

Summary
We present a high-content analysis (HCA) protocol for monitoring the outgrowth capacity of human neurons derived from induced pluripotent stem cells (iPSCs). We describe steps to perform HCA imaging, followed by quantifying the morphology of dendrites and axons within a high-throughput system to evaluate neurons obtained through various differentiation approaches. This protocol can be used to screen for modulators of neuronal morphogenesis or neurotoxicity. The approach can be applied to patient-derived iPSCs to identify patient-specific defects and possible therapeutic strategies.
For complete details on the use and execution of this protocol, please refer to Zink et al. (2020) and Inak et al. (2021). The protocol can be used in combination with Zink et al. (2022).
Subject areas: High Throughput Screening, Microscopy, Neuroscience, Stem Cells, Cell Differentiation
Graphical abstract
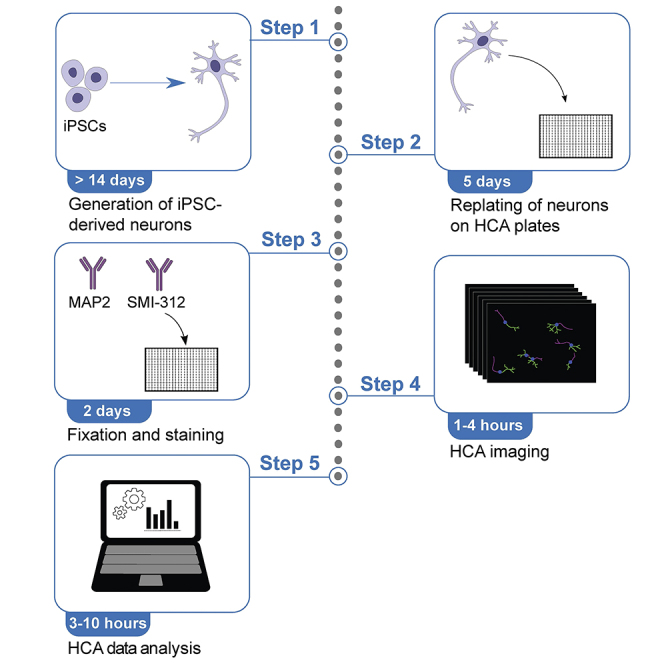
Highlights
-
•
High-content analysis of human iPSC-derived neurons
-
•
Automated microscopy to quantify neuronal morphology
-
•
Quantification of axonal and dendritic outgrowths
-
•
Data analysis based on open-source software CellProfiler
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
We present a high-content analysis (HCA) protocol for monitoring the outgrowth capacity of human neurons derived from induced pluripotent stem cells (iPSCs). We describe steps to perform HCA imaging, followed by quantifying the morphology of dendrites and axons within a high-throughput system to evaluate neurons obtained through various differentiation approaches. This protocol can be used to screen for modulators of neuronal morphogenesis or neurotoxicity. The approach can be applied to patient-derived iPSCs to identify patient-specific defects and possible therapeutic strategies.
Before you begin
The protocol described here enables to assess the morphology of human neurons derived from induced pluripotent stem cell (iPSCs). Before starting the protocol, it is important to obtain iPSC-derived neurons that can be robustly grown and visually monitored. Here, we describe specific steps for neurons differentiated from a human iPSC line engineered to express the pan-neuronal transcription factor Neurogenin 2 (NGN2) construct in the AAVS1 locus in an inducible manner. Using this iPSC line (BIHi005-A-24), it is possible to activate the expression of NGN2 upon doxycycline (DOX) exposure which converts the iPSCs into a pure neuronal population in about 14 days (Zhang et al., 2013) (Figure 1). However, the HCA protocol described here can also be reliably applied to iPSC-derived neurons obtained using other differentiation approaches. For example, we successfully used this protocol for dopaminergic neurons obtained by exposing iPSCs to specific growth factors and small molecules (Inak et al., 2021; Zink et al., 2020).
Figure 1.

Neurons differentiated from human induced pluripotent stem cells (iPSCs)
Upper panel, step-by-step protocol for the NGN2 induction from the engineered iPSC line BIHi005-A-24. Lower panel, exemplary images of the differentiating neurons at different time points. Scale bar: 200 μm.
