

Abstract
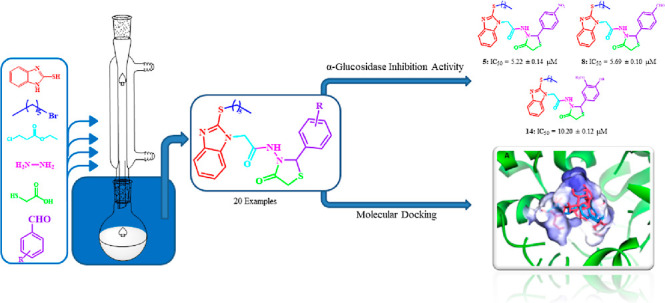
In this research work, we have focused our efforts to synthesize a series of 2-mercaptobenzimidazole-based 1,3-thiazolidin-4-ones (5–24) following a multistep reaction strategy and characterization of the synthesized derivatives with the help of various spectroscopic techniques. To find the antidiabetic potentials of the synthesized compounds (5–24), in vitro alpha-glucosidase inhibitory activity was performed using acarbose (IC50 = 873 ± 1.2 μM) as the reference standard. The results of the antidiabetic assay were very encouraging because compounds 5, 8, and 14 showed excellent inhibitions with IC50 values of 5.22 ± 0.14, 5.69 ± 0.10, and 10.20 ± 0.12 μM, respectively. The experimental results of anti-alpha-glucosidase activity prompted us to investigate and propose a possible mechanism of how the active molecules will interact with the target enzyme. For this purpose, molecular docking with the AutoDock Vina (an open-source and reliable docking platform) gave us an insight into the binding interactions of the active compounds to different amino acid residues of the enzyme.
Introduction
Small heterocycles have been a source of great fascination for organic and medicinal chemists because of their structural and therapeutic diversity. Benzimidazole is one of the most important bioactive aromatic heterocycles and is found naturally in the form of N-ribosyl-dimethylbenzimidazole in vitamin B-12. This nucleus has largely been employed in designing drugs and their synthesis.1,2 In recent times, through de novo transcriptome and gene analysis, a series of new derivatives of benzimidazole were found active against microbes (specifically Staphylococcus aureus).3 Benzimidazole-2-thiol (B2T) is among the most puissant derivatives of benzimidazole and is an honored structure in medicinal and organic chemistry.4 A recent study reported a series of amide derivatives of B2T. The synthesized compounds were found active against numerous strains of microorganisms such as Escherichia coli, Candida albicans, and S. aureus and cytotoxic against human colorectal cell line (HCT116).5 Similarly, another study also reported high anticonvulsant activity of B2T derivatives.6 In another report, antimicrobial and antiproliferative activities enhancement was also recorded through the presence of α-bromophenyallylidene pharmacophore in B2T imines.7
Research studies have shown medicinal properties of structures such as B2T acyclic nucleosides as antibacterial agents8 and aminoacetylenic-5-ethoxy-2-mercaptobenzimidazoles as antifungal agents.9 Furthermore, B2T-based thiazolidinone and isoxazole heterocycles as effective anti-inflammatory, anticonvulsant, and analgesic agents,10 triazolylacetohydrazides as an anti-inflammatory, analgesic, and antimicrobial agents,11 afobazole as an effective anxiolytic drug,12 and oxadiazoles as antidiabetic agents.13
Recently, 4-thiazolidinones have been among the most thoroughly investigated organic compounds and a variety of biological activities have been reported for their derivatives. In nature, the presence of the thiazolidine ring in penicillin and its related derivatives was the first recognition of its medicinal characteristics.14 It has some very fascinating activity profiles similar to COX-1 inhibitors,15 inhibitors of the bacterial enzyme MurB, a precursor acting during the biosynthesis of peptidoglycan,16 non-nucleoside inhibitors of HIV-RT,17 and anti-histaminic agents.18 Some derivatives such as chromenyl-based 2-imino thiazolidin-4-one derivatives acted as tubulin polymerization inhibitors and were evaluated for their in vitro cytotoxicity against HepG2 (liver cancer), A549 (lung cancer), MDA-MB-231 and BT471 (breast cancer), and HCT-116 (colon cancer) cell lines by the MTT assay.19 An innovative series of 5-benzylidene-2-arylimino-4-thiazolidinone derivatives consisting of a phenylaminopyrimidine core were tested for their anticancer activity on K562 (chronic myeloid leukemia), PC3 (prostate cancer), and SHSY-5Y (neuroblastoma) cells.20 4-Chloro-2-(2-fluorophenoxy)-N-(4-oxo-2-(p-tolyl)thiazolidin-3-yl)benzamide exhibited considerable benzodiazepine agonists, possessing anticonvulsant effects.21 Thiazolidinone-based compounds were proved to be promising drugs and acted as single-targeted and multi-targeted agents for controlling hyperglycemia in patients suffering from type 2 diabetes mellitus (DM) and associated complications by evaluating their inhibitory effects against PPA and α-glucosidase.
