

Summary
IL11 initiates fibroblast activation but also causes epithelial cell dysfunction. The mechanisms underlying these processes are not known. We report that IL11-stimulated ERK/P90RSK activity causes the phosphorylation of LKB1 at S325 and S428, leading to its inactivation. This inhibits AMPK and activates mTOR across cell types. In stromal cells, IL11-stimulated ERK activity inhibits LKB1/AMPK which is associated with mTOR activation, ⍺SMA expression, and myofibroblast transformation. In hepatocytes and epithelial cells, IL11/ERK activity inhibits LKB1/AMPK leading to mTOR activation, SNAI1 expression, and cell dysfunction. Across cells, IL11-induced phenotypes were inhibited by metformin stimulated AMPK activation. In mice, genetic or pharmacologic manipulation of IL11 activity revealed a critical role of IL11/ERK signaling for LKB1/AMPK inhibition and mTOR activation in fatty liver disease. These data identify the IL11/mTOR axis as a signaling commonality in stromal, epithelial, and cancer cells and reveal a shared IL11-driven mesenchymal program across cell types.
Subject areas: Cell biology, Functional aspects of cell biology
Graphical abstract
Highlights
-
•
How IL11 activates the stroma but causes epithelial dysfunction is not known
-
•
IL11 stimulates ERK/P90RSK to inhibit LKB1/AMPK and activate mTOR
-
•
In disease, IL11 causes EMT: damaging the epithelium and activating the stroma
-
•
Activation of AMPK with metformin reduces IL11-induced disease phenotypes
Cell biology; Functional aspects of cell biology
Introduction
Interleukin 11 (IL11) is a little studied cytokine that was recently found to cause ERK-dependent fibroblast-to-myofibroblast transformation and fibrosis across organs (Cook and Schafer, 2020; Schafer et al., 2017; Widjaja et al., 2019). Unexpectedly, during studies of alcoholic hepatitis and non-alcoholic steatohepatitis (NASH), IL11 was found to have ERK-driven metabolic and NOX4/JNK-related toxic effects in hepatocytes (Dong et al., 2021b; Effenberger et al., 2022; Widjaja et al., 2019). IL11-induced hepatotoxicity was also described following drug-induced hepatocyte injury, which was also ERK-dependent and related to NOX4/JNK activation (Dong et al., 2021a; Widjaja et al., 2021a). IL11-induced epithelial cell dysfunction has been described more generally with cellular dedifferentiation implicated in its pathobiology (Strikoudis et al., 2019; Widjaja et al., 2022).
We hypothesized that there is a conserved role for IL11-stimulated ERK activity in stromal cells and epithelial cells that leads to both myofibroblast transformation and hepatocyte/epithelial cell dysfunction. In support of this, ERK activation in the liver has previously been linked with diet-induced metabolic perturbation (Jiao et al., 2013; Zheng et al., 2009b) and ERK is a recognized pathway for the activation of fibroblasts (Du et al., 2008; Schafer et al., 2017) and hepatic stellate cells (HSCs) (Foglia et al., 2019; Widjaja et al., 2019). Intriguingly, IL11 activates ERK-dependent, rapamycin-sensitive gene translation in fibroblasts, implicating mTORC1 for IL11 effects in fibroblasts (Widjaja et al., 2021b). This stimulated the question: could the IL11/ERK/mTOR axis represent a common pathway for stromal cell activation and hepatocyte dysfunction? And, if so, what is the full signaling pathway and is it more generalizable?
In this study, we investigate whether IL11-stimulated ERK/P90RSK can phosphorylate LKB1 (STK11) to inactivate LKB1/AMPK and increase mTOR activity. Fittingly, LKB1 and AMPK are important for fibroblast activation (Rangarajan et al., 2018; Thakur et al., 2015; Vaahtomeri et al., 2008), diet-induced liver steatosis (Shaw et al., 2005; Woods et al., 2017) and renal tubular epithelial cell dysfunction (Han et al., 2016). LKB1, a master kinase and tumor suppressor, forms a heterotrimeric complex with MO25 (mouse protein 25) and STRAD (Ste20-related adaptor) and is thought to be constitutively active in the absence of phosphorylation (Zeqiraj et al., 2009). However, the inactivation of LKB1 has been reported following dual phosphorylation by ERK/P90RSK in a melanoma cell line (Zheng et al., 2009a).
Here, we use a range of techniques to dissect the effects of IL11 on the ERK//LKB1/AMPK/mTOR axis in primary human fibroblasts, HSCs, hepatocytes, renal epithelial, and lung epithelial cancer cells. Furthermore, we examine the signaling effects of genetic or pharmacologic inhibition or activation of IL11 signaling in livers of mice with diet-induced NASH.
Results
An axis of IL11/ERK/P90RSK inhibits LKB1 to activate fibroblasts
We have shown previously that TGFβ1 or IL11 stimulation of human cardiac fibroblasts (HCFs) over a 24-h time course causes variable STAT3 activation but shared biphasic activation of ERK (Widjaja et al., 2021b). We examined whether this pattern was linked with phosphorylation (p-) of LKB1 (S428; putative inactive LKB1), AMPK (T172; active AMPK), or mTOR (S2448; active mTOR) (Figures 1A and S1). Following IL11 or TGFβ1 stimulation, LKB1 was phosphorylated at the P90RSK site (S428) in a biphasic manner over the time course. In these experiments, we were not able to assess the phosphorylation of endogenous LKB1 (S325), the specific ERK site, as detection was only possible following LKB1 overexpression, as shown later in discussion. In stimulated cells, AMPK activity (p-AMPK) was inverse to p-LKB1 levels, with progressive loss in AMPK activity up to 24 h post-stimulation. Reciprocal to the decrease in p-AMPK, p-mTOR expression increased over the IL11 and TGFβ1 time courses (Figures 1A and S1).
Figure 1.

An axis of IL11/ERK/P90RSK activity leads to the inhibition of LKB1
(A) Western blots (WB) and densitometry analyses of phosphorylated (p-) and total LKB1, AMPK, and mTOR in human cardiac fibroblasts (HCFs) stimulated with IL11 over a time course; one-way ANOVA with Dunnett’s correction (n = 3 biological replicates).
(B) WB of p- and total ERK, P90RSK, LKB1, AMPK, mTOR, P70S6K, S6RP, ɑSMA, and GAPDH levels in HCFs stimulated with IL11 or TGFβ1 alone or in presence of anti-IL11 (×203), antiI-IL11RA (×209) or an IgG control (n = 2 biological replicates).
(C) WB of p- and total AMPK, mTOR, P70S6K, S6RP, ɑSMA, and GAPDH levels in IL11-stimulated HCFs in the presence of DMSO, AICAR, or 991 (n = 3 biological replicates).
(D and E) IL11 (D) and MMP2 (E) concentration in the supernatant of HCFs stimulated with (D) HyperIL11, (E) IL11, or (D-E) TGFβ1 in the presence of DMSO, metformin, AICAR, or 991; one-way ANOVA with Sidak’s correction (n = 4 biological replicates).
(F) WB of p- ERK, P90RSK, LKB1, AMPK, mTOR, total LKB1, and GAPDH in A549 cells infected with adenovirus-driven expression of wild-type (WT) LKB1 stimulated with IL11 across time (total protein levels are shown in Figure S5, n = 2 biological replicates).
(G and H) WB of p- and total ERK, P90RSK, LKB1, AMPK, mTOR, P70S6K, S6RP, and GAPDH levels from adenovirus-driven expression of either null control vector, WT-LKB1 or DM-LKB1-infected A549 cells that are stimulated with IL11 (G) alone (n = 2 biological replicates) or (H) with the addition of DMSO, U0126 or BI-D1870 (n = 1 biological replicate) for 2 h.
(A–H) IL11, TGFβ1, HyperIL11 (10 ng/mL), IgG, ×203, ×209 (2 μg/mL), AICAR (1 mM), BI-D1870, U0126 (10 μM), DMSO (0.1%), 991 (1 μM), metformin (1 mM); 24-h stimulation unless otherwise specified. (A) Data are shown as mean ± SD, (D-E) data are shown as box-and-whisker with median (middle line), 25th–75th percentiles (box) and min-max percentiles (whiskers); ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001,∗∗∗∗p < 0.0001. BL: Baseline; FC: Fold change. See also Figures S1–S6.