Institutional permissions
Studies with control iPSCs were performed in accordance with the approval by the Ethic Committee of the Medical Faculty of Heinrich Heine University (study number: 2020-967_2).
Preparation of cell culture materials and buffers
Required media and solutions used in this protocol can either be prepared in advance and stored as indicated, or can be made fresh on the day of the experiment. Please refer to materials and equipment for a complete list of recipes.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Guinea Pig IgG (H+L), highly cross-adsorbed, CF™ 488A antibody produced in donkey (1: 1000) | Sigma-Aldrich | Cat#SAB4600033 |
| Anti-MAP2 polyclonal antibody (guinea pig) (1: 1000) | Synaptic Systems | Cat#188004 |
| Anti-Neurofilament mixture of monoclonal antibodies, SMI312 (mouse) (1:500) | BioLegend | Cat#837904 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (1: 1000) | Invitrogen | Cat#A-11031 |
| Chemicals, peptides, and recombinant proteins | ||
| 16% Paraformaldehyde (PFA) | Thermo Fisher Scientific | Cat#28906 |
| B27 | Gibco | Cat#17504044 |
| BDNF | MACS Miltenyi | Cat#130-096-811 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A9647 |
| Cytosin-1-β-D-arabinofuranosid (AraC) | Sigma-Aldrich | Cat#C1768 |
| DMEM/F-12 | Gibco | Cat#31330095 |
| Doxycycline hydrochloride (DOX) | Sigma-Aldrich | Cat#D3072 |
| Geltrex | Gibco | Cat#A1413201 |
| GlutaMAX | Gibco | Cat#35050061 |
| Hoechst 33342 | Invitrogen | Cat#H3570 |
| Laminin | Sigma-Aldrich | Cat#L2020 |
| MycoZap | Lonza | Cat#VZA-2012 |
| Neurobasal | Gibco | Cat#21103-049 |
| NT-3 | Biozol/PeproTech | Cat#450-03 |
| PBS w/o calcium and magnesium | Gibco | Cat#14190144 |
| ROCK inhibitor Y-27632 | Enzy Life Sciences | Cat#ALX-270-333-M005 |
| Sodium Azide | Sigma-Aldrich | Cat#71289 |
| StemPro Accutase | Thermo Fisher Scientific | Cat#A1110501 |
| Triton X-100 | Bio-Rad | Cat#161-0407 |
| Deposited data | ||
| CellProfiler pipeline neurite outgrowth | This paper | https://doi.org/10.5281/zenodo.6642365 |
| Experimental models: Cell lines | ||
| Human: BIHi005-A-24 cell line | Berlin Institute of Health | N/A |
| Software and algorithms | ||
| CellProfiler 4.2.1. | Carpenter et al. (2006) | https://cellprofiler.org/ |
| Other | ||
| 384 well Cell Culture Microplate, PS, F-Bottom, black TC, μCLEAR | Greiner Bio-One | Cat#781091 |
| CytoSmart Cell Counter | Greiner Bio-One | Cat#6749 |
| Operetta® CLS™ | PerkinElmer | Cat#HH16000000 |
Materials and equipment
Maturation medium for NGN2 neurons
| Reagent | Final concentration | Amount |
|---|---|---|
| Neurobasal medium | n/a | 48.3 mL |
| B-27 | 1 × | 1 mL |
| GlutaMAX | 1 × | 0.5 mL |
| BDNF (10 μg/mL) | 10 ng/mL | 50 μL |
| NT-3 (10 μg/mL) | 10 ng/mL | 50 μL |
| Laminin (10 μg/mL) | 0.2 μg/mL | 10 μL |
| MycoZap | 1 × | 100 μL |
| DOX (3 mg/mL, add directly before use) | 3 μg/mL | n/a |
| AraC (add only from day 6) | 2 μM | 25 μL |
| Total | n/a | 50 mL |
Store at 2°C–8°C for up to 2 weeks. Always add fresh DOX directly before use to the medium.
Note: Thawed doxycycline (DOX) aliquots should be kept at 2°C–8°C in the dark and used for maximal 1 week.