Nowadays, molecular hybridization methodologies are used as an effective tool for the design and development of new chemical entities with enhanced pharmacological activities in drug discovery research. Keeping in view, the outstanding pharmacological activities of 2-mercaptobenzimidazole and thiazolidinone scaffolds, we synthesized B2T-based thiazolidin-4-one derivatives and speculated further boasting of activities from 2-mercaptobenzimidazole-thiazolidinone hybrids.
Results and Discussion
Chemistry
To synthesize thiazolidinone derivatives of benzimidazole-2-thiol, it was S-alkylated with bromohexane followed by esterification with chloroethylacetate and then hydrazinated with hydrazine hydrate. Acetohydrazide obtained was treated with different aromatic aldehydes to afford a series of Schiff’s bases that on cyclocondensation with mercaptoacetic acid in the presence of anhydrous ZnCl2 as a catalyst gave a series of thiazolidinones (5–24). The structures of the products (Scheme 1) were inferred from the spectral data (Table 1).
Scheme 1. Synthetic Pathway for Compounds (1–24).
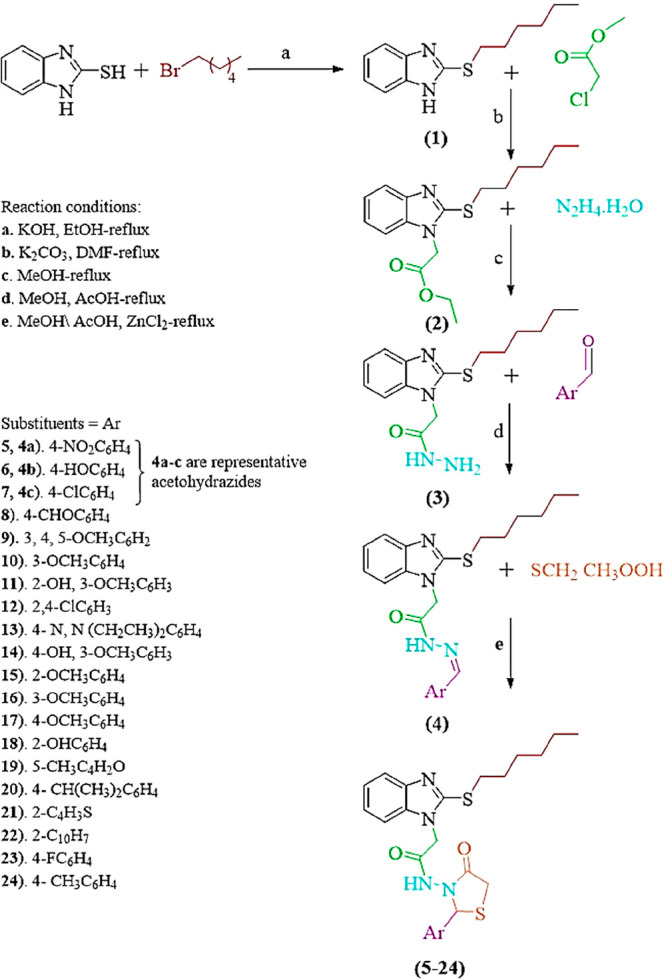
Table 1. In Vitro α-Glucosidase Inhibition Activity of the Synthesized Compounds (5–24).
| compounds | substituents attached | IC50 ± μM (SEM) |
|---|---|---|
| 5 | 4-NO2 C6H4 | 5.22 ± 0.14 |
| 6 | 4-OH C6H4 | N/Aa |
| 7 | 4-Cl C6H4 | N/A |
| 8 | 4-CHO C6H4 | 5.69 ± 0.10 |
| 9 | 3,4,5-OCH3C6H2 | N/A |
| 10 | 3-OHC6H4 | N/A |
| 11 | 2-OH,3-OCH3C6H3 | 24.10 ± 0.30 |
| 12 | 2,4-ClC6H3 | 33.39 ± 0.38 |
| 13 | 4-N,N(CH2CH3)2C6H4 | 189.89 ± 0.53 |
| 14 | 4-OH,3-OCH3C6H3 | 10.20 ± 0.12 |
| 15 | 2-OCH3C6H4 | 89.28 ± 0.52 |
| 16 | 3-OCH3C6H4 | 71.64 ± 0.58 |
| 17 | 4-OCH3C6H4 | 63.68 ± 0.52 |
| 18 | 2-OHC6H4 | N/A |
| 19 | 5-CH3C4H2O | 39.06 ± 0.27 |
| 20 | 4-CH(CH3)2C6H4 | 65 ± 1.55 |
| 21 | 2-C4H2S | 54.43 ± 0.65 |
| 22 | 2-C10H7 | 41.00 ± 0.36 |
| 23 | 4-FC6H4 | 26.38 ± 0.18 |
| 24 | 4-CH3C6H4 | 23.75 ± 0.25 |
| acarbose | standard | 873 ± 1.2 μM |
N/A (not active).
