

Abstract
Elevated O-GlcNAcylation is emerging as a general characteristic of most cancers. Although O-GlcNAcylation can regulate many cell biological pathways, recent evidence suggests that it is a key regulator of metabolic pathways including glycolysis in cancer cells. This review summarizes our current understanding of how O-GlcNAcylation regulates glycolytic pathways and contributes to alterations in cancer cell metabolism.
Keywords: O-GlcNAcylation, O-GlcNAc transferase, Glycolysis, Cancer, Metabolism
Introduction
The hexosamine biosynthetic pathway (HBP) is a shunt off from glycolysis where approximately 2–5% of all glucose enters (Bond and Hanover 2015; Marshall et al. 1991). Inputs such as glutamine, acetyl-CoA, and uridine-5′-triphosphate (UTP) from major metabolic pathways are then enzymatically incorporated along with glucose to generate the negatively charged, amino sugar UDP-GlcNAc (Ruan et al. 2013). Intracellular concentrations of UDP-GlcNAc vary based on the availability of the precursor nutrients, implicating the synthesis of UDP-GlcNAc as a means to sense the nutritional state of the cell (Marshall et al. 2004; Wellen et al. 2010; Weigert et al. 2003; Wang et al. 1998a). Following its synthesis, UDP-GlcNAc is then utilized by enzymes localized in golgi or endoplasmic reticulum for either the N-linked or O-linked glycosylation of secreted or membrane-bound proteins. UDP-GlcNAc can also be used for intracellular glycosylation of nuclear or cytoplasmic proteins that is catalyzed by the activity of the glycosyltransferase O-GlcNAc transferase or OGT (Haltiwanger et al. 1990; Hart and Akimoto 2009), a function exclusive to this enzyme. Conversely, this covalently linked sugar moiety is removed, exclusively, through the enzymatic activity of O-GlcNAcase or OGA (Overdijk et al. 1981; Gao et al. 2001). The O-linked addition of a β-N-acetyl glucosamine (O-GlcNAc) monosaccharide to the serine and/or threonine residues of target proteins is one of the most abundant post-translational modifications regulating the proteome (Hart et al. 2011). The biological manifestations of O-GlcNAcylation are analogous to phosphorylation such that O-GlcNAc modified proteins display altered interactions and stability. Moreover, there is a complex interplay between O-GlcNAcylation and phosphorylation on the same sites or same protein that is described in more detail in other reviews (Love and Hanover 2005) (Hart et al. 2011). Thus far, over 4000 O-GlcNAc modified proteins have been identified (Ma and Hart 2014), thereby possessing an expansive portfolio of regulatory mechanisms on essential signaling networks and cellular metabolism.
O-GlcNAcylation has been implicated to be critical for development and homeostasis of normal cell growth. In 2000, Shafi and colleagues generated a mouse model expressing Cre under the ZP3 promoter, driving its expression specifically in the female germline. These mice were then crossed to their previously generated OGTfl/fl mice where they observed that no offspring were produced bearing the OGT deleted allele, revealing that OGT is required for embryogenesis (Shafi et al. 2000). Later, in 2004, O’Donnell et al. expanded on these observations by performing several tissue-specific knockouts and studying its effect on somatic cells, where they observed T-cell apoptosis, hyperphosphorylation of tau in neurons, and growth arrest in fibroblasts in response to OGT deletion (O’Donnell et al. 2004). Taken together, these experiments highlight the necessity for O-GlcNAcylation during development and in maintaining normal cell behavior. Additionally, changes to OGTexpression and O-GlcNAcylation have been implicated in neurodegenerative diseases such as Parkinson’s (Gong et al. 2012; Marotta et al. 2015; Wani et al. 2017), amyotrophic lateral sclerosis (ALS) (Shan et al. 2012; Ludemann et al. 2005), and Alzheimer’s (Liu et al. 2004; Yuzwa et al. 2014a, 2014b; Yuzwa and Vocadlo 2014; Zhu et al. 2014; Liu et al. 2009), as well as other metabolic disorders including obesity (Lagerlof et al. 2016; Yang et al. 2015a; Ruan et al. 2014), diabetes (Federici et al. 2002; Vosseller et al. 2002; Brownlee 2001), and cancer (Zhu et al. 2014; Ferrer et al. 2016; Caldwell et al. 2010; Lynch et al. 2012; Yang et al. 2015b; Shi et al. 2010).
Under oxygenated conditions, healthy cells utilize glycolysis to metabolize glucose into pyruvate for the production of energy via the tricarboxylic cycle (TCA) cycle and oxidative phosphorylation. However, while studying carcinoma cells in the 1920’s, Otto Warburg observed that the metabolism of these cancer cells was different from that of normal adult cells (Warburg et al. 1927). Instead of opting for the more energy efficient pathway of oxidative phosphorylation, Warburg noticed that cancer cells continuously employed glycolysis (Warburg et al. 1927; Warburg 1956). Today, this is referred to as the “Warburg Effect” and it describes the process in which cancer cells reprogram their metabolism to enhance glucose uptake and reduce pyruvate to lactate and oxidized nicotinamide adenine dinucleotide (NAD+) and reduce TCA cycling, leading to the accumulation of biosynthetic precursor molecules used to sustain cell growth and survival (Warburg et al. 1927; Warburg 1956; Liberti and Locasale 2016; Vander Heiden et al. 2009; DeBerardinis et al. 2008). In addition to the accumulation of macromolecule precursors, the fermentation to lactate has gained the attention of researchers as this not only results in an indefinite glycolytic cycle via the production of NAD+, but also locally reduces the pH of the microenvironment (Griffiths 1991; Webb et al. 2011). Not surprisingly, it has been shown that tumors can adapt and thrive under these acidic conditions through the elevation of inflammatory signaling (Xu and Fidler 2000), epigenetic regulation (McBrian et al. 2013; Corbet et al. 2016), and further rewiring their lipid metabolism (Corbet et al. 2016; Kondo et al. 2017). Moreover, the products from glycolysis feed downstream pathways that are commonly mutated and/or hyperactive in most cancers (Vander Heiden et al. 2009; Hirschey et al. 2015; Schulze and Harris 2012). In this review, we will highlight the latest discoveries into the role of O-GlcNAcylation in the regulation of glycolytic metabolism with a focus on cancer.