Alternatives: If using a different neuronal differentiation approach, other media might be necessary.
8% Paraformaldehyde (PFA)
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% PFA | 8% | 10 mL |
| PBS w/o calcium and magnesium | n/a | 10 mL |
| Total | n/a | 20 mL |
Store at 2°C–8°C for up to 1 week or at −20°C for several months.
CRITICAL: PFA is flammable, toxic, carcinogenic, and it is an irritant for the skin, eyes, and respiratory tract. A lab coat, face mask, and safety goggles should be worn when handling PFA. All steps should be performed inside a fume hood; waste should be disposed in special containers in accordance with applicable regional and national laws and regulations.
Blocking solution for immunofluorescence staining
| Reagent | Final concentration | Amount |
|---|---|---|
| Bovine Serum Albumin 10% (w/v) | 3% | 9 mL |
| Triton-x-100 | 0.5% | 150 μL |
| PBS w/o calcium and magnesium | n/a | 21 mL |
| Total | n/a | 30 mL |
Store at −20°C for several months.
Alternatives: Antibodies.
We use neuronal markers SMI312 to identify axons and MAP2 to identify dendrites (Hoffmann et al., 2021). However, other markers may also be used. This protocol can be adapted to screen using a general neurite staining, for example beta-III-tubulin (TUJ1).
Alternatives: High-content analysis imaging platform.
Here, we describe the use of HCA system Operetta® CLS™ (Perkin Elmer). However, we successfully applied the same HCA protocol also to the HCA system CellInsight High-Content Screening (Thermo Scientific).
Step-by-step method details
Cell seeding and culturing on HCA plates
Timing: 5 days
In this step, iPSC-derived neurons are seeded onto coated HCA plates and cultured for several days to allow for neurite outgrowth (Figure 1).
-
1.Preparation of HCA plates.
-
a.Coat a black wall, clear flat-bottom 384-well plate suited for imaging using 50 μL geltrex coating solution (1:100 dilution in DMEM-F/12) per well, and incubate for 1 h at 37°C.
-
a.
Note: If HCA plates are not directly used after coating, they can be wrapped with parafilm and stored for up to two weeks at 2°C–8°C.
Alternatives: Instead of geltrex, coating with matrigel is also possible. In our hands, the two coating materials give comparable results and provide good neuronal attachment and outgrowth. However, matrigel is not a defined formula, and thus it may lead to potential batch-to-batch variability. Other coating substances can also be employed, depending on the neural cell type of use.
-
2.Seeding of iPSC-derived neurons (Figure 1).
-
a.For 6-well plates, split iPSC-derived neurons by applying 0.5 mL accutase® (an enzyme mixture with proteolytic and collagenolytic activity) onto the cells for 3 min at room temperature (RT, 20°C–25°C).
-
b.Dilute accutase activity by adding 1 mL of culture media and wash off the cells. Collect the cells and transfer them into a 15 mL falcon tube with 7.5 mL PBS to further dilute accutase activity. Wash the well with 1 mL of culture media and transfer any of the remaining cells into the same 15 mL falcon tube.
-
c.Centrifuge for 5 min at 100 × g, remove the supernatant, and re-suspend the cells in maturation media. Count the cells using a hemocytometer or a cell counter.
-
d.Mix the cell suspension with maturation medium containing fresh DOX and 10 μM ROCK-inhibitor to a concentration of around 40,000 cells/mL. Pipette 50 μL of the cell suspension per well (384-well plate).
-
e.Incubate the cells in a tissue culture incubator at 37°C and 5% CO2.
-
a.
Note: Optimal seeding density might need to be adapted depending on the specific neuronal type and iPSC line used for differentiation. Low density helps with downstream image analysis, while high density usually improves cell survival. In our hands 2,000–5,000 cells per well (40,000–100,000 cells/mL) yield the best results in this HCA pipeline.
-
3.Culturing of iPSC-derived neurons.
-
a.Every second day, remove half of the culture medium and replace it with maturation medium containing fresh DOX.
-
a.
Note: If there are many dead cells, you can remove the culture medium completely and wash the cells with PBS. However, this should only be done if the cells attach strongly to the bottom surface.