1H NMR spectrum of compound 1 exhibited broad singlets at δ 0.81–0.94 ppm and δ 3.24–3.30 ppm due to methyl (CH3CH2−) and methylene (−SCH2CH2−) protons, respectively. A multiplet of eight protons [8H, −(CH2)4−] at δ 1.24–1.83 ppm in 1HNMR spectra and carbon peaks in the range of δ 14.1–38.4 ppm in 13C NMR spectra represented the aliphatic chain, and therefore, confirming S-alkylation of benzimidazole-2-thiol.
In compound 2, protons of the two methylene groups (−NCH2CO– and −COOCH2CH3) resonated as a singlet at δ 4.85 ppm, and a quartet at δ 4.16 ppm (J = 7.6 Hz, 2H), respectively. A triplet of three protons at δ 1.21 ppm represented methyl protons of the ester (J = 7.6 Hz, −CH2CH3). The 13CNMR spectrum exhibited a carbon peak at δ 160 ppm corresponding to CO-acyclic, and therefore, showing the formation of the ester.
Two singlets appearing at δ 8.50 (−N=CH) and δ 4.10 ppm (HN–NH2) in 1H NMR spectra and a carbon peak at δ 162 ppm in 13C NMR spectra indicated the amide linkage (−COHN−) of compound 3.
1H NMR spectra of compound 4 not only showed the absence of NH2 protons but also the presence of −N=CH proton (azomethine) at δ 8.85 ppm. The carbon signal at δ 145.1 ppm in 13CNMR spectra for −N=CH–Ar indicated hydrazone formation.
Formation of N-(2-(substituted benzylidene)-4-oxothiazolidin-3-yl-2-(2-(hexylthio)-1H-benzo[d]imidazole-1-yl) acetamides (5–24) was confirmed from spectral data. Characteristic 1H NMR peaks for compounds 5–24 included two singlets for methylene protons (−COCH2S) and CH proton (−SCHN) at δ 3.65–3.90 and δ 4.90–5.28 ppm of the 4-thiazolidinone ring, respectively. In 13CNMR analysis, two signals for methylene and methine of the thiazolidinone ring appeared at around δ 40 and δ 65–84 ppm, respectively. The signals for aromatic carbons resonated at δ 110–153 ppm. The appearance of carbon peaks at δ 160–164 and δ 170–175 ppm confirmed the presence of carbonyl carbons both CO-acyclic and CO-cyclic, respectively.
The synthesized compounds were purified using different purification techniques and purity was confirmed through TLC performed in n-hexane:ethyl acetate solvent system and the spots visualized under UV light. The structures of the products formed were confirmed with the help of spectroscopic techniques such as HR–MS (ESI), 1H NMR, and 13C NMR.
α-Glucosidase Inhibitory Activity of the Synthesized Compounds (5–24)
The synthesized compounds were evaluated for their α-glucosidase inhibitory activity in comparison to standard acarbose. IC50 values are also shown in Table 1. Solutions of the synthesized compounds at various concentrations were prepared, and their α-glucosidase inhibitory effects were evaluated in comparison to acarbose (IC50 = 873 ± 1.2 μM) which is a standard α-glucosidase inhibitor.22 Compounds 5–24 showed variable degrees of α-glucosidase inhibitory potential compared to the standard. The IC50 values shown by compounds 5–24 ranged from 5.22 ± 0.14 to 189.89 ± 0.53 μM. Among others in the series, the three most active compounds 5 (IC50 = 5.22 ± 0.14 μM), 8 (IC50 = 5.69 ± 0.10 μM), and 14 (IC50 = 10.20 ± 0.12 μM) demonstrated significantly high degrees of inhibition against α-glucosidase enzyme even superior to the standard acarbose.
Molecular Modeling Studies
Homology Modeling of Alpha-Glucosidase
The synthesized compounds were evaluated against alpha-glucosidase from Saccharomyces cerevisiae (Baker’s yeast). So far, the crystal structure for alpha-glucosidase is not known yet. For the purpose to perform molecular docking studies of synthesized compounds, the crystal structure of S. cerevisiae α-glucosidase was built by using the Modeller 10.1 software package (http://salilab.org/modeller/). In FASTA format, the sequence of the target was fetched directly from UniProt (code p53341). The target sequence was used to BLAST search the PDB for standard structures that were used as templates for homology/comparative modeling, that is, as a template sequence, the crystallographic structure of isomaltase from S. cerevisiae with PDB ID: 3AJ7 and having similarity of 72.4% was selected. Among the five models, the top model was chosen and modification of the constructed model (97.3% amino acids were positioned in favored regions, 1.7% in allowed regions, and 1.0% in outlier region) was confirmed by the Ramachandran plot (Figure 1).23 ERRAT was also used for assessment of the prepared model24 using QMEAN with Z-score for energy minimization (Qualitative Model Energy Analysis, https://swissmodel.expasy.org/qmean/).25
Figure 1.

