

Abstract
Hypomethylating agents (HMAs), such as azacitidine and decitabine, induce cancer cell death by demethylating DNAs to promote the expression of tumor-suppressor genes. HMAs also reactivate the transcription of endogenous double-stranded RNAs (dsRNAs) that trigger the innate immune response and subsequent apoptosis via viral mimicry. However, the expression patterns of endogenous dsRNAs and their relevance in the efficacy of HMAs remain largely uninvestigated. Here, we employ amidine-conjugated spiropyran (Am-SP) to examine the dynamic expression pattern of total dsRNAs regulated by HMAs. By analyzing the bone-marrow aspirates of myelodysplastic syndrome or acute myeloid leukemia patients who received the HMAs, we find a dramatic increase in total dsRNA levels upon treatment only in patients who later benefited from the therapy. We further apply our approach in solid tumor cell lines and show that the degree of dsRNA induction correlates with the effectiveness of decitabine in most cases. Notably, when dsRNA induction is accompanied by increased expression of nc886 RNA, decitabine becomes ineffective. Collectively, our study establishes the potential application of monitoring the total dsRNA levels by a small molecule as an analytical method and a dynamic marker to predict the clinical outcome of the HMA therapy.
Keywords: MT: Noncoding RNAs, hypomethylating agents, spiropyran, double-stranded RNAs, dynamic biomarker, PKR, nc886, innate immune response
Graphical abstract
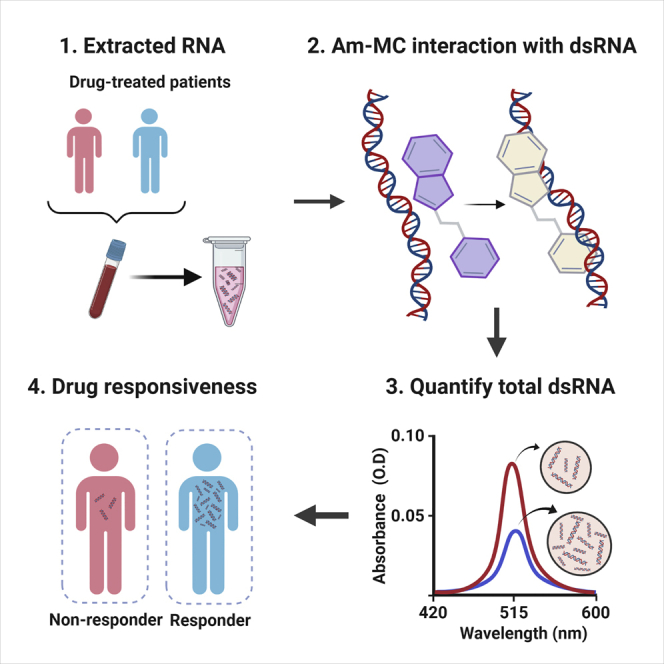
HMAs reactivate the transcription of endogenous dsRNAs that trigger antiviral signaling. An amidine-conjugated spiropyran was employed to monitor the collective expression of all dsRNAs. The induction of dsRNAs measured by the spiropyran could be used as a dynamic marker to predict the clinical outcome of the HMA therapy.
Introduction
Chemotherapy constitutes a major axis of today’s cancer treatment scene.1 One of the most important aspects of anti-cancer drug navigation is the timely prediction of patients' response to therapy and adequate monitoring of chemoefficacy. Hypomethylating agents (HMAs) are the cytosine analogs that were developed over 50 years ago as cytostatic agents.2 Two HMAs, 5-azacytidine (azacitidine [AZA]) and 2′-deoxy-5-azacytidine (decitabine [DAC]), have been incorporated as standard treatment in patients with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) of the elderly.3,4 Due to their effectiveness in inhibiting DNA methylation and significant clinical benefits, substantial efforts are ongoing to develop more effective epigenetic cancer therapies and to understand the exact action mechanism of these HMAs.5
The key rationale behind the HMA-based chemotherapy is the reactivation of hypermethylated tumor-suppressor genes that were silenced during tumorigenesis.6,7 Recently, numerous studies have shown that demethylation by HMAs also results in the transcription of endogenous double-stranded RNAs (dsRNAs), including endogenous retrovirus (ERV), short-interspersed nuclear element (SINE), and long-interspersed nuclear element (LINE) RNAs.8, 9, 10, 11, 12 These dsRNAs are recognized by dsRNA sensors of the innate immune response systems, such as protein kinase R (PKR) and melanoma differentiation-associated protein 5 (MDA5), which leads to global suppression of translation and induces apoptosis by transforming the cells into the viral mimicry state.9,11 The strength of immune activation by HMAs is regulated at multiple levels. Vitamin C increases the transcriptional activity, which results in higher dsRNA expression and, subsequently, stronger immune response.13 Post-transcriptionally, dsRNA-binding protein STAUFEN1 and long noncoding RNA TINCR form a complex to stabilize SINE and ERV RNAs activated by DAC to ensure downstream innate immune activation.9 Notably, the patients with low expression of STAUFEN1 and TINCR showed inferior response to the HMA therapy.9 In an effort to develop markers to predict the effectiveness of HMAs, a number of high-throughput studies characterized the demethylation by HMAs,9,10,14,15 but we still do not fully understand the extent of dsRNA induction required for sufficient downstream effect of HMAs to take place. If available, such information will greatly assist in predicting the efficacy of the HMA therapy.
In general, the expression of dsRNAs is analyzed by a polymerase chain reaction method, which can detect a specific dsRNA in a sequence-dependent manner. However, the human genome contains thousands of endogenous dsRNAs that are regulated by HMAs, and examining their expressions individually is impractical. Currently, dsRNAs longer than 40 bp can be detected using J2 or K1 antibody.16, 17, 18, 19 However, these antibodies are used mostly for the visualization of dsRNAs through imaging. Some quantitative analysis can be done through sandwich ELISA, but its clinical application is still limited.18,20 Alternatively, dsRNA-mediated immune activation can be measured indirectly by measuring the interferons, but the interferon response can occur by stimuli that may not rely on dsRNAs.21
In an effort to develop a detection method that utilizes a small molecule, a spiropyran (SP)-based approach has recently been employed to measure total dsRNA concentration based on changes in absorbance of the compound due to interaction with dsRNAs.22 The main principle is the intercalating activity of a planar structured isoform of SP known as a merocyanine (MC).22, 23, 24 The efficient intercalation of MC shifts its own absorbance by protonation of the phenolate oxygen in a dsRNA concentration-dependent manner.23,25 However, conventional SP-based dsRNA detection is insufficient to apply in a clinical setting due to low sensitivity from the weak electrostatic interaction with dsRNAs.
Herein, we employed a derivative of SP with amidine moiety (Am-SP and Am-MC for closed and open form, respectively) for enhanced dsRNA detection. After characterizing the interaction between dsRNAs and Am-SP/MC, we applied our sensor to bone-marrow aspirates from MDS or AML patients who received the HMA therapy. Moreover, we extended the application of Am-SP/MC to solid tumor cell lines that responded differently to DAC. Collectively, the current study provides an improved and clinically applicable platform for the detection of dsRNAs and introduces dsRNA induction as a potential dynamic biomarker to monitor the effect of HMAs.
Results
Am-MC interacts with dsRNAs with enhanced affinity
The spiropyran derivative used in this study has three associated forms, each of which displays a distinct absorbance spectrum (Figure 1A). Reversible isomerization between SP and MC can be induced using UV and visible light while MC can be converted to MCH+ via protonation. To enhance the water solubility of the spiropyran, we attached a trimethylammoniopropyl moiety to the indolenium nitrogen (Figure S1). Previously, we showed that a derivative of this spiropyran could interact with dsRNAs and undergo protonation to MCH+ in pH 7 solution with low ionic strength.22 This change from MC to MCH+ was the key sensing principle that enabled the quantification of total dsRNA concentrations.
Figure 1.