TGFβ-stimulated ERK activity in fibroblasts is IL11-dependent (Schafer et al., 2017; Widjaja et al., 2021b). We used neutralizing antibodies against IL11 (×203) or IL11RA (×209) to assess whether TGFβ-stimulated p-LKB1 (24 h) was similarly dependent on IL11 (Figure 1B). IL11 and TGFβ increased p-ERK, p-P90RSK (S380), and p-LKB1, which was associated with diminished p-AMPK and increased p-mTOR. We extended downstream analyses and observed levels of p-P70S6K (T389) and its substrate S6RP (S235/236) were increased, as were the mesenchymal marker (ɑSMA) levels, in IL11 or TGFβ stimulated cells. These signaling events were collectively inhibited by ×203 or ×209 in TGFβ stimulated cells (Figure 1B).
mTOR activity is regulated by complex mechanisms and LKB1 controls multiple (>12) AMP-related protein kinases (Shackelford and Shaw, 2009). We sought to determine the specific importance of AMPK for mTOR activation by stimulating fibroblasts with IL11 in the presence or absence of the AMPK activating compounds, AICAR or 991 (Figure 1C) (Lai et al., 2014). IL11 stimulation (10ng/mL, 24 h) caused the expected decrease in p-AMPK and increase in p-mTOR/p-P70S6k/p-S6RP that was associated with higher ACTA2 (ɑSMA) expression, indicative of fibrogenesis. In cells incubated with either AICAR or 991, p-AMPK levels were maintained, mTOR/P70RSK remained inactive, and expression of the mesenchymal marker (ɑSMA) was inhibited.
We then examined if AMPK activity is involved in the self-amplifying loop of IL11 secretion from fibroblasts by stimulating cells with an IL11RA:IL11 fusion protein (HyperIL11) or TGFβ in the presence or absence of AMPK activating compounds (Figure 1D). HyperIL11 and TGFβ induced IL11 secretion, as expected, and this effect was significantly inhibited by metformin, AICAR, or 911. Similarly, IL11- or TGFβ-induced MMP2 secretion from fibroblasts was reduced by AMPK activation, with a similar profile of inhibition (magnitude of inhibition: 911 > AICAR > metformin) (Figure 1E). We have shown previously that IL11 effects in cardiac fibroblasts are critically dependent on ERK (Schafer et al., 2017), which we confirmed for IL11-induced IL11 or MMP2 secretion in the primary fibroblast cultures used here (Figure S2).
Phosphorylation of both S325 and S428 of LKB1 is required for its inactivation
To study the relationship between IL11 stimulated p-ERK/p-P90RSK and LKB1 activity, we used LKB1-deficient A549 cells for heterologous, adenovirus-driven expression of wild-type LKB1 (WT-LKB1) or mutant LKB1 (LKB1 S325A and S428A, designated as double mutant LKB1 (DM-LKB1)). We first confirmed the expression of IL11RA in A549 cells, a lung epithelial cancer cell line (Figure S3).
After determining the optimal multiplicity of infection (MOI) of adenovirus encoding WT-LKB1 or DM-LKB1 (Figure S4), we stimulated WT-LKB1-infected A549 cells with IL11 over a time course. IL11 increased p-ERK levels in WT-LKB1-transduced A549 cells (Figure 1F). In these overexpression experiments, the p-LKB1 (S325) antibody detected a band co-migrating with LKB1 and, following IL11 stimulation, LKB1 was first (at 15 min) phosphorylated at S325, coincident with increased p-ERK levels, and later (at 2-h time point) phosphorylated at S428, along with P90RSK activation. AMPK activity (p-AMPK) was maintained until both LKB1 sites were phosphorylated at which point p-AMPK levels diminished and p-mTOR levels increased (Figures 1F and S5).
A549 cells were then infected with adenovirus encoding either WT-LKB1 or DM-LKB1 and stimulated with IL11. Cells expressing WT-LKB1 showed the phosphorylation of both S325 and S428, whereas those expressing DM-LKB1 did not, confirming antibody specificity (Figure 1G). As compared to control cells, p-ERK and p-P90RSK levels were increased in IL11 stimulated cells expressing WT-LKB1 but to a lesser extent in those expressing DM-LKB1, suggesting crosstalk (Du et al., 2008). In cells expressing WT-LKB1, IL11 stimulation reduced p-AMPK levels and increased p-mTOR/p-P70S6K and p-S6RP levels. In contrast, cells expressing DM-LKB1, which cannot be inactivated by ERK/P90RSK, had high levels of p-AMPK expression and undetectable p-mTOR following IL11 stimulation.
Our data suggested that, following IL11 stimulation, there is sequential phosphorylation of LKB1 by ERK and P90RSK (Figure 1F) and that dual phosphorylation is needed to inhibit LKB1 (Figures 1F and 1G). We examined this premise experimentally using pharmacologic inhibition of either ERK (U0126) or P90RSK (BI-D1870) in cells stimulated with IL11 (Figure 1H). A549 cells expressing WT-LKB1 showed the expected increases in the phosphorylation of ERK/P90RSK/LKB1, decreased p-AMPK and elevated p-mTOR. In cells expressing DM-LKB1, p-ERK/p-P90RSK was increased but the downstream signaling changes were not seen. Cells stimulated with IL11 in the presence of U0126 had no detectable p-ERK/p-P90RSK, no LKB1 phosphorylation, preserved p-AMPK, and low p-mTOR (Figure 1H). On the other hand, cells that were stimulated with IL11 in the presence of BI-D1870 had increased p-ERK but not p-P90RSK, elevated p-LKB1 (S325) but not p-LKB1 (S428) and preserved p-AMPK and low p-mTOR (Figure 1H). These data confirm that LKB1 is sequentially phosphorylated by ERK/P90RSK and that both S325 and S428 both need to be phosphorylated to fully inhibit LKB1.
To confirm that the IL11/LKB1/mTOR pathway is also relevant in cancer cell lines where LKB1 is expressed, we performed a separate set of IL11 time course experiment in H1792 cells, an LKB1-expressing lung epithelial cancer cell line (Figure S6A). Consistent with the observed signaling effects in A549 cells, IL11-induced activation of ERK and P90RSK, phosphorylation of LKB1, inactivation of AMPK, and increased p-mTOR p-P70S6K, and p-S6RP levels in H1792 cells (Figure S6A). As seen in A549, all signaling changes downstream of ERK (P90RSK, LKB1, AMPK, mTOR, P70S6K, S6RP) were abolished in the presence of U0126 (Figure S6B).
IL11/ERK activity inhibits LKB1/AMPK to activate hepatic stellate cells
Pursuing the hypothesis that IL11/ERK is important for LKB1/AMPK activity across cell types, we next studied HSCs. We first compared the effects of IL6 and IL11 as these cytokines are sometimes portrayed as having overlapping effects in the liver (Schmidt-Arras and Rose-John, 2016). Primary human HSCs were stimulated with a dose range of IL6 or IL11 for 24 h (Figure 2A). This showed dose-dependent IL6-mediated phosphorylation of STAT3 but not ERK. In contrast, IL11 stimulation for 24h had no effect on p-STAT3 but dose-dependently increased p-ERK. Across the dose range (1.25–20 ng/mL) IL6 had no effect on p-LKB1 or p-AMPK levels, whereas IL11 dose-dependently phosphorylated LKB1 and inhibited AMPK (Figure 2A).
Figure 2.

IL11/ERK activity inhibits LKB1/AMPK to activate hepatic stellate cells
(A) WB of p- and total STAT3, ERK, LKB1, and AMPK in HSCs stimulated with increasing doses of either IL6 or IL 11 (n = 1 biological replicate, except for BL (n = 2)).
(B and C) WB of p- and total ERK, P90RSK, LKB1, AMPK, mTOR, P70S6K, and S6RP in HSCs stimulated with (B) IL11 or TGFβ1 in the presence of IgG, ×203, or ×209 (n = 2 biological replicates) or (C) IL11 in the presence of U0126 or DMSO (n = 3 biological replicates).
(D and E) (D) IL11 or (E) MMP2 levels in the supernatant from (D) HyperIL11 or (E) IL11-stimulated HSCs in the presence of DMSO or U0126; one-way ANOVA with Tukey’s correction (n = 3 biological replicates).
(F) WB of p- and total AMPK, mTOR, P70S6K, S6RP, and GAPDH in IL11-stimulated HSCs with the addition of DMSO, AICAR, or 991 (n = 3 biological replicates).
(G and H) (G) IL11 (n = 3 biological replicates) and (H) MMP2 (n = 4 biological replicates) concentrations in the (G) HyperIL11, (H) IL11, or (G-H) TGFβ-stimulated HSC supernatant in the presence of DMSO, metformin, AICAR, or 991; one-way ANOVA with Sidak’s corrections.
(I) WB of phospho and total LKB1 and GAPDH in adenovirus-driven expression of null control vector, WT-LKB1 or DM-LKB1-infected HSCs at BL or stimulated with IL11 (n = 2 biological replicates). (A–I) HyperIL11, IL11, TGFβ1 (10 ng/mL), IgG, ×203, ×209 (2 μg/mL), AICAR (1 mM), DMSO (0.1%), 991 (1 μM), metformin (1 mM), U0126 (10 μM); 24-h stimulation. (D–E, G–H) Data are shown as box-and-whisker with median (middle line), 25th–75th percentiles (box) and min-max percentiles (whiskers); ∗∗∗∗p < 0.0001). BL: Baseline. See also Figure S7.