Ramachandran plot of homology modeled α-glucosidase generated by Rampage.
Molecular Docking
Molecular docking is the computational study of searching for an appropriate ligand that fits both geometrically and energetically into the active site of a protein. Different software packages can be used for performing docking studies such as GOLD, Autodock Vina, MOE, Glide, and so forth. Autodock Vina in USCF Chimera was used to carry out molecular docking studies of our synthesized compounds against homology modeled alpha-glucosidase. The active site of receptor protein was found by selecting residues of the catalytic triad. Among the synthesized structures/compounds (5–24), only compounds 5, 8, and 14 were tested in silico for their binding toward determined as the α-glucosidase. 3D studies of the highest-scoring complexes gave an insight into the mechanism of deactivation/inhibition for the three most active compounds 5, 8, and 14.
Compound 5 exhibited a very good accommodation of the docked ligand in the active gorge of homology modeled α-glucosidase with a Vina score of −8.7. Various non-bonding interactions can be seen between the docked ligand and active site residues upon visual analysis. These interactions include pi-sulfur with Phe167, pi-donor hydrogen bonding with His279, conventional H-bond with His239 and Arg312, carbon-H bond with Arg349, and pi–sigma interaction with Tyr71, as shown in Figure 2.
Figure 2.

(A) Superimposition of 5 (pink stick model) on acarbose (sky blue stick model). (B) Stereoview of ligand–receptor interaction. (C) 2D diagram of ligand 5 interactions with amino acid residues. (D) Close-up view of the highest-scoring pose of 5 (pink stick model).
Compound 8 formed a stable receptor–ligand complex with a Vina score of −8.8. To evaluate the occupancy of the docked ligand in the active pocket of the enzyme, the complex was further analyzed. The best-docked pose of 8 showed different interactions with four amino acid residues of the target enzyme. The docked ligand can be witnessed for its pi–sigma interaction with Ala278 and Tyr71, pi–sulfur with His239, and conventional H-bond with Arg312, as shown in Figure 3.
Figure 3.

(A) Superimposition of 8 (purple stick model) on acarbose (light green stick model). (B) Stereoview of ligand–receptor interaction. (C) 2D diagram of ligand 8 interactions with amino acid residues. (D) Close-up view of the highest-scoring pose of 8 (purple stick model).
Compound 14 occupied the active cavity of the receptor protein with a docking score of −8.2. Analysis of the best docked pose for compound 14 displayed interactions with several amino acid residues such as carbon H-bond with Asp349 and pi–anion interaction with Asp408. His239 is involved in the pi–sulfur interaction. Ala278 and Tyr71 show pi–sigma interactions, as shown in Figure 4.
Figure 4.

(A) Superimposition of 14 (dark green stick model) on acarbose (pink stick model). (B) Stereoview of ligand–receptor interaction. (C) 2D diagram of ligand 14 interactions with amino acid residues. (D) Close-up view of the highest-scoring pose of 14 (dark green stick model).
Experimental Section
General
Solvents and chemicals of synthetic grade were obtained from BDH, Daejung, Alfa Aesar, Merck, and Sigma-Aldrich. The Silica-gel glass plates with dimensions 3 × 8 cm (Merck) were employed for the TLC profile study under UV lamps. 1H NMR and 13C NMR spectra were recorded in ppm via a Bruker AVANCE-400 spectrometer (400 MHz, Made in Germany) in DMSO-d6 with TMS as internal standard. The HR-ESIMS spectra were recorded on JEOL JMS600 (made in USA).
Synthesis of 2-(Hexylthio)-1H-benzo[d]imidazole (1)
2-Mercaptobenzimidazole (33.3 mmol, 5 g) was added to 50 mL of ethanol which was made alkaline by adding KOH (33.3 mmol, 1.86 g). Then, 33.3 mmol (5.50 g, 5.0 mL) of bromohexane was added dropwise to the reaction mixture and refluxed for about 7 h. After completion, the reaction mixture was filtered. The filtrate was cooled, and the solvent (ethanol) was evaporated. The white, shiny needle-like crystals of 2-(hexylthio)-1H-benzo[d]imidazole formed were and recrystallized from ethanol with mp 112–114 °C.26
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.27–1.38) m, overlapped peaks, 6H, −CH2–CH2–(CH2)2–, (1.67) p, J = 4.8 Hz, 2H, −CH2–CH2–CH2–, (3.21) broad singlet, 2H, −S–CH2–CH2–, (7.27–7.50) dd, J = 4.8 Hz, 4H, Ar–H, (7.16) t, J = 2.4 Hz, 2H, Ar–H, (12.17) s, 1H, −CNHC–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.7 (−CH2−), 29.0 (−CH2−), 30.7 (−CH2−), 32.1 (−CH2S−), 152.0 (−SNCN−), 142.8 (−NCCH−), 137.0 (−NCCH−), 121.5 (2CH–Ar), 117.6 (CH–Ar),109.6 (CH–Ar).