Note: The optimal time point for fixation might need to be adapted depending on the specific neuronal type and iPSC line used. We recommend fixing the iPSC-derived neurons 4 days after seeding them to ensure sufficient level of neurite outgrowth for effective classification by the HCA software.
Fixation and staining of iPSC-derived neurons
In this step, iPSC-derived neurons are fixed and immunostained for axonal and dendritic markers.
-
4.Fixation of iPSC-derived neurons.
-
a.Add 50 μL of the 8% PFA solution directly to the culture medium for a final 4% PFA solution. Incubate for 15 min at RT under a fume hood.
-
b.Aspirate the culture medium / PFA solution and carefully wash each well three times with 50 μL PBS. After the last washing step, leave 50 μL of 0.05% sodium azide in PBS in each well to improve conservation, or add the blocking solution for the subsequent staining steps.
-
c.If the cells will not be stained directly after fixation, seal the plate with parafilm and store it at 2°C–8°C.
-
a.
Note: The fixative is added directly into the medium to avoid detachment of the cells during aspiration of the culture medium. It is possible to add 4% PFA to the culture medium (for a final PFA concentration of 2%), but then the incubation time doubles. If cells adhere well, it is also possible to aspirate the culture medium and add 4% PFA directly to the well.
Pause point: The plate can be stored at 2°C–8°C for a few weeks before staining.
-
5.Staining of iPSC-derived neurons.
-
a.Aspirate the PBS and add 50 μL of the blocking solution to each well. Incubate at RT for 1 h or overnight (12–24 h) at 2°C–8°C.
-
b.Prepare the primary antibody solution in blocking buffer.
-
i.Dilute the SMI312 antibody 1:500 and the MAP2 antibody 1:1000.
-
i.
-
c.After the incubation time, aspirate the blocking solution and add 20 μL of the primary antibody solution per well. Seal the plate with parafilm and incubate overnight (12–24 h) at 2°C–8°C.
-
d.The next day, aspirate the primary antibody solution, and wash each well three times with 50 μL PBS.
-
e.Prepare the secondary antibody solution in blocking buffer including Hoechst 3342.
-
i.Dilute both secondary antibodies 1:1000 and Hoechst 33342 1:2500.
-
i.
-
f.Add 20 μL of the secondary antibody solution per well. Incubate in the dark at RT for at least 1 h.
-
g.After the incubation time, aspirate the secondary antibody solution and wash each well three times with 50 μL PBS. After the last washing step, leave 80 μL of 0.05% sodium azide in PBS in each well.
-
h.For long-term storage of the plate, seal the plate with parafilm and keep at 2°C–8°C. Check regularly that there is sufficient PBS inside the wells of the plate to prevent the fixed cells from drying out.
-
a.
Note: The fluorescent dyes on secondary antibodies are light-sensitive and should not be exposed to incident light. After incubation with secondary antibodies, the plates should also always be protected from the light.
Note: The exact antibody dilutions might need to be adapted.
Note: Instead of Hoechst, other nuclear dyes can also be used (e.g., DAPI).
HCA imaging
In this step, using a HCA imaging platform, images of the wells are acquired in an automated fashion. The described settings are specific for the Operetta® CLS™ together with its software Harmony (Perkin Elmer). Comparable settings exist for other HCA imaging platforms as well.
-
6.Set up the HCA platform and microscope settings.
-
a.Place the plate in the HCA imaging platform.
-
b.Select the plate format.
-
i.Check manufacturers’ comments on plate usage.
-
i.
-
c.Select the 20× Air, NA 0.4 objective.
-
d.Choose the non-confocal operational mode.
-
e.Choose a binning of ‘1’.
-
f.Define the plate layout.
