

Abstract
New antibiotics with unique biological targets are desperately needed to combat the growing number of resistant bacterial pathogens. ATP synthase, a critical protein found in all life, has recently become a target of interest for antibiotic development due to the success of the anti-tuberculosis drug bedaquiline, and while many groups have worked on developing drugs to target bacterial ATP synthase, few have been successful at inhibiting Pseudomonas aeruginosa (PA) ATP synthase specifically. PA is one of the leading causes of resistant nosocomial infections across the world and is extremely challenging to treat due to its various antibiotic resistance mechanisms for most commonly used antibiotics. Herein, we detail the synthesis and evaluation of a series of C1/C2 quinoline analogues for their ability to inhibit PA ATP synthase and act as antibiotics against wild-type PA. From this survey, we found six compounds capable of inhibiting PA ATP synthase in vitro showing that bulky/hydrophobic C1/C2 substitutions are preferred. The strongest inhibitor showed an IC50 of 10 μg/mL and decreased activity of PA ATP synthase to 24% relative to the control. While none of the compounds were able to inhibit wild-type PA in cell culture, two showed improved inhibition of PA growth when permeability of the outer membrane was increased or efflux was knocked out, thus demonstrating that these compounds could be further developed into efficacious antibiotics.
Introduction
Multidrug-resistant bacterial infections are a growing health crisis that could lead to up to 10 million deaths worldwide by 2050 if recent trends continue.1,2 Currently, the leading causes of drug-resistant nosocomial infections across the world are the ESKAPE pathogens [Enterococcus facium, Staphylococcus aureus (SA), Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa (PA), and Enterobacter spp.], some of which have no clinical treatments available.3−5 Multidrug-resistant P. aeruginosa (MDRPA), specifically, has been classified by the CDC as a “Serious Threat”1 due to the lack of available treatments. Current treatments for PA and MDRPA infections, including β-lactams, aminoglycosides, cephalosporins, fluoroquinolones, and polymyxins, all have known resistance mechanisms that can affect an entire class of drugs with the same mechanism of action,6 thus necessitating new antibiotics targeting novel cellular mechanisms.7 To this end, bacterial bioenergetic pathways are emerging targets for antibiotic development, especially ATP synthase, which is essential for most cells and is the target for the anti-tubercular drug bedaquiline (BDQ) (1).8
ATP synthase is a ubiquitous bioenergetic enzyme found in all domains of life. In its simplest bacterial form, ATP synthase is localized in the plasma membrane and is made up of two multi-subunit domains, F1 and Fo. The Fo domain uses the H+ electrochemical gradient present across the cell membrane to drive rotation of the oligomeric c-ring, which in turn drives rotation within the F1 motor to catalyze the phosphorylation of ADP to produce ATP (Figure 1A). Additionally, in some bacteria, ATP synthase can act in reverse as an ATP-driven proton pump responsible for maintaining the cytoplasmic pH.9−14
Figure 1.

(A) Cartoon structure of bacterial ATP synthase, where the H+ translocating c-ring of Fo is shown in green with red dots, indicating locations of the H+ binding sites (Glu65 in MT). (B) Structure of BDQ, 1, highlighting substituents at positions C1 and C2 on the quinoline core. (C) Crystal structure of BDQ bound to the MT c-ring (PDB 4V1F). Residues interacting with BDQ are highlighted and correspond to highlighted residues in panel (D). (D) Amino acid sequence alignment of two regions of subunit c from Mycobacterium tuberculosis (MT) and Pseudomonas aeruginosa (PA); MT numbering.
BDQ (Figure 1B), a diarylquinoline, was the first new class of antibiotics to be developed for the treatment of MDR Mycobacterium tuberculosis (MT) in decades. BDQ is highly potent against various drug-resistant MT strains [minimum inhibitory concentrations (MIC) ranging from 0.003 to 0.060 μg/mL]8 while also being highly selective for MT over human ATP synthase.15 The primary binding site for BDQ is on the Foc-subunit at the H+-binding Glu65 where it blocks H+-driven rotation of the c-ring.15,16 BDQ binding to the MT c-ring was captured in high-resolution by X-ray crystallography (Figure 1C),15 and a recent cryo electron microscopy study showed that subunit a contributes to high affinity BDQ binding at this site by creating a hydrophobic pocket for the quinoline moiety.17 Building from BDQ, many laboratories have explored ATP synthase as a potential target for antibiotic18−21 and adjuvant22−25 development against Gram-positive and Gram-negative bacteria. A screen of C3-substituted quinolines correlated hydrophilicity (cLog P) and antibacterial activity (MIC) against Escherichia coli,21 and a screen of >700 BDQ derivatives with novel lateral chains against a panel of Gram-positive and -negative pathogens identified an analog active against Streptococcus pneumoniae.18 However, no quinoline-based compounds were active against PA except at high concentrations, and some have attributed this lack of efficacy in Gram-negative pathogens to efflux mechanisms and poor diffusion across the outer membrane.18
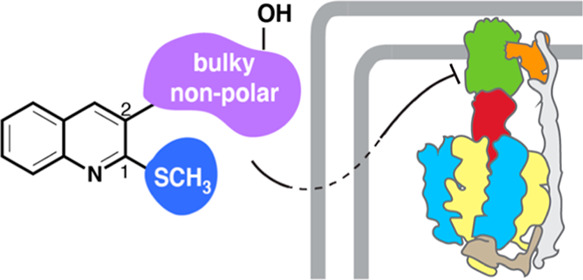
Herein, we describe the synthesis and evaluation of an initial set of 16 C1 and C2 quinoline analogs (2 known and 14 novel) for their ability to inhibit PA ATP synthase in vitro and act as antibiotics against PA in liquid culture. This set of molecules was chosen based on prior structure–activity relationship (SAR) studies18,21 and an evaluation of the amino acid differences between the BDQ binding site on subunit c of MT ATP synthase15,16 and the same region of PA subunit c, which overall is less sterically congested and more hydrophobic (Figure 1D). Of this initial survey, six compounds were found to inhibit PA ATP synthase, which demonstrates the potential to exploit PA ATP synthase as an antibiotic target.
Results and Discussion
Compound Design
The goal of this initial survey was to first determine whether quinoline derivatives could be developed that inhibit PA ATP synthase, since to date none have been disclosed, and then to examine whether those molecules could act as antibiotics. Based on the binding of BDQ to the c-ring of MT ATP synthase (Figure 1C), the C1 and C2 substituents are responsible for most of the interactions with the pocket surrounding the proton-binding site (Glu65). The most important interaction for BDQ binding to subunit c is the hydrogen bond between the C2 dimethylamino (DMA) group and the carboxylate of Glu65, which is the residue responsible for reversible proton binding and shuttling on the protein.15−17 He and co-workers determined that, for MT ATP synthase inhibition, the bromine, hydroxy, and stereocenters are non-essential, and that although the DMA and aryl groups are essential, their locations and connectivities can be modified, including being attached to the C1 rather than C2 position of the quinoline.16 Furthermore, in the intact complex, C1 and C2 substituents determine BDQ binding to the secondary “lagging” site at the interface between subunits a and c.17
We probed the chemical space, using both novel and known compounds, at the C1 and C2 positions of the quinoline core, focusing on functional groups that would likely interact with the equivalent binding pocket on the c-ring of PA ATP synthase (Figure 1D). Briefly, this pocket is more non-polar in PA ATP synthase, due to the presence of Phe in place of Asp32, and also less sterically congested, due to the replacement of Tyr68, Phe69, and Leu72 with Thr, Met, and Val, respectively. To accommodate these differences, we hypothesized that increased hydrophobic surface area and additional π-stacking interactions would promote binding. At the C1 position, alkoxy groups, like the methoxy present in BDQ, have been shown to be effective inhibitors of both MT8,16 and Gram-positive bacterial ATP synthase,18 but bulkier methyl sulfides have not been evaluated. At the C2 position, we chose to investigate a series of synthetically accessible, variably sized, primarily non-polar substitutions. The resulting series described herein consists of a mixture of known (9 and 11) and novel (8, 10, and 12–23) compounds.
