.Chen P, et al. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019;38:254. doi: 10.1186/s13046-019-1234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee J-Y, et al. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: the versatile adjuvant for gefitinib therapy. PLoS ONE. 2011;6:e23756–e23756. doi: 10.1371/journal.pone.0023756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen A, Xu J, Johnson AC. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene. 2006;25:278–287. doi: 10.1038/sj.onc.1209019. [DOI] [PubMed] [Google Scholar]
- 95.Doumiati S, Haupt K, Rossi C. Autophosphorylation activation and inhibition by curcumin of the epidermal growth factor receptor reconstituted in liposomes. J. Mol. Recognit.: JMR. 2012;25:623–629. doi: 10.1002/jmr.2194. [DOI] [PubMed] [Google Scholar]
- 96.Starok M, et al. EGFR Inhibition by curcumin in cancer cells: A dual mode of action. Biomacromol. 2015;16:1634–1642. doi: 10.1021/acs.biomac.5b00229. [DOI] [PubMed] [Google Scholar]
- 97.Ali A, et al. Molecular engineering of curcumin, an active constituent of Curcuma longa L. (Turmeric) of the family Zingiberaceae with improved antiproliferative activity. Plants. 2021 doi: 10.3390/plants10081559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Velpula KK, et al. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget. 2017;8:35639. doi: 10.18632/oncotarget.16767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Struve N, et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene. 2020;39:3041–3055. doi: 10.1038/s41388-020-1208-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kobayashi Y, et al. EGFR Exon 18 mutations in lung cancer: Molecular predictors of augmented sensitivity to afatinib or Neratinib as compared with first- or third-generation TKIs. Clin. Cancer Research: An Off. J. Am. Assoc. Cancer Res. 2015;21:5305–5313. doi: 10.1158/1078-0432.ccr-15-1046. [DOI] [PubMed] [Google Scholar]
- 101.Frega S, Conte P, Fassan M, Polo V, Pasello G. A triple rare E709K and L833V/H835L <em>EGFR</em> Mutation Responsive to an irreversible Pan-HER inhibitor: A Case report of lung adenocarcinoma treated with Afatinib. J. Thorac. Oncol. 2016;11:e63–e64. doi: 10.1016/j.jtho.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 102.McGovern SL, et al. Extracellular domain mutations in EGFR occur uniquely in glioblastoma and favor ligand-independent formation of the active state. Int. J. Radiat. Oncol. Biol. Phys. 2012;84:S177. doi: 10.1016/j.ijrobp.2012.07.459. [DOI] [Google Scholar]
- 103.Xu H, et al. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017;14:512–516. doi: 10.3892/ol.2017.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brüchle NO, et al. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J. Investig. Dermatol. 2008;128:791–796. doi: 10.1038/sj.jid.5701088. [DOI] [PubMed] [Google Scholar]
- 105.Kim K-A, et al. R-Spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell. 2008;19:2588–2596. doi: 10.1091/mbc.e08-02-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nam J-S, Turcotte TJ, Smith PF, Choi S, Yoon JK. Mouse cristin/R-spondin family proteins are novel ligands for the Frizzled 8 and LRP6 receptors and activate β-catenin-dependent gene expression. J. Biol. Chem. 2006;281:13247–13257. doi: 10.1074/jbc.M508324200. [DOI] [PubMed] [Google Scholar]
- 107.Wang D, et al. Structural basis for R-spondin recognition by LGR4/5/6 receptors. Genes Dev. 2013;27:1339–1344. doi: 10.1101/gad.219360.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee JC, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3:e485. doi: 10.1371/journal.pmed.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kancha RK, Peschel C, Duyster J. The epidermal growth factor receptor-L861Q mutation increases kinase activity without leading to enhanced sensitivity toward epidermal growth factor receptor kinase inhibitors. J. Thorac. Oncol. 2011;6:387–392. doi: 10.1097/JTO.0b013e3182021f3e. [DOI] [PubMed] [Google Scholar]
- 110.Yang M, et al. Case report: Osimertinib achieved remarkable and sustained disease control in an advanced non-small-cell lung cancer harboring EGFR H773L/V774M mutation complex. Lung Cancer (Amsterdam, Netherlands) 2018;121:1–4. doi: 10.1016/j.lungcan.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 111.Chen L-C, Shih J-Y, Yu C-J, Yang C-Y. A rare epidermal growth factor receptor H773L/V774M compound mutation in advanced non-small-cell lung cancer with poor response to epidermal growth factor receptor tyrosine kinase inhibitor. Respirol. Case Rep. 2019;7:e00425–e00425. doi: 10.1002/rcr2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nayuta H, et al. Detection of EGFR Mutation Distribution and Transcriptional Variants in IDH-wildtype High-grade Gliomas using a Next-generation Sequencing Oncopanel. J. Neuro-Oncol. 2021 doi: 10.21203/rs.3.rs-861386/v1. [DOI] [Google Scholar]