-
i.Select the wells and the fields within each well that should be imaged.Note: These are the setting we determined to work most effectively for us. However, each of the settings might need to be adapted to your specific neuronal cell type, iPSC line, or HCA platform.Note: It is possible to use confocal operation mode instead of non-confocal. Confocal imaging can provide higher resolution especially if cells adhere not as a monolayer but partially stack upon each other in multiple layers. However, confocal imaging requires higher exposure time and LED power, thus leading to prolonged acquisition time.Note: A higher binning is also possible, which increases signal-to-noise ratio, shortens exposure time and thus reduces acquisition time. However, this can lead to decreased spatial resolution.Note: Determine which and how many fields need to be imaged to represent the well. In our experience, neurons are likely to attach more closely to the border of the well than in the middle. Therefore, it is useful to include outer fields in the imaging set-up. It is important to point out that outer fields include the border of the well. Thus, individual field comparisons should be avoided, as it is difficult to compare fields with borders (i.e., outer fields) to fields without borders. We recommend to always perform statistical tests on all fields of each well combined.
-
i.
-
g.Create a channel for each fluorophore you are using and determine the exposure time, LED power and height (z-off) for each channel.
-
i.Select one well and field of a well and click snapshot.
-
ii.On the image control panel, use the histogram to verify the maximum value. We recommend decreasing exposure time or LED power to keep the maximum value below 60,000.
-
iii.To determine the height, acquire a z-stack (click ‘Test’, not ‘Snapshot’) and identify the image with the sharpest focus. Enter the respective height for each channel.Note: You can set a different height for each channel. This can be helpful as neurites sometimes tend to be at a different focus plane than the nuclei.
-
i.
-
a.
-
7.HCA Imaging.
-
a.Go to ‘Run Experiment’, name it and click ‘Start’.
-
b.After the run is finished, export the data as .tiff files.
-
a.
Note: When considering the settings, it is important to find a balance between high-quality images and the time needed to acquire them. As an example, image acquisition with 3 channels of one plate with 100 wells selected and 13 fields of view per well takes about 25 min. With the 20 × objective, it is possible to acquire up to 25 fields of view. We recommend to capture at least 12 fields of view per image to allow robust data collection. However, before starting the acquisition, we suggest imaging some wells with all 25 fields in order to examine whether the cells grow uniformly throughout the well.
Note: If cells are not in focus, include additional Z-planes. This however increases the acquisition time.
Note: If working with neurons derived from patients or exposed to treatments, always set up the image acquisition based on a well containing control/untreated neurons. But before starting the analysis, remember to also image the wells containing the diseased/treated neurons. This is important to avoid out-of-focus images and inadequate exposure times.
HCA image analysis
This step describes the HCA image analysis for the quantification of neurite outgrowth using the open-source software CellProfiler, which allows the processing of images from high-throughput experiments (Figures 2A and 2B, and Methods video S1).
Figure 2.

HCA pipeline for neurite quantification
(A) Schematic representation of the individual steps of the HCA protocol with iPSC-derived neurons.
(B) Stepwise exemplary images of the HCA protocol generated by CellProfiler analysis. Scale bar: 50 μm.
(C) Merged color image obtained by HCA microscopy. Scale bar: 50 μm.
(D) Image showing the CellProfiler mask superimposed over the merged grayscale image obtained by HCA microscopy. Scale bar: 50 μm.
The HCA pipeline, example images to test the pipeline and a video of all the steps can be downloaded here: https://github.com/StemCellMetab/neurite-outgrowth.
-
8.Setting up the CellProfiler analysis pipeline.
-
a.Open the CellProfiler analysis pipeline.
-
b.Go to Images.
-
i.Add images by dragging and dropping them into CellProfiler.
-
i.
-
c.Go to Metadata. Enter the extraction method and metadata source.
-
i.To set an expression to enable CellProfiler to interpret metadata from a file name or folder, click on the magnifying glass. The test text should reflect the name correctly. In case it does not, click ‘Guess’ to update the text or edit it manually.
-
ii.Click ‘Submit’.
-
iii.Click ‘Update’ to extract and display the metadata in the CellProfiler window.
-
i.
-
d.Go to NameAndTypes and adjust the settings according to your staining and the naming of your images.
-
i.In the default settings of our pipeline, channel 1 is set as Hoechst (staining of nuclei), channel 2 is set as MAP2 (dendritic staining), and channel 3 is set as SMI312 (axonal staining).
-
i.
-
e.Set the default input and output folders.
-
i.Select the ‘File’ dropdown menu and select ‘preferences’. Select ‘Browse’ next to the ‘Default Output Folder’ and ‘Default Input Folder’.