Of the known compounds, tetrahydropyrimidine 9 has been previously synthesized;26 however, no biological evaluation has been conducted to date despite its structural similarity to BDQ-related ATP synthase inhibitors. Hydrazines 11 and 12 were developed and evaluated by Hamama et al. for their activity as both antibacterial and antifungal agents. Hydrazine 11 was found to inhibit PA with an MIC of 80 μg/mL, whereas 12 showed weaker activity against PA.27 The authors proposed that the antibiotic activity of this series was due to their ability to act as radical scavengers, but because of their quinoline structures, we probed 11, 12, and related compounds 10 and 13, as ATP synthase inhibitors.
Synthesis of Inhibitors
All C1/C2 quinoline analogs were synthesized from commercially available 2-chloroquinoline-3-carbaldehyde (2, Scheme 1). First, the C1 position of 2 was derivatized under nucleophilic aromatic substitution conditions. As seen in Scheme 1, treatment of 2 with either sodium methoxide in anhydrous methanol or benzyl alcohol, potassium carbonate, and N,N-dimethylformamide produced methoxy quinoline 3 and benzyloxy quinoline 4, respectively. Treatment of 2 with sodium sulfide produced thiol 5, which was then further transformed in situ to methyl sulfide 6 with the addition of methyl iodide under the same conditions in high yield.
Scheme 1. Synthesis of C1 Derivatives.

Analogs 3, 4, and 6 were then further derivatized at the C2 position through a variety of carbonyl addition reactions (Schemes 2 and 3). To produce carbamate 8, the aldehyde of 3 was reduced to alcohol 7 using sodium borohydride in methanol in quantitative yield. Treatment of 7 with (4-aminophenyl)methanol, triphosgene, and pyridine (pyr) in anhydrous dichloromethane (DCM) afforded carbamate 8 in 29% yield (Scheme 2). Tetrahydropyrimidine 9 was synthesized from 3 using the published one pot cyclocondensation method developed by Shastri et al.26 Hydrazines 10–12 were produced from 3 using previously published condensation methods by Hamama et al. (Scheme 2).27 Thiazole 13 was also produced in high yield using a variation of Hamama’s method by treating 12 with phenacyl bromide and acetic anhydride.27 Reductive amination of 3, 4, and 6 with a variety of amino acid methyl esters (l-Ala, l-Val, d-Met, l-Trp, l-Tyr, and d-His) followed by sodium borohydride provided analogs 14–23 in moderate to good yields, as seen in Scheme 3. This series of amino acids was chosen for this preliminary evaluation because they are hydrophobic, except for histidine, which is partially positively charged at pH 7, and are wide ranging in size. We also chose to leave the methyl esters in place for 14–23 due to the hydrophobic nature of the c-subunit of PA ATP synthase.
Scheme 2. Synthesis of Carbamate, Heterocyclic, and Hydrazine C2 Derivatives 8–13.

Scheme 3. Synthesis of Amino Acid C2 Derivatives 14–23 via Reductive Amination.

PA ATP Synthase Inhibition
Compounds 8–23 were evaluated for their ability to inhibit PA ATP synthase activity using a luminescence-based assay and to inhibit wild-type PA cell growth in liquid culture. ATP synthesis activity of inverted membrane vesicles of PA was measured by energizing the endogenous electron transport system with β-d-nicotinamide adenine dinucleotide (NADH) to generate a proton gradient and subsequently drive ATP synthesis from ADP and phosphate. ATP synthesized after 10 min (Figure S1) was detected using a luciferin–luciferase assay system. Due to the known interference of similar compounds with luciferase,28 samples removed from the ATP synthesis reaction were diluted 500-fold before addition into the ATP detection reaction. To account for residual ATP in inverted membrane vesicles and/or enzymatic sources of ATP synthesis other than ATP synthase, control reactions were conducted in the presence of a protonophore (Figure S1). After an initial screen of all compounds (Figure S2), additional replicates were completed with compounds demonstrating significant dose-dependent inhibition. Compounds 8, 10, 15, 17, 18, 19, and 22 all showed dose-dependent inhibition of ATP synthesis (Figure 2) with relative IC50 values in the 2–17 μg/mL range. Compound 10 was excluded from further evaluation due to significant inhibition of H+ gradient formation (Figure S3). While the remaining six compounds caused dose-dependent inhibition, the maximum level of inhibition varied among compounds. Compounds 18 and 22 caused complete inhibition of ATP synthesis activity at 128 μg/mL, while inhibition caused by 8, 15, 17, and 19 was not complete even at the highest concentration of compound.
Figure 2.

Inhibition of ATP synthesis activity by selected compounds. Relative activity was calculated by normalizing luminescence values to a control with no inhibitor. Activity at each concentration of the inhibitor is shown as mean ± standard error (n ≥ 3). Dose–response curves (red) were fitted using GraphPad Prism 9. The dotted line indicates the mean background activity that can be attributed to ATP sources other than ATP synthase (Figure S1).
Based on amino acid sequence alignment, the assumed binding region on PA ATP synthase is expected to be relatively hydrophobic and spacious compared to the analogous BDQ binding region of MT ATP synthase. By evaluating the structural similarities between molecules in this study that are capable of inhibiting PA ATP synthase compared to those that are not, some conclusions can be drawn to help guide future compound development. First, steric bulk is required at either the C1 or C2 position of the quinoline but not both. For example, 22, which has the methyl sulfide at C1 and the bulky tyrosine substituent at C2, is active, but 21 with the same C2 substitution and the additional bulky C1 benzyloxy group is not active. Additionally, 15, which has a C2 valine and C1 benzyloxy, is active but 14, which has a C2 alanine and C1 methoxy, is not. Second, functional groups capable of π-stacking, possibly with the phenylalanine in the binding region, also increased activity (compounds 8, 18, 19, and 22). Third, the presence of the methyl sulfide improved activity compared to the methoxy, as seen in the C2 methionine (16vs17), tryptophan (18vs19), and tyrosine (20vs22) series. Fourth, stereochemistry of the amino acid at the C2 position may not affect activity, but more evaluation is needed to confirm this observation. Finally, hydrogen bonding, possibly with the aspartic acid present in the binding region, could play a role in binding based on the activity of 8 and 22 both of which have a hydroxyl group.
Antibacterial Activity
All compounds were evaluated in a standard broth microdilution MIC assay against both methicillin susceptible SA (MSSA) and wild-type PA (WTPA). Interestingly, despite the inhibition of ATP synthase activity by 8, 15, 17, 18, 19, and 22 and the previously reported activity by 11 and 12,27 none of the series showed whole cell antibiotic activity against PA (Tables 1 and S1). C1 methyl sulfide quinoline 6 (Table S1) and methionine C1 methyl sulfide 17 did have activity against MSSA, both with MIC = 64 μg/mL. However, this activity may not be due to ATP synthase inhibition.
Table 1. Antibacterial Activity of 8–23 against S. aureus and P. aeruginosa.
| compound | MSSA MIC (μg/mL)a | WTPA MIC (μg/mL)a |
|---|---|---|
| 8 | >128 | >256 |
| 9 | >128 | >128 |
| 10 | >128 | >128 |
| 11 | >128 | >128 |
| 12 | >128 | >128 |
| 13 | >128 | >128 |
| 14 | >128 | >128 |
| 15 | >128 | >256 |
| 16 | >128 | >128 |
| 17 | 64 | >256 |
| 18 | >128 | >256 |
| 19 | >128 | >256 |
| 20 | >128 | >128 |
| 21 | >128 | >128 |
| 22 | >128 | >256 |
| 23 | >128 | >128 |
n = 3, MIC = minimum inhibitory concentration of >85% reduction in pathogen growth with compound compared to pathogen alone (DMSO only) at OD 590 nm (no visible growth).
PA Accumulation Evaluation
As stated and seen in other ATP synthase inhibitor development work,18 one of the major challenges of developing antibiotics that target Gram-negative bacteria is the accumulation problem where molecules cannot penetrate the tightly packed, polyanionic lipopolysaccharide outer membrane and/or are quickly shuttled out of the cell by promiscuous efflux pumps. Therefore, we assessed whether our active ATP synthase inhibitors could be suffering from poor accumulation due to one or both mechanisms.