- 113.Zhang W, et al. Mutation and polymorphism in the EGFR-TK domain associated with lung cancer. J. Thoracic Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer. 2006;1:635–647. [PubMed] [Google Scholar]
- 114.Meiyanto E, et al. Curcumin and its analogues (PGV-0 and PGV-1) enhance sensitivity of resistant MCF-7 cells to doxorubicin through inhibition of HER2 and NF-kB activation. Asian Pac. J. Cancer Prevent.: APJCP. 2014;15:179–184. doi: 10.7314/apjcp.2014.15.1.179. [DOI] [PubMed] [Google Scholar]
- 115.Wang Q, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32:42–56.e46. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 2018;9:1004–1004. doi: 10.3389/fimmu.2018.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Behnan J, Finocchiaro G, Hanna G. The landscape of the mesenchymal signature in brain tumours. Brain. 2019;142:847–866. doi: 10.1093/brain/awz044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martinez-Lage M, et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019;7:203–203. doi: 10.1186/s40478-019-0803-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 2018 doi: 10.3389/fimmu.2018.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rahimi Koshkaki H, et al. Immunohistochemical characterization of immune infiltrate in tumor microenvironment of glioblastoma. J. Personal. Med. 2020;10:112. doi: 10.3390/jpm10030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tang G, Yin W. Development of an immune infiltration-related prognostic scoring system based on the genomic landscape analysis of glioblastoma multiforme. Front. Oncol. 2020 doi: 10.3389/fonc.2020.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu S, et al. The prognostic landscape of tumor-infiltrating immune cells and immune checkpoints in glioblastoma. Technol. Cancer Res. Treat. 2019;18:1533033819869949. doi: 10.1177/1533033819869949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rick J, et al. TMIC-22. Identification of cancer-associated fibroblasts in glioblastoma and defining their protumoral effects. Neuro Oncol. 2019;21:vi252–vi252. doi: 10.1093/neuonc/noz175.1056. [DOI] [Google Scholar]
- 125.Trylcova J, et al. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumour Biol.: The J. Int. Soc. Oncodev. Biol. Med. 2015;36:5873–5879. doi: 10.1007/s13277-015-3259-8. [DOI] [PubMed] [Google Scholar]
- 126.Mu L, et al. CD4+ and perivascular Foxp3+ T cells in glioma correlate with angiogenesis and tumor progression. Front. Immunol. 2017;8:1451. doi: 10.3389/fimmu.2017.01451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Francis JM, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4:956–971. doi: 10.1158/2159-8290.Cd-13-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu F, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol. Cell. 2015;60:307–318. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guillaudeau A, et al. EGFR soluble isoforms and their transcripts are expressed in meningiomas. PLoS ONE. 2012;7:e37204. doi: 10.1371/journal.pone.0037204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kuo W-T, et al. Quantitative analysis of ligand-EGFR interactions: A platform for screening targeting molecules. PLoS ONE. 2015;10:e0116610. doi: 10.1371/journal.pone.0116610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weinholdt C, et al. Prediction of regulatory targets of alternative isoforms of the epidermal growth factor receptor in a glioblastoma cell line. BMC Bioinform. 2019;20:434. doi: 10.1186/s12859-019-2944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rutkowska A, Stoczyńska-Fidelus E, Janik K, Włodarczyk A, Rieske P. EGFR(vIII): An oncogene with ambiguous role. J. Oncol. 2019;2019:1092587. doi: 10.1155/2019/1092587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. Febs J. 2013;280:5350–5370. doi: 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 134.Fontanilles M, et al. Simultaneous detection of EGFR amplification and EGFRvIII variant using digital PCR-based method in glioblastoma. Acta Neuropathol. Commun. 2020;8:52. doi: 10.1186/s40478-020-00917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data produced by this study are disclosed in the manuscript and additional files. The raw data of gene expression can be accessed at the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), using accession number GSE206241.