O-GlcNAc regulation of cancer
The O-linked addition of a β-N-acetylglucosamine sugar moiety was first identified in 1983 when Torres et al. isolated lymphocytes and utilized the intrinsic galactosylation processes to label N- or O-linked sugar modified proteins (Torres and Hart 1984). Since its discovery, O-GlcNAcylation and the enzymes responsible for its addition and removal, OGT and OGA respectively, have emerged as key regulators of the various “Hallmarks of Cancer” (Hanahan and Weinberg 2000, 2011), which was comprehensively summarized by Ferrer et al. (2016); here, we will focus on the most current concepts surrounding the role of O-GlcNAc in regulating cancer metabolism.
Perhaps one of the most promiscuous proto-oncogenes, c-MYC is estimated to be elevated and/or deregulated in approximately 70% of cancers (Dang 2012) and has even been shown to maintain a gene signature penetrant to a plethora of cancers, which largely enriches for protein translation machinery (Dang 2012; Ji et al. 2011). With many downstream transcriptional targets, MYC has a role in regulating a wide variety of biological processes (Dang 1999). In 1995, Chou et al. identified an O-GlcNAc modification on the threonine-58 residue of c-Myc (Chou et al. 1995a; Chou et al. 1995b). This site is also phosphorylated by GSK3β consequently resulting in the degradation of c-Myc (Preston et al. 2015). Building on the correlation of OGT and c-Myc expression, the Mills group established (Itkonen et al. 2013) OGT regulation of c-Myc in prostate cancer cells and found tight correlation in human prostate cancer tissue. Recently, Swamy and colleagues examined the role of O-GlcNAcylation in T cell function utilizing cytotoxic T cells isolated from a tamoxifen-inducible OGT knockout mouse model, where they observed a loss in c-Myc expression corresponding with the loss of OGT expression. Swamy et al. also employed a c-Myc knock-in mouse and analyzed c-Myc expression in activated T cells that were treated with either an OGT or OGA inhibitor, where they observed c-Myc expression to be decreased with OGT inhibition and increased with OGA inhibition, indicating the O-GlcNAc modification as a stabilizing factor for c-Myc (Swamy et al. 2016). Interestingly, OGT itself is regulated in a non-transcriptional manner by c-Myc in breast cancer cells. Mechanistically, c-Myc transcriptionally regulates the chaperone protein HSP90 that regulates OGT stability in cancer cells (Sodi et al. 2015). Thus, a model of reciprocal regulation between OGT and c-Myc has been proposed in which a feed-forward-regulatory loop may be present in cancer cells (Sodi et al. 2015). OGT has been linked, directly and indirectly, to nearly all of the “Hallmarks of Cancer” except for avoiding immune destruction and enabling replicative immortality (Ferrer et al. 2016; Hanahan and Weinberg 2000, 2011). Since O-GlcNAcylation stabilizes c-Myc (Itkonen et al. 2013; Swamy et al. 2016) and c-Myc has been shown to activate telomerase (Wang et al. 1998b), one could hypothesize that the activation of telomerase may at least partially depend on OGT expression, promoting replicative immortality. Likewise, c-Myc has been shown to downregulate MHC class I (Versteeg et al. 1988) and class II (God et al. 2015) antigens in melanoma and B-cell lymphoma, respectively, contributing to the ability of these tumors to evade immune detection. It would be interesting to evaluate whether OGT through c-Myc and its regulation of telomerase (Dang 1999; Wang et al. 1998b) and MHC class I and class II antigens (Chou et al. 1995a; Versteeg et al. 1988; God et al. 2015) indeed regulate these two critical features of cancer biology.
Providing another example of OGT manipulating a key regulator of tumorigenesis, Bullen et al. showed OGT directly modifies the extensively characterized tumor suppressor, AMP-activated protein kinase (AMPK) and inhibits its activation (Bullen et al. 2014). Furthermore, suppression of OGT with RNAi can induce metabolic stress and lead to the activation of AMPK (Ferrer et al. 2014). Also functioning as a nutrient-sensor (Hardie et al. 2012), AMPK is activated by a low energy state and in response phosphorylates substrates to shift metabolism from anabolic to catabolic (Hardie et al. 2012; Jorgensen et al. 2004; Carling et al. 1989; Suzuki et al. 2013). The downstream network of biosynthetic processes inhibited by AMPK activation includes the sterol regulatory element binding protein 1 (SREBP-1), the master transcriptional regulator of fatty acid synthesis and lipid metabolism that is intimately involved in maintaining cancer cell growth (Eberle et al. 2004; Lewis et al. 2015). Recently, we have demonstrated that OGT regulates SREBP-1 and lipid metabolism in breast cancer by downregulating AMPK activity (Sodi et al. 2017). The elevated expression and activity of SREBP-1 and the enzymes involved in the free fatty acid synthesis pathway are well known to be elevated and required for tumor growth (Rohrig and Schulze 2016) (Baenke et al. 2013). In our studies, we found that suppressing OGT expression in triple-negative breast cancer (TNBC) cells results in a significant decrease in 43% of the total lipid metabolites as determined by a metabolomics profiling. Targeting OGT either genetically or pharmacologically in breast cancer cell lines reduced free fatty acids levels and lipid droplets. Intriguingly, protein expression analysis from the transgenic mouse breast cancer model MMTV-PyMT, which closely mimics human breast cancer tumorigenesis by undergoing each stage of progression, revealed a correlative increase in global O-GlcNAcylation, OGT, SREBP-1, and the lipogenic enzymes as disease severity progressed. Previously, our lab has shown that reducing OGT expression or activity can block breast cancer growth both in vitro and in vivo (Caldwell et al. 2010) (Lynch and Reginato 2011). To test role of SREBP-1 in OGT-mediated oncogenesis, we performed a series of rescue experiments in which we targeted OGT expression and then we exogenously supplemented these cells with lipids or reconstituted the expression of SREBP-1, each producing a partial rescue of cancer cell growth with the latter also being confirmed in vivo. Surprisingly, SREBP-1 overexpression in context of OGT knockdown reversed GLUT1 downregulation and glucose transport in a HIF-1α independent manner suggesting key role of glucose uptake in OGT-mediated cancer growth phenotype. Since we were unable to detect direct O-GlcNAcylation of SREBP-1 by OGT, we examined the role of AMPK since it has been shown to phosphorylate and inhibit the proteolytic activation of SREBP-1 (Li et al. 2011). To confirm that the regulation of SREBP-1 and lipid metabolism by OGT was mediated through AMPK, we performed the OGT RNAi experiments in AMPK−/− MEFs or in breast cancers cells treated with Compound C, an inhibitor of AMPK; in both cases, we observed a diminished impact on the protein levels of SREBP-1 and its transcriptional target ACLY (Sodi et al. 2017). Together, these data describe a novel mechanism in which OGT regulates breast cancer growth by upregulating SREBP-1 and lipid metabolism via suppressing AMPK activation. In addition to these findings, we have also recently shown that OGT regulates the NAD+-dependent deacetylase SIRT1 via AMPK and that by downregulating AMPK and SIRT1, the previously described effect on FOXM1 signaling (Caldwell et al. 2010; Lynch et al. 2012) is enhanced and promotes breast cancer metastasis (Ferrer et al. 2017). Thus, OGT lies at the crux of many key oncogenic signaling pathways regulating, both, cancer growth and invasion. Since cancer cells are more dependent on OGT for growth and survival compared to non-transformed epithelial cells (Sodi et al. 2015; Ferrer et al. 2014), a therapeutic window may exist to treat cancers with OGT inhibitors.