HSCs were incubated TGFβ1 in the presence or absence of anti-IL11 (×203) or anti-IL11RA (×209) or stimulated with IL11 (Figure 2B). IL11 stimulation increased p-ERK/p-LKB1, diminished p-AMPK and increased p-mTOR/p-P70S6K, p-S6RP and TGFβ1 stimulation caused similar activation. Consistent with the requirement for an autocrine loop of IL11 activity downstream of TGFβ1 for HSC activation, anti-IL11 or anti-IL11RA1 prevented TGFβ1-induced ERK activation, restored AMPK activity, and reduced levels of the myofibroblast marker, ɑSMA (Figure 2B). The critical and apical role of IL11-stimulated ERK activity was shown using U0126, which inhibited IL11-mediated ERK activation as well as all downstream signaling, and also HSC-to-myofibroblast transformation (Figure 2C).
We stimulated HSCs with HyperIL11, which induced IL11 secretion that was inhibited by U0126, demonstrating the autocrine loop of IL11 activity again (Figure 2D). IL11 induces the secretion of MMP2 from HSCs (Widjaja et al., 2019), which we reaffirmed and also showed to be ERK dependent (Figure 2E).
We then examined whether the restoration of AMPK activity following IL11-induced LKB1 inactivation could rescue downstream events. Cells stimulated with IL11 (10 ng/mL, 24 h) and incubated with AMPK-activating compounds had normal p-AMPK levels, did not activate the mTOR, and did not undergo myofibroblast transformation (Figure 2F). Metformin, AICAR, or 991 all reduced TGFβ1- or HyperIL11-induced secretion of both IL11 and MMP2 from HSCs with similar profiles of effect (Figures 2G and 2H). Although endogenous p-LKB1 (S325) could not be detected in HSCs protein extracts, we confirmed this event occurred following optimal MOI determination and subsequent transduction of HSCs with adenovirus encoding WT-LKB1 or DM-LKB1 in the presence or absence of IL11 stimulation (Figures 2I and S7).
IL11/ERK activity inhibits LKB1/AMPK that is associated with hepatotoxicity
Primary human hepatocytes were stimulated with a dose range of IL6 or IL11 for 24 h. As seen in HSCs (Figure 2A), there was dose-dependent IL6-mediated phosphorylation of STAT3 but not ERK, whereas IL11 dose-dependently increased p-ERK but not p-STAT3 (Figure 3A). Across the dose range (1.25–20 ng/mL), IL6 had no effect on p-LKB1 or p-AMPK levels. In contrast, IL11 dose-dependently increased p-LKB1 which was accompanied by a reciprocal reduction in p-AMPK.
Figure 3.

IL11/ERK activity inhibits LKB1/AMPK to cause hepatocyte dysfunction
(A) WB of p- and total STAT3, ERK, LKB1, and AMPK in hepatocytes following a dose range stimulation with either IL6 or IL11 (n = 1 biological replicate, except for BL (n = 2)).
(B and C) WB of p- and total ERK, P90RSK, LKB1, AMPK, ULK, ACC, mTOR, P70S6K, and S6RP in hepatocytes stimulated with (B) IL11 or palmitate in the presence of IgG, ×203, or ×209 (n = 2 biological replicates),or (C) IL11 in the presence of U0126 or DMSO (n = 3 biological replicates).
(D and E) Secreted (D) IL11 or (E) ALT in the supernatant of (D) HyperIL11 or (E) IL11-stimulated hepatocytes in the presence of DMSO or U0126; one-way ANOVA with Tukey’s correction (n = 3 biological replicates).
(F–H) (F) WB of p- and total AMPK, ULK, ACC, mTOR, P70S6K, S6RP, and GAPDH, (G) representative fluorescence images (scale bars, 100 μm and (H) quantification of DCFDA staining (one-way ANOVA with Dunnett’s correction) for ROS detection from 100× field images in IL11-stimulated hepatocytes in the presence of DMSO, AICAR, 991, or metformin (n = 3 biological replicates).
(I and J) (I) IL11 (n = 3 biological replicates) and (J) ALT (n = 4 biological replicates) concentrations in the supernatant of palmitate-loaded hepatocytes in the presence of DMSO, metformin, AICAR, 991, or U0126; one-way ANOVA with Dunnett’s correction.
(K) WB of p- and total LKB1 and GAPDH in adenovirus-driven expression of null control vector, WT-LKB1 or DM-LKB1-infected hepatocytes at BL or stimulated with IL11. (A-K) IL11, HyperIL11 (10 ng/mL), IgG, ×203, ×209 (2 μg/mL), AICAR (1 mM), DMSO (0.1%), 991 (1 μM), metformin (1 mM), U0126 (10 μM); 24-h stimulation. (D-E, H-J) Data are shown as box-and-whisker with median (middle line), 25th–75th percentiles (box) and min-max percentiles (whiskers) (∗∗∗∗p < 0.0001). BL: Baseline. See also Figures S8–S10.
We have described previously an autocrine loop of IL11 activity in steatotic hepatocytes that causes metabolic dysfunction and ERK/NOX4-driven lipotoxicity (Dong et al., 2021b; Widjaja et al., 2019). To explore these further, primary human hepatocytes were loaded with palmitate in the presence or absence of anti-IL11 (×203) or anti-IL11RA (×209) or stimulated with IL11 (Figure 3B). Hepatocytes stimulated with IL11 had increased p-ERK/p-P90RSK/p-LKB1, diminished p-AMPK, and increased p-mTOR. Similar to IL11-stimulated hepatocytes, steatotic hepatocytes had lesser LKB1/AMPK activity that was accompanied by a decrease in the corresponding activation of AMPK substrates (p-ACC and p-ULK) (Figure 3B) (Egan et al., 2011; Ha et al., 1994). Inhibition of IL11 signaling in steatotic hepatocytes with either ×203 or ×209 abrogated ERK activation restored p-AMPK levels and increased the corresponding levels p-ULK and p-ACC (Figure 3B), further confirming the autocrine loop of IL11 activity. As seen for HSCs (Figure 2C), the critical and initiating role of IL11-stimulated ERK activity for all downstream events in hepatocytes was demonstrated using U0126 (Figure 3C). We stimulated hepatocytes with HyperIL11 that induced ERK-dependent IL11 secretion, reconfirming the autocrine IL11 loop in hepatocytes (Figure 3D) and we confirmed here that IL11 hepatotoxicity was ERK-dependent (Figure 3E).
In hepatocytes, we again assessed the specific role of AMPK downstream of LKB1 in IL11-stimulated hepatocytes (Figure 3F). IL11 caused decreased p-AMPK, p-ACC, and p-ULK levels and increased p-mTOR. However, in cells stimulated with IL11 and incubated with either AICAR or 991, p-AMPK, p-ACC, and p-ULK levels were maintained, the mTOR/P70RSK/S6RP axis was not activated and pathogenic NOX4/JNK activity was mitigated (Figure 3F). Furthermore, ALT levels as well as IL11-induced ROS, which is in part NOX4 dependent (Widjaja et al., 2021a), were diminished by AMPK activation in IL11-stimulated hepatocytes (Figures 3G–3H and S8).
In steatotic hepatocytes, metformin, AICAR, or 991 significantly reduced IL11 secretion and cell death, with similar profiles of effect (metformin < AICAR <911) (Figures 3I and 3J). As expected, U0126 also inhibited IL11 secretion and cell death (Figures 3I and 3J). Endogenous p-LKB1 (S325) was not detected in hepatocytes, and we therefore confirmed this by infecting hepatocytes with WT-LKB1 or DM-LKB1, IL11 stimulation, and immunoblotting (Figure 3K).
We then examined the effects of IL11 in renal tubular epithelial cells (TECs) that dedifferentiate to become steatotic and dysfunctional following the genetic inhibition of LKB1 (Boehlke et al., 2010; Han et al., 2016). We stimulated TECs with IL11 and observed increased p-ERK, p-P90RSK, and p-LKB1, along with the inactivation of AMPK and ACC and concomitant activation of mTOR (Figure S9). TEC dedifferentiation occurs via an epithelial-to-mesenchymal transition (EMT) related process (partial EMT (pEMT)) that is critically dependent on SNAI1 upregulation and E-Cadherin suppression (Han et al., 2016; Lovisa et al., 2015). TECs stimulated with IL11 upregulated SNAI1 while E-Cadherin expression was downregulated (Figure S9). The importance of ERK activity in IL11-stimulated TECs was shown using U0126 which inhibited downstream signaling events as well as pEMT (Figure S9).