For antibiotics to readily diffuse across the membrane, they typically must follow the experimentally determined “Rules of Entry” for accumulation in Gram-negative bacteria, namely: (i) molecular weight < 500 g/mol; (ii) cLog D7.4 between −2 and 0; (iii) ≤5 rotatable bonds; (iv) high polar surface area (average 165 Å); (v) low globularity; and (vi) presence of a 1° amine or guanidinium.29−31 Unsurprisingly, our active compounds, 8, 15, 17, 18, 19, and 22, break many of these rules since they were designed to target the membrane-embedded-binding pocket (see Table S2). While all have molecular weights <500 g/mol and low globularity, all also have too many rotatable bonds, with carbamate 8 having the least at 6 and valine 15 and methionine 17 having the most at 9, and all are too hydrophobic based on their cLog P and total polar surface areas. Carbamate 8 does not have a primary amine, but 15, 17, 18, 19, and 22 have 2° amines.
To examine experimentally whether penetration of the outer membrane could be improved, 8, 15, 17, 18, 19, and 22 were co-dosed with 50 and 100 μM of pentamidine, a known antibiotic adjuvant capable of increasing the uptake of antibiotics in Gram-negative bacteria.32 Compounds 8, 17, 18, 19, and 22 all showed a statistically significant reduction of PA growth in liquid culture when co-dosed with pentamidine at both concentrations, with 100 μM of pentamidine being more effective than 50 μM of pentamidine, compared to compound alone (Figure S4). Tryptophan 18 had the largest reduction in PA growth of the set when co-administered with either 50 or 100 μM of pentamidine. Unsurprisingly, valine 15, which is the most hydrophobic of all, showed no significant reduction in growth at either concentration of pentamidine. While the co-administration of pentamidine did not allow for complete PA growth inhibition with any of the inhibitors, these observations indicated that our inhibitors could act as antibiotics if they can be delivered past the membrane.
To examine whether efflux was also contributing to the lack of whole cell activity with our inhibitors, we evaluated the antibacterial activity of 8, 15, 17, 18, 19, and 22 against PA efflux knockout strain, GKCW120 (PΔ6).31 As seen in Table 2, carbamate 8 and methionine 17 were able to inhibit PΔ6 with MICs of 64 and 256 μg/mL respectively, which indicates that efflux could also be a contributing factor to the lack of antibiotic activity seen against WTPA.
Table 2. Antibacterial Activity of 8, 15, 17, 18, 19, and 22 against P. aeruginosa Efflux Knockout PΔ6.
| compound | PΔ6 MIC (μg/mL)a |
|---|---|
| 8 | 64 [>4] |
| 15 | >256 [0] |
| 17 | 256 [>2] |
| 18 | >256 [0] |
| 19 | >256 [0] |
| 22 | >256 [0] |
Conclusions
We have detailed an initial evaluation of a series of C1 and C2 quinoline derivatives for their ability to inhibit PA ATP synthase and act as antibiotics against PA, a Gram-negative pathogen. Six of the initial set of 16 compounds surveyed were capable of inhibiting PA ATP synthase in vitro with compound 22, which has a methyl sulfide at C1 and tyrosine at C2, showing the strongest activity. Consistent with our hypothesis, increasing hydrophobicity, especially with functional groups capable of π-stacking, and having steric bulk at either the C1 or C2 position increased activity. Although none of the compounds capable of inhibiting ATP synthase displayed whole cell antibiotic activity against WTPA, further evaluation of these compounds indicated that they may be suffering from poor accumulation due to them being unable to cross the outer membrane to reach ATP synthase in the inner membrane and/or due to efflux. In conclusion, this initial evaluation of quinoline-based compounds demonstrates that targeted PA ATP synthase inhibitors capable of acting as antibiotics can be developed, which will help treat this problematic drug-resistant pathogen.
Experimental Section
Synthesis and Spectroscopic Data
General
Reagents and solvents purchased were of reagent grade and used without further purification. All reactions were performed in flame-dried glassware under an Ar or N2 atmosphere. Evaporation and concentration in vacuo were performed at 40 °C. TLC was conducted using precoated SiO2 60 F254 glass plates from EMD Milliepore with visualization by UV light (254 or 366 nm). NMR (1H or 13C) were recorded on an Varian INOVA-400 MHz spectrometer at 298 K. Residual solvent peaks were used as an internal reference. Coupling constants (J) (H, H) are given in Hz. Coupling patterns are designated as singlet (s), doublet (d), triplet (t), quadruplet (q), quintuplet (qu), doublet of doublets (dd), multiplet (m), or broad singlet (br). IR spectra were recorded on a Shimadzu IRSpirit FT-IR spectrophotometer and measured neat. Low-resolution mass spectral data were acquired on a Shimadzu single quadrupole LCMS-2020. High-resolution mass spectral data were acquired on a Thermo Scientific Q Exactive Plus mass spectrometer coupled to a Waters Acquity UPLC, and the detected masses are given as m/z with m representing the molecular ion.
2-Methoxyquinoline-3-carbaldehyde (3)
2-Chloroquinoline-3-carbaldehyde (5 g, 26.1 mmol) was dissolved in anhydrous methanol (260 mL, 0.1 M) under an inert atmosphere and anhydrous conditions. To the mixture, a solution of sodium methoxide in methanol (15–20 mL, 30% by wt solution) was added, and the reaction was warmed to reflux for 16 h. The solution was then cooled, and approximately 200 mL of methanol was removed under reduced pressure. 1 M aqueous HCl was then added, and the resulting precipitate was filtered and dried providing 3 (4.32 g, 88%) as an off-white solid. 1H NMR (CDCl3, 400 MHz): δ 10.48 (s, 1H), 8.61 (s, 1H), 7.89–7.86 (m, 2H), 7.75 (t, J = 8.8 Hz, 1H), 7.45 (t, J = 8 Hz, 1H), 4.20 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 189.54, 161.45, 148.99, 140.42, 132.90, 129.99, 127.39, 125.34, 124.58, 120.26, 54.31. IR (film) νmax: 2869, 2831, 2745, 1680, 1619, 1600, 1554, 1499, 1436, 1385, 1341, 1257, 1155, 1107, 1007, 899, 762, 751, 693, 606, 472 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C11H9NO2, 188.0712; found, 188.0708.
2-(Benzyloxy)quinoline-3-carbaldehyde (4)
2-Chloroquinoline-3-carbaldehyde (2.56 g, 13.38 mmol) was dissolved in N,N-dimethyl formamide (25 mL, 0.54 M) under an inert atmosphere and anhydrous conditions. Benzyl alcohol (2.75 mL, 26.45 mmol) and potassium carbonate (3.68 g, 26.66 mmol) were added, and the reaction was heated to reflux for 4 h. The solution was then cooled to room temperature and diluted with ethyl acetate (50 mL). The solution was then washed with deionized (DI) H2O (3 × 25 mL). The organic layer was then dried over Na2SO4 and concentrated. A column dry load was prepared by dissolving the mixture in ethyl acetate, adding 1.0 g of silica gel, and then evaporating the mixture under reduced pressure. Flash chromatography (SiO2, 1 × 10 cm, 10% EtOAc/hexanes gradient elution) afforded 4 (162.2 mg, 4.6%) as a yellow solid. 1H NMR (CDCl3, 400 MHz): δ 10.54 (s, 1H), 8.61 (s, 1H), 7.90 (d, J = 8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.76 (t, J = 8.8 Hz, 1H), 7.58 (d, J = 8 Hz, 2H), 7.47–7.35 (m, 4H), 5.68 (s, 2H). 13C NMR (CDCl3, 100 MHz): δ 189.43, 160.91, 149.09, 140.12, 136.89, 132.82, 129.99, 128.77, 128.42, 128.32, 127.52, 125.36, 124.71, 120.21, 68.31. IR (film) νmax: 2954, 2921, 2861, 1694, 1616, 1599, 1501, 1424, 1401, 1340, 1260, 1112 cm–1. C17H13NO2 264.1019; found, 264.1009.