Interaction of the Am-MC with dsRNAs
(A) Schematic illustration of Am-SP, Am-MC, and Am-MCH+ used in this study. Interaction between Am-MC and dsRNA stabilizes Am-MCH+, resulting in decreased absorbance at 515 nm. (B, C) The absorbance spectra change of Am-MC by different concentrations of 20-bp poly AU (B) or poly GC (C) dsRNAs. (D) Quantification of the absorbance change at 515 nm by 20-bp poly AU or poly GC dsRNAs. (E) The absorbance spectra of Am-MC when poly AU dsRNAs with different lengths were added; 14 μM 100-bp poly AU, 70 μM 20-bp poly AU, and 140 μM 10-bp poly AU dsRNAs were used.
To increase the affinity between MC and negatively charged dsRNAs, we replaced the nitro group in the spiropyran with a positively charged amidine group, which resulted in a greater net positive charge of the molecule (Figures 1A and S1). Indeed, such spiropyran derivative was shown to exhibit enhanced affinity toward DNA duplexes.26 We denote the amidine-conjugated spiropyran as Am-SP. We then used UV-visible (UV-vis) spectroscopy and circular dichroism spectroscopy to characterize the interaction between the Am-SP and dsRNAs. These methods allowed us to confirm the mode and strength of interaction by quantifying the shifts in absorbance.27,28 First, we characterized the interaction between the closed Am-SP with dsRNAs. We prepared 100 μM 20-bp poly AU and poly GC dsRNAs in 9 mM Na+ and 1 mM cacodylate buffer at pH 7 and added 12 μM Am-SP. No change in the absorbance spectrum was observed, which indicated that the closed Am-SP did not interact with dsRNAs, potentially due to its non-planar structure hindering its association with RNA base pairs (bp) (Figures S2A and S2B). We also performed the same experiment in triple-distilled water (TDW) at pH 7 without Na+ ions and obtained the same result (Figures S2C and S2D).
Next, we irradiated Am-SP with UV light for 30 s to convert it to the open Am-MC and incubated it with poly AU or poly GC dsRNAs in 9 mM Na+ and 1 mM cacodylate buffer at pH 7. Upon adding dsRNAs, we observed a sharp decrease in the MC absorbance at 515 nm and an increase at 420 nm. Both the addition of poly AU and poly GC dsRNAs resulted in a similar change, although a slightly greater change was observed for poly GC (Figure S3). The shift from 515 to 420 nm could be described as hypsochromic, or blueshift, a common indicator of intercalation of the planar MC between the base pairs of the duplex nucleic acids.24 The degree of the change was greater in Am-MC compared with that of the conventional MC with a nitro group, indicating that the affinity between spiropyran and dsRNA was enhanced. We further increased the change in absorbance by reducing the ionic strength of the solution by using TDW at pH 7, which resulted in far greater (nearly 90%) changes in the absorbance at 515 nm (Figures 1B and 1C). There was also a consistent increase in absorbance peak at 420 nm, indicating the conversion of MC to MCH+ at a constant ratio during intercalation (Figure S4). Using the Am-MC, a 100 μM 20-bp dsRNA was enough to saturate the protonation of MC to MCH+, which was about five times more sensitive than our previous work.
Last, we used circular dichroism spectroscopy to characterize the mode of interaction and to examine whether intercalation resulted in any conformational changes to the nucleic acid duplex. Before the addition of Am-MC, the dsRNAs adopted a traditionally observed A-form helical conformation. Of note, we conducted the experiment in TDW because the maximum change in the absorbance of Am-MC was observed when TDW was used as the solvent. Our result indicated that, even without Na+ ions, dsRNAs could maintain their A-form helical structure (Figure S5). Upon introduction of Am-MC, we found increased base-pair stacking at ∼260 nm and a decreased helicity at ∼240 nm, which were representative changes associated with intercalative mode of binding (Figure S5). The increased stacking indicated that the Am-MC intercalated between the RNA bp, while a moderate decrease in helicity indicated a slight unwinding of the helix due to the intercalation of the Am-MC. These signature effects of intercalation were observed for both poly GC and poly AU dsRNAs.
Spectral change of Am-MC can be used to infer the dsRNA expression
For 20-bp dsRNAs, increasing the concentration of dsRNAs resulted in a greater change in the absorbance of Am-MC. By quantifying the absorbance change at 515 nm, we could clearly observe that the magnitude of the change was correlated with the concentration of dsRNAs (Figure 1D). Moreover, the change in the absorbance followed nearly a linear behavior initially, which became saturated at ∼100 μM in pH 7 TDW for both poly GC and poly AU dsRNAs.
Next, we asked whether the length of dsRNA plays a role in the interaction with Am-MC. First, we found that individual RNA nucleotides (rATP, rUTP, rGTP, and rCTP) did not interact with Am-MC (Figure S6). We then synthesized 10-bp, 20-bp, and 100-bp poly AU dsRNAs and analyzed their interaction with Am-MC. To compare dsRNAs with different lengths, we used equal base-pair concentration rather than equal molar concentration. In other words, 10-bp poly AU dsRNAs have a 10 times higher number of molecules than 100-bp poly AU dsRNAs, but they have the same number of RNA bases. We found that, although 10-bp poly AU dsRNA resulted in a slightly weaker change in the absorbance, 20-bp and 100-bp dsRNAs at the same base-pair concentration yielded an indistinguishable shift in the absorbance (Figure 1E). This indicates that a single 100-bp dsRNA produces the same shift as five molecules of 20-bp dsRNA.
Last, we compared the interaction of Am-MC with single-stranded RNA (ssRNA), dsRNA, or dsDNA. For ssRNA, we used in vitro transcribed luciferase mRNA with a length of 1,929 nucleotides. For dsRNA, we utilized pcDNA3 EGFP plasmid and transcribed sense and antisense EGFP mRNA using T7 RNA polymerase, which yielded approximately 717-bp-long dsRNA. Considering that Am-MC interacts with dsRNAs through intercalation, we compared equal bp concentrations of ssRNA and dsRNA. In other words, we used twice the amount of dsRNA because it exists in a double-stranded form. We found that an equal base-pair concentration of dsRNA yielded a greater spectral change of Am-MC than that of ssRNA (Figures S7A and S7B). To our surprise, ssRNA also altered the spectra of Am-MC, but the degree was smaller than that of dsRNA. In addition, we compared the interaction of Am-MC with dsDNA by utilizing the 717-bp EGFP dsDNA that we used as a template to transcribe the dsRNA. We found that Am-MC could also interact with dsDNA, resulting in a spectral change similar to dsRNA (Figure S7C). Although the degree of the change was smaller for dsDNA than that of dsRNA, our data indicate that removing genomic DNA and long ssRNA would be necessary for an accurate assessment of dsRNA expression.
Am-MC can monitor the expression of endogenous dsRNAs
Having established the interaction between synthetic oligonucleotides and Am-SP/MC, we applied our approach to the cellular scale to quantify the endogenous dsRNA expressions in HCT116 colorectal cancer cells. Specifically, we aimed to establish an experimental condition to capture the increase in total dsRNA expression when HCT116 cells were treated with HMAs such as DAC. We extracted DNA-free total RNAs from HCT116 cells and examined their effect on the absorbance of Am-MC. We found that 2 μg of total RNAs induced about 40% change in the absorbance, which was likely attributed to all of the secondary structured regions of various RNA species (Figure 2A). Treating total RNAs with RNase A to digest ss- and dsRNAs resulted in a significant increase in absorbance at 515 nm, indicating the original absorbance change was due to the interaction between Am-MC and various RNA species. Furthermore, to enrich and examine only the dsRNAs, we treated total RNAs with RNase T1 followed by phenol-chloroform extraction to remove ssRNAs and short structured RNAs. When 2 μg of RNase T1 digested RNAs were incubated with Am-MC, about ∼10% change in the absorbance was observed (Figure 2A). Of note, we confirmed the removal of short hairpin RNAs by RNase T1 digestion by utilizing a recently published primary microRNA (pri-miRNA) library.29 These pri-miRNA hairpin RNAs were the ideal samples as they contain imperfect stems with multiple bulges and internal loops and have a mean length of 35 ± 1 bp.29, 30, 31 Specifically, we transcribed over 1,800 pri-miRNA hairpins in vitro using T7 RNA polymerase, incubated them with RNase T1, and analyzed its effect through gel electrophoresis. RNase T1 completely removed these RNAs, and we could not detect any RNA signal (Figure S8).