The finding that IL11 stimulates SNAI1-related EMT in TECs prompted us to revisit the effects of IL11 in hepatocytes that can also undergo SNA1-related dedifferentiation causing their dysfunction (Grant Rowe et al., 2011; Sekiya and Suzuki, 2011). In hepatocytes, IL11 stimulation upregulated SNAI1 which was also seen in palmitate-loaded hepatocytes, where SNAI1 expression was found to be IL11 dependent (Figure S10).
IL11-dependent activation of ERK/P90RSK and inhibition of LKB1/AMPK in liver disease
To translate our findings to the in vivo setting, we took advantage of samples that we had kept from our previous NASH studies (Dong et al., 2021b; Widjaja et al., 2019). We first examined samples from WT mice or mice with conditional knockout (CKO) of Il11ra1 in hepatocytes maintained on a western diet with fructose (WDF, 16 weeks) (Dong et al., 2021b). We initially probed liver extracts for p-ERK, which was previously shown to be elevated in livers of WT mice but not CKO mice on WDF, and substantiated this result, confirming sample integrity (Figure 4A).
Figure 4.

IL11-mediated inhibition of LKB1 occurs in the livers of mice with non-alcoholic steatohepatitis
WB showing hepatic levels of p- and total ERK, P90RSK, LKB1, AMPK, mTOR, P70S6K, S6RP, ɑSMA, and GAPDH from (A) NC or WDF fed-wild-type (WT) or hepatocyte-specific Il11ra1 knockout mice (CKO).
(B) NC or WDF-fed Il11ra1+/+ (WT) and WDF-fed Il11ra1−/− (KO) that were infected with AAV8-ALB-Null, and WDF-fed KO mice that were previously infected with AAV8-ALB-mbIl11ra1 (full-length membrane-bound Il11ra1) in order to specifically restore the expression of Il11ra1 in hepatocytes.
(C) WDF-fed mice that were on therapeutic reversal dosing experiment; mice were treated with 10 mg/kg (2×/week; IP) of either IgG or ×209 16 weeks after the start of WDF for a duration of 8 weeks while they were still on continuous WDF feeding (n = 3 mice/group).
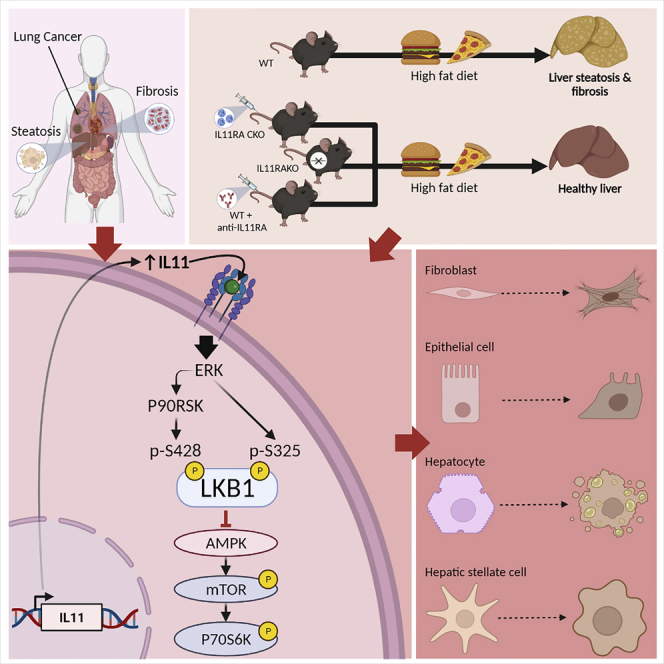
(D) Schematic of the proposed role of ERK/P90RSK activity downstream of IL11 stimulation for the phosphorylation of LKB1 at both S325 and S428 sites, leading to LKB1 inactivation and consequent effects in stromal, epithelial, and cancer cells.
To generate new insights, we then assessed the LKB1/AMPK/mTOR axis and related signaling. This revealed that WT mice on WDF had elevated p-P90RSK/p-LKB1, lesser p-AMPK, and increased p-mTOR/p-P70S6K/p-S6RP along with evidence for HSC-to-myofibroblast transformation (ɑSMA upregulation) (Figure 4A). This recapitulates the in vitro data in hepatocytes (Figure 2) and shows a role for the IL11/ERK/LKB1/AMPK axis in NASH.
We next studied liver samples from WT mice or mice globally null for Il11ra1 infected with control AAV8 (Null) or AAV8 encoding albumin-driven Il11ra1 expression, which restores IL11RA1 expression in hepatocytes only (Dong et al., 2021b) (Figure 4B). We determined sample integrity by examining p-ERK in liver extracts from WT mice on WDF (16 weeks), which we confirmed was increased. In WT mice on WDF, we again observed elevated p-P90RSK/p-LKB1, lesser p-AMPK, and increased p-mTOR and ɑSMA levels. Il11ra1 null mice on WDF infected with control AAV8 were protected from NASH and activation of the ERK/LKB1/AMPK axis was not apparent. In contrast, hepatocyte-specific restoration of IL11RA1 expression was associated with increased p-ERK/p-P90RSK/p-LKB1, lesser p-AMPK, and these mice developed NASH and metabolic derangement (Dong et al., 2021b). Of note, the restoration of IL11 signaling in hepatocytes only increased ɑSMA expression (in the absence of HSC activation or fibrosis (Dong et al., 2021b)) in keeping with the activation of a mesenchymal program in hepatocytes.
We then studied liver samples from mice with NASH, secondary to WDF for 16 weeks, that were administered a neutralizing IL11RA antibody (×209) or an IgG control antibody for a further 8 weeks, while the mice were maintained on WDF (Widjaja et al., 2019) (Figure 4C). WT mice on WDF for 16 weeks and those receiving IgG by study end (24 weeks) had increased p-ERK, as previously reported, and the expected phosphorylation changes in LKB1/AMPK/mTOR and other proteins. ɑSMA was upregulated after 16 weeks of WDF and increased further at 24 weeks in mice receiving IgG, showing disease progression. In contrast, the IL11/ERK pathway, and associated downstream changes, were inhibited by the administration of ×209 from week 16 to 24 of WDF and ɑSMA levels were lower than at 16 weeks, showing disease reversal. We schematically summarize our study findings across cell types, in the context of published IL11-associated phenotypes, in Figure 4D.
Discussion
It is generally accepted that LKB1, an important tumor suppressor and master kinase (Hardie and Alessi, 2013), is constitutively active (Shackelford and Shaw, 2009; Zeqiraj et al., 2009). Dual phosphorylation (S325, S428) and inactivation of LKB1 have been described in a B-RAF V600E expressing melanoma cell line (Zheng et al., 2009a) but other studies found that the phosphorylation of these sites did not affect LKB1 activity (Denison et al., 2009; Sapkota et al., 2002). Our study shows that LKB1 can be phosphorylated and inactivated in cells exposed to an extrinsic signaling factor and that this holds true across several primary human cell types, as well as in cancer cells that lack the V600E mutation.
Some tumor cells, such as A549 (Rapoza et al., 2006), secrete IL11, as do tumor subclones (Janiszewska et al., 2019; Marusyk et al., 2014) and the tumor stroma (Nishina et al., 2021). In Peutz Jeghers syndrome, heterozygous loss-of-function in LKB1 is associated with high levels of IL11 in intestinal polyps and an increased risk of cancers (Ollila et al., 2017). There is extensive literature on IL11-induced activation of JAK/STAT3 in cancer cells (Ernst and Putoczki, 2014; To et al., 2022) and on the association of IL11 with chemotherapy resistance and cancer metastasis (Ernst and Putoczki, 2014; Janiszewska et al., 2019; Marusyk et al., 2014). We suggest that IL11/ERK-mediated inactivation of LKB1/AMPK in cancer cells, which has been overlooked, underlies some of the effects of IL11 in oncogenesis, perhaps specifically cancers associated with SNAI1 expression (Barrallo-Gimeno and Nieto, 2005; Su et al., 2020).
In fibroblasts, the inhibition of LKB1/AMPK causes myofibroblast transformation and metformin, an indirect AMPK activator, reverses fibrosis (Thakur et al., 2015; Vaahtomeri et al., 2008). In hepatocytes, the LKB1/AMPK axis is critical for metabolism, with AMPK inactivation causing NASH (Long and Zierath, 2006; Shaw et al., 2005; Woods et al., 2017). We surmise that the dual effect of IL11 on LKB1/AMPK in the stroma and epithelium may explain why its inhibition in NASH reverses not only fibrosis, owing to HSC transformation, but also steatosis, owing to hepatocyte dysfunction (Dong et al., 2021b; Widjaja et al., 2019). We speculate that some of the beneficial effects of metformin for metabolism, fibrosis, cancer, and diseases of aging may relate to its activation of AMPK in the context of IL11/ERK/P90RSK-mediated LKB1 inhibition in disease (Mohammed et al., 2021).