O-GlcNAc regulation of glucose uptake
The glucose transporter (GLUT) family of proteins is a group of transmembrane proteins that facilitate the uptake of glucose (Thorens and Mueckler 2010). In the context of direct regulation, GLUT4, the transporter expressed in skeletal muscle and adipose tissue, and its transport vesicle proteins have been shown to be O-GlcNAc modified (Park et al. 2005; Buse et al. 2002). While it remains elusive as to how O-GlcNAcylation of GLUT4 alters the transporter’s ability to facilitate glucose uptake, Slawson et al. speculate that O-GlcNAc modification may alter the translocation of GLUT4 (Park et al. 2005), which is critical in maintaining glucose uptake (Vosseller et al. 2002), possibly by blocking GLUT4 phosphorylation, modulating downstream signaling, or through the direct regulation of vesicular proteins (Slawson et al. 2006). The most ubiquitously expressed glucose transporter (Augustin 2010), GLUT1, has been shown to be indirectly regulated by OGT in cancer cells (Ferrer et al. 2014). Mechanistically, overexpression of OGT and enhancing global O-GlcNAcylation increased the transcription of GLUT1 and glucose uptake (Ferrer et al. 2014). This effect on GLUT1 was dependent on OGT regulation of hypoxia inducible factor 1 alpha (HIF-1α), a key regulator of GLUT1 transcription (Iyer et al. 1998). In this way, elevated O-GlcNAcylation may further orchestrate the flux of glucose into the hexosamine biosynthetic pathway and enhance synthesis of UDP-GlcNAc; this is an important consideration, as the GLUT family of transporters are a class of proteins that are heavily N-glycosylated (Augustin 2010). Specifically, the N-linked addition of the β-N-acetylglucosamine sugar moiety to GLUT1 has been implicated in maintaining the appropriate structure and function of this transporter (Asano et al. 1991). Moreover, overexpression of SREBP-1 (Sodi et al. 2017), HIF-1α, and GLUT1 (Ferrer et al. 2014) are able to rescue glucose uptake in OGT depleted cancer cells and reverse metabolic stress and cell death. Thus, OGT regulation of GLUT1 levels and activity in cancer cells is critical for tumor promoting functions of O-GlcNAcylation. Whether OGT can increase glucose uptake via direct or indirect regulation of GLUTs needs further investigation.
In a key step to ensure that the glucose transported into the cell proceeds through to glycolysis, it is essential for glucose to be phosphorylated by hexokinase (HK) to generate the charged product glucose-6-phosphate (G-6-P), which can no longer passively cross the plasma membrane. Coinciding with the indirect regulation of GLUT1 by OGT, HK is also positively regulated by the HIF-1α transcriptional program (Iyer et al. 1998; Semenza 2010) and Yi et al. were able to show that HK activity was enhanced with OGT overexpression (Yi et al. 2012). More recently, Baldini et al. demonstrated that hexokinase IV, or glucokinase (GCK), the main enzyme in liver that regulates the flux of glucose, is O-GlcNAc modified both in vitro and in vivo, positively regulating its expression (Baldini et al. 2016). In sum, this data suggests OGT and O-GlcNAcylation regulate early stages of glucose metabolism.
O-GlcNAc regulation of glycolysis
Once glucose enters the cell and is trapped via HK-mediated phosphorylation, its metabolic fate is then determined by the needs of the cell (Vander Heiden et al. 2009). In the scenario where biosynthetic demands and energy state of the cell are in equilibrium, glucose will be converted into glycogen and stored for later use (Mulukutla et al. 2016). However, if the cell requires more energy, TCA cycle intermediates, nucleic, amino, or fatty acids the glucose can be metabolized via the pentose phosphate pathway or by glycolysis (Mulukutla et al. 2016). In this section, we will cover recent discoveries linking O-GlcNAcylation to the regulation of glycolysis.
Catalyzing the committed step of phosphorylating fructose-6-phosphate (F-6-P) to produce fructose-1,6-bisphosphate (F-1,6-BP), phosphofructokinase 1 (PFK1) serves as a major regulator of flux through glycolysis (Li et al. 2015). Depending on nutrient availability and the needs of the cell, controlling the function of PFK1 works in a manner similar to a valve in a pipeline; when the valve is open and PFK1 is active, F-6-P is phosphorylated into F-1,6-BP and continues on with glycolytic processing, but if the valve is closed by PFK1 inhibition, F-6-P can convert back to G-6-P and enter the pentose phosphate pathway (Jiang et al. 2014; Stincone et al. 2015) (Fig. 1), which we discuss in the next section of this review. PFK1 is well known to be allosterically regulated by substrate availability and the nutrient status of the cell (Li et al. 2015). Recently, the Hsieh-Wilson lab showed that PFK1 is negatively regulated by O-GlcNAcylation (Yi et al. 2012). They demonstrated that upon treating the cells with the OGA inhibitor, PUGNAC, thereby preventing the removal of the O-linked β-N-acetylglucosamine moiety from modified proteins, they observed a decrease in PFK1 activity. Furthermore, upon overexpressing OGT in 293T cells they were able to show a similar decrease in PFK1 activity. Using a proximity-based biotinylation immunoprecipitation assay they showed that PFK1 is O-GlcNAcylated. Additionally, they mapped the site and identified Serine 529 to be the site of O-GlcNAcylation on PFK1and showed that mutation of this O-GlcNAc site does not respond to increases or decreases in overall O-GlcNAcylation levels when compared to wild type protein. They also successfully generated a homologous model of rabbit PFK1 complexed with fructose-2,6-bisphosphate and found that the S530 (O-GlcNAcylation site for rabbit homolog) forms hydrogen bond with the two phosphates. This was an important finding as it suggests that the addition of O-GlcNAc to this site on PFK1 will block the interaction with its allosteric activator. In addition they showed that O-GlcNAcylation hinders oligomerization of PFK1 and modulating its activity. When the wild type PFK1 was subjected to increased glycosylation, there was a reduction in glycolytic rate measured by lactate production but the same result was not seen in S529A mutated protein.