Figure 2.

Detection of total dsRNA from HCT116 cells
(A) The absorbance spectra of Am-MC when mock-treated (HCT116), RNase T1-treated (RNase T1), or RNase A-treated (RNase A) HCT116 total RNA was added. Quantified percentage change at 515-nm peak is shown on the right. (B) Recovery of the indicated amount of EGFP dsRNAs spiked in to 4 μg of total RNA from HCT116 cells. The absorbance spectra of Am-MC for one set of experiment is shown on the left. The middle shows quantified absorbance change for three biological replicates with linear fitting; 400 ng of RNase T1-treated samples were used for the analysis. The right shows the dot blot using a J2 antibody for one set of experiments and the quantified intensity of the biological triplicates; 1 μg of RNase T1-treated RNA was used for the analysis. (C) Selected ERV RNA expression in total RNAs extracted from HCT116 cells after treating them with DAC for the indicated number of days. (D) Dot-blot assay to detect elevated dsRNA expression after DAC treatment; 1 μg of total RNA from HCT116 cells 5 days after the DMSO (DAC−) or DAC treatment was used for the analysis. (E) Western blotting of the PKR signaling pathway on HCT116 cell lysates 5 days after the DMSO (DAC−) or DAC treatment. TUBB was used as a loading control. (F) The absorbance spectra (left) and quantified 515-nm peak (right) of 2 μg of RNase T1-treated total RNAs from HCT116 cells after DAC treatment for the indicated duration. (G) The absorbance spectra of Am-MC when RNAs from DAC-treated HCT116 cells (5 days after the treatment) were digested with RNase III; 2 μg of RNase III digested RNA was used. In all plots, an average of three biological replicates are shown with error bars indicating SD. A Student’s t test was used for pairwise comparison, and one-way ANOVA was also used to analyze the data in (F). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further test the ability of Am-MC to detect changes in cellular dsRNA expression, we performed a spike-in and recovery experiment where we added known amounts of synthetic dsRNA and recovered them through the spectral change of Am-MC at 515 nm. We utilized EGFP dsRNA synthesized above and spiked in 0–4 μg of EGFP dsRNA into 4 μg of total RNAs extracted from HCT116 cells. We then treated these RNAs with RNase T1 and incubated 400 ng of enriched dsRNAs with Am-MC. We observed a clear linear correlation (r2 = 0.981) between the spectral change of Am-MC at 515 nm and the amount of dsRNAs added (Figure 2B). As a comparison, we performed a dot-blot assay using a J2 antibody on the same dsRNA spike-in samples. The detection using the J2 antibody required at least 2 μg of spiked in dsRNAs, and the linear correlation between the signal intensity and the amount of dsRNAs added was not as robust (r2 = 0.875) (Figure 2B).
We then applied this approach to examine the increase in total dsRNA levels by DAC. We treated HCT116 cells with 500 nM DAC for 24 h and harvested cells 0, 1, 3, 5, and 7 days later. We extracted DNA-free total RNAs and examined dsRNA induction. Using RT-qPCR, we examined the expression of selected ERV RNAs, which clearly showed strong induction, especially 3 days after the treatment (Figure 2C). Notably, not all ERV RNAs showed increased expression, and individual ERV RNAs showed very different degrees of induction. This further calls for the need to examine the total dsRNA expression in order to assess the effectiveness of DAC in reactivating endogenous dsRNAs to trigger antiviral signaling. To complement ERV RT-qPCR data, the induction of long dsRNAs by DAC was analyzed through the dot-blot assay using a J2 antibody on RNAs harvested 5 days after the DAC treatment (Figure 2D). Moreover, we also showed that DAC treatment resulted in the phosphorylation of PKR and its downstream substrate eIF2α, indicating immune activation due to an increased level of long dsRNAs (Figure 2E).
To analyze total dsRNA expression, we treated total RNAs with RNase T1 and incubated them with Am-MC. We found a clear decreasing trend in the absorbance peak at 515 nm over time (ANOVA, p < 0.05), indicating that the total dsRNA expression was increased continuously from 0 to 7 days after DAC treatment (Figure 2F). Of note, compared with our previous approach using a spiropyran with a nitro group, the current method with Am-MC yielded a greater and more consistent change in absorbance. In addition, Am-MC required about one-fifth of the sample amount in order to obtain a reliable change in the absorbance. To further confirm that the spectral changes of Am-MC was indeed due to increased expression of dsRNAs, we digested total RNAs from DAC-treated cells with RNase III instead of RNase T1. We found that the difference in the spectral properties of Am-MC for control (DAC−) and DAC-treated samples became negligible, indicating that the decrease in 515 nm peak of Am-MC was due to increased expression of dsRNAs that were sensitive to RNase III digestion (Figure 2G).
Total dsRNA induction is related to the patient response to HMAs
AZA and DAC are US Food and Drug Administration-approved HMAs for the treatment of elderly patients with MDS and AML.3,4 Using our Am-SP/MC, we asked whether dsRNA induction can be used as a dynamic biomarker to predict the therapy outcome (Figure 3A). We investigated 10 consecutive MDS or AML patients who were treated with AZA or DAC as the first-line therapy from July 2017 to December 2019 in Seoul National University Hospital (Seoul, Korea) and had paired cryopreserved samples of bone-marrow aspirates. Detailed information of the patients is summarized in Table S1. A pair comprises one baseline sample and one sample acquired at the time of response evaluation. Of note, we strictly limited this follow-up period to two to four cycles after initiation of the HMA therapy in order to investigate the potential of using dsRNA induction as an early marker for therapy outcome. We categorized them into two groups; five who did not benefit from either AZA or DAC (non-responder) and five who showed a response to either AZA or DAC (responder), based on response criteria for MDS32 and AML.33 We extracted DNA-free total RNAs from these paired bone-marrow aspirates and analyzed their dsRNA expression using the Am-MC. We found a significant change in the absorbance of the Am-MC in all patients of the responder group after receiving HMAs, while none from the non-responder group showed significant absorbance change (Figures 3B and S9). This indicates strong induction of dsRNAs in patients who benefited from the HMA therapy. Interestingly, the absorbance changes between the two groups before receiving therapy were indistinguishable (Figure 3B). In other words, the basal level of dsRNA could not predict the therapy outcome, while the increased dsRNA expression after the therapy could clearly predict the long-term clinical benefit from the therapy. Previously, we also showed that the expression of a number of ERV RNAs from bone-marrow aspirates of patients before receiving the HMA therapy could not predict the treatment outcome.9 Indeed, when we examined the basal expression of eight selected ERV RNAs via RT-qPCR, none of the genes showed any significant differences between the two groups (Figure 3C).
Figure 3.

dsRNA induction as a dynamic marker for the HMA therapy
(A) Schematic of the process for analyzing dsRNA expression in patient bone-marrow aspirates. (B) The total dsRNA expression in bone-marrow aspirates measured by the absorbance change at 515 nm of Am-MC. A total of 10 paired patient (five non-responders and five responders) samples were analyzed. (C) Expression of eight selected ERV RNAs in bone-marrow aspirates from patients prior to receiving HMAs. Each ERV expression was normalized by the median. (D) The induction of eight selected ERV genes after receiving HMAs was analyzed by RT-qPCR. All ERV RNA expressions were normalized to that of GAPDH mRNA. A paired Student’s t test was used to compare Am-MC spectral change before and after the therapy. A non-paired Student’s t test was used to compare the Am-MC spectral change between responder and non-responder groups. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We further examined the expression of a number of ERV RNAs from patients’ bone-marrow aspirates. We could clearly see strong induction of ERV RNAs in patients in the responder group, while the degree of induction was much smaller for patients in the non-responder group (Figure 3D). Of note, the amount of total RNAs available for patient #9 was too little for both Am-MC and RT-qPCR analyses, and, consequently, RT-qPCR data for patient #9 are missing. Moreover, we could not perform statistical analysis for individual ERV RNA induction because we could analyze only one sample per patient without biological replicates. Regardless, the difference in the degree of ERV induction between the two groups was apparent. Interestingly, most patients in the non-responder group still showed weak induction of ERV RNAs. However, the total dsRNA expression measured by Am-MC did not change significantly. Patient #6 did not show strong induction of selected ERV RNAs, but did show a dramatic change in the spectrum of Am-MC. Moreover, the type and magnitude of ERV induction were very different among patients. For example, patient #10 showed about a 90-fold increase in ERVL RNA expression upon receiving HMAs, while, for patient #8, ERVL expression was not induced at all (Figure 3D). Nevertheless, both patients showed a clear increase in the total dsRNA level assessed by Am-MC.