The importance of LKB1/AMPK for renal epithelial cells is known and their deficiency for LKB1 causes pEMT resulting in a non-proliferative, dedifferentiated mesenchymal state, which is SNAI1-dependent (Boehlke et al., 2010; Han et al., 2016; Lovisa et al., 2015). To add to this, we show here that IL11 stimulates the dedifferentiation and mesenchymal transition of TECs, and also of hepatocytes, which is associated with SNAI1 upregulation (Grant Rowe et al., 2011; Sekiya and Suzuki, 2011). Thus, IL11-related pathology might more generally be characterized by the particular combination of stromal cell activation and epithelial cell pEMT (Strikoudis et al., 2019; Widjaja et al., 2022).
IL11 evolved some 600m years ago and plays a critical role in fin regeneration in fishes, tail regrowth in tadpoles, and limb renewal in axolotl (Allanki et al., 2021; Darnet et al., 2019; Tsujioka et al., 2017; Wang et al., 2005). In these species, IL11 is required for blastema formation at the site of injury (Allanki et al., 2021; Tsujioka et al., 2017). Blastemas are collagen rich, TGFβ dependent (Chablais and Jazwinska, 2012), and consist of migrating stromal and dedifferentiated polarized (e.g. epithelial) cells, which create the new appendage (Darnet et al., 2019). Thus, IL11-induced activation of a shared mesenchymal program in both stromal and polarized cells, which we identified here, could be relevant for appendage regeneration in some species.
In conclusion, we show that IL11 stimulates sustained ERK activity across cell types that lead to P90RSK activation and the sequential phosphorylation of LKB1 at S325 (by ERK) and S428 (by P90RSK), causing its inactivation. This inhibits AMPK and activates mTOR. This signaling axis causes both epithelial cell dysfunction and stromal cell activation, which is seen in a number of diseases associated with increased IL11 which include pulmonary fibrosis, inflammatory bowel disease, NASH, and kidney disease.
Limitations of the study
While we examined the effects of IL11-induced LKB1 inactivation on AMPK/mTOR, LKB1 regulates up to 12 other kinases that were not studied here. The activation of ERK/P90RSK is centrally important for IL11-driven phenotypes; however, the activation of this axis can also lead to GSK3β inactivation and SNA1 upregulation, which requires study. In NASH, we were not able to dissect the relative contributions of IL11-stimulated STAT3 versus ERK activation for disease phenotypes and use these experiments only to confirm the effects of IL11 on ERK/P90RSK/LKB1 signaling in vivo. The specific role for the dual phosphorylation of LKB1 (S325, S428) in signaling and disease requires new genetic models, which we are in the process of generating.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-phospho ACC Ser79 (Rabbit monoclonal) | Cell Signaling | Cat# 11818 |
| Anti-ACC (Rabbit monoclonal) | Cell Signaling | Cat# 3676 |
| Anti-phospho AMPK Thr172 (Rabbit monoclonal) | Cell Signaling | Cat# 2535 |
| Anti-AMPK (Rabbit monoclonal) | Cell Signaling | Cat# 5832 |
| Anti-Cleaved Caspase 3 Asp175 (Rabbit polyclonal) | Cell Signaling | Cat# 9661 |
| Anti-Caspase 3 (Rabbit polyclonal) | Cell Signaling | Cat# 9662 |
| Anti-E-Cadherin (Rabbit monoclonal) | Cell Signaling | Cat# 3195 |
| Anti-phospho-ERK1/2 Thr202/Tyr204 (Rabbit monoclonal) | Cell Signaling | Cat# 4370 |
| Anti-ERK1/2 (Rabbit monoclonal) | Cell Signaling | Cat# 4695 |
| Anti-GAPDH (Rabbit monoclonal) | Cell Signaling | Cat# 2118 |
| IgG1 (Mouse monoclonal) | Genovac | Clone 11E10 |
| Anti-IL11 (Mouse monoclonal) | Genovac (Widjaja et al., 2019) | Clone ×203 |
| Anti-IL11RA (Mouse monoclonal) for neutralising study | Genovac (Widjaja et al., 2019) | Clone ×209 |
| Anti-IL11RA (Rabbit monoclonal) for IF | Abcam | Cat# ab125015 |
| Anti-phospho JNK Thr183/Tyr185 (Rabbit monoclonal) | Cell Signaling | Cat# 4668 |
| Anti-JNK (Rabbit monoclonal) | Cell Signaling | Cat# 9258 |
| Anti-phospho LKB1 Ser325 (Goat polyclonal) | Gifted by Prof. Dario Alessi, University of Dundee | N/A |
| Anti-phospho LKB1 Ser428 (Rabbit monoclonal) | Cell Signaling | Cat# 3482 |
| Anti-phospho-mTOR Ser2448 (Rabbit polyclonal) | Cell Signaling | Cat# 2971 |
| Anti-mTOR (Rabbit polyclonal) | Cell Signaling | Cat# 2972 |
| Anti-NOX4 (Rabbit monoclonal) | Thermo Fisher Scientific | Cat# MA5-32090 |
| Anti-phospho p70S6K Thr389 (Rabbit monoclonal) | Cell Signaling | Cat# 9234 |
| Anti-p70S6K (Rabbit monoclonal) | Cell Signaling | Cat# 2708 |
| Anti-phospho P90RSK Ser380 (Rabbit monoclonal) | Cell Signaling | Cat# 11989 |
| Anti-RSK (Rabbit monoclonal) | Cell Signaling | Cat# 9355 |
| Anti-phospho-S6 ribosomal protein Ser235/236 (Rabbit monoclonal) | Cell Signaling | Cat# 4858 |
| Anti-S6 ribosomal protein (Rabbit monoclonal) | Cell Signaling | Cat# 2217 |
| Anti-ɑ-SMA (Rabbit monoclonal) | Cell Signaling | Cat# 19245 |
| Anti-SNAI1 (Rabbit monoclonal) | Cell Signaling | Cat# 3879 |
| Anti-phospho-STAT3 Tyr705 (Mouse monoclonal) | Cell Signaling | Cat# 4113 |
| Anti-STAT3 | Cell Signaling | Cat# 4904 |
| Anti-phospho ULK1 Ser555 | Cell Signaling | Cat# 5869 |
| Anti-ULK1 | Cell Signaling | Cat# 8054 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | Cat# 7076 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat# 7074 |
| Bacterial and virus strains | ||
| AAV8-ALB-Null | Vector Biolabs (Dong et al., 2021b) | N/A |
| AAV8-ALB(1.9)-iCre | Vector Biolabs | Cat# VB1570 |
| AAV8-ALB(1.9)-Il11ra1 | Vector Biolabs (Dong et al., 2021b) | N/A |
| Ad-CMV-Null | Vector Biolabs | Cat# 1300 |
| Ad-h-STK11 | Vector Biolabs | Cat# ADV-224574 |
| Ad-h-STK11 (S325A/S428A) | Vector Biolabs (this manuscript) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| AICAR | Sigma | Cat# A9978 |
| BI-D1870 | Selleck Chemicals | Cat# S2843 |
| Bovine serum albumin | Sigma | Cat# A8806 |
| D(−)Fructose | Sigma | Cat# F0127 |
| DMEM | Gibco | Cat# 11995065 |
| DMSO | Sigma | Cat# D2650 |
| Epithelial cell growth medium | ScienCell | Cat# 4101 |
| Epithelial cell growth supplement | ScienCell | Cat# 4152 |
| ex229 (compound 991) | Selleck Chemicals | Cat# S8654 |
| Fetal bovine serum | ScienCell | Cat# 0500 |
| Fetal bovine serum | Gibco | Cat# 10500064 |
| Fibroblasts growth supplement-2 | ScienCell | Cat# 2382 |
| Fibroblasts medium-2 | ScienCell | Cat# 2331 |
| Metformin | Sigma | Cat# PHR1084 |
| Palmitate | Sigma | Cat# P5585 |
| Penicillin-streptomycin | ScienCell | Cat# 0513 |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Pierce™ Phosphatase Inhibitor Mini Tablets | Thermo Fisher Scientific | Cat# A32957 |
| Pierce™ Protease Inhibitor Tablets, EDTA-free | Thermo Fisher Scientific | Cat# A32965 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat# 89901 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34096 |
| U0126 | Cell Signaling | Cat# 9903 |
| Recombinant HyperIL-11 | Genscript (Dams-Kozlowska et al., 2012) | N/A |
| Recombinant human IL6 | R&D Systems | Cat# 206-IL |
| Recombinant human IL11 | Genscript | (Dong et al., 2021b) |
| Recombinant human TGFβ1 | Bio-Rad | Cat# PHP143B |
| RPMI-1640 | Gibco | Cat# 11875093 |
| Critical commercial assays | ||
| ALT Activity Assay Kit | Abcam | Cat# ab105134 |
| DCFDA Cellular Assay Kit | Abcam | Cat# ab113851 |
| Human IL11 Quantikine ELISA kit | R&D Systems | Cat# D1100 |
| Experimental models: Cell lines | ||
| A549 (LKB1-deficient lung epithelial cancer cell line) | ATCC | Cat# CCL-185 |
| H1792 (LKB1-expressing lung epithelial cancer cell line) | ATCC | Cat# CRL-5895 |
| Primary human cardiac fibroblasts | ScienCell | Cat# 6330 |
| Primary human hepatic stellate cells | ScienCell | Cat# 5300 |
| Primary human hepatocytes | ScienCell | Cat# 5200 |
| Primary human tubular epithelial cells | ScienCell | Cat# 4100 |
| Experimental models: Organisms/strains | ||
| C57BL/6N mice | InVivos | N/A |
| Il11ra1−/− mice | (Schafer et al., 2017) | https://www.jax.org/strain/004621 |
| Il11ra1loxP/loxP | (Widjaja et al., 2021a) | https://www.jax.org/strain/034465 |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad Software Inc. | https://www.graphpad.com/ |
| ImageJ | National Institutes of Health (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Others | ||
| Normal chow (Irradiated rat and mouse diet) | Specialty Feeds | N/A |
| Western diet | Research Diet | Cat# D12079B |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stuart A. Cook (stuart.cook@duke-nus.edu.sg).