HR-MS (ESI) (m/z) [M + H]+: 235.1208, mass calcd for [C13H19N2S]+, 235.1623; found, 235.1208.
Synthesis of Ethyl-2-[2-(hexylthio)-1H-benzo[d]imidazole-1-yl]acetate (2)
The thioether, 2-(hexylthio)-1H-benzo[d]imidazole 1 (25.9 mmol, 5.70 g), and anhydrous K2CO3 (25.9 mmol, 3.57 g) were added to 20 mL of DMF and agitated. Chloroethylacetate (25.9 mmol, 3.17 g, 3 mL) was added dropwise and refluxed at ambient temperature for 10 h. The oily filtrate obtained was condensed over a water bath.27
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.25–1.38) m, overlapped peaks, 6H, −CH2–CH2–(CH2)2–, (1.67) p, J = 4.8 Hz, 2H, −CH2–CH2–CH2–, (3.21) broad singlet, 2H, −S–CH2–CH2–, (7.47–7.55) dd, J = 4.8 Hz, 2H, Ar–H, (7.16) t, J = 2.4 Hz, 2H, Ar–H, (5.07) s, 2H, −NCH2CO, (4.17) q, J = 4.8 Hz, 2H, −CH2–CH3, (1.20) t, J = 4.8 Hz, 3H, −CH2–CH3. 13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.7 (−CH2−), 29.0 (−CH2−), 30.7 (−CH2−), 32.1 (−CH2S−), 151.8 (−SNCN−), 142.8 (−NCCH−), 136.4 (−NCCH−), 121.8 (2CH–Ar), 117.6 (CH–Ar),109.6 (CH–Ar), 44.7 (−NCH2CO–Aliphatic), 167.6 (−NCH2CO–Aliphatic), 61.3 (−OCH2−), 14.0 (−CH3).
HR-MS (ESI) (m/z) [M + H]+: 321.1608, mass calcd for [C17H25N2O2S]+, 321.1631; found, 321.1608.
Synthesis of 2-(2-Hexylthio)-1H-benzo[d]imidazole-1-yl)acetohydrazide (3)
Ethyl-2-(2-(hexylthio)-1H-benzo[d]imidazole-1-yl) acetate 2 (36 mmol, 11 g) was taken in methanol and stirred for 10 min. N2H4·H2O (36 mmol, 1.8 g, 2.0 mL) was added dropwise into the reaction mixture and refluxed for 10 h at 80 °C. The reaction mixture was decanted into distilled ice-cooled water. The precipitated product was collected by filtration, washed with water, and dried in the open air. It was further recrystallized from ethanol.28 Off-white crystalline solid, mp 116–118 °C (yield 90%) was obtained.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.26–1.38) m, overlapped peaks, 6H, −CH2–CH2–(CH2)2–, (1.70) t, J = 4.8 Hz, 2H, −CH2–CH2–CH2–, (3.33) broad singlet, 2H, −S–CH2–CH2–, (7.37–7.53) dd, J = 4.8 Hz, 2H, Ar–H, (7.14) d, J = 4.4 Hz, 2H, Ar–H, (4.74) s, 2H, −NCH2CO, (9.47) s, 1H, −NH–NH2, (4.33) s, 1H, −NH–NH2.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.7 (−CH2−), 29.0 (−CH2−), 30.7 (−CH2−), 32.1 (−CH2S−), 152.0 (−SNCN−), 142.9 (−NCCH−), 136.6 (−NCCH−), 121.5 (2CH–Ar), 117.5 (CH–Ar),109.6 (CH–Ar), 44.6 (−NCH2CO–Aliphatic), 165.4 (−NCH2CO–Aliphatic).
HR-MS (ESI) (m/z) [M + H]+: 307.1510, mass calcd for [C15H23N4OS]+, 307.1587; found, 307.1510.