Spectral change of Am-MC correlates with DAC response in solid cancer cell lines
Based on the working mechanism of HMAs, they can be applied to cancer types other than MDS and AML. Indeed, numerous studies utilized solid tumor cell lines such as colorectal cancer and ovarian cancer to study the action mechanism of HMAs.8,9,11 Therefore, we asked whether we could apply our approach of using the Am-MC to monitor the kinetics of the total dsRNA induction as a marker to predict the efficacy of HMAs in various solid cancer cell lines.
We used seven different cancer cell lines and examined cell death, total dsRNA induction, and eight individual ERV RNA expressions upon DAC treatment. We treated cells with a fixed concentration of DAC for 24 h and examined the cell viability 4 days later. Consistent with a previous report,34 we found that cells responded differently to DAC. HCT116 colorectal cancer cells showed about a 50% decrease in cell viability, while A375 melanoma cells did not respond to DAC (Figure 4A). Moreover, increasing the concentration of DAC resulted in decreased cell proliferation in all cancer cells analyzed (Figure 4A). We then extracted total RNAs from these cancer cells at 1, 3, 5, and 7 days after 500 nM DAC treatment and analyzed dsRNA induction by Am-MC and RT-qPCR. Consistent with the cell viability result, HCT116 showed a continuous increase in the absorbance change over time, indicating elevated expression of total dsRNAs (Figure 4B). When individual ERV RNA expression was examined via RT-qPCR, we found that ERVL showed a 200-fold increase by day 3, and most of the other ERV RNAs showed increasing expression patterns over time (Figure 4C). In addition to HCT116, MCF-7 breast cancer cells also showed strong induction of dsRNAs 3 days after DAC treatment (Figure 4B). Analyzing individual ERV RNAs also revealed the induction of many ERV RNAs starting 3 days after drug treatment. Notably, MLT1C49 RNA showed a 150-fold induction by day 7, which is very different from the response in HCT116 cells (Figure 4C). This result further strengthens our argument that monitoring the total dsRNA level rather than analyzing the expression of individual dsRNAs provides a more relevant marker for the effect of DAC. Interestingly, despite the dsRNA induction, the cell viability did not decrease significantly in MCF-7 cells (Figure 4A). We further investigate the possible factors that might account for this phenomenon in the next section.
Figure 4.

Analysis of dsRNA induction by DAC in solid tumor cell lines
(A) Heatmap of cell viability of different cancer cell lines 4 days after the DAC treatment. (B) Percentage absorbance change of 515-nm peak of Am-MC after incubating with 2 μg of RNase T1-treated RNAs from indicated cells. The average of three biological replicates is shown with error bars indicating SD. (C) Expression change of eight selected ERV RNAs measured by RT-qPCR. All ERV RNA expressions were normalized to that of GAPDH mRNA. The average of three biological replicates were shown with error bars denoting SD. (D) The correlation between cell viability and the absorbance change of Am-MC from day 0 to day 5. (E) The correlation between cell viability and the basal Am-MC absorbance at 515 nm.
PANC-1 pancreatic cancer, CAOV3 ovarian cancer, and HeLa cervical cancer cells showed a moderate increase in total dsRNA expression and a moderate decrease in cell proliferation, particularly for PANC-1 and CAOV3 (Figures 4A and 4B). Examining individual ERV RNAs also showed only a small increase in HeLa and a moderate increasing trend in CAOV3 cells (Figure 4C). For PANC-1, ERVL expression was increased over 100-fold by day 7, but the rest of ERV RNAs only showed a small change (Figure 4C). This result again indicates that examining a single or a few ERV genes as a marker for the effect of DAC can be misleading.
Last, A549 lung cancer and A375 melanoma cells did not show any induction in total dsRNA expression, and their proliferation was unaffected by DAC treatment (Figures 4A and 4B). Interestingly, when we examined individual ERV RNA expressions, MER21C showed a 150-fold induction 7 days after the DAC treatment in A549 cells (Figure 4C). Nevertheless, the overall dsRNA induction measured by Am-MC was marginal, and the cell proliferation did not decrease significantly in A549 cells. In A375 cells, there was no induction in any of the eight ERV genes examined, and there was no change in total dsRNA expression by DAC (Figures 4A–4C).
When we compared the cell proliferation with the degree of total dsRNA induction 5 days after the DAC treatment in different cancer cell lines, we could clearly observe a negative correlation that increased dsRNA induction resulted in decreased cell viability (Pearson’s r = −0.755 and p < 0.05) (Figure 4D). Interestingly, when we plotted the basal dsRNA expression measured by Am-MC’s absorbance change at day 0 versus cell viability, we saw a weak positive correlation (Figure 4E). A375 cells showed high basal absorbance change at day 0, indicating that perhaps the basal endogenous dsRNA expression was high in these cells, but they still did not respond to DAC treatment. At the same time, HCT116 cells with low basal dsRNA level showed strong dsRNA induction by DAC treatment, resulting in a significant decrease in cell viability.
Previous investigations on DAC categorized CAOV3 and A375 cells in a group with a good response to DAC.34 We also observed a moderate increase in the total dsRNA expression as well in many individual ERV RNAs in CAOV3 cells, but the magnitude was too small to be effective in triggering apoptosis. To understand the discrepancy, we modified the DAC treatment condition and examined its effect on dsRNA induction and cell viability. For CAOV3, we increased the DAC concentration to 1 μM and the duration of treatment from 1 to 7 days. Under the modified treatment condition, we found increased dsRNA induction by day 5, which was accompanied by a significant decrease in cell viability (Figures 5A–5C). When compared with the previous condition, the absorbance change of Am-MC between day 0 and day 7 was increased from 2% to 15%, while the cell viability was decreased from 95% to 32%. Similarly, we increased the DAC treatment concentration to 5 μM and the duration of treatment to 9 days in A375 cells. Clearly, we found increased total dsRNA expression (absorbance change of Am-MC from 0% to 16%), which was accompanied by a decreased cell viability from 93% to 41% (Figures 5A–5C).
Figure 5.

Optimizing DAC treatment condition using Am-MC
(A) Absorbance change of Am-MC upon addition of 2 μg of RNase T1-treated RNAs extracted from CAOV3 (left) or A375 (right) cells treated with DAC (1 μM for CAOV3 and 5 μM for A375). The average of three biological replicates is shown with error bars indicating SD. (B) Expression of eight selected ERV RNAs quantified by RT-qPCR. The average of three biological replicates is shown with error bars indicating SD. (C) The correlation between the cell viability and the absorbance change of Am-MC for two different treatment conditions. Vertical error bars (n = 4) and horizontal error bars (n = 3) were expressed as SD.