Materials availability
Ad-h-STK11 (S325A/S428A) can be commercially obtained from Vector Biolabs.
Experimental models and subject details
Ethics statements
All experimental protocols involving human subjects (commercial primary human cell lines) have been performed in accordance with the ICH Guidelines for Good Clinical Practice. As written in their respective datasheets, ethical approvals have been obtained by the relevant parties and all participants gave written informed consent to either ATCC or ScienCell from which A549, H1792, primary human cardiac fibroblasts, primary human hepatocytes, and primary human hepatic stellate cells were commercially sourced.
Animal studies were carried out in compliance with the recommendations in the Guidelines on the Care and Use of Animals for Scientific Purposes of the National Advisory Committee for Laboratory Animal Research (NACLAR). All experimental procedures were approved (SHS/2014/0925 and SHS/2019/1482) and conducted in accordance with the SingHealth Institutional Animal Care and Use Committee.
Cell culture
The cell lines were obtained commercially and therefore no additional cell authentication was performed. All cells were grown and maintained at 37 °C and 5% CO2. The culture of primary human hepatic stellate cells (HSCs) and primary human hepatocytes were described previously (Viswanathan et al., 2021). Primary human cardiac fibroblasts (HCFs (52-year-old male), ScienCell) were grown and maintained in complete fibroblasts medium-2 (ScienCell) which contains 5% fetal bovine serum (FBS, ScienCell), 1% fibroblasts growth supplement-2 (FGS-2, ScienCell) and 1% penicillin-streptomycin (P/S, ScienCell). Primary human renal proximal tubular epithelial cells (TECs (20-year-old female), ScienCell) were grown and maintained in complete epithelial cell growth medium (ScienCell) containing 10% FBS (ScienCell), epithelial cell growth supplement (EpiCGS, ScienCell) and 1% P/S (ScienCell). LKB-deficient human lung epithelial carcinoma cells (A549 (58-year-old male), ATCC) were grown and maintained in DMEM (Gibco) complete medium which contains 10% FBS (Gibco) and 1% P/S (Gibco). LKB-expressing human lung epithelial carcinoma cells (H1792 (50-year-old male, ATCC) were grown and maintained in RPMI-1640 (Gibco) complete medium containing 10% FBS (Gibco) and 1% P/S (Gibco). The growth medium was renewed every 2–3 days and cells were passaged at 80% confluence, using standard trypsinization techniques. All experiments with primary cells were carried out at low cell passage (<P3). Cells were serum-starved overnight in basal media prior to stimulation. Cells were stimulated with different treatment conditions and durations, as outlined in the main text or figure legends. Stimulated cells were compared to unstimulated cells that have been grown for the same duration under the same conditions, but without the stimuli. AICAR, BI-D1870, 991, and metformin were dissolved in 100% DMSO and added to the media in 1:1000 dilution to get a final DMSO concentration of 0.1%. Palmitate was conjugated in fatty acids free BSA in the ratio of 6:1; 0.5% BSA was used as control (Dong et al., 2021b). The culture methods for primary human hepatocytes and hepatic stellate cells are briefly summarised as follows:
Human hepatocytes (HH)
ScienCell product data sheet for HH (22-week-old foetus, ScienCell) from which all experimental data in this manuscript were generated, states: “HH from ScienCell Research Laboratories are isolated from human liver. HH are cryopreserved immediately after purification and delivered frozen. HH are not recommended for expanding or long-term cultures since the cells do not proliferate in culture.” Once HH have been recovered from the initial thaw cycle, HH are seeded at a density 4 × 105 cells/well of a collagen-coated 6-well plate and used directly for downstream experiments within 48 h of seeding. We have noted that when HH are seeded at low densities (<4 × 105 cells/well of a 6-well plate), HH will de-differentiate and fail to replicate in vivo cellular physiology. Therefore, it is important to follow the counting and seeding guidelines to ensure that hepatocyte functions are preserved ex vivo.
Human hepatic stellate cells (HSC)
Likewise, ScienCell product data sheet for HSC (50-year-old male,ScienCell) states: “HSC from ScienCell Research Laboratories are isolated from the human liver. HSC are cryopreserved at passage 1 (P1) and delivered frozen. HSC are guaranteed to further expand for 15 population doublings under the condition provided by ScienCell Research Laboratories”. All the HSC experiments were carried out at low cell passage (P3). Cryopreserved HSC P1 are first thawed and passaged twice in poly-L-Lysine-coated flask and once they reach P3, HSC are cryopreserved and stored in liquid nitrogen. For experiments, we directly thaw and seed P3 cells at a density of 2.5 × 105 cells/well of a 6-well plate. Controlled population doublings are achieved by plating HSC at the cell densities specified in this protocol, which also ensures that the cells are maintained at an exponential growth phase throughout the primary culture. Within 24 h of seeding, HSC are then serum-starved overnight prior to stimulation.
Animal models
Animal models were first described in detail in (Widjaja et al., 2019) and (Dong et al., 2021b). Mice were housed in temperatures of 21–24 °C with 40–70% humidity on a 12 h light/12 h dark cycle and provided with food and water ad libitum.
Mouse models of NASH
8-week-old male C57BL/6N, Il11ra1−/− mice, and Il11ra1loxP/loxP and their respective control were fed western diet (D12079B, Research Diets) supplemented with 15% weight/volume fructose in drinking water (WDF) for 16–24 weeks as outlined in the main text/figure legends. Control mice received normal chow (NC) and tap water.
Il11ra-floxed (Il11ra1loxP/loxP) mice
6-8-week-old male homozygous Il11ra1-floxed mice (Dong et al., 2021b) were intravenously injected with 4 × 1011 genome copies (gc) of AAV8-ALB-iCre virus (CKO) or a similar amount of AAV8-ALB-Null virus. Both controls and CKO mice were fed WDF or NC three weeks following virus administration.
Il11ra1-deleted mice (KO)
6-8-week-old male Il11ra1−/− mice (Schafer et al., 2017) were intravenously injected with 4 × 1011 gc of AAV8-ALB-Il11ra1 virus to induce hepatocyte specific expression of mouse Il11ra1. As controls, both Il11ra1−/− mice and their wildtype littermates (Il11ra1+/+) were intravenously injected with 4 × 1011 gc AAV8-ALB-Null virus. 3 weeks after virus injection, mice were fed with WDF or NC.
In vivo administration of anti-IL-11 or anti-IL11RA monoclonal antibodies
Male C57BL/6N mice were fed with WDF for 16 weeks to induce NASH and then treated with either 10 mg/kg (2×/week via intraperitoneal injection) of anti-IL11RA (×209) or IgG isotype control (11E10) for 8 weeks while they were on continuous WDF feeding.