Synthesis of N-(Substituted benzylidene)-2-(2-(hexylthio)-1H-benzo[d]imidazole-1-yl) Acetohydrazides (4)
A mixture of equimolar acyl hydrazide 3 (163 mmol, 0.50 g) and aromatic substituted benzaldehyde (163 mmol) was taken in methanol (30 mL). A catalytic amount of glacial acetic acid was added and refluxed for 3–4 h. The hot reaction mixture was decanted into distilled ice-cold water. The product precipitated was filtered and recrystallized from ethanol.29
Physical characteristics and NMR data (both 1H and 13C) of some of the representative acetohydrazides (4a and 4b) are given below.
(E)-2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N′-(4-hydroxybenzylidene) Acetohydrazide (4a)
White amorphous solid; yield: 75%; mp 178–180 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.27–1.88) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.30) broad singlet, 2H, −S–CH2–CH2–, (7.45–7.58) m, overlapped peaks, 4H, Ar–H, (4.91) s, 2H, −NCH2CO–, (9.99) s, 1H, −CONHN–, (8.13) s, 1H, Ar–CH=N–, azomethine, (6.81–7.15) dd, J = 6.8, 5.6 Hz, 4H, Ar–H, (9.92) s, 1H, OH–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.7 (−CH2−), 29.0 (−CH2−), 30.8 (−CH2−), 32.1 (−CH2S−), 152.0 (−SNCN−), 142.5 (−NCCH−), 134.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 44.5 (−NCH2CO–Aliphatic), 159.3 (−NCH2CO–Aliphatic), 144.9 (−N=CH–Ar, azomethine carbon), 135.3 (C–Ar), 146.9 (OH–C–Ar), 128.8 (2CH–Ar), 117.7 (2CH–Ar).
HR-MS (ESI) (m/z) [M + H]+: 411.2008, mass calcd for [C22H27N4O2S]+, 411.1849; found, 411.2008.
(E)-2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N′-(4-chlorobenzylidene) Acetohydrazide (4b)
Brown amorphous solid; yield: 75%; mp 160–162 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.83) broad singlet, 3H, −CH2CH3, (1.22–1.84) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.29) broad singlet, 2H, −S–CH2–CH2–, (7.47–7.55) m, overlapped peaks, 4H, Ar–H, (5.40) s, 2H, −NCH2CO–, (11.49) s, 1H, −CONHN–, (8.08) s, 1H, Ar–CH=N–, azomethine proton, (7.15–7.80) dd, J = 4.0, 8.0 Hz, 4H, Ar–H.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.7 (−CH2−), 29.0 (−CH2−), 30.8 (−CH2−), 32.1 (−CH2S−), 152.1 (−SNCN−), 137.0 (−NCCH−), 134.4 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 44.5 (−NCH2CO–Aliphatic), 167.7 (−NCH2CO–Aliphatic), 143.1 (−N=CH–Ar, azomethine carbon, 132.9 (C–Ar), 142.9 (Cl–C–Ar), 123.1 (2CH–Ar), 128.8 (2CH–Ar).
HR-MS (ESI) (m/z) [M + H]+: 429.1438, mass calcd for [C22H26ClN4OS]+, 429.1510; found, 429.1438.
Synthesis of 2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(substituted benzylidene)-4-oxothiazolidin-3-yl] Acetamides (5–24)
Acetohydrazides (0.22 mmol) and 0.1 g of anhydrous ZnCl2 (catalyst) was taken in 20 mL of methanol and stirred for 10 min. Then, freshly distilled mercaptoacetic acid (0.22 mol) was added and the mixture was heated under reflux from 10 to 12 h. The reaction mixture was then cooled to ambient temperature and neutralized with 5% sodium bicarbonate solution. The solid precipitate was filtered, washed, dried, and recrystallized from an appropriate solvent. The products formation was confirmed by TLC and chemical structures of the synthesized compounds were found by their HR-ESIMS, 1H NMR, and 13C NMR spectra.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-nitrophenyl)-4-oxothiazolidin-3-yl]acetamide (5)
Pale yellow amorphous solid; yield: 70%; mp 145–147 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.85) broad singlet, 3H, −CH2CH3, (1.33–1.62) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.26) broad singlet, 2H, −S–CH2–CH2–, (6.29–8.40) m, overlapped peaks, 8H, Ar–H, (4.31) s, 2H, −NCH2CO–, (10.01) s, 1H, −CONHN–, (4.78) s, 2H, −COCH2S–, (5.24) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 25.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.0.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 174.5 (−CO-cyclic), 35.6 (−COCH2S−), 76.6 (−SCHN−), 135.3 (C–Ar), 146.6 (NO2–C–Ar), 130.1 (2CH–Ar), 115.8 (2CH–Ar).