nc886 induction counters the effect of DAC
One notable result from our investigation is that MCF-7 cells showed strong dsRNA induction measured by both Am-MC and RT-qPCR, but the effect of dsRNAs on cell viability was marginal. A possible explanation is that DAC treatment also resulted in increased expression of RNAs that inhibited the dsRNA sensors of the innate immune response system. Therefore, we investigated the expression of a noncoding RNA nc886, which is a potent inhibitor of PKR.35
nc886, also known as pre-miR-886 or vault RNA 2-1 (vtRNA2-1), is a 101-nucleotide-long noncoding RNA that is transcribed by RNA polymerase III.36 nc886 forms a short hairpin structure that binds to PKR and inhibits its activation by preventing PKR-dsRNA interaction.35,37 nc886 expression is also closely related to drug response as doxorubicin treatment suppresses nc886 expression to trigger apoptosis mediated by PKR.38 Considering that the nc886 promoter is hypermethylated in breast cancer, we asked whether DAC treatment results in nc886 reactivation, which might counter the effect of endogenous dsRNAs. Using RT-qPCR, we examined the level of nc886 in MCF-7 cells upon DAC treatment and found that its expression was increased over 10-fold within 3 days after the treatment (Figure 6A). To test whether downregulating nc886 could enhance the effect of DAC, we transfected cells with siRNAs that could bind to nc886 and decreased its expression by 50% (Figure 6B). Notably, we found significantly reduced cell viability when nc886-depleted cells were treated with DAC (Figure 6C). Similar effects were observed when we used a different siRNA to downregulate nc886 (Figure S10). In addition, we examined the effect of nc886 knockdown on the PKR signaling pathway. First, we confirmed PKR and eIF2α phosphorylation in nc886-depleted cells. This effect is consistent with previous reports, which reported that nc886 depletion decreases proliferation and activates the PKR signaling pathway.39,40 More importantly, while DAC treatment alone only weakly activated PKR, treating DAC in nc886-depleted cells resulted in increased pPKR and peIF2α levels (Figure 6D). Of note, the additive effect of nc886 knockdown on DAC-treated cells was not as dramatic due to the strong induction of nc886 by DAC. Indeed, the remaining nc886 expression was even higher than that of the dimethyl sulfoxide (DMSO)-treated control cells (Figure 6B). Nevertheless, lowering the expression of nc886 resulted in stronger activation of the PKR signaling pathway, indicating that targeting nc886 could enhance the downstream effect of dsRNA induction by DAC.
Figure 6.

Effect of nc886 in preventing DAC-mediated cell death
(A) Expression of nc886 RNA quantified by RT-qPCR in DAC-treated MCF-7 cells. nc886 RNA expressions were normalized to that of vtRNA1-1. (B) The downregulation of nc886 expression after transfecting cells with siRNAs against nc886. (C) The effect of knockdown of nc886 on cell viability with or without DAC treatment. MCF-7 cells 4 days after DMSO (DAC−) or DAC treatment were used. (D) Western blotting of the PKR signaling pathway when nc886 was knocked down with or without DAC treatment. MCF-7 cells 5 days after DMSO (DAC−) or DAC treatment were used for the analysis. siLuc was used as a transfection control (sinc886−), and TUBB was used as a loading control. Quantified data for biological triplicates is shown below. In all plots, the average of three biological replicates is shown with error bars indicating SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We then examined the nc886 expression in other cell lines used in this study. We found that PANC-1 and CAOV3 showed strong induction of nc886, but the degree of dsRNA induction was only moderate in these cells (Figure S11). In HeLa and A549 cells, neither dsRNA nor nc886 was reactivated by DAC treatment. Notably, nc886 expression did not change in HCT116 cells, which was consistent with our data that DAC treatment resulted in a strong downstream immune activation in these cells (Figure S11).
Discussion
In this study, we report the application of a spiropyran derivative, Am-MC, with an enhanced affinity toward dsRNAs for the examination of total dsRNAs expressed in cells. By quantifying the dsRNA-mediated absorbance change of Am-MC, we could accurately monitor changes in the total dsRNA expression. We applied this approach to show that MDS and AML patients who benefited from the HMA therapy exhibited increased total dsRNA expression within the first two to four cycles of the HMA therapy. Solid tumor cell lines also showed a good correlation between total dsRNA induction and cell viability in response to DAC treatment. Notably, in both cases, only the change in dsRNA expression rather than the basal expression of dsRNAs before HMA treatment provided a powerful predictive marker for the treatment.
To date, quantification of dsRNAs has mostly relied on RT-qPCR and next-generation sequencing, which is costly and labor intensive. The dot-blot assay using the J2 antibody requires vacuum-based sample preparation apparatus and suffers from low sensitivity. The need to seek an improved analytical diagnosis system with clinical applicability is driven by the development of a chemical-based indicator. Spiropyran-based sensors have been widely used to detect nucleic acids.23,24,41 We also applied the traditional nitro-spiropyran derivative in the colorimetric detection of dsRNAs.22 By conjugating a positively charged amidine group, our Am-SP/MC resulted in a 5-fold increase in the binding affinity toward dsRNAs compared with traditional nitro-spiropyran. In addition, interaction with dsRNAs rather than with ssRNAs or dsDNAs resulted in the largest change in the absorbance spectra of Am-MC. To our surprise, luciferase mRNA also affected the absorbance spectra of Am-MC. One possibility is the generation of untemplated dsRNA byproducts during the in vitro transcription, as reported previously.42,43 Alternatively, luciferase mRNAs may contain hairpin structures that are long enough to interact with Am-MC. This potential interaction with short hairpin structures requires the RNase T1 digestion, which poses a limitation to our approach. Moreover, our Am-MC still required at least 500 ng of total RNAs for reliable measurement. Although the current Am-MC was sufficient in monitoring dsRNA induction in bone-marrow aspirates of MDS and AML patients, further modifications may improve the sensitivity and specificity of the spiropyran-based dsRNA sensor.
One advantage of using spiropyran is that many functional moieties can be conjugated to the compound to enhance its affinity toward a specific target. Indeed, spiropyran derivatives are used to detect different cations, anions, acids, solvents, and even G-quadruplex DNAs and aptamers.23 In our case, conjugating additional positively charged moiety may further enhance the affinity toward dsRNAs, resulting in increased sensitivity. Moreover, the current Am-MC could reliably detect dsRNAs longer than 20 bp. However, in order to induce immune response, dsRNAs should be at least 33 bp and preferably longer than 79 bp.44,45 Although our Am-MC can detect long dsRNAs much more efficiently than the short dsRNAs, it needs further modification to specifically recognize long dsRNAs for a more reliable assessment of immune-activating dsRNAs present in cells. One possibility is making a spiropyran oligomer, which may preferentially bind to long dsRNAs. Synthesizing these spiropyran derivatives and examining their potential application as a long dsRNA-specific sensor may further strengthen the clinical application of dsRNA measurement.
Analysis of dsRNA expression in bone-marrow aspirates of MDS and AML patients demonstrated the potential clinical application of our Am-SP/MC. Strikingly, all five patients who benefited from the HMA therapy showed a dramatic change in the absorbance of Am-MC, indicating strong induction of dsRNA expression. More importantly, we analyzed patients whose follow-up samples were obtained two to four cycles after initiation of the HMA therapy. Although such strict selection criteria limited the number of samples we could analyze, our approach clearly demonstrated the potential of using the dsRNA induction as an early indicator to predict the overall treatment response at the time of the first bone-marrow response evaluation. Notably, the basal dsRNA expression before the therapy did not provide any information about the long-term outcome of the therapy. Analyzing individual ERV elements also failed to provide a common gene that could be used as a predictive marker. In fact, some of the patients from the non-responder group showed induction of a number of ERV elements, but the overall magnitude might be too low to have a benefit. These encouraging results need to be confirmed in a prospective study with a larger number of patients. Evaluating the dsRNA induction earlier after initiation of HMA treatment (e.g., after the first cycle of treatment) for the early prediction of treatment response could be considered. The correlation between dsRNA induction measured by Am-MC and the downstream immune response, such as the expression of interferon-stimulated genes, should be examined. Moreover, dsRNA expression in the peripheral blood instead of bone-marrow aspirate may provide a more attractive approach if they show a good correlation of dsRNA induction with each other since bone-marrow study is an invasive and painful procedure. Such an approach will be particularly useful in examining the effect of HMAs in solid tumors. Our study provides the scientific foundation that the change in total dsRNA expression accessed by Am-SP can be developed as a potential dynamic biomarker to predict the outcome of the current HMA therapy in clinical practice.