Method details
Adenoviral transduction of A549 and HSCs
A549 cells were seeded in 6-well plates at a density of 2 × 105 cells/well. To determine the optimal multiplicity of infection (MOI), A549 (LKB-deficient) and HSC (LKB-expressing) were first infected with Ad-CMV-Null (Vector Biolabs), Ad-WT-LKB1 (Ad-h-STK11, Vector Biolabs) or Ad-DM-LKB1 (Ad-h-STK11 (S325A/S428A), a custom-made double phosphosite mutant construct generated from the Ad-h-STK11 background) at an MOI of 5, 10 or 20 and incubated for 2 or 24 h at 37 °C in serum-free DMEM. Cells were then washed with PBS and the efficiency of gene expression was determined by immunoblotting for LKB1. Two hours incubation at an MOI of 20 was chosen for A549 whereas 24 h incubation with MOI of 20 was selected for HSC. Viral transduction experiments in hepatocytes, an LKB-expressing cell, were performed according to the HSC protocol (24 h at MOI of 20).
Reactive oxygen species (ROS) detection
Primary human hepatocytes were seeded on 96-well Cellcarrier Black plate (60055501, Perkin Elmer) at a density of 1 × 104/well. For this experiment, cells were not serum-starved prior to IL11 stimulation. Twenty-four hours following stimulation, media were removed, and cells were rinsed with ROS buffer followed by incubation with 100 μL/well of 20 μM of DCFDA solution (ab113851, abcam) for 45 min at 37 °C in the dark according to the manufacturer’s protocol. Fluorescence fold changes were quantified and normalised to baseline controls. Live cells with positive 2′, 7’ -dichlorofluorescein (DCF) staining were imaged with a filter set appropriate for fluorescein (FITC) using a fluorescence microscope (Leica) and captured images at 20× magnification.
Colorimetric assays and enzyme-linked immunosorbent assay (ELISA)
Alanine Aminotransferase (ALT) activity in the cell culture supernatant was measured using ALT Activity Assay Kit ( Abcam). The levels of IL11 in equal volumes of cell culture media were quantified using Human IL11 Quantikine ELISA kit (R&D Systems). Both assays were performed according to the manufacturer’s protocol.
Immunoblotting
Western blots were carried out on total protein extracts from hepatocytes and liver tissues. Hepatocyte and liver tissue lysates were homogenised in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) containing protease and phosphatase inhibitors (Roche). Protein lysates were separated by SDS-PAGE and transferred to PVDF membranes. Protein bands were visualised using the Femto ECL detection system (Thermo Fisher Scientific) with the appropriate secondary antibodies: anti-mouse HRP or anti-rabbit HRP. Densitometry analyses were performed by ImageJ software and are provided either in the figures or in Datafile S1. Uncropped western blot images are provided in the Data S2.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software (version 9). Datasets with n ≥ 3 were tested for normality with Shapiro-Wilk tests and all were found to follow normal distribution. One-way ANOVA with Dunnett’s correction was used when several conditions were compared to one condition, one-way ANOVA with Tukey’s correction was used when several conditions were compared to each other. One-way ANOVA with Sidak’s correction was used when different stimulants and corresponding inhibitors were conducted concurrently and compared against one common baseline condition. The criterion for statistical significance was set at p < 0.05. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
We thank Prof. Lewis Cantley for the discussion relating to this article and Prof. Dario Alessi for kindly supplying the p-LKB1 (S325) antibody. We would also like to acknowledge the technical expertise and support of Dong Jinrui and Benjamin George. Funding: This research is supported by the National Medical Research Council (NMRC), Singapore STaR awards (NMRC/STaR/0029/2017), NMRC Centre Grant to the NHCS, MOH-CIRG18nov-0002, GOH Foundation, Tanoto Foundation. A.A.W. is supported by Khoo Foundation.
Author contributions
S.A.C. and A.A.W. conceived and designed the study. A.A.W., J.G.W.T., S.V., and J.T. performed in vitro cell culture, cell biology, and molecular biology experiments. J.G.W.T. and S.G.S. performed in vivo studies. A.A.W., D.C., L.W.W., and S.A.C. analyzed the data. A.A.W., L.W.W., and S.A.C. prepared the article with input from co-authors.
Declaration of interests
A.A.W. and S.A.C. are co-inventors of the patent applications: US Patent App. 16/865,259 (Treatment and prevention of metabolic diseases), US Patent 11319368 (treatment of hepatotoxicity with IL-11 antibody), and US Patent 11339216 (treatment of kidney injury) . S.A.C is a co-inventor of the patent: WO/2017/103108 (treatment of fibrosis), WO/2018/109174 (IL11 ANTIBODIES), WO/2018/109170 (IL11RA antibodies) and a co-founder and shareholder of Enleofen Bio PTE LTD, a company that developed anti-IL11 therapeutics, which were acquired for further development by Boehringer Ingelheim. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Published: August 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104806.
Contributor Information
Anissa A. Widjaja, Email: anissa.widjaja@duke-nus.edu.sg.
Stuart A. Cook, Email: stuart.cook@duke-nus.edu.sg.
Supplemental information
Data and code availability
-
•
All data are provided in the manuscript or in the supplementary materials. Source data are provided as Data S1. The densitometry analyses are provided as Data S2. Uncropped Western blots are provided as Data S3.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Allanki S., Strilic B., Scheinberger L., Onderwater Y.L., Marks A., Günther S., Preussner J., Kikhi K., Looso M., Stainier D.Y.R., Reischauer S. Interleukin-11 signaling promotes cellular reprogramming and limits fibrotic scarring during tissue regeneration. Sci. Adv. 2021;7:eabg6497. doi: 10.1126/sciadv.abg6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo-Gimeno A., Nieto M.A. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- Boehlke C., Kotsis F., Patel V., Braeg S., Voelker H., Bredt S., Beyer T., Janusch H., Hamann C., Gödel M., et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chablais F., Jazwinska A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development. 2012;139:1921–1930. doi: 10.1242/dev.078543. [DOI] [PubMed] [Google Scholar]
- Cook S.A., Schafer S. Hiding in plain sight: interleukin-11 emerges as a master regulator of fibrosis, tissue integrity, and stromal inflammation. Annu. Rev. Med. 2020;71:263–276. doi: 10.1146/annurev-med-041818-011649. [DOI] [PubMed] [Google Scholar]
- Dams-Kozlowska H., Gryska K., Kwiatkowska-Borowczyk E., Izycki D., Rose-John S., Mackiewicz A. A designer hyper interleukin 11 (H11) is a biologically active cytokine. BMC Biotechnol. 2012;12:8. doi: 10.1186/1472-6750-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnet S., Dragalzew A.C., Amaral D.B., Sousa J.F., Thompson A.W., Cass A.N., Lorena J., Pires E.S., Costa C.M., Sousa M.P., et al. Deep evolutionary origin of limb and fin regeneration. Proc. Natl. Acad. Sci. USA. 2019;116:15106–15115. doi: 10.1073/pnas.1900475116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison F.C., Hiscock N.J., Carling D., Woods A. Characterization of an alternative splice variant of LKB1. J. Biol. Chem. 2009;284:67–76. doi: 10.1074/jbc.M806153200. [DOI] [PubMed] [Google Scholar]
- Dong J., Viswanathan S., Adami E., Schafer S., Kuthubudeen F.F., Widjaja A.A., Cook S.A. The pro-regenerative effects of HyperIL6 in drug induced liver injury are unexpectedly due to competitive inhibition of IL11 signaling. Elife. 2021;10:e68843. doi: 10.7554/eLife.68843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Viswanathan S., Adami E., Singh B.K., Chothani S.P., Ng B., Lim W.W., Zhou J., Tripathi M., Ko N.S.J., et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat. Commun. 2021;12:66. doi: 10.1038/s41467-020-20303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Guan T., Zhang H., Xia Y., Liu F., Zhang Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2008;368:402–407. doi: 10.1016/j.bbrc.2008.01.099. [DOI] [PubMed] [Google Scholar]
- Effenberger M., Widjaja A.A., Grabherr F., Schaefer B., Grander C., Mayr L., Schwaerzler J., Enrich B., Moser P., Fink J., et al. Interleukin-11 drives human and mouse alcohol-related liver disease. Gut. 2022:2021–326076. doi: 10.1136/gutjnl-2021-326076. [DOI] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R., et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M., Putoczki T.L. Molecular pathways: IL11 as a tumor-promoting cytokine-translational implications for cancers. Clin. Cancer Res. 2014;20:5579–5588. doi: 10.1158/1078-0432.CCR-13-2492. [DOI] [PubMed] [Google Scholar]