Fig. 1. O-GlcNAcylated proteins involved in glycolysis, PPP, TCA cycle and glutamine metabolism.
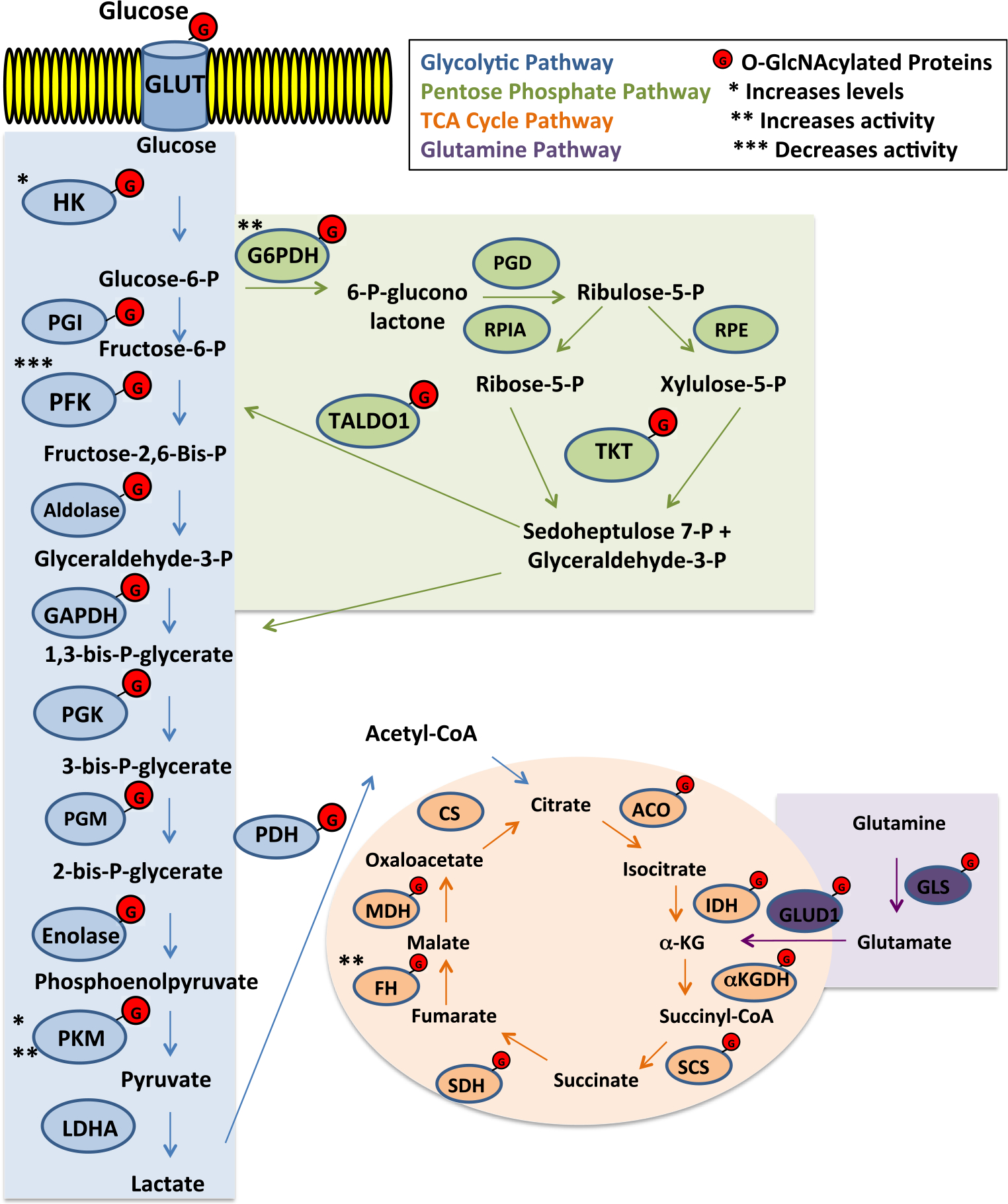
All enzymes marked with G have been shown to be O-GlcNAcylated
Glycolytic metabolism in cancer is critical as it regulates the flux of metabolites into various biosynthetic pathways to support the growth of tumor. To understand the role of PFK1 O-GlcNAcylation on regulating glycolytic activity, the authors measured flux through glycolysis and the pentose phosphate pathway and observed that when PFK1 O-GlcNAcylation levels were elevated, flux through pentose phosphate pathway was increased and there was no observable difference with expression of the S529A mutant. Interestingly, they also show that elevated O-GlcNAc induces cell growth in H1299 lung cancer cells and that this could be attributed to PFK1. Thus, the regulation of PFK1 by O-GlcNAcylation plays a pivotal role in supporting cell growth by allocating the metabolic flux of glucose.
Pyruvate kinase M2 (PKM2), another key glycolytic enzyme that catalyzes the final reaction of glycolysis, was recently showed to be O-GlcNAcylated by the Champattanachai lab (Chaiyawat et al. 2015). In their studies, the group analyzed the post-translational modification landscape by immunoprecipitating PKM2 from HEK-293T cells under conditions where O-GlcNAcylation levels were either reduced with siRNA targeting OGT or increased by treatments with the OGA inhibitor, Thiamet-G (TMG) and confirmed that PKM2 was O-GlcNAcylated via mass-spectrometry. Computational predictions showed that there are 11 potential O-GlcNAcylation sites on PKM2 and 3 out of those 11 sites were also sites of phosphorylation. In addition, they also showed that the transcription of PMK2 was responsive to O-GlcNAcylation, as PKM2 mRNA levels were significantly downregulated in OGT RNAi targeted cells and upregulated in cells treated with TMG, a result that was mirrored at the level of protein expression and kinase activity. Interestingly, when the group compared PKM2 O-GlcNAcylation and the subsequent effect on kinase activity in metastatic versus non-metastatic colorectal cancer cell lines, they noted an elevation in O-GlcNAcylated PKM2 and reduced specific-activity in the metastatic cell line. These results provide another example where cancer cells dynamically manipulate protein O-GlcNAcylation to regulate key glycolytic enzymes. It will be interesting to expand on their results and determine if the O-GlcNAcylation of PKM2 confers an advantage to the metastatic cell in comparison to the primary tumor and test whether the metastatic cell prefers an unmodified PKM2 to promote glycolysis, where the primary tumor might prefer an O-GlcNAcylated PKM2 to downregulate kinase activity and fuel the pentose phosphate pathway.