Our results also provide the foundation to monitor the effectiveness of HMAs in solid tumors. By analyzing several cancer cell lines, we showed that the dsRNA induction measured by Am-MC showed a good correlation with the cell viability in DAC-treated cells. Similar to the case of MDS/AML patients, the basal expression of dsRNAs before DAC treatment failed to provide the treatment outcome. It is the change in the total dsRNA expression that could reliably translate into the effectiveness of DAC in inducing cell death. We further demonstrated that Am-MC could be used as a tool that complemented cell viability data in studying the downstream effect of DAC. Through this approach, we found an interesting phenomenon where reactivation of PKR inhibitor nc886 countered the effect of dsRNA induction by DAC. By monitoring changes in inhibitors of dsRNA-mediated immune activation such as TAR RNA-binding protein (TRBP), adenosine deaminase acting on RNA (ADAR), and P58IPK, we may obtain a more comprehensive picture to predict the effect of DAC accurately.46, 47, 48 Such a diagnostic platform will significantly enhance the clinical applicability of HMAs in cancer.
Although HMA monotherapy is the standard treatment for MDS and AML, it is not considered a curative option due to limitations in terms of efficacy. However, HMA is still attractive as a partner for combination treatment with newer anti-cancer agents, including BCL-2 inhibitors and immune checkpoint inhibitors, attributed to its immunomodulatory and possible undefined mechanism.49, 50, 51, 52 A study to evaluate whether an increase in dsRNA expression in response to HMA exposure can also predict the synergistic effect of HMA-containing combination therapy could be considered not only in MDS or AML but also in solid tumors.
Materials and methods
Synthesis
Am-SP was designed by conjugating an amidine moiety to the spiropyran described previously.22 The detailed synthesis steps are provided in the supplemental information. The NMR spectroscopy of the final Am-SP product is shown in Figure S1.
Characterization
Am-SP was dissolved in TDW to produce the desired concentration of 12 μM. The Am-SP was exposed to 254-nm UV light using a UV lamp (7 mW/cm2) for 30 s to convert it to the open Am-MC form with an absorbance of ∼0.080 at 515 nm. The experiment was kept in the dark to avoid any reverse isomerization back to the Am-SP. RNA samples prepared at a specific concentration were added directly by pipetting into a cuvette containing Am-MC, and the absorbance (A) was measured within 2 min using a spectrophotometer (Eppendorf, USA) with a path length of 1 mm. Since the duration of the experiment was much shorter than the time required for the thermal equilibrium of Am-MC (∼6 h), thermal conversion was neglected. To determine the thermal equilibrium of Am-MC, the time-dependent absorbance at 515 nm was measured using Varioskan Lux microplate reader (Thermo Fisher Scientific, USA). Absorbance was measured with the baseline and background correction in order to scale and establish consistent isosbestic points at ∼395 and ∼480 nm. The percentage absorbance change at 515 nm was calculated with the following equation: (Asample − Acontrol)/Acontrol × 100 (%).
Synthetic DNA and RNA samples
Ten- or 20-bp synthetic dsRNAs were ordered in dialyzed lyophilized powder from Bioneer (Korea) and resuspended in a suitable amount of TDW at pH 7 and stored at −20°C. The 100-bp poly AU RNA was purchased from Santa Cruz Biotechnology (USA) and handled in the same way. For long ssRNA, luciferase mRNA (Promega, USA) was used and dissolved in a suitable amount of TDW. For long dsRNA, pcDNA3 EGFP plasmid was used as a template to synthesize sense and antisense RNA using MEGAscript T7 transcription kit (Thermo Fisher Scientific, USA). For the dsDNA sample, EGFP PCR product, used as a template for dsRNA, was utilized, and pri-miRNA hairpin RNAs were synthesized from cDNA library using MEGAscript T7 transcription kit following the previous publication.29 Briefly, cDNA library was amplified using forward (5′-TAATACGACTCACTATAGGGCCTATTCAGTTACAGCG-3′) and reverse (5′-GTTGCTAGCTTCAGTACG-3′) primers. The PCR product was used as a template for in vitro transcription. Transcribed RNA was column purified using the MEGAclear Transcription Clean-Up kit (Thermo Fisher Scientific, USA).
RNase treatment
RNase T1 (Worthington Biochemical, USA), RNase III (Thermo Fisher Scientific, USA), and RNase A (Worthington Biochemical, USA) were used to remove ssRNAs, dsRNA, or total RNAs, respectively. RNase stock solutions were prepared by dissolving 100 mg of RNase T1 (activity of 346,000 ku/mg) or 1 g of RNase A (activity of 6,800 ku/mg) in 1 mL of TDW. RNase III was supplied in a solution and did not require reconstitution. One to 5 μg of total RNAs in TDW were incubated with 30 μL of RNase T1 or RNase A for 30 min at 37°C. For RNase III, 5 μg of total RNA was mixed with 5 μL of 10× RNase III reaction buffer, 15 μL of RNase III (15 U), and TDW to a final volume of 50 μL. The mixture was incubated at 37°C for 1 h. The RNase digested RNAs were cleaned through phenol-chloroform extraction. Twelve micromolar Am-MC was added to the sample, and the absorbance was measured and analyzed as above.
Cell lines and cell culture
Human colorectal cancer (HCT116), breast cancer (MCF-7), cervical cancer (HeLa), ovarian cancer (CAOV3), pancreatic cancer (PANC-1), lung cancer (A549), and melanoma cancer (A375) cell lines were purchased from Korea Cell Line Bank (KCLB, Korea) or American Type Culture Collection (ATCC, USA). HCT116, MCF-7, A375, and A549 cells were cultured in RPMI-1640 (Welgene, Korea) supplemented with 10% fetal bovine serum (FBS; GIBCO, USA). HeLa, CAOV3, and PANC-1 were cultured in DMEM (Welgene, Korea) supplemented with 10% FBS. All cancer cells were incubated at 37°C in 5% CO2 humidified incubator.
MTT assay
Cells were seeded into a 96-well plate at a density of ∼1 × 104 cells/well. After 24 h, cells were treated with 500 nM DAC (Sigma-Aldrich, USA) for 24 h and cultured for three additional days. Then 10 μL of MTT solution (2.5 mg/mL in PBS, Thermo Fisher Scientific, USA) was added, and the cells were incubated for 3 h. The formed formazan was solubilized with 100 μL of DMSO (Santa Cruz Biotechnology, USA), and the absorbance at 590 nm was measured using a Varioskan Lux microplate reader (Thermo Fisher Scientific, USA).
Total RNA extraction, sample preparation, and RT-qPCR
Cells were seeded at a density of ∼5 × 105 cells in a 100-mm culture dish. The cells were treated with 500 nM DAC for 24 h, and total RNA was extracted using TRIsure (Meridian Bioscience, UK) at 0, 1, 3, 5, or 7 days after treatment. DNA was removed using DNase I (Takara, Japan), and RNA was purified via phenol-chloroform extraction. For RT-qPCR analysis, total RNA was reverse transcribed using RevertAid reverse transcriptase (Thermo Fisher Scientific, USA) with random hexamers (Thermo Fisher Scientific, USA). Synthesized cDNA was amplified using the SensiFAST SYBR Lo-Rox Kit (Meridian Bioscience, UK) in AriaMx Real-Time PCR system (Agilent Technologies, USA). The sequences of the primers used in this study are provided in Table S2. To measure the total dsRNA level using Am-SP, 1–5 μg of DNA-free total RNA was further treated with RNase T1 and cleaned through phenol-chloroform extraction. From 0.2 to 2 μg of RNase-T1-treated samples were incubated with 12 μM Am-MC, and the absorbance was measured and analyzed as described above.
Transfection
To modulate nc886 levels, MCF-7 cells were transfected with sinc886 (Bioneer, Korea) using Lipofectamine 3000 (Thermo Fisher Scientific, USA) following the manufacturer’s instruction. MCF-7 cells were transfected with siLuc (control) or sinc886 twice, 24 h before and 24 h after DAC treatment. The siRNA sequences used in this study are available in Table S3.