-
ii.Enter a filename for saving.
-
i.
-
f.Go to the ExportToSpreadsheet Module at the end of the pipeline.
-
i.Enter a name at ‘filename prefix’.
-
i.
-
a.
Note: If you have a very large dataset, CellProfiler might not be able to export to a spreadsheet. In that case, replace the ‘ExportToSpreadsheet’ module with the ‘ExportToDatabase’ module and adjust the settings to your needs.
-
9.Testing and adjusting the pipeline.
-
a.Click on ‘Start Test Mode’.
-
b.A green check-mark will appear next to the modules that contain no errors. A red ‘x’ will appear next to the modules that do contain errors. The ‘ExportToSpreadsheet’ module always has a yellow triangle during the test mode. If you hover with the mouse over the red ‘x’, CellProfiler will spell out the exact error.
- c.
-
d.Test the settings on multiple image sets. Select the ‘Test’ dropdown menu to select specific image sets to adjust the settings for images from controls as well as treated cells.
-
a.
-
10.Starting the analysis.
-
a.Exit the test mode (click ‘Exit Test Mode’), disable all windows and click the ‘Analyze Images’ button.
-
b.CellProfiler will now analyze the image sets and export the results to a spreadsheet on the selected drive and folder. The pipeline will also save a spreadsheet containing the used settings (filename prefixExperiment).
-
a.
Note: MAP2 and SMI-312 staining distinguish between dendrites and axons, respectively (Figures 2C and 2D). The HCA protocol can be adapted also to capture neurites in general, which can be visualized using an antibody against beta-III-tubulin (TUJ1). In this case, change ‘NamesAndTypes’ accordingly, and delete the modules that are duplicated.
Note: Further statistical analysis of the data and graphical representations might be necessary, and can be carried out using different software (e.g., Microsoft Excel or GraphPad Prism).
Table 1.
Description of each module of the CellProfiler pipeline
| Module | Description |
|---|---|
| IdentifyPrimaryObjects | Identifies the nuclei via Hoechst staining. Determine the typical diameter empirically by testing different image sets. Nuclei touching the border of the image are removed. |
| Threshold | Converts the SMI312 grayscale image into a binary image for masking the nuclei in the next step. |
| MaskObjects | Keeps only those nuclei with at least 20% overlap with the SMI312 staining. This way only neuronal nuclei will remain. This might be necessary for primary cultures with a mixed assortment of cell types. |
| IdentifySecondaryObjects | Identifies axons (+cell body) in the SMI312 image around the previously identified and masked nuclei. |
| IdentifySecondaryObjects | Identify dendrites (+cell body) by using the MAP2 image around the previously identified and masked nuclei. |
| MaskObjects | Both SMI312 and MAP2 stain the cell body next to axons and dendrites, respectively. By masking the SMI312 staining with the MAP2 staining the overlap is considered the cell body. |
| IdentifyTertiaryObjects | Removes the cell body from the ‘axons+cell body’ identified before so that only the axons will remain. |
| IdentifyTertiaryObjects | Removes the cell body from the ‘dendrites+cell body’ identified before so that only the dendrites will remain. |
| MeasureObjectSizeShape | Measures the size and shape of axons and dendrites. |
| ConvertObjectsToImage | The skeleton module requires a binary picture, so this module converts the identified axons to a binary image. |
| ConvertObjectsToImage | The skeleton module requires a binary picture, so this module converts the identified dendrites to a binary image. |
| MeasureObjectSkeleton | Measures the skeletal features (branches, endpoints, trunks) for the axons starting from the cell body. |
| MeasureObjectSkeleton | Measures the skeletal features (branches, endpoints, trunks) for the dendrites starting from the cell body. |
| ExportToSpreadsheet | Exports the data (number of objects, measurements, etc.). |
Expected outcomes
The protocol described here enables monitoring neuronal morphology and quantifying outgrowth capacity of human neurons derived from iPSCs using high-content analysis (HCA). Upon re-plating onto a 384-well-plate, iPSC-derived neurons should attach and form neurites within 1–5 days. This HCA analysis pipeline allows quantification of both axonal and dendritic length, number of branch points, staining signal areas, and other parameters (Figure 2). Here, we applied the HCA protocol to human neurons generated through the over-expression of NGN2 (Zhang et al., 2013) using engineered iPSCs. Our protocol can be employed to address the impact on neurite outgrowth and branching of iPSC-derived neurons exposed to different drugs, siRNAs, or guide RNAs. The HCA pipeline can also be applied to neurons differentiated from patient-derived iPSCs to possibly unveil disease-specific neuronal defects (Inak et al., 2021). In combination with the assessment of live mitochondrial activity (Zink et al., 2022), this protocol can also enable the identification of modulators of human neuronal and mitochondrial toxicity (Zink et al., 2020).