Recent work by the Ju lab established an association between key glycolytic enzymes and overall O-GlcNAcylation (Zhang et al. 2017). The authors in this paper show that there is an increased overall O-GlcNAcylation and expression of glycolytic components like GLUT1, HK2, lactate dehydrogenase (LDHA), and PFK1 in pre-B-acute lymphoblastic leukemia (pre-B-ALL) patients compared to healthy donors and expression of some of these genes required OGT. When the group evaluated overall O-GlcNAcylation present in pre-B-ALL patients with varying levels of LDHA expression, they found that a positive correlation exists between plasma LDHA concentrations and levels of O-GlcNAcylation.
Apart from the independent studies mentioned above, in a proteomics screening on cells isolated from skeletal muscle, Claude Michalski’s group identified that fructose bisphosphate aldolase, triose phosphate isomerase, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are all O-GlcNAcylated (Cieniewski-Bernard et al. 2004). Although the functional consequence of the modification on some of these enzymes remains elusive, collectively, these data suggest O-GlcNAcylation is both a direct and indirect regulator of nearly every glycolytic enzyme (Fig. 1).
O-GlcNAc regulation of the pentose phosphate pathway
Shunting off from glycolysis, the pentose phosphate pathway (PPP) presents an immediate alternative metabolic fate for glucose-6-phosphate. The utilization of the PPP serves to produce pentose sugars and reduced nicotinamide adenine dinucleotide phosphate (NADPH) to support nucleotide and lipid synthesis respectively as well as help scavenge reactive oxygen species (ROS) (Stincone et al. 2015). While the canonical regulatory mechanisms involving oxidative stress or inhibition of PFK1 activity have been extensively studied (Jiang et al. 2014; Stincone et al. 2015), efforts to elucidate the role of O-GlcNAcylation in controlling flux through the PPP have only recently been examined. In 2012, when characterizing O-GlcNAcylation in primary and metastatic colorectal cancers (CRC) and the effect of O-GlcNAcase (OGA) inhibition, Yehezkel et al. performed a transcriptome analysis comparing metastatic CRC clones that had been subjected to OGA silencing to their control counterparts They found the pentose phosphate pathway was elevated nearly 3-fold in response to globally increased O-GlcNAcylation (Yehezkel et al. 2012) (Fig. 1). Upon OGA silencing, they found that the rate-limiting enzyme of PPP glucose-6-phosphate dehydrogenase (G6PD) was elevated at the RNA level. Conversely, in response to reduced O-GlcNAcylation in breast cancer cells, PPP metabolites including ribulose 5-phosphate, xylulose 5-phosphate, and sedoheptulose 7-phosphate were found to be significantly decreased (Ferrer et al. 2014). Although these studies suggest a dynamic role for O-GlcNAcylation in the regulation of the pentose phosphate pathway, it was not clear whether this regulation was directly regulating key enzymes of the PPP or other key enzymes involved in glycolysis as mentioned above.
Recent studies have shown enzymes involved in the pentose phosphate pathway to be O-GlcNAcylated (Fig. 1). More recently G6PD was shown to be O-GlcNAc modified on serine 84 using a chemoenzymatic labeling methods (Rao et al. 2015). In an elegant study, Rao et al. showed that overexpressing OGT or inhibiting OGA to increase O-GlcNAcylation enhanced G6PD activity and these results could be nullified by mutating the modified site to a valine (Rao et al. 2015). Additionally, by using radioisotope labeling they were able to show that this modification led to the enhanced production of NADPH and other PPP metabolites, thereby promoting the activity of the pathway (Rao et al. 2015). Strikingly, they demonstrated the importance of this modification to G6PD by analyzing its presence in human lung tumor samples compared to the adjacent tissue, where tumors exhibited elevated O-GlcNAcylated G6PD that intensified with disease progression (Rao et al. 2015). In addition to G6PD, it has recently been shown in a proteomic analysis comparing invasive ductal carcinoma (IDC) tumors with or without lymph node metastases that two additional PPP enzymes, transaldolase (TALDO1) and transketolase (TKT), are differentially O-GlcNAcylated with TALDO1 found only to be modified in lymph node metastases, while TKT was found only to be modified in IDC without metastasis (Jiang et al. 2016). Thus, it is becoming more evident that O-GlcNAc is directly involved in the regulation of flux through the pentose phosphate pathway and thus may help promote nucleotide and lipid synthesis as well as combat oxidative stress during cancer progression (Yi et al. 2012; Yehezkel et al. 2012; Rao et al. 2015; Jiang et al. 2016).
O-GlcNAc regulation of TCA metabolites
As a segue between glycolysis and the electron transport chain (ETC), the TCA cycle is a metabolic hub of enzymatic reactions that yields essential precursor metabolites for macromolecule biosynthesis and reducing molecules that fuel energy production via the ETC (Akram 2014). The regulatory role for O-GlcNAcylation on the TCA cycle is now beginning to be fully appreciated and understood. In 2014, the Slawson Lab performed a SILAC-labeling analysis on neuroblastoma cells overexpressing either OGT or OGA to alter intracellular O-GlcNAcylation, where they identified altered expression of components in the pyruvate dehydrogenase complex (PDH), aconitase, isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase complex, and succinyl-CoA ligase (Tan et al. 2014). Corroborating with these results, we performed LC-MS analysis to measure the concentrations of TCA cycle intermediates in MDA-MB-231 triple negative breast cancer cells where OGT had been suppressed with shRNA (Ferrer et al. 2014). Under conditions of reduced O-GlcNAcylation, we observed an increase in TCA cycle metabolite production, which was consistent with previous findings that targeting OGT in cancer reverses the Warburg Effect (Caldwell et al. 2010; Itkonen et al. 2013). Together, these discoveries show a dynamic response to altered O-GlcNAcylation and suggest that the role of O-GlcNAcylation in the regulation of the TCA cycle enzymes warrants a deeper analysis.