Human bone-marrow samples
Ten paired (a sample at the time of initial diagnosis and the other at the time of response evaluation) cryopreserved bone-marrow aspirates from 10 MDS or AML patients treated with AZA or DAC as the first-line therapy at Seoul National University Hospital (SNUH) were analyzed. Patients were selected using the following criteria. First, patients underwent follow-up analysis of their bone-marrow aspirates after receiving the HMA therapy. Second, the follow-up period was limited to two to four cycles after initiation of the therapy to test the potential for using early dsRNA induction to predict the overall therapy outcome. Third, patients were excluded if they had inadequate bone-marrow samples to interpret or ambiguous bone-marrow findings that made it hard to define either responder or non-responder according to the International Working Group standardized criteria for MDS32 and 2017 European LeukemiaNet criteria for AML.33 Only patients who received a complete remission (CR) or its variants (for example, marrow CR or CR with incomplete blood recovery) after HMA treatment were classified as responders. DNA-free total RNA was purified from the bone-marrow aspirates using NucleoSpin RNA Kit (Macherey-Nagel, Germany) and analyzed using Am-SP and RT-qPCR as described above. The use of clinical data and preserved samples from the biorepository was approved by the Institutional Review Board (IRB) of SNUH, Seoul, Republic of Korea (IRB no. H 2012-185-1185). Detailed information of the patients is summarized in Table S1.
Dot blot
One microgram of total RNAs was blotted on a positively charged nylon membrane (Cytiva, USA) using a Bio-Dot SF microfiltration apparatus (Bio-Rad, USA). Nylon membrane was activated in TDW and placed on the membrane support. A vacuum was applied for 30 min, and the membrane was air dried. RNAs blotted onto the nylon membrane were crosslinked using a microprocessor-controlled UV crosslinker (Spectrolinker XL-1000) with the “optimal crosslink” mode. After UV crosslinking, membranes were washed with Tris-buffered saline solution (TBST, 1.36 M NaCl, 0.2 M Tris base, 0.5% Tween 20). After blocking the membranes with 5% skim milk for 1 h, membranes were incubated overnight at 4°C with J2 antibody (Jena Bioscience, Germany) at 1:500 dilution. After three washes with TBST, membranes were incubated for 2 h with a horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson Laboratory, USA) at 1:5,000 dilution.
Western blot
Cell lysates were prepared by resuspending cell pellets in the lysis buffer (50 mM Tris-HCl pH 8.0, 100 mM KCl, 0.5% NP-40, 10% glycerol, and 1 mM DTT) supplemented with a protease inhibitor cocktail (Merck, USA). For complete lysis, cells were sonicated using a Bioruptor sonicator (Diagenode, Belgium). Then 40 μg of protein samples were separated on a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride (PVDF membrane using an Amersham semi-dry transfer system (GE Healthcare, USA). The membrane was blocked in 5% skim milk for 1 h and incubated overnight with a primary antibody in 1% skim milk at 1:1,000 dilution. Membranes were washed three times in PBS supplemented with 0.1% Tween 20 (PBST). HRP-conjugated secondary antibody (Thermo Fisher Scientific, USA) was used at 1:5,000 dilution in 1% skim milk. The following primary antibodies were used: anti-PKR (Santa Cruz Biotechnology, USA), anti-pPKR (Abcam, USA), anti-eIF2α (Cell Signaling Technology, USA), anti-peIF2α (Cell Signaling Technology, USA), and anti-β-tubulin (TUBB; Cell Signaling Technology, USA).
Statistical analysis
Statistical analysis was carried out using Student’s t test (compared with the control group) or one-tailed ANOVA. The detailed statistical analysis is described in each figure caption.
Acknowledgments
We thank Professor V. Narry Kim at Seoul National University for providing the pri-miRNA cDNA library. We thank the members of our laboratories and all of the people who helped with the research, especially Ryeongeun Cho. This work was financially supported by the Basic Research Laboratory Program through the National Research Foundation of Korea (grant number NRF-2021R1A4A3032789) funded by the Korean government’s Ministry of Science and ICT, and KAIST Interdisciplinary Research Group project (grant number N11220037) funded by KAIST.
Author contributions
M.K., R.K., J.M.B., J.J., S.-J.V.L., J.H., and Y.K. designed the research. M.K., R.K., J.M.B., J.J., A.A.A., D.K., J.Y., J.S., and Y.K. performed the research. R.K. synthesized Am-SP. J.M.B., D.-Y.S., Y.K., S.-S.Y., and J.H. collected and analyzed patient samples. M.K., R.K., J.M.B., J.J., A.A.A., J.H., and Y.K. wrote the paper. All authors analyzed the data.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.07.014.
Contributor Information
Junshik Hong, Email: hongjblood@snu.ac.kr.
Yoosik Kim, Email: ysyoosik@kaist.ac.kr.
Supplemental information
Data availability
All data and resources used in this study will be made available upon written request to the corresponding authors.
References
- 1.Chabner B.A., Roberts T.G. Chemotherapy and the war on cancer. Nat. Rev. Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Šorm F., Pískala A., Cihák A., Veselý J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. 1964;20:202–203. doi: 10.1007/BF02135399. [DOI] [PubMed] [Google Scholar]
- 3.Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Lai C., Doucette K., Norsworthy K. Recent drug approvals for acute myeloid leukemia. J. Hematol. Oncol. 2019;12:100. doi: 10.1186/s13045-019-0774-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly T.K., De Carvalho D.D., Jones P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwon M.J., Shin Y.K. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int. J. Mol. Sci. 2011;12:983–1008. doi: 10.3390/ijms12020983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng J.M.-K., Yu J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int. J. Mol. Sci. 2015;16:2472–2496. doi: 10.3390/ijms16022472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiappinelli K.B., Strissel P.L., Desrichard A., Li H., Henke C., Akman B., Hein A., Rote N.S., Cope L.M., Snyder A., et al. Inhibiting DNA methylation Causes an interferon response in cancer via dsRNA including endogenous Retroviruses. Cell. 2017;169:361. doi: 10.1016/j.cell.2017.03.036. [DOI] [PubMed] [Google Scholar]
- 9.Ku Y., Park J.H., Cho R., Lee Y., Park H.M., Kim M., Hur K., Byun S.Y., Liu J., Lee Y.S., et al. Noncanonical immune response to the inhibition of DNA methylation by Staufen1 via stabilization of endogenous retrovirus RNAs. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016289118. e2016289118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehdipour P., Marhon S.A., Ettayebi I., Chakravarthy A., Hosseini A., Wang Y., de Castro F.A., Loo Yau H., Ishak C., Abelson S., et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. 2020;588:169–173. doi: 10.1038/s41586-020-2844-1. [DOI] [PubMed] [Google Scholar]
- 11.Roulois D., Loo Yau H., Singhania R., Wang Y., Danesh A., Shen S.Y., Han H., Liang G., Jones P.A., Pugh T.J., et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolff F., Leisch M., Greil R., Risch A., Pleyer L. The double-edged sword of (re)expression of genes by hypomethylating agents: from viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun. Signal. 2017;15:13. doi: 10.1186/s12964-017-0168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M., Ohtani H., Zhou W., Ørskov A.D., Charlet J., Zhang Y.W., Shen H., Baylin S.B., Liang G., Grønbæk K., Jones P.A. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA. 2016;113:10238–10244. doi: 10.1073/pnas.1612262113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klco J.M., Spencer D.H., Lamprecht T.L., Sarkaria S.M., Wylie T., Magrini V., Hundal J., Walker J., Varghese N., Erdmann-Gilmore P., et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. 2013;121:1633–1643. doi: 10.1182/blood-2012-09-459313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai H.C., Li H., Van Neste L., Cai Y., Robert C., Rassool F.V., Shin J.J., Harbom K.M., Beaty R., Pappou E., et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhir A., Dhir S., Borowski L.S., Jimenez L., Teitell M., Rötig A., Crow Y.J., Rice G.I., Duffy D., Tamby C., et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560:238–242. doi: 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y., Park J., Kim S., Kim M., Kang M.-G., Kwak C., Kang M., Kim B., Rhee H.-W., Kim V.N. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell. 2018;71:1051–1063.e6. doi: 10.1016/j.molcel.2018.07.029. e1056. [DOI] [PubMed] [Google Scholar]