2-Mercaptoquinoline-3-carbaldehyde (5)
2-Chloroquinoline-3-carbaldehyde (1.65 g, 8.66 mmol) and Na2S (1.6 g, 20.8 mmol) were dissolved in N,N-dimethyl formamide (43 mL, 0.2 M) and allowed to stir at 23 °C for 2 h. The reaction was then cooled to 0 °C, and three drops of acetic acid were added, and the precipitate that formed was filtered and dried to yield 5 (1.00 g, 62%) as an orange solid. 1H NMR (DMSO-d6, 400 MHz): δ 14.0 (br, 1H) 10.72 (s, 1H), 8.38 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.77 (t, J = 8.4 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 191.90, 181.01, 141.06, 137.07, 134.39, 131.84, 130.76, 125.08, 121.73, 116.30. IR (film) νmax: 3093, 2981, 2953, 1688, 1620, 1287, 1225, 1157, 1083, 742 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C10H7NOS, 190.0319; found, 190.0319.
2-(Methylthio)quinoline-3-carbaldehyde (6)
2-Chloroquinoline-3-carbaldehyde (1.65 g, 8.66 mmol) and Na2S (1.6 g, 20.8 mmol) were dissolved in N,N-dimethyl formamide (43 mL, 0.2 M) and allowed to stir at 23 °C for 2 h. Then, methyl iodide (0.55 mL, 8.8 mmol) was added, and the reaction was allowed to stir at 23 °C for 1 h. The reaction was then cooled to 0 °C, and the precipitate that formed was filtered and dried to yield 6 (1.60 g, 91%) as a brown solid. 1H NMR (CDCl3, 400 MHz): δ 10.33 (s, 1H), 8.48 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 8 Hz, 1H), 7.81 (t, J = 6.8 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 2.74 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 190.29, 159.87, 149.84, 142.68, 133.21, 129.43, 128.33, 127.43, 126.35, 124.67, 13.28. IR (film) νmax: 3058, 2984, 2917, 1692, 1619, 1396, 1222, 1064, 741 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C11H9NOS, 204.0474; found, 204.0474.
(2-Methoxyquinolin-3-yl)methanol (7)
2-Methoxyquinoline-3-carbaldehyde (3, 250 mg, 1.33 mmol) was dissolved in anhydrous methanol (14 mL, 0.09 M) and cooled to 0 °C under inert conditions. Sodium borohydride was added, and the reaction was allowed to naturally warm to room temperature over 1 h. The reaction was quenched with DI H2O (50 mL) and then extracted with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with saturated aqueous NaCl, dried over Na2SO4, and concentrated under reduced pressure, which yielded 7 (250 mg, quant.) as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 8.35 (s, 1) 7.89 (d, J = 8.2 Hz, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.68 (t, J = 7.0 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 4.83 (s, 2H), 4.05 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): δ 159.51, 145.72, 139.03, 130.28.127.89, 126.47, 124.66, 124.56, 121.69, 53.69, 41.43. IR (film) νmax: 3321, 3232, 2920, 2851, 1632, 1474, 1439, 1402, 1347, 1262, 1040, 1015, 903, 751 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C11H11NO2, 190.0868; found, 190.0874.
(2-Methoxyquinolin-3-yl)methyl(4-(hydroxymethyl)phenyl)carbamate (8)
(2-Methoxyquinolin-3-yl)methanol (7, 82 mg, 0.43 mmol), (4-aminophenyl)methanol (53 mg, 0.43 mmol), and anhydrous pyridine (173 mL, 2.15 mmol) were dissolved in 7 mL of anhydrous DCM under inert conditions and cooled to 0 °C. Triphosgene (51 mg, 0.17 mmol) in 7 mL of anhydrous DCM was then added dropwise to the reaction over 10 min. The reaction was then allowed to warm naturally and stir for 18 h. The reaction was quenched with saturated aqueous NaHCO3 (15 mL) and extracted with ethyl acetate. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. Flash chromatography (SiO2, 1 × 10 cm, 20–50% EtOAc/hexanes gradient elution) afforded 8 (42 mg, 29%) as an off-white solid. 1H NMR (CDCl3, 400 MHz): δ 8.00 (s, 1H), 7.84 (d, J = 8.4, 1H), 7.69 (d, J = 7.6, 1H), 7.61 (t, J = 7.2, 1H), 7.38–7.26 (m, 5H), 6.99 (br, 1H), 5.30 (s, 2H), 4.61 (s, 2H), 4.10 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 160.23, 146.41, 137.51, 137.33, 136.26, 129.85, 129.60, 128.12, 127.70, 127.07, 125.07, 124.43, 120.57, 118.90, 65.03, 62.33, 53.87. IR (film) νmax: 3389, 3069, 2949, 1706, 1603, 1535, 1446, 1403, 1221, 1057 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C19H18N2O4, 339.1345; found, 339.1344.
Ethyl 4-(2-Methoxyquinolin-3-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (9)
Following the procedure from Chemical Science Transactions, 2018, 7(1), 89–94, 2-methoxyquinoline-3-carbaldehyde (3, 50 mg, 0.26 mmol), ethyl acetoacetate (67 mL, 0.53 mmol), urea (31 mg, 0.53 mmol), and p-toluenesulfonic acid monohydrate (15 mg, 30%) were dissolved in anhydrous ethanol (2.6 mL, 0.1 M) under inert conditions and warmed to reflux for 5 h. The reaction was then cooled to 0 °C and upon addition of chilled DI H2O, a precipitate formed. The precipitate was filtered and dried to provide 9 (46 mg, 52%) as a white solid, matching the previously reported spectral data.
(E)-N′-((2-Methoxyquinolin-3-yl)methylene)benzohydrazide (10)
Following the procedure from J. Heterocyclic Chem.2018, 55, 2623, 2-methoxyquinoline-3-carbaldehyde (3, 1.00 g, 5.3 mmol) and benzoic hydrazine (0.72 g, 5.3 mmol) were dissolved in anhydrous methanol (25 mL, 0.21 M) and warmed to 65 °C for 20 h. The precipitate was filtered, dried, and recrystallized in ethanol to provide 10 (414 mg, 47%) as a yellow crystalline solid. 1H NMR (acetone-d6): δ 11.15 (br, 1H), 8.68, (d, J = 16.8 Hz, 2H), 7.93 (m, 3 H), 7.70 (d, J = 8.2 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.51–7.34 (m, 4H) 3.99 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): δ 163.17, 159.43, 146.23, 141.95, 134.55, 133.19, 132.04, 130.81, 128.87, 128.63, 127.77, 126.61, 124.91, 124.85, 118.69, 53.83. IR (film) νmax: 3268, 3238, 3053, 3028, 2981, 1649, 1620, 1543, 1356, 1277, 1014, 698 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C18H15N3O2, 306.1243; found, 306.123.
(1E,2E)-1,2-Bis((2-methoxyquinolin-3-yl)methylene)hydrazine (11)
Following the procedure from J. Heterocyclic Chem.2018, 55, 2623, 2-methoxyquinoline-3-carbaldehyde (3) (502 mg, 2.68 mmol) was dissolved in 12.5 mL of anhydrous methanol (0.21 M) and warmed to 65 °C. Hydrazine (0.67 mL, 13.4 mmol) and catalytic acetic acid were then added, and the reaction was allowed to stir at reflux for 20 h. Upon completion, the crude reaction mixture was concentrated under reduced pressure and purified using flash chromatography (SiO2, 4 × 20 cm, 75% EtOAc/hexane), which provided 11 (1.00 g, quant.) as an off white solid, matching the previously reported spectral data.
(E)-2-((2-Methoxyquinolin-3-yl)methylene)hydrazine-1-carbothioamide (12)
Following the procedure from J. Heterocyclic Chem.2018, 55, 2623, 2-methoxyquinoline-3-carbaldehyde (3) (2.00 g, 10.7 mmol) and thiosemicarbazide (0.988 g, 10.7 mmol) were dissolved in ethanol (100 mL, 0.1 M). Five drops of glacial acetic acid were added to the mixture, which was then warmed to reflux for 1 h. The precipitate that formed was filtered, dried, and recrystallized in methanol to provide 12 (1.21 g, 43%) as a light yellow crystalline solid, matching the previously reported spectral data.