Click chemistry labeling and β-elimination/Michael Addition with DTT (BEMAD) methods have helped progress our ability to detect O-GlcNAcylated proteins and are being increasingly employed to identify substrates (Khidekel et al. 2003; Xu et al. 2017; Vosseller et al. 2005). Both of these techniques have been instrumental in identifying how O-GlcNAcylation is involved in the regulation of TCA cycle enzymes (Clark et al. 2008; Cao et al. 2013) (Fig. 1). Utilizing the UDP-GalNAz labeling approach, Clark et al. were able to show that both the alpha and beta subunits of PDH are O-GlcNAc modified in their cortical neuron model (Clark et al. 2008). Another proteomic study employing the BEMAD method in mitochondria isolated from rat livers, identified malate dehydrogenase (MDH) as a substrate for O-GlcNAcylation (Cao et al. 2013). Expanding on these analyses, recent work out of the Hart Lab where they isolated cardiac mitochondria from streptozoticin-induced diabetic rats and performed a large-scale O-GlcNAc-modified proteome analysis with the BEMAD technique they identified every enzyme in the TCA cycle, with the exception of citrate synthase and fumarate hydratase, as being O-GlcNAcylated, with several having multiple modified sites (Ma et al. 2016). Additionally, they followed up and validated the PDHA1 (alpha subunit) modification by performing an immunoprecipitation with the RL-2 (O-GlcNAc antibody) and were able to detect PDHA1, which could be reduced when in the presence of a recombinant homologue of human OGA (Ma et al. 2016). Wang et al. recently demonstrated with click chemistry labeling that fumarate hydratase (FH) is indeed O-GlcNAc modified in pancreatic cancer cells and this modification assists in overcoming nutrient deprivation and to support cell survival and proliferation (Wang et al. 2017). Specifically, they describe a competition between two well-studied nutrient sensors AMPK and OGT and their respective modifications on the Ser75 residue of FH, in which under glucose-deprived conditions, O-GlcNAcylation at this site impedes the ability of AMPK to interact with the ATF2, which would normally drive expression of genes involved in growth arrest via the local production of fumarate and epigenetic regulation of histone H3 (Wang et al. 2017). In this context, the authors found the modification of FH to be more important for a DNA repair mechanism, rather than for regulating its role in the TCA cycle. Moving forward, it would be interesting to utilize different models and conditions to see if the O-GlcNAc modification of FH serves additional purposes in regulating the TCA cycle.
O-GlcNAc regulation of glutaminolysis
Glutamine serves cellular metabolism as another major bioenergetic substrate that contributes to a variety of processes such as the TCA cycle, amino acid synthesis, and is a critical component for synthesizing UDP-GlcNAc (Altman et al. 2016). In the aforementioned proteomics analysis, Clark et al. also identified O-GlcNAc modifications on glutamine synthetase (GLS), glutamine dehydrogenase (GLUD1) (Fig. 1), and both isoforms of aspartate aminotransferase (Got1 and Got2) (Clark et al. 2008). However, functional studies to examine the impact of directly modifying these enzymes on their activity have yet to be performed. It’s established that c-Myc (Altman et al. 2016; Dang 2010), HIF-1α (Sun and Denko 2014; Stegen et al. 2016), and SIRT1 (Ren et al. 2017; Corbet et al. 2014) can regulate glutamine metabolism and each of these proteins are subjected to regulation via O-GlcNAcylation (Ferrer et al. 2014, 2016; Sodi et al. 2015); therefore, it would be interesting to determine both direct and indirect regulation of O-GlcNAcylation on glutamine metabolism.
Future directions
O-GlcNAcylation plays a key role in the regulation of major metabolic pathways and many of the enzymes involved in these pathways have been identified as being O-GlcNAc modified (Jiang et al. 2016; Ma et al. 2016; de Queiroz et al. 2014). Moving forward, understanding how these modifications individually regulate each of the metabolic enzymes identified from the proteomic screens (Cieniewski-Bernard et al. 2004; Jiang et al. 2016; Xu et al. 2017; Clark et al. 2008; Ma et al. 2016) will add to our understanding of how O-GlcNAcylation can regulate cellular metabolism in both normal and disease states.
O-GlcNAcylation is a critical regulator of both glucose and glutamine metabolism, yet little is known whether this nutrient-sensitive modification plays a role in acetate metabolism. In recent years, acetate has become a focal point of several research groups where they have shown certain cancers, such as glioblastoma and brain metastases, preferentially utilize acetate as a bioenergetic substrate (Mashimo et al. 2014). Schug, et al. showed that under conditions of metabolic stress, such as nutrient deprivation and hypoxia, acetyl-CoA synthetase 2 (ACSS2) promotes acetate utilization to maintain breast cancer cell growth (Schug et al. 2015). These studies suggest that acetate metabolism is responsive to the nutrient status and oxygen supply of cells, both of which are a common link shared with O-GlcNAcylation.
While the detection methods for O-GlcNAcylation have drastically improved, this is an area that still presents technical challenges and lacks in real-time analysis capabilities. Efforts have been made to develop fluorescent analogues of UDP-GlcNAc and track the targeting of substrate proteins (Yeager and Finney 2005); however, we have yet to be able to move this into human subjects. The DeBarardinis lab has been infusing cancer patients with isotopomers of glucose, lactate, and acetate to trace the utilization of these metabolites in each unique situation (Pichumani et al. 2016; Maher et al. 2012) to further understand cancer metabolism utilization in vivo. In 2013, the Taniguchi lab developed an isotopomer for UDP-GlcNAc to trace its utilization for both N- and O-glycosylation (Nakajima et al. 2013); future studies could attempt to utilize this tool in patients with cancer or diabetes to see if we can track changes occurring at real-time in human patient tissue.
Finally, several groups have been working to develop inhibitors of both OGT (Gloster et al. 2011; Ortiz-Meoz et al. 2015) and OGA (Yuzwa et al. 2008) activity, which have been demonstrated to be effective in targeting phenotypes in vitro (Caldwell et al. 2010; Sodi et al. 2015; Ferrer et al. 2014; Yuzwa et al. 2008). In order to continue to progress and one day reach the clinic, it is essential that we move to testing these compounds in vivo to characterize the true potential as a therapy for metabolic diseases.
Acknowledgements
We thank Neha Manjari Akella for critical reading of this manuscript.