Limitations
This protocol does not allow to determine the functional properties of iPSC-derived neurons. To assess these properties, calcium imaging or electrophysiological measurement should be carried out. In order to investigate functional neuronal aspects, co-culture with glia cells might also be required, as pure neurons alone may not develop functional synapses (Klapper et al., 2019).
With this protocol, it is also not possible to perform Scholl analysis, which is a widely used approach for assessing dendritic arborization. In fact, even though a Scholl analysis plugin for CellProfiler is available, its functions appear quite limited, and so far we were not able to implement it into our HCA pipeline. One specific challenge is that Scholl analysis requires neurons to be imaged as individual cells. However, this is not easy to achieve for human iPSC-derived neurons that tend to grow in dense cultures to maintain their survival. Perhaps, one can envision that the use of sparsely fluorescently labeled cultures, for example obtained through low-titer virus transfection using a reporter gene such as GFP, might limit the neuronal signals that need to be resolved, thereby allowing to visualize single neurons for Scholl analysis. We hope that these issues can be resolved soon to enable the inclusion of Scholl analyses in future HCA outgrowth pipelines of human iPSC-derived neurons.
Another limitation of our protocol is that, given the complex morphology of neurons, axonal/dendritic crossing-overs from two or more neighboring neurons may take place. This phenomenon likely adds ‘noise’ to the estimation of both lengths and ramifications. By using overlapping SMI312 and MAP2 staining to determine the cell body, neurite crossing and overlap of neighboring cells might falsely be labeled as cell body and thus be excluded from the neurite analysis. This is a problem inherent to automated imaging approaches of cultured neurons. In our opinion, it is therefore essential to first optimize the assay to determine the best seeding density and fixation time point for each neuronal cell type. Careful, manual evaluation of the images is key, and experimenters should not rely solely on automated processing and quantification.
Troubleshooting
Problem 1
Neurons are too confluent or too sparse to be properly visualized and analyzed (see cell seeding and culturing on HCA plates, step 2).
Potential solution
Determine the optimal cell density in a pilot experiment. Neuronal seeding should be sufficiently dense to ensure survival, but also sparse enough to allow proper imaging and downstream quantification analysis.
Problem 2
The staining does not highlight distinct neurite structures (see cell seeding and culturing on HCA plates, step 3).
Potential solution
In case neuritic structures are not yet fully formed, they may exhibit lower staining intensities because they do not possess a mature protein composition. This problem could be overcome by fixing the differentiating neurons at later time points or by using higher antibody concentrations. We recommend to test multiple time points in pilot experiments, and check that there is sufficient neurite outgrowth. At the same time, the neurite network should not be too dense, as this will compromise the efficient detection in image analyses. In our hands, allowing iPSC-derived neurons to grow for 4 days after splitting on the HCA plate is sufficient to ensure proper neurite outgrowth. However, the optimal fixation time point might need to be determined for different neuronal types. In our experience, iPSC-derived neurons grown for up to 7 days can still be readily assessed by this HCA pipeline. At longer time points, the complex structure of neurons may prevent obtaining reliable HCA results.
Problem 3
Neurons detach after washing steps or after fixation (see fixation and staining of iPSC-derived neurons).
Potential solution
-
•
Try different coatings or increase the concentration of the coating material.
-
•
Reduce the washing steps.
-
•
Seed a higher number of cells to compensate for cell loss during fixation and staining.
Problem 4
Cells are out-of-focus (see HCA imaging, step 6, g).