(E)-2-(2-((2-Methoxyquinolin-3-yl)methylene)hydrazineyl)-4-phenylthiazole (13)
Modification of the procedure from J. Heterocyclic Chem.2018, 55, 2623, (E)-2-((2-methoxyquinolin-3-yl)methylene)hydrazine-1-carbothioamide 12 (0.5 g, 1.9 mmol), phenacyl bromide (0.38 g, 1.9 mmol), and acetic anhydride (2 mL) were dissolved in hot N,N-dimethylformamide (10 mL, 0.2 M) and then stirred at room temperature for 18 h. The precipitate was filtered and washed successively with DI H2O and hot ethanol to provide 13 (0.64 g, 92%) as a yellow solid. 1H NMR (DMSO-d6): δ 9.64 (s, 1H), 8.91 (s, 1H), 8.11–8.09 (m, 2H) 8.00 (d, J = 7.6 Hz, 2H), 7.83 (d, J = 8.4 Hz, 1H). 7.75 (t, J = 8 Hz, 1H), 7.53–7.47 (m, 3H), 7.38 (t, J = 7.2, 1H). 4.09 (s, 3H), 2.66 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): δ 172.30, 159.57, 155.52, 149.34, 146.65, 145.94, 135.57, 133.67, 131.21, 128.99, 128.91, 128.25, 126.65, 125.66, 124.94, 124.69, 118.01, 113.11, 109.57, 53.86, 22.72. IR (film) νmax: 3657, 3090, 2972, 2889, 1695, 1623, 1570, 1488, 1445, 1372, 1275, 1238, 946 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C22H18N4O2S, 403.1223; found, 403.1205.
Amino Acid Series
General Procedure A: Reductive Amination of Methoxy and Methyl Sulfide Quinoline Carbaldehydes
The starting aldehyde (1 equiv) and amino acid methyl ester (1.2 equiv) were dissolved in anhydrous methanol (0.09 M) under inert conditions. N,N-Diisopropylethylamine (3 equiv) was then added, and the reaction was allowed to stir at 23 °C for 18–20 h. NaBH4 (2 equiv) was then added. After 1 h, the reaction was diluted with DI H2O and extracted with ethyl acetate. The organic layer was washed with saturated aqueous NaCl, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography of the crude extracts (SiO2, 1 × 10 cm, 15–100% EtOAc/hexane or 0–5% CH3OH/CH2Cl2 gradient elution) provided the desired products.
Methyl ((2-Methoxyquinolin-3-yl)methyl)-l-alaninate (14)
Using general procedure A, from 2-methoxyquinoline-3-carbaldehyde (3) (50 mg, 0.27 mmol) and l-alanine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 10% EtOAc/hexane) provided 14 as a white solid (8 mg, 11%). 1H NMR (CDCl3, 400 MHz): δ 7.95 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.2 Hz, 1H), 7.37 (t, J = 8.4 Hz, 1H), 4.11 (s, 3H), 3.92–3.81 (m, 2H), 3.65 (s, 3H), 3.44 (q, J = 7.2 Hz, 1H), 1.38 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 176.15, 160.86, 145.91, 136.55, 129.14, 127.37, 126.99, 125.43, 124.22, 123.95, 56.30, 53.71, 52.02, 47.21, 19.36. IR (film) νmax: 3222, 2902, 2520, 2053, 1600, 1501, 1330, 1030, 817 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C15H18N2O3, 275.1396; found, 275.1389.
Methyl ((2-Methoxyquinolin-3-yl)methyl)-d-methioninate (16)
Using general procedure A, from 2-methoxyquinoline-3-carbaldehyde (3) (50 mg, 0.27 mmol) and d-methionine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 10 EtOAc/hexane) provided 16 as a white solid (35 mg, 40%). 1H NMR (CDCl3, 400 MHz): δ 7.95 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 6.8 Hz, 1H), 7.59 (t, J = 8.8 Hz, 1H), 7.37 (t, J = 8.4 Hz, 1H), 4.10 (s, 3H), 3.84 (m, 2H), 3.61 (s, 3H), 3.48 (q, J = 5.6 Hz, 1H), 2.65 (t, J = 7.2 Hz, 2H) 2.1 (s, 3H), 2.05–1.88 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 175.59, 160.86, 145.90, 136.45, 129.13, 127.37, 127.00, 125.42, 124.20, 124.07, 59.96, 53.69, 52.05, 47.48, 33.01, 30.69, 15.60. IR (film) νmax: 2948, 2916, 2360, 2343, 1732, 1626, 1442, 1399, 1258, 1160, 1101, 755 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C17H22N2O3S, 335.1429; found, 335.1444.
Methyl ((2-(Methylthio)quinolin-3-yl)methyl)-d-methioninate (17)
Using general procedure A, from 2-(methylthio)quinoline-3-carbaldehyde (3) (50 mg, 0.25 mmol) and d-methionine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 8% EtOAc/hexane) provided 17 as a white solid (22 mg, 26%). 1H NMR (CDCl3, 400 MHz): δ 7.95 (m, 2H), 7.73 (d, J = 8.2 Hz, 1H), 7.62 (t, J = 8.2 Hz, 1H), 7.42 (t, J = 7.4 Hz, 1H), 4.01–3.77 (m, 2H), 3.71 (s, 3H), 3.51 (q, J = 5.1 Hz, 1H), 2.75–2.67 (m, 5H), 2.09 (s, 3H) 2.02–1.88 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 176.49, 160.42, 148.26, 134.51, 131.88, 130.02, 128.56, 128.30, 126.65, 126.08, 60.22, 52.45, 49.05, 33.20, 30.98, 15.68, 13.28. IR (film) νmax: 3343, 2981, 2343, 1734 1397, 1136, 752 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C17H22N2O2S2, 351.1195; found, 351.1180.
Methyl ((2-Methoxyquinolin-3-yl)methyl)-l-tryptophanate (18)
Using general procedure A, from 2-methoxyquinoline-3-carbaldehyde (3) (50 mg, 0.27 mmol) and l-tryptophan methyl ester, flash chromatography (SiO2, 1 × 10 cm, 10–40% EtOAc/hexane) provided 18 as a brown solid (46 mg, 44%). 1H NMR (CDCl3, 400 MHz): δ 8.21 (s, 1H), 7.79 (d, J = 8.8, 1H), 7.71 (s, 1H), 7.58–7.49 (m, 3H), 7.48–7.30 (m, 2H), 7.19 (t, J = 8.0, 1H), 7.08–7.04 (m, 2H), 3.93–3.69 (m, 6H), 3.64 (s, 3H), 3.41–3.10 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 175.32, 160.75, 145.80, 136.47, 136.40, 129.04, 127.52, 127.40, 126.87, 125.34, 124.09, 123.67, 123.13, 122.35, 119.72, 118.93, 111.40, 111.36, 61.28, 53.39, 52.06, 47.42, 29.59. IR (film) νmax: 3377, 2981, 2948, 2360, 1731, 1442, 1241, 739 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C23H23N3O3, 390.1818; found, 390.1817.
Methyl ((2-(Methylthio)quinolin-3-yl)methyl)-l-tryptophanate (19)
Using general procedure A, from 2-(methylthio)quinoline-3-carbaldehyde (3) (50 mg, 0.25 mmol) and l-tryptophan methyl ester, flash chromatography (SiO2, 1 × 10 cm, 8% EtOAc/hexane) provided 19 as a white solid (30 mg, 30%). 1H NMR (CDCl3, 400 MHz): δ 8.15 (s, 1H), 7.91 (d, J = 7.6, 1H), 7.70 (s, 1H), 7.62–7.57 (m, 2H),7.48 (d, J = 6.8, 1H), 7.40–7.36 (m, 2H), 7.20 (t, J = 8.0, 1H), 7.09–7.05 (m, 2H), 4.00–3.73 (m, 3H), 3.68 (s, 3H), 3.32–3.14 (m, 2H), 2.63 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 175.24, 159.19, 147.25, 136.36, 133.43, 130.80, 129.16, 127.66, 127.62, 127.58, 125.87, 125.21, 123.20, 122.32, 119.70, 118.97, 111.34, 61.32, 52.10, 48.33, 29.53, 13.04. IR (film) νmax: 3370, 2925, 2850, 1732, 1597, 1456, 1203, 1045, 742 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C23H23N3O2S, 406.1589; found, 406.1579.