References
- Akram M (2014) Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys 68(3):475–478 [DOI] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16(11):749. [DOI] [PubMed] [Google Scholar]
- Asano T et al. (1991) The role of N-glycosylation of GLUT1 for glucose transport activity. J Biol Chem 266(36):24632–24636 [PubMed] [Google Scholar]
- Augustin R (2010) The protein family of glucose transport facilitators: it’s not only about glucose after all. IUBMB Life 62(5):315–333 [DOI] [PubMed] [Google Scholar]
- Baenke F et al. (2013) Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6(6):1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini SF et al. (2016) Glucokinase expression is regulated by glucose through O-GlcNAc glycosylation. Biochem Biophys Res Commun 478(2):942–948 [DOI] [PubMed] [Google Scholar]
- Bond MR, Hanover JA (2015) A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol 208(7):869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820 [DOI] [PubMed] [Google Scholar]
- Bullen JW et al. (2014) Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J Biol Chem 289(15):10592–10606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buse MG et al. (2002) Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am J Physiol Endocrinol Metab 283(2):E241–E250 [DOI] [PubMed] [Google Scholar]
- Caldwell SA et al. (2010) Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29(19):2831–2842 [DOI] [PubMed] [Google Scholar]
- Cao W et al. (2013) Discovery and confirmation of O-GlcNAcylated proteins in rat liver mitochondria by combination of mass spectrometry and immunological methods. PLoS One 8(10):e76399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D et al. (1989) Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem 186(1–2):129–136 [DOI] [PubMed] [Google Scholar]
- Chaiyawat P et al. (2015) Alteration of O-GlcNAcylation affects serine phosphorylation and regulates gene expression and activity of pyruvate kinase M2 in colorectal cancer cells. Oncol Rep 34(4):1933–1942 [DOI] [PubMed] [Google Scholar]
- Chou TY, Dang CV, Hart GW (1995a) Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci U S A 92(10):4417–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TY, Hart GW, Dang CV (1995b) c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem 270(32):18961–18965 [DOI] [PubMed] [Google Scholar]
- Cieniewski-Bernard C et al. (2004) Identification of O-linked N-acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol Cell Proteomics 3(6):577–585 [DOI] [PubMed] [Google Scholar]
- Clark PM et al. (2008) Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J Am Chem Soc 130(35):11576–11577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C et al. (2014) The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res 74(19):5507–5519 [DOI] [PubMed] [Google Scholar]
- Corbet C et al. (2016) Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab 24(2):311–323 [DOI] [PubMed] [Google Scholar]
- Dang CV (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19(1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2010) Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 70(3):859–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012) MYC on the path to cancer. Cell 149(1):22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz RM, Carvalho E, Dias WB (2014) O-GlcNAcylation: the sweet side of the cancer. Front Oncol 4:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ et al. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20 [DOI] [PubMed] [Google Scholar]
- Eberle D et al. (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86(11):839–848 [DOI] [PubMed] [Google Scholar]
- Federici M et al. (2002) Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 106(4):466–472 [DOI] [PubMed] [Google Scholar]
- Ferrer CM et al. (2014) O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell 54(5):820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Sodi VL, Reginato MJ (2016) O-GlcNAcylation in cancer biology: linking metabolism and signaling. J Mol Biol 428(16):3282–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM et al. (2017) O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene 36(4):559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y et al. (2001) Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem 276(13):9838–9845 [DOI] [PubMed] [Google Scholar]
- Gloster TM et al. (2011) Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7(3):174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- God JM et al. (2015) Elevation of c-MYC disrupts HLA class II-mediated immune recognition of human B cell tumors. J Immunol 194(4):1434–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Liu F, Iqbal K (2012) O-GlcNAc cycling modulates neurodegeneration. Proc Natl Acad Sci U S A 109(43):17319–17320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JR (1991) Are cancer cells acidic? Br J Cancer 64(3):425–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltiwanger RS, Holt GD, Hart GW (1990) Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J Biol Chem 265(5):2563–2568 [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13(4):251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Akimoto Y (2009) The O-GlcNAc modification. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (eds). Essentials of glycobiology, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), Chapter 18 [Google Scholar]
- Hart GW et al. (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80:825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD et al. (2015) Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol 35(Suppl):S129–S150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkonen HM et al. (2013) O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res 73(16):5277–5287 [DOI] [PubMed] [Google Scholar]
- Iyer NV et al. (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12(2):149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H et al. (2011) Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One 6(10):e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Wu M (2014) Regulation of the pentose phosphate pathway in cancer. Protein Cell 5(8):592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K et al. (2016) Proteomic analysis of O-GlcNAcylated proteins in invasive ductal breast carcinomas with and without lymph node metastasis. Amino Acids 48(2):365–374 [DOI] [PubMed] [Google Scholar]
- Jorgensen SB et al. (2004) The alpha2–5’AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53(12):3074–3081 [DOI] [PubMed] [Google Scholar]
- Khidekel N et al. (2003) A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J Am Chem Soc 125(52):16162–16163 [DOI] [PubMed] [Google Scholar]
- Kondo A et al. (2017) Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep 18(9):2228–2242 [DOI] [PubMed] [Google Scholar]
- Lagerlof O et al. (2016) The nutrient sensor OGT in PVN neurons regulates feeding. Science 351(6279):1293–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CA et al. (2015) SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34(40):5128–5140 [DOI] [PubMed] [Google Scholar]
- Li Y et al. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13(4):376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XB, Gu JD, Zhou QH (2015) Review of aerobic glycolysis and its key enzymes - new targets for lung cancer therapy. Thorac Cancer 6(1):17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41(3):211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F et al. (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A 101(29):10804–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F et al. (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132(Pt 7):1820–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Hanover JA (2005) The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Science’s STKE : signal transduction knowledge environment 2005(312):re13. [DOI] [PubMed] [Google Scholar]
- Ludemann N et al. (2005) O-glycosylation of the tail domain of neurofilament protein M in human neurons and in spinal cord tissue of a rat model of amyotrophic lateral sclerosis (ALS). J Biol Chem 280(36):31648–31658 [DOI] [PubMed] [Google Scholar]
- Lynch TP, Reginato MJ (2011) O-GlcNAc transferase: a sweet new cancer target. Cell Cycle 10(11):1712–1713 [DOI] [PubMed] [Google Scholar]
- Lynch TP et al. (2012) Critical role of O-linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 287(14):11070–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Hart GW (2014) O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics 11(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J et al. (2016) Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J Proteome Res 15(7):2254–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher EA et al. (2012) Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25(11):1234–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta NP et al. (2015) O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson’s disease. Nat Chem 7(11):913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S, Bacote V, Traxinger RR (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 266(8):4706–4712 [PubMed] [Google Scholar]