- 18.Ku J., Kim S., Park J., Kim T.S., Kharbash R., Shin E.C., Char K., Kim Y., Li S. Reactive polymer targeting dsRNA as universal virus detection platform with enhanced sensitivity. Biomacromolecules. 2020;21:2440–2454. doi: 10.1021/acs.biomac.0c00379. [DOI] [PubMed] [Google Scholar]
- 19.Schönborn J., Oberstrass J., Breyel E., Tittgen J., Schumacher J., Lukacs N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 1991;19:2993–3000. doi: 10.1093/nar/19.11.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita M., Adachi K., Nagasawa M. Development of a homogeneous time-resolved fluorescence assay for detection of viral double-stranded RNA. Anal. Biochem. 2019;566:46–49. doi: 10.1016/j.ab.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen P., Chung M.T., McHugh W., Nidetz R., Li Y., Fu J., Cornell T.T., Shanley T.P., Kurabayashi K. Multiplex serum cytokine immunoassay using nanoplasmonic biosensor microarrays. ACS Nano. 2015;9:4173–4181. doi: 10.1021/acsnano.5b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali A.A., Kang M., Kharbash R., Kim Y. Spiropyran as a potential molecular diagnostic tool for double-stranded RNA detection. BMC Biomed. Eng. 2019;1:6. doi: 10.1186/s42490-019-0008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali A.A., Kharbash R., Kim Y. Chemo- and biosensing applications of spiropyran and its derivatives - a review. Anal. Chim. Acta. 2020;1110:199–223. doi: 10.1016/j.aca.2020.01.057. [DOI] [PubMed] [Google Scholar]
- 24.Andersson J., Li S., Lincoln P., Andréasson J. Photoswitched DNA-binding of a photochromic spiropyran. J. Am. Chem. Soc. 2008;130:11836–11837. doi: 10.1021/ja801968f. [DOI] [PubMed] [Google Scholar]
- 25.Kortekaas L., Browne W.R. The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome. Chem. Soc. Rev. 2019;48:3406–3424. doi: 10.1039/c9cs00203k. [DOI] [PubMed] [Google Scholar]
- 26.Hammarson M., Nilsson J.R., Li S., Lincoln P., Andréasson J. DNA-Binding properties of amidine-substituted spiropyran photoswitches. Chemistry. 2014;20:15855–15862. doi: 10.1002/chem.201405113. [DOI] [PubMed] [Google Scholar]
- 27.Kapuscinski J. Interactions of nucleic acids with fluorescent dyes: spectral properties of condensed complexes. J. Histochem. Cytochem. 1990;38:1323–1329. doi: 10.1177/38.9.1696951. [DOI] [PubMed] [Google Scholar]
- 28.Hegde A.H., Seetharamappa J. Fluorescence and circular dichroism studies on binding and conformational aspects of an anti-leukemic drug with DNA. Mol. Biol. Rep. 2014;41:67–71. doi: 10.1007/s11033-013-2838-2. [DOI] [PubMed] [Google Scholar]
- 29.Kim K., Baek S.C., Lee Y.Y., Bastiaanssen C., Kim J., Kim H., Kim V.N. A quantitative map of human primary microRNA processing sites. Mol. Cell. 2021;81:3422–3439.e11. doi: 10.1016/j.molcel.2021.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Han J., Lee Y., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 31.Ma H., Wu Y., Choi J.G., Wu H. Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proc. Natl. Acad. Sci. USA. 2013;110:20687–20692. doi: 10.1073/pnas.1311639110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheson B.D., Greenberg P.L., Bennett J.M., Lowenberg B., Wijermans P.W., Nimer S.D., Pinto A., Beran M., de Witte T.M., Stone R.M., et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 33.Döhner H., Estey E., Grimwade D., Amadori S., Appelbaum F.R., Büchner T., Dombret H., Ebert B.L., Fenaux P., Larson R.A., et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart M.L., Tamayo P., Wilson A.J., Wang S., Chang Y.M., Kim J.W., Khabele D., Shamji A.F., Schreiber S.L. KRAS genomic status predicts the sensitivity of ovarian cancer cells to decitabine. Cancer Res. 2015;75:2897–2906. doi: 10.1158/0008-5472.CAN-14-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee E.K., Hong S.-H., Shin S., Lee H.-S., Lee J.-S., Park E.J., Choi S.S., Min J.W., Park D., Hwang J.-A., et al. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget. 2016;7:75000–75012. doi: 10.18632/oncotarget.11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y.S. A novel type of non-coding RNA, nc886, implicated in tumor sensing and suppression. Genomics Inform. 2015;13:26–30. doi: 10.5808/GI.2015.13.2.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K.-S., Park J.-L., Lee K., Richardson L.E., Johnson B.H., Lee H.-S., Lee J.-S., Kim S.-B., Kwon O.-H., Song K.S., et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget. 2014;5:3944–3955. doi: 10.18632/oncotarget.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunkeaw N., Lee Y.-S., Im W.R., Jang J.J., Song M.-J., Yang B., Park J.-L., Kim S.-Y., Ku Y., Kim Y., et al. Mechanism mediated by a noncoding RNA, nc886, in the cytotoxicity of a DNA-reactive compound. Proc. Natl. Acad. Sci. USA. 2019;116:8289–8294. doi: 10.1073/pnas.1814510116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golec E., Lind L., Qayyum M., Blom A.M., King B.C. The noncoding RNA nc886 regulates PKR signaling and cytokine production in human cells. J. Immunol. 2019;202:131–141. doi: 10.4049/jimmunol.1701234. [DOI] [PubMed] [Google Scholar]
- 40.Lee E.K., Hong S.H., Shin S., Lee H.S., Lee J.S., Park E.J., Choi S.S., Min J.W., Park D., Hwang J.A., et al. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget. 2016;7:75000–75012. doi: 10.18632/oncotarget.11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brieke C., Heckel A. Spiropyran photoswitches in the context of DNA: synthesis and photochromic properties. Chemistry. 2013;19:15726–15734. doi: 10.1002/chem.201302640. [DOI] [PubMed] [Google Scholar]
- 42.Gholamalipour Y., Karunanayake Mudiyanselage A., Martin C.T. 3' end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-Seq analyses. Nucleic Acids Res. 2018;46:9253–9263. doi: 10.1093/nar/gky796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mu X., Greenwald E., Ahmad S., Hur S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018;46:5239–5249. doi: 10.1093/nar/gky177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim S., Ku Y., Ku J., Kim Y. Evidence of aberrant immune response by endogenous double-stranded RNAs: attack from within. Bioessays. 2019;41:1900023. doi: 10.1002/bies.201900023. [DOI] [PubMed] [Google Scholar]
- 45.Hur S. Double-stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol. 2019;37:349–375. doi: 10.1146/annurev-immunol-042718-041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanghvi V.R., Steel L.F. The cellular TAR RNA binding protein, TRBP, promotes HIV-1 replication primarily by inhibiting the activation of double-stranded RNA-dependent kinase PKR. J. Virol. 2011;85:12614–12621. doi: 10.1128/JVI.05240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samuel C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA–triggered innate immune responses. J. Biol. Chem. 2019;294:1710–1720. doi: 10.1074/jbc.TM118.004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodman A.G., Smith J.A., Balachandran S., Perwitasari O., Proll S.C., Thomas M.J., Korth M.J., Barber G.N., Schiff L.A., Katze M.G. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007;81:2221–2230. doi: 10.1128/JVI.02151-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DiNardo C.D., Jonas B.A., Pullarkat V., Thirman M.J., Garcia J.S., Wei A.H., Konopleva M., Döhner H., Letai A., Fenaux P., et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020;383:617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 50.Wong K.K., Hassan R., Yaacob N.S. Hypomethylating agents and immunotherapy: therapeutic synergism in acute myeloid leukemia and myelodysplastic syndromes. Front. Oncol. 2021;11:624742. doi: 10.3389/fonc.2021.624742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daver N., Garcia-Manero G., Basu S., Boddu P.C., Alfayez M., Cortes J.E., Konopleva M., Ravandi-Kashani F., Jabbour E., Kadia T., et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9:370–383. doi: 10.1158/2159-8290.CD-18-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu G., Wu Y., Wang W., Xu J., Lv X., Cao X., Wan T. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell. Mol. Immunol. 2019;16:401–409. doi: 10.1038/s41423-018-0026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and resources used in this study will be made available upon written request to the corresponding authors.