HR-MS (ESI) (m/z): 558.9307 [M + 2Na – H]+, mass calcd for [C24H27N5O4S2], 513.1505; found, 513.9596.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-hydroxyphenyl)-4-oxothiazolidin-3-yl] Acetamide (6)
Yellowish white amorphous solid; yield: 70%; mp 138–140 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.86) broad singlet, 3H, −CH2CH3, (1.27–1.88) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.30) broad singlet, 2H, −S–CH2–CH2–, (7.15–7.54) m, overlapped peaks, 8H, Ar–H, (4.32) s, 2H, −NCH2CO–, (9.99) s, 1H, −CONHN–, (3.64) s, 2H, −COCH2S–, (4.75) s, 1H, CH-cyclic, −SCHN–, (9.48) s, 1H, Ar–OH.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 25.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.0.7 (−CH2S−), 152.6 (−SNCN−), 142.9 (−NCCH−), 134.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 170.4 (−CO-cyclic), 35.6 (−COCH2S−), 87.6 (−SCHN−), 135.3 (C–Ar), 156.9 (OH–C–Ar), 130.1 (2CH–Ar), 115.8 (2CH–Ar).
HR-MS (ESI) (m/z): 504.2165 [M + NH4 + 2H]+3; mass calcd for [C24H28N4O3S2], 484.1608; found, 484.1827.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-chlorophenyl)-4-oxothiazolidin-3-yl]acetamide (7)
White amorphous solid; yield: 70%; mp 140–142 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.88) broad singlet, 3H, −CH2CH3, (1.33–1.84) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (7.14–7.47) m, overlapped peaks, 8H, Ar–H, (4.74) s, 2H, −NCH2CO–, (11.49) s, 1H, −CONHN–, (3.64) s, 2H, −COCH2S–, (5.20) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 25.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.9 (−CH2S−), 152.9 (−SNCN−), 142.9 (−NCCH−), 136.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.3 (−NCH2CO–Aliphatic), 166.2 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 45.0 (−COCH2S−), 63.7 (−SCHN−), 132.3 (C–Ar), 144.3 (Cl–C–Ar), 123.1 (2CH–Ar), 125.8 (2CH–Ar).
HR-MS (ESI) (m/z): 542.7307 [M + K + H]+2; mass calcd for [C24H27ClN4O2S2], 502.1264; found, 502.7502.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-formylphenyl)-4-oxothiazolidin-3-yl]acetamide (8)
White crystalline amorphous solid; yield:77%; mp 160–162 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.85) broad singlet, 3H, −CH2CH3, (1.33–1.62) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.26) broad singlet, 2H, −S–CH2–CH2–, (6.29–8.40) m, overlapped peaks, 8H, Ar–H, (4.28) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.40) s, 2H, −COCH2S–, (4.95) s, 1H, CH-cyclic, −SCHN–, (9.90) s, 1H, COH–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.0 (−CH2S−), 152.6 (−SNCN−), 142.9 (−NCCH−), 134.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 35.6 (−COCH2S−), 83.0 (−SCHN−), 135.3 (C–Ar), 145.0 (CAr–COH), 130.1 (2CH–Ar), 125.8 (2CH–Ar), 183.0 (CHO–Ar).
HR-MS (ESI) (m/z): 574.9091 [M + 2K]2+; mass calcd for [C25H28N4O3S2], 496.1602; found, 496.9828.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(3,4,5-trimethoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (9)
Off white amorphous solid; yield: 70%; mp 109–111 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.26–1.62) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.23) broad singlet, 2H, −S–CH2–CH2–, (7.11–7.51) m, overlapped peaks, 6H, Ar–H, (4.45) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.74) s, 2H, −COCH2S–, (5.94) s, 1H, CH-cyclic, −SCHN−), (3.62) s, 9H, 3OCH3–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 152.8 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 121.9 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.4 (−CO-cyclic), 35.6 (−COCH2S−), 82.0 (−SCHN−), 133.5. (C–Ar), 137.6, 56.1 (2OCH3–Ar), 60.8 (OCH3–Ar).
HR-MS (ESI) (m/z): 564.9981 [M + Li]+; mass calcd for [C27H34N4O5S2], 558.1970; found, 558.0571.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(3-hydroxyphenyl)-4-oxothiazolidin-3-yl]acetamide (10)
White amorphous solid; yield: 75%; mp 196–198 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, −CH2CH3, (1.26–1.68) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.23) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.50) m, overlapped peaks, 8H, Ar–H, (4.34) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (4.66) s, 2H, −COCH2S–, (4.96) s, 1H, CH-cyclic, −SCHN−), (8.47) s, 1H, OH–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0(−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 142.8 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 168.0 (−NCH2CO–Aliphatic), 175.0 (−CO-cyclic), 35.6 (−COCH2S−), 80.0 (−SCHN−), 140.6 (C–Ar), 156.9 (CAr–OH), 114.4 (2CH–Ar), 119.5 (CH–Ar), 130.0 (CH–Ar).