Methyl ((2-Methoxyquinolin-3-yl)methyl)-l-tyrosinate (20)
Using general procedure A, from 2-methoxyquinoline-3-carbaldehyde (3) (50 mg, 0.27 mmol) and l-tyrosine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 10–40% EtOAc/hexane) provided 20 as a white solid (85 mg, 87%). 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.63 (d, J = 7.2 Hz, 1H), 7.58 (t, J = 5.6 Hz, 1H), 7.36 (t, J = 6.8 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.73 (d, J = 8.8 Hz, 2H), 3.97 (s, 3H), 3.95–3.75 (m, 2H), 3.62 (s, 3H), 3.50 (m, 1H), 3.00–2.84 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 174.98, 169.84, 154.96, 145.84.136.95, 139.52, 129.28, 128.96, 127.46, 126.82, 125.32, 124.28, 123.32, 115.64, 62.34, 53.70, 52.09, 47.40, 38.85. IR (film) νmax: 3271, 3140, 3083, 2917, 1726, 1630, 1573, 1512, 1399, 1260, 1169, 1011 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C21H22N2O4, 367.1658; found, 367.1661.
Methyl ((2-(Methylthio)quinolin-3-yl)methyl)-l-tyrosinate (22)
Using general procedure A, from 2-(methylthio)quinoline-3-carbaldehyde (3) (50 mg, 0.25 mmol) and l-tyrosine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 8% EtOAc/hexane) provided 22 as a white solid (27 mg, 29%). 1H NMR (DMSO-d6, 400 MHz): δ 9.27 (s, 1H), 7.87–7.84 (m, 2H), 7.72 (d, J = 6.8 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.46 (t, J = 6.4 Hz, 1H), 7.01 (d, J = 10 Hz, 2H), 6.68 (d, J = 8.8 Hz, 2H), 3.86–3.63 (m, 2H), 3.56 (s, 3H), 3.39 (m, 1H), 2.844–2.75 (m, 2H), 2.61 (s, 3H). 13C NMR (DMSO-d6, 100 MHz): δ 174.43, 158.40, 155.90, 146.23, 132.57, 131.44, 130.21, 129.15, 127.87, 127.60, 127.03, 125.44, 125.24, 114.92, 62.45, 51.34, 46.48, 38.05, 12.28. IR (film) νmax: 3321, 2981, 2360, 1724, 1512, 1399, 1166, 747 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C21H22N2O3S, 383.1429; found, 383.1421.
Methyl ((2-Methoxyquinolin-3-yl)methyl)-d-histidinate (23)
Using general procedure A, from 2-methoxyquinoline-3-carbaldehyde (3) (50 mg, 0.27 mmol) and d-histidine methyl ester, flash chromatography (SiO2, 1 × 10 cm, 0–5% CH3OH/CH2Cl2) provided 23 as a white solid (46 mg, 51%). 1H NMR (CDCl3, 400 MHz): δ 7.83–7.81 (m, 2H), 7.66 (d, J = 6.8 Hz, 1H), 7.58 (t, J = 6.0 Hz, 1H), 7.53 (s, 1H), 7.35 (t, J = 6.0 Hz, 1H), 6.80 (s, 1H), 4.08 (s, 3H), 3.87–3.80 (m, 2H), 3.63 (s, 3H), 3.60–3.57 (m, 1H), 3.07–2.84 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 174.59, 160.80, 146.00, 137.22, 134.78, 129.36, 127.38, 127.00, 125.31, 124.36, 123.32, 60.85, 53.70, 52.22, 47.71, 29.64. IR (film) νmax: 2948, 2886, 2360, 2343, 1735, 1474, 1443, 1399, 1258, 1010, 758 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C18H20N4O3, 341.1614; found, 341.1623.
Methyl ((2-(Benzyloxy)quinolin-3-yl)methyl)-l-valinate (15)
2-(benzyloxy)quinoline-3-carbaldehyde 4 (27.8 mg, 0.106 mmol) and l-valine methyl ester hydrochloride (31.3 mg, 0.187 mmol) were dissolved in tetrahydrofuran (3 mL, 0.035 M) and stirred for 24 h under an inert atmosphere and anhydrous conditions. Sodium borohydride (17.6 mg, 0.465 mmol) was added, and the reaction was stirred for 1.5 h. Tetrahydrofuran was evaporated under reduced pressure, and the reaction mixture was dissolved in ethyl acetate (15 mL). The organic layer was washed with DI water (3 × 20 mL). The organic layer was dried with Na2SO4 and concentrated under reduced pressure. Flash chromatography (SiO2, 1 × 10 cm, 5% EtOAc/hexanes gradient elution) afforded 15 (25.5 mg, 63%) as an off-white solid. 1H NMR (CDCl3, 400 MHz): δ 7.99 (s, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.42–7.32 (m, 4H), 5.60 (s, 2H), 3.94–3.78 (m, 2H), 3.54 (s, 3H), 3.06 (d, J = 5.6 Hz, 1H), 1.98–1.90 (m, 1H). 0.96–0.80 (m, 6H). 13C NMR (CDCl3, 100 MHz): δ 175.62, 160.37, 145.83, 137.64, 136.79, 129.16, 128.62, 128.27, 127.95, 127.41, 127.06, 125.58, 124.40, 124.31, 110.23, 67.83, 67.25, 51.58, 48.02, 31.99, 29.91, 19.42, 18.91. IR (film) νmax: 2958, 2929, 2855, 1734, 1625, 1507, 1457, 1419, 1345, 1260 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C23H26N2O3, 379.2022; found, 379.203.
Methyl ((2-(Benzyloxy)quinolin-3-yl)methyl)-l-tyrosinate (21)
2-(Benzyloxy)quinoline-3-carbaldehyde 4 (31.5 mg, 0.120 mmol) and l-tyrosine methyl ester hydrochloride (40.2 mg, 0.173 mmol) were dissolved in tetrahydrofuran (3 mL, 0.040 M) and stirred for 24 h under an inert atmosphere and anhydrous conditions. Sodium borohydride (19.8 mg, 0.523 mmol) was added, and the reaction was stirred for 1.5 h. Tetrahydrofuran was evaporated under reduced pressure, and the reaction mixture was dissolved in ethyl acetate (15 mL). The organic layer was washed with DI water (3 × 20 mL). The organic layer was dried with Na2SO4 and concentrated under reduced pressure, affording 21 (36.1 mg, 68%) as an off-white solid. 1H NMR (CDCl3, 400 MHz): δ 8.03 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 8 Hz, 1H), 7.52 (d, J = 7.4 Hz, 2H), 7.43–7.33 (m, 4H), 7.05 (d, J = 8.4 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 5.62 (s, 2H), 4.83 (s, 2H), 3.74–3.65 (m, 4H), 3.05–2.79 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 175.72, 159.82, 154.77, 137.29, 136.01, 130.63, 129.53, 128.77, 128.29, 128.20, 127.65, 127.15, 125.54, 125.03, 124.59, 115.67, 67.97, 61.56, 56.03, 40.25, 29.93. IR (film) νmax: 3293, 2954, 2925, 2853, 1734, 1515, 1457, 1421, 1343, 1259, 1109 cm–1. HRMS (ESI) m/z: [M + H]+ calcd for C27H26N2O4, 443.1971; found, 443.1964.
Biological Evaluation
All compounds evaluated in biological assays were >95% pure via 1H NMR.
General Sterilization Procedure
The following are general steps, unless otherwise noted. All steps were completed with aseptic techniques. All media and glassware were sterilized via autoclave at 121 °C for 60 min. All agitation occurred at 160 rpm in a temperature-controlled console shaker (Excella E25) at 37 °C. Full strength tryptic soy broth (TSB) was made by dissolving 30 g of BD Bacto TSB powder in 1 L of DI water. Purchased and acquired bacterial strains used were S. aureus (SA, ATCC 29213), P. aeruginosa (PA, ATCC 9027), and P. aeruginosa efflux knockout GKCW120 (PΔ6)31 provided by Zgurskaya and co-workers at the University of Oklahoma.