- Marshall S, Nadeau O, Yamasaki K (2004) Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes: differential effects on glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J Biol Chem 279(34):35313–35319 [DOI] [PubMed] [Google Scholar]
- Mashimo T et al. (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159(7):1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrian MA et al. (2013) Histone acetylation regulates intracellular pH. Mol Cell 49(2):310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulukutla BC et al. (2016) Regulation of glucose metabolism - a perspective from cell bioprocessing. Trends Biotechnol 34(8):638–651 [DOI] [PubMed] [Google Scholar]
- Nakajima K et al. (2013) Mass isotopomer analysis of metabolically labeled nucleotide sugars and N- and O-glycans for tracing nucleotide sugar metabolisms. Mol Cell Proteomics 12(9):2468–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell N et al. (2004) Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol 24(4):1680–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Meoz RF et al. (2015) A small molecule that inhibits OGTactivity in cells. ACS Chem Biol 10(6):1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overdijk B et al. (1981) Isolation and further characterization of bovine brain hexosaminidase C. Biochim Biophys Acta 659(2):255–266 [DOI] [PubMed] [Google Scholar]
- Park SY, Ryu J, Lee W (2005) O-GlcNAc modification on IRS-1 and Akt2 by PUGNAc inhibits their phosphorylation and induces insulin resistance in rat primary adipocytes. Exp Mol Med 37(3):220–229 [DOI] [PubMed] [Google Scholar]
- Pichumani K et al. (2016) Hepatic gluconeogenesis influences (13)C enrichment in lactate in human brain tumors during metabolism of [1, 2-(13)C]acetate. Neurochem Int 97:133–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston GC et al. (2015) Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J 34(15):2008–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao X et al. (2015) O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nat Commun 6:8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren NS et al. (2017) Haploinsufficiency of SIRT1 enhances glutamine metabolism and promotes cancer development. Curr Biol 27(4):483–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrig F, Schulze A (2016) The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16(11):732–749 [DOI] [PubMed] [Google Scholar]
- Ruan HB et al. (2013) Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab 24(6):301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB et al. (2014) O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159(2):306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT et al. (2015) Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27(1):57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, Harris AL (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491(7424):364–373 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20(1):51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi R et al. (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 97(11):5735–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Vocadlo DJ, Krieger C (2012) Reduced protein O-glycosylation in the nervous system of the mutant SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurosci Lett 516(2):296–301 [DOI] [PubMed] [Google Scholar]
- Shi Y et al. (2010) Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 24(9):1588–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Housley MP, Hart GW (2006) O-GlcNAc cycling: how a single sugar post-translational modification is changing the way we think about signaling networks. J Cell Biochem 97(1):71–83 [DOI] [PubMed] [Google Scholar]
- Sodi VL et al. (2015) mTOR/MYC Axis regulates O-GlcNAc transferase expression and O-GlcNAcylation in breast cancer. Molecular cancer research : MCR 13(5):923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodi VL et al. (2017) Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene. 10.1038/onc.2017.395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen S et al. (2016) HIF-1alpha promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support Postimplantation bone cell survival. Cell Metab 23(2):265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincone A et al. (2015) The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90(3):927–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RC, Denko NC (2014) Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19(2):285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T et al. (2013) Inhibition of AMPK catabolic action by GSK3. Mol Cell 50(3):407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy M et al. (2016) Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol 17(6):712–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EP et al. (2014) Altering O-linked beta-N-acetylglucosamine cycling disrupts mitochondrial function. J Biol Chem 289(21):14719–14730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Mueckler M (2010) Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab 298(2):E141–E145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres CR, Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 259(5):3308–3317 [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg R et al. (1988) c-myc down-regulates class I HLA expression in human melanomas. EMBO J 7(4):1023–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosseller K et al. (2002) Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A 99(8):5313–5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosseller K et al. (2005) Quantitative analysis of both protein expression and serine / threonine post-translational modifications through stable isotope labeling with dithiothreitol. Proteomics 5(2):388–398 [DOI] [PubMed] [Google Scholar]
- Wang J et al. (1998a) A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 393(6686):684–688 [DOI] [PubMed] [Google Scholar]
- Wang J et al. (1998b) Myc activates telomerase. Genes Dev 12(12):1769–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T et al. (2017) O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat Cell Biol 19(7):833–843 [DOI] [PubMed] [Google Scholar]
- Wani WY et al. (2017) O-GlcNAc regulation of autophagy and alpha-synuclein homeostasis; implications for Parkinson’s disease. Mol Brain 10(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956) On respiratory impairment in cancer cells. Science 124(3215):269–270 [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J Gen Physiol 8(6):519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb BA et al. (2011) Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 11(9):671–677 [DOI] [PubMed] [Google Scholar]
- Weigert C et al. (2003) Palmitate-induced activation of the hexosamine pathway in human myotubes: increased expression of glutamine: fructose-6-phosphate aminotransferase. Diabetes 52(3):650–656 [DOI] [PubMed] [Google Scholar]
- Wellen KE et al. (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24(24):2784–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Fidler IJ (2000) Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res 60(16):4610–4616 [PubMed] [Google Scholar]
- Xu Z et al. (2017) Systematic identification of the protein substrates of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase-T1/T2/T3 using a human proteome microarray. Proteomics 17(11). 10.1002/pmic.201600485 [DOI] [PubMed] [Google Scholar]
- Yang YR et al. (2015a) Obesity resistance and increased energy expenditure by white adipose tissue browning in Oga(+/−) mice. Diabetologia 58(12):2867–2876 [DOI] [PubMed] [Google Scholar]
- Yang YR et al. (2015b) Elevated O-GlcNAcylation promotes colonic inflammation and tumorigenesis by modulating NF-kappaB signaling. Oncotarget 6(14):12529–12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager AR, Finney NS (2005) Synthesis of fluorescently labeled UDP-GlcNAc analogues and their evaluation as chitin synthase substrates. J Organomet Chem 70(4):1269–1275 [DOI] [PubMed] [Google Scholar]
- Yehezkel G et al. (2012) O-linked beta-N-acetylglucosaminylation (O-GlcNAcylation) in primary and metastatic colorectal cancer clones and effect of N-acetyl-beta-D-glucosaminidase silencing on cell phenotype and transcriptome. J Biol Chem 287(34):28755–28769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W et al. (2012) Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337(6097):975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Vocadlo DJ (2014) O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem Soc Rev 43(19):6839–6858 [DOI] [PubMed] [Google Scholar]
- Yuzwa SA et al. (2008) A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4(8):483–490 [DOI] [PubMed] [Google Scholar]
- Yuzwa SA et al. (2014a) O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol 426(8):1736–1752 [DOI] [PubMed] [Google Scholar]
- Yuzwa SA et al. (2014b) Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol Neurodegener 9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B et al. (2017) Bitterness in sugar: O-GlcNAcylation aggravates pre-B acute lymphocytic leukemia through glycolysis via the PI3K/Akt/c-Myc pathway. Am J Cancer Res 7(6):1337–1349 [PMC free article] [PubMed] [Google Scholar]
- Zhu Y et al. (2014) The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem 289(50):34472–34481 [DOI] [PMC free article] [PubMed] [Google Scholar]