- Foglia B., Cannito S., Bocca C., Parola M., Novo E. ERK pathway in activated, myofibroblast-like, hepatic stellate cells: a critical signaling crossroad sustaining liver fibrosis. Int. J. Mol. Sci. 2019;20:E2700. doi: 10.3390/ijms20112700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant Rowe R., Lin Y., Shimizu-Hirota R., Hanada S., Neilson E.G., Greenson J.K., Weiss S.J. Hepatocyte-derived Snail1 propagates liver fibrosis progression. Mol. Cell Biol. 2011;31:2392–2403. doi: 10.1128/MCB.01218-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J., Daniel S., Broyles S.S., Kim K.H. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- Han S.H., Malaga-Dieguez L., Chinga F., Kang H.M., Tao J., Reidy K., Susztak K. Deletion of Lkb1 in renal tubular epithelial cells leads to CKD by altering metabolism. J. Am. Soc. Nephrol. 2016;27:439–453. doi: 10.1681/ASN.2014121181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D.G., Alessi D.R. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 2013;11:36. doi: 10.1186/1741-7007-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M., Tabassum D.P., Castaño Z., Cristea S., Yamamoto K.N., Kingston N.L., Murphy K.C., Shu S., Harper N.W., Del Alcazar C.G., et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol. 2019;21:879–888. doi: 10.1038/s41556-019-0346-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao P., Feng B., Li Y., He Q., Xu H. Hepatic ERK activity plays a role in energy metabolism. Mol. Cell. Endocrinol. 2013;375:157–166. doi: 10.1016/j.mce.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.-C., Kviklyte S., Vertommen D., Lantier L., Foretz M., Viollet B., Hallén S., Rider M.H. A small-molecule benzimidazole derivative that potently activates AMPK to increase glucose transport in skeletal muscle: comparison with effects of contraction and other AMPK activators. Biochem. J. 2014;460:363–375. doi: 10.1042/BJ20131673. [DOI] [PubMed] [Google Scholar]
- Long Y.C., Zierath J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovisa S., LeBleu V.S., Tampe B., Sugimoto H., Vadnagara K., Carstens J.L., Wu C.-C., Hagos Y., Burckhardt B.C., Pentcheva-Hoang T., et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A., Tabassum D.P., Altrock P.M., Almendro V., Michor F., Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed I., Hollenberg M.D., Ding H., Triggle C.R. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front. Endocrinol. 2021;12:718942. doi: 10.3389/fendo.2021.718942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina T., Deguchi Y., Ohshima D., Takeda W., Ohtsuka M., Shichino S., Ueha S., Yamazaki S., Kawauchi M., Nakamura E., et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat. Commun. 2021;12:2281. doi: 10.1038/s41467-021-22450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila S., Domènech-Moreno E., Laajanen K., Wong I.P., Tripathi S., Pentinmikko N., Gao Y., Yan Y., Niemelä E.H., Wang T.C., et al. Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. J. Clin. Invest. 2017;128:402–414. doi: 10.1172/JCI93597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan S., Bone N.B., Zmijewska A.A., Jiang S., Park D.W., Bernard K., Locy M.L., Ravi S., Deshane J., Mannon R.B., et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018;24:1121–1127. doi: 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoza M.L., Fu D., Sendak R.A. Development of an in vitro potency assay for therapeutic TGFbeta antagonists: the A549 cell bioassay. J. Immunol. Methods. 2006;316:18–26. doi: 10.1016/j.jim.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Sapkota G.P., Boudeau J., Deak M., Kieloch A., Morrice N., Alessi D.R. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz–Jeghers cancer syndrome. Biochem. J. 2002;362:481–490. doi: 10.1042/0264-6021:3620481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S., Viswanathan S., Widjaja A.A., Lim W.-W., Moreno-Moral A., DeLaughter D.M., Ng B., Patone G., Chow K., Khin E., et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. doi: 10.1038/nature24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Arras D., Rose-John S. IL-6 pathway in the liver: from physiopathology to therapy. J. Hepatol. 2016;64:1403–1415. doi: 10.1016/j.jhep.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S., Suzuki A. Glycogen synthase kinase 3β-dependent Snail degradation directs hepatocyte proliferation in normal liver regeneration. Proc. Natl. Acad. Sci. USA. 2011;108:11175–11180. doi: 10.1073/pnas.1016122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford D.B., Shaw R.J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J., Lamia K.A., Vasquez D., Koo S.-H., Bardeesy N., Depinho R.A., Montminy M., Cantley L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strikoudis A., Cieślak A., Loffredo L., Chen Y.-W., Patel N., Saqi A., Lederer D.J., Snoeck H.-W. Modeling of fibrotic lung disease using 3D organoids derived from human pluripotent stem cells. Cell Rep. 2019;27:3709–3723.e5. doi: 10.1016/j.celrep.2019.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J., Morgani S.M., David C.J., Wang Q., Er E.E., Huang Y.-H., Basnet H., Zou Y., Shu W., Soni R.K., et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. 2020;577:566–571. doi: 10.1038/s41586-019-1897-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur S., Viswanadhapalli S., Kopp J.B., Shi Q., Barnes J.L., Block K., Gorin Y., Abboud H.E. Activation of AMP-activated protein kinase prevents TGF-β1-induced epithelial-mesenchymal transition and myofibroblast activation. Am. J. Pathol. 2015;185:2168–2180. doi: 10.1016/j.ajpath.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To S.Q., Dmello R.S., Richards A.K., Ernst M., Chand A.L. STAT3 signaling in breast cancer: multicellular actions and therapeutic potential. Cancers. 2022;14:429. doi: 10.3390/cancers14020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujioka H., Kunieda T., Katou Y., Shirahige K., Fukazawa T., Kubo T. interleukin-11 induces and maintains progenitors of different cell lineages during Xenopus tadpole tail regeneration. Nat. Commun. 2017;8:495. doi: 10.1038/s41467-017-00594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaahtomeri K., Ventelä E., Laajanen K., Katajisto P., Wipff P.-J., Hinz B., Vallenius T., Tiainen M., Mäkelä T.P. Lkb1 is required for TGFbeta-mediated myofibroblast differentiation. J. Cell Sci. 2008;121:3531–3540. doi: 10.1242/jcs.032706. [DOI] [PubMed] [Google Scholar]
- Viswanathan S., Ng B., Widjaja A.A., Pua C.J., Tham N., Tan J., Cook S.A., Schafer S. Critical conditions for studying interleukin-11 signaling in vitro and avoiding experimental artefacts. Curr. Protoc. 2021;1:e251. doi: 10.1002/cpz1.251. [DOI] [PubMed] [Google Scholar]
- Wang T., Holland J.W., Bols N., Secombes C.J. Cloning and expression of the first nonmammalian interleukin-11 gene in rainbow trout Oncorhynchus mykiss. FEBS J. 2005;272:1136–1147. doi: 10.1111/j.1742-4658.2005.04544.x. [DOI] [PubMed] [Google Scholar]
- Widjaja A., Shekeran S.G., Adami E., Ting J.G.W., Tan J., Viswanathan S., Lim S.Y., Tan P.H., Hübner N., Coffman T., Cook S.A. A neutralizing IL-11 antibody improves renal function and increases lifespan in a mouse model of alport syndrome. J. Am. Soc. Nephrol. 2022;33:718–730. doi: 10.1681/ASN.2021040577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja A.A., Singh B.K., Adami E., Viswanathan S., Dong J., D’Agostino G.A., Ng B., Lim W.W., Tan J., Paleja B.S., et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of non-alcoholic steatohepatitis. Gastroenterology. 2019;157:777–792.e14. doi: 10.1053/j.gastro.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Widjaja A.A., Dong J., Adami E., Viswanathan S., Ng B., Pakkiri L.S., Chothani S.P., Singh B.K., Lim W.W., Zhou J., et al. Redefining IL11 as a regeneration-limiting hepatotoxin and therapeutic target in acetaminophen-induced liver injury. Sci. Transl. Med. 2021;13:eaba8146. doi: 10.1126/scitranslmed.aba8146. [DOI] [PubMed] [Google Scholar]
- Widjaja A.A., Viswanathan S., Jinrui D., Singh B.K., Tan J., Wei Ting J.G., Lamb D., Shekeran S.G., George B.L., Schafer S., et al. Molecular dissection of pro-fibrotic IL11 signaling in cardiac and pulmonary fibroblasts. Front. Mol. Biosci. 2021;8:740650. doi: 10.3389/fmolb.2021.740650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A., Williams J.R., Muckett P.J., Mayer F.V., Liljevald M., Bohlooly-Y M., Carling D. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 2017;18:3043–3051. doi: 10.1016/j.celrep.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E., Filippi B.M., Deak M., Alessi D.R., van Aalten D.M.F. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–1711. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B., Jeong J.H., Asara J.M., Yuan Y.-Y., Granter S.R., Chin L., Cantley L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Zhang W., Pendleton E., Leng S., Wu J., Chen R., Sun X.J. Improved insulin sensitivity by calorie restriction is associated with reduction of ERK and p70S6K activities in the liver of obese Zucker rats. J. Endocrinol. 2009;203:337–347. doi: 10.1677/JOE-09-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data are provided in the manuscript or in the supplementary materials. Source data are provided as Data S1. The densitometry analyses are provided as Data S2. Uncropped Western blots are provided as Data S3.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.