Antimicrobial Susceptibility Assay Procedure
Susceptibility testing was performed in biological triplicate, using the broth microdilution method as outlined by the Clinical and Laboratory Standards Institute. Briefly, MIC determinations were carried out in 96-well microtiter plates with two-fold serial dilutions of the compounds from 0 to 256 μg/mL (final assay concentrations) in dimethyl sulfoxide (DMSO). Briefly, to each well, 1 μL of compound in DMSO, 89 μL of TSB, and 10 μL of bacterial inoculum, grown from a single colony in 10 mL of TSB for 4–6 h, were added. After incubation for 12–15 h at 37 °C, absorbance at 590 nm was read on a Biotek Synergy HTX Multi-mode plate reader. Data were processed by background subtracting the media absorbance and then normalizing the data to full bacterial growth with only vehicle. MIC is defined as the lowest concentration of antibiotic or antibiotic/adjuvant combination that achieves ≥85% growth inhibition, which corresponds to no visible growth.
Adjuvant Assay Procedure with Pentamidine
Growth inhibition was determined by broth microdilution according to the CLSI M100-S23 guidelines. Master plates were prepared for both pentamidine (50 and 100 μM in DMSO) and evaluated compounds (0 μg/mL to 128 μg/mL in DMSO). Liquid cultures of PA were grown for 4–6 h by inoculating a single bacterial culture into 10 mL of TSB. In each well of rows A–G, 178 mL of full-strength TSB, 20 μL of PA culture, and 1 μL from each master plate were added. Controls for each assay included pentamidine only (H1–H3), vehicle (DMSO) in PA (H4–H6), and media only (H7–H12). Plates were covered and incubated at 37 °C for 12–18 h. Optical density (OD590) was measured on a Biotek Synergy HTX-Multimode plate reader, and data were processed by background subtracting the media absorbance (H7–H12) and then normalizing the data to full bacteria growth with only vehicle (H4–H6). MIC is defined as the lowest concentration of antibiotic or antibiotic/adjuvant combination that achieves ≥85% growth inhibition.
Preparation of Inverted Membrane Vesicles
PA cultures were scaled up to 1 L LB broth grown for 8 h at 37 °C with shaking. Cells were harvested by centrifugation, resuspended in TMG buffer (50 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 10% (v/v) glycerol) including 1 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride, and lysed by four passages through an Avestin B-15 homogenizer operating at 19,000 psi. The lysate was cleared by centrifugation at 9000g for 10 min, and the supernatant was further centrifuged at 193,000g to collect the membrane vesicles. Vesicles were resuspended in TMG buffer with a Dounce homogenizer and stored at −80 °C. The protein concentration in the vesicles was determined using modified Lowry assay.33
ATP Synthesis Assay
Measurement of ATP synthesis activity of PA vesicles in the presence of inhibitors was adapted from Hards et al.34 In a 96-well microplate, 2 μL of the test compound or DMSO alone was diluted into reaction buffer, which contained 5 mM Tricine, pH 8.0, 50 mM KCl, 2.5 mM MgCl2, 3.75 mM potassium phosphate, 0.1 mM adenosine diphosphate, and 2.5 mM reduced NADH at final dilution. The synthesis reaction was initiated by mixing in 50 μL of 0.2 mg/mL vesicles (200 μL of final reaction volume) and then incubated 10 min at room temperature. The reaction was stopped by diluting a 50 μL reaction sample into 1% trichloroacetic acid, 10 mM ethylenediaminetetraacetic acid, and 2 mM carbonyl cyanide 3-chlorophenylhydrazone. From the stop mix, samples were diluted 100-fold in water to dilute test compounds and stop components, and ATP concentration was determined by luminescence using a luciferin–luciferase system (ATP Determination Kit, Molecular Probes). Luminescence was measured in an opaque white microplate using a BioTek Synergy H1 microplate reader. Data were normalized to DMSO control values, and dose–response curves were fitted using GraphPad Prism 9 to determine relative IC50 values.
Determination of Electron Transport Chain Activity
In a black 96-well microplate, PA vesicles were mixed with HMK buffer (50 mM HEPES-KOH, pH 7.5, 2.5 mM MgCl2, and 300 mM KCl) including 0.3 μg/mL 9-amino-6-chloro-2-methoxyacridine and 64 μg/mL test compound or an equivalent volume of DMSO. Fluorescence (λex = 415 nm; λem = 485 nm) was measured using a BioTek Synergy H1 microplate reader. A fluorescence baseline was measured for 30 s. The electron transport chain (ETC) was initiated by addition of NADH to 0.83 mM, and fluorescence was monitored for 90 s. Finally, nigericin was added to 0.5 μg/mL to equilibrate the proton gradient, and fluorescence return was monitored for 30 s.
Acknowledgments
The authors thank Dr. Helen Zgurskaya and co-workers at the University of Oklahoma for providing the PA GKCW120 (PΔ6) strain. The authors would like to acknowledge Rebecca Shaffer for preparation of PA membrane vesicles and all the UNC Asheville CHEM 312 students from Fall 2019, Fall 2020, and Fall 2021 who worked on projects related to this manuscript.
Glossary
Abbreviations
- PA
Pseudomonas aeruginosa
- MT
Mycobacterium tuberculosis
- SA
Staphylococcus aureus
- MS
methicillin-susceptible
- MDR
multi-drug resistant
- MIC
minimum inhibitory concentration
- IC50
inhibitory concentration of 50%
- SAR
structure activity relationship
- DMA
dimethyl amino
- DMSO
dimethyl sulfoxide
- DCM
dichloromethane
- THF
tetrahydrofuran
- TSB
tryptic soy broth
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03127.
Inactive compound data, control experiments, and data on antibiotic accumulation (PDF)
Author Contributions
A.J.E.B., L.L.C., S.A.D., D.C., R.S.M., D.J.P., O.L.P., A.B.R., M.L.R., and G.E.V. contributed equally and are listed in alphabetical order. J.F.C. synthesized compounds 4, 15, and 21. S.A.D. and D.C. synthesized compounds 5 and 6. D.J.P. and A.B.R. synthesized compounds 7 and 8. G.E.V., A.J.E.B., and L.L.C. synthesized compound 9. O.L.P. and R.S.M. synthesized compounds 10–12. A.L.W. synthesized compound 3, 14, 16–20, 22, and 23. M.L.R. and A.L.W. performed the antibiotic activity, adjuvant, and efflux assays. M.L.R. and P.R.S. performed the PA ATP synthase and ETC inhibition assays. P.R.S. and A.L.W. designed experiments and analyzed data. J.F.C., P.R.S., and A.L.W. wrote the manuscript and Supporting Information. All authors contributed to manuscript revision.
The authors would like to gratefully acknowledge the financial support from the Research Corporation for Science Advancement Cottrell Scholar Award (23975), the NIH NIAID grant (1R15AI163474-01), the North Carolina GlaxoSmithKline Foundation, and the University of North Carolina Asheville Department of Chemistry and Biochemistry.
The authors declare no competing financial interest.