Potential solution
-
•
Try confocal mode or different Z-stacks.
-
•
Optimize the seeding conditions. Ideally, cells should grow as individual cells.
-
•
Check if the plate used is suitable for HCA imaging and whether the optical transparency is sufficient. We suggest to check the web page of the manufacturer of the HCA instrument to identify recommended plates suitable for imaging on that instrument.
Problem 5
The CellProfiler pipeline does not generate results (see HCA image analysis).
Potential solution
-
•
Try to determine the point of the pipeline where an error may have occurred. CellProfiler shows a red ‘x’ if there is a problem within one module.
-
•
Check that you changed ‘NamesAndTypes’ according to the naming system of your images.
-
•
Check if the default input and output folders are set correctly.
-
•
Start ‘Test Mode’ and check on multiple image sets that all settings (size, threshold, etc.) match your images.
-
•
For other more specific errors, check CellProfiler’s forum, where many issues are discussed and covered.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alessandro Prigione (alessandro.prigione@hhu.de).
Materials availability
This study did not generate new unique materials.
Acknowledgments
The authors acknowledge support from the Center for Advanced Imaging (CAi) at the Heinrich Heine University Düsseldorf where HCA imaging was performed. A.P. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG) (#PR1527/5-1 and PR 1527/6-1), the European Joint Programme for Rare Diseases (EJPRD) and German Federal Ministry of Education and Research (BMBF) (#01GM2002A), the United Mitochondrial Disease Foundation (UMDF), People Against Leigh syndrome (PALS), Fondation Maladies Rare and Association AMMi, and the University Hospital Düsseldorf (Forschungskommission UKD). S.C. acknowledges support of the University Hospital Düsseldorf (Forschungskommission UKD). M.B. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG) (INST 208/760-1FUGG).
Author contributions
Conceptualization, S.L., A.Z., and A.P.; methodology, S.L., A.Z., M.B., A.P., S.C., and C.M.; investigation, S.L., N.S.T., S.D., and C.M.; writing – original draft, S.L.; writing – review and editing, S.L., A.P., and S.C.; funding acquisition, A.P., S.C., and M.B.; resources, M.B., N.S.T., and S.D.; supervision, A.P. and S.C.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101567.
Data and code availability
The CellProfiler pipeline for neurite outgrowth generated during this study is available at Github: https://github.com/StemCellMetab/neurite-outgrowth and has been deposited at Zenodo: https://doi.org/10.5281/zenodo.6642365.
References
- Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann P.C., Giandomenico S.L., Ganeva I., Wozny M.R., Sutcliffe M., Lancaster M.A., Kukulski W. Electron cryo-tomography reveals the subcellular architecture of growing axons in human brain organoids. Elife. 2021;10:e70269. doi: 10.7554/elife.70269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inak G., Rybak-Wolf A., Lisowski P., Pentimalli T.M., Jüttner R., Glažar P., Uppal K., Bottani E., Brunetti D., Secker C., et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021;12:1929. doi: 10.1038/s41467-021-22117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper S.D., Garg P., Dagar S., Lenk K., Gottmann K., Nieweg K. Astrocyte lineage cells are essential for functional neuronal differentiation and synapse maturation in human iPSC-derived neural networks. Glia. 2019;67:1893–1909. doi: 10.1002/glia.23666. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink A., Conrad J., Telugu N.S., Diecke S., Heinz A., Wanker E., Priller J., Prigione A. Assessment of ethanol-induced toxicity on iPSC-derived human neurons using a novel high-throughput mitochondrial neuronal health (MNH) assay. Front. Cell Dev. Biol. 2020;8:590540. doi: 10.3389/fcell.2020.590540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink A., Haferkamp U., Wittich A., Beller M., Pless O., Prigione A. High-content screening of mitochondrial polarization in neural cells derived from human pluripotent stem cells. STAR Protocols. 2022 doi: 10.1016/j.xpro.2022.101602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CellProfiler pipeline for neurite outgrowth generated during this study is available at Github: https://github.com/StemCellMetab/neurite-outgrowth and has been deposited at Zenodo: https://doi.org/10.5281/zenodo.6642365.