Supplementary Material
References
- CDC . Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention, 2019, p 148. [Google Scholar]
- de Kraker M. E. A.; Stewardson A. J.; Harbarth S. Will 10 Million People Die a Year Due to Antimicrobial Resistance by 2050?. PLoS Med. 2016, 13, e1002184 10.1371/journal.pmed.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGowan A.; Macnaughton E. Antibiotic Resistance. Medicine 2017, 45, 622–628. 10.1016/j.mpmed.2017.07.006. [DOI] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Santajit S.; Indrawattana N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. 10.1155/2016/2475067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti M.; Vena A.; Croxatto A.; Righi E.; Guery B. How to Manage Pseudomonas Aeruginosa Infections. Drugs Context 2018, 7, 212527. 10.7573/dic.212527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. Platforms for Antibiotic Discovery. Nat. Rev. Drug Discovery 2013, 12, 371–387. 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Göhlmann H. W. H.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; et al. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium Tuberculosis. Science 2005, 307, 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W.; Davies K. M. Rotary ATPases: A New Twist to an Ancient Machine. Trends Biochem. Sci. 2016, 41, 106–116. 10.1016/j.tibs.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Allegretti M.; Klusch N.; Mills D. J.; Vonck J.; Kühlbrandt W.; Davies K. M. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature 2015, 521, 237–240. 10.1038/nature14185. [DOI] [PubMed] [Google Scholar]
- Zhou A.; Rohou A.; Schep D. G.; Bason J. V.; Montgomery M. G.; Walker J. E.; Grigorieff N.; Rubinstein J. L. Structure and Conformational States of the Bovine Mitochondrial ATP Synthase by Cryo-EM. Elife 2015, 4, e10180 10.7554/eLife.10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobti M.; Walshe J. L.; Ishmukhametov R.; Stewart A. G.. Visualizing Movements in E. Coli F1Fo ATP Synthase Indicates How the F1 and Fo Motors Are Coupled. 2019, bioRxiv:622084. [Google Scholar]
- Sobti A. G.; Ishmukhametov A.; Bouwer C.; Ayer J. C.; Suarna M.; Smith M.; Christie N. J.; Stocker R.; Duncan R.; Stewart T. M. Cryo-EM Reveals Distinct Conformations of E. Coli ATP Synthase on Exposure to ATP. eLife 2016, 8, e43864 10.7554/eLife.43864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H. Regulation and Function of Versatile Aerobic and Anaerobic Respiratory Metabolism in Pseudomonas Aeruginosa. Front. Microbiol. 2011, 2, 1–13. 10.3389/fmicb.2011.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss L.; Langer J. D.; Yildiz Ö.; Eckhardt-Strelau L.; Guillemont J. E. G.; Koul A.; Meier T. Structure of the mycobacterial ATP synthase F o rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C.; Preiss L.; Wang B.; Fu L.; Wen H.; Zhang X.; Cui H.; Meier T.; Yin D. Structural Simplification of Bedaquiline: The Discovery of 3-(4-(N,N-Dimethylaminomethyl)Phenyl)Quinoline-Derived Antitubercular Lead Compounds. ChemMedChem 2017, 12, 106–119. 10.1002/cmdc.201600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Courbon G. M.; Bueler S. A.; Mai J.; Liu J.; Rubinstein J. L. Structure of Mycobacterial ATP Synthase Bound to the Tuberculosis Drug Bedaquiline. Nature 2020, 589, 143–147. 10.1038/s41586-020-3004-3. [DOI] [PubMed] [Google Scholar]
- Balemans W.; Vranckx L.; Lounis N.; Pop O.; Guillemont J.; Vergauwen K.; Mol S.; Gilissen R.; Motte M.; Lançois D.; De Bolle M.; Bonroy K.; Lill H.; Andries K.; Bald D.; Koul A. Novel Antibiotics Targeting Respiratory ATP Synthesis in Gram-Positive Pathogenic Bacteria. Antimicrob. Agents Chemother. 2012, 56, 4131–4139. 10.1128/AAC.00273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zeng Y.; Sheng L.; Larson P.; Liu X.; Zou X.; Wang S.; Guo K.; Ma C.; Zhang G.; et al. A Cinchona Alkaloid Antibiotic That Appears to Target ATP Synthase in Streptococcus Pneumoniae. J. Med. Chem. 2019, 62, 2305–2332. 10.1021/acs.jmedchem.8b01353. [DOI] [PubMed] [Google Scholar]
- Lamontagne Boulet M.; Isabelle C.; Guay I.; Brouillette E.; Langlois J.-P.; Jacques P.-É.; Rodrigue S.; Brzezinski R.; Beauregard P. B.; Bouarab K.; et al. Tomatidine Is a Lead Antibiotic Molecule That Targets Staphylococcus Aureus ATP Synthase Subunit C. Antimicrob. Agents Chemother. 2018, 62, e02197-17 10.1128/AAC.02197-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumaillé P.; Dassonville-Klimpt A.; Peltier F.; Mullié C.; Andréjak C.; Da-Nascimento S.; Castelain S.; Sonnet P. Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs. Pharmaceuticals 2019, 12, 91. 10.3390/ph12020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard M.; Nøhr-Meldgaard K.; Bojer M. S.; Krogsgård Nielsen C.; Meyer R. L.; Slavetinsky C.; Peschel A.; Ingmer H. Inhibition of the ATP Synthase Eliminates the Intrinsic Resistance of Staphylococcus Aureus towards Polymyxins. mBio 2017, 8, e01114-17 10.1128/mBio.01114-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nøhr-Meldgaard K.; Ovsepian A.; Ingmer H.; Vestergaard M. Resveratrol Enhances the Efficacy of Aminoglycosides against Staphylococcus Aureus. Int. J. Antimicrob. Agents 2018, 52, 390–396. 10.1016/j.ijantimicag.2018.06.005. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Medina R.; Wright G. D. A. Venturicidin A, A Membrane-active Natural Product Inhibitor of ATP synthase Potentiates Aminoglycoside Antibiotics. Sci. Rep. 2020, 10, 8134. 10.1038/s41598-020-64756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgrom Y. M.; Duncan T. M. Complex effects of macrolide venturicidins on bacterial F-ATPases likely contribute to their action as antibiotic adjuvants. Sci. Rep. 2021, 11, 13631. 10.1038/s41598-021-93098-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri R. A.; Joshi P. P. A Green Approach for the Synthesis of 2,4-Dihydro-pyrimidinones Using PTSA. Chem. Sci. Trans. 2018, 7, 89–94. 10.7598/cst2018.1441. [DOI] [Google Scholar]
- Hamama W. S.; Ibrahim M. E.; Gooda A. A.; Zoorob H. H. Efficient Synthesis, Antimicrobial, Antioxidant Assessments and Geometric Optimization Calculations of Azoles- Incorporating Quinoline Moiety. J. Heterocycl. Chem. 2018, 55, 2623–2634. 10.1002/jhet.3322. [DOI] [Google Scholar]
- Auld D. S.; Inglese J.. Interferences with Luciferase Reporter Enzymes. In Assay Guidance Manual [Internet]; Markossian S., Grossman A., Brimacombe K., Arkin M., Auld D., Austin C. P., Baell J., Chung T. D. Y., Coussens N. P., Dahlin J. L., Devanarayan V., Foley T. L., Glicksman M., Hall M. D., Haas J. V., Hoare S. R. J., Inglese J., Iversen P. W., Kales S. C., Lal-Nag M., Li Z., McGee J., McManus O., Riss T., Saradjian P., Sittampalam G. S., Tarselli M., Trask O. J., Wang Y., Weidner J. R., Wildey M. J., Wilson K., Xia M., Xu X.; ed. ; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda (MD), Jul 1, 2016. [updated 2018 Jul 1]. [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter S. J.; Geddes E. J.; Drown B. S.; Motika S. E.; Lee M. R.; Hergenrother P. J. Compound Uptake into E. Coli Can Be Facilitated by N-Alkyl Guanidiniums and Pyridiniums. ACS Infect. Dis. 2021, 7, 162–173. 10.1021/acsinfecdis.0c00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S. J.; Krishnamoorthy G.; Wolloscheck D.; Walker J. K.; Rybenkov V. V.; Parks J. M.; Zgurskaya H. I. Molecular Properties That Define the Activities of Antibiotics in Escherichia Coli and Pseudomonas Aeruginosa. ACS Infect. Dis. 2018, 4, 1223–1234. 10.1021/acsinfecdis.8b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Xia L.; Huang W.; Xu Y.; Gu Y.; Liu C.; Ji L.; Li W.; Wu Y.; Zhou K.; Feng X. Pentamidine Sensitizes FDA-Approved Non-Antibiotics for the Inhibition of Multidrug-Resistant Gram-Negative Pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1771–1779. 10.1007/s10096-020-03881-0. [DOI] [PubMed] [Google Scholar]
- Fillingame R. Identification of the dicyclohexylcarbodiimide-reactive protein component of the adenosine 5’-triphosphate energy-transducing system of Escherichia coli. J. Bacteriol. 1975, 124, 870–883. 10.1128/jb.124.2.870-883.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hards K.; Cheung C. Y.; Waller N.; Adolph C.; Keighley L.; Tee Z.; Harold L. K.; Menorca A.; Bujaroski R. S.; Buckley B. J.; et al. An amiloride derivative is active against the F1Fo-ATP synthase and cytochrome bd oxidase of Mycobacterium tuberculosis. Commun. Biol. 2022, 5, 166. 10.1038/s42003-022-03110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.