HR-MS (ESI) (m/z): 542.7310 [M + K + NH4 + H]3+; mass calcd for [C24H28N4O3S2], 484.1608; found, 484.7266.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[-2-(2-hydroxy-3-methoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (11)
Yellow amorphous solid; yield: 85%; mp 194–196 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.27–1.70) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.26) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.47) m, overlapped peaks, 7H, Ar–H, (4.54) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (4.30) s, 2H, −COCH2S–, (5.05) s, 1H, CH-cyclic, −SCHN−), (10.65) s, 1H, OH–Ar, (3.43) s, 3H, CH3O–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.0 (−CO-cyclic), 35.6 (−COCH2S−), 80.0 (−SCHN−), 140.6 (C–Ar), 147.9 (CAr–OH), 145.1 (CH3O–CAr), 121.4 (2CH–Ar), 110.2 (CH–Ar), 58.1 (OCH3–Ar).
HR-MS (ESI) (m/z): 532.7002 [M + NH4]+; mass calcd for [C25H30N4O4S2], 514.1708; found, 514.7664.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(2,4-dichlorophenyl)-4-oxothiazolidin-3-yl]acetamide (12)
White amorphous solid; yield: 80%; mp 144–146 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.85) broad singlet, 3H, −CH2CH3, (1.27–1.69) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.26) broad singlet, 2H, −S–CH2–CH2–, (7.08–7.45) m, overlapped peaks, 7H, Ar–H, (4.50) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (4.31) s, 2H, −COCH2S–, (4.85) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 166.4 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 35.6 (−COCH2S−), 80.0 (−SCHN−), 140.6 (C–Ar), 135.4 (CAr–Cl), 134.1 (CAr–Cl), 131.5 (2CH–Ar), 126.2 (CH–Ar).
HR-MS (ESI) (m/z): 542.9428 [M + Li]+; mass calcd for [C24H26Cl2N4O2S2], 536.0874; found, 536.0042.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-(diethylaminophenyl]-4-oxothiazolidin-3-yl]acetamide (13)
Orange–white amorphous solid; yield: 74%; mp 110–112 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.85) broad singlet, 3H, −CH2CH3, (1.11–1.57) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (6.01–7.51) m, overlapped peaks, 8H, Ar–H, (4.34) s, 2H, −NCH2CO–, (8.51) s, 1H, −CONHN–, (4.56) s, 2H, −COCH2S–, (4.94) s, 1H, CH-cyclic, −SCHN−), (1.11–1.57) m, 6H, N(CH2–CH3)2, (3.25) m, overlapped peaks, 4H, 2NCH2–CH3).
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 14.0 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.0 (−CO-cyclic), 35.6 (−COCH2S−), 80.0 (−SCHN−), 128.7 (C–Ar), 127.4 (2CH–Ar), 112.8 (2CH–Ar), 148.0 (ArC–N), 12.5 (2CH3), 48.1 (2CH2).
HR-MS (ESI) (m/z): 581.2722 [M + ACN + H]+; mass calcd for [C28H37N5O2S2]: 539.2388; found, 540.2388.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-hydroxy-3-methoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (14)
White crystalline solid; yield: 85%; mp 192–194 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.27–1.74) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (6.99–7.45) m, overlapped peaks, 7H, Ar–H, (4.43) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (4.79) s, 2H, −COCH2S–, (5.10) s, 1H, CH-cyclic, −SCHN–, (8.53) s, 1H, OH–Ar, (3.47) s, 3H, CH3O–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 42.0 (−CH2S−), 152.6 (−SNCN−), 136.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 35.6 (−COCH2S−), 84.0 (−SCHN−), 132.6 (C–Ar), 144.9 (CAr–OH), 138.1 (CH3O–CAr), 121.4 (2CH–Ar), 110.2 (CH–Ar), 63.1 (OCH3–Ar).
HR-MS (ESI) (m/z): 558.9367 [M + ACN + 3H]+3; mass calcd for [C25H30N4O4S2]: 514.1708; found, 514.8793.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(2-methoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (15)
White crystalline solid; yield: 70%; mp 142–144 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.85) broad singlet, 3H, −CH2CH3, (1.19–1.70) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.23) broad singlet, 2H, −S–CH2–CH2–, (7.08–7.51) m, overlapped peaks, 8H, Ar–H, (4.34) s, 2H, −NCH2CO–, (10.10) s, 1H, −CONHN–, (4.79) s, 2H, −COCH2S–, (5.22) s, 1H, CH-cyclic, −SCHN−), (3.73) s, 3H, CH3O–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 44.0 (−NCH2CO–Aliphatic), 167.4 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 35.6 (−COCH2S−), 83.0 (−SCHN−), 140.6 (C–Ar), 144.3(CH3O–CAr), 128.4 (2CH–Ar), 110.2 (CH–Ar), 53.1 (OCH3–Ar).
HR-MS (ESI) (m/z): 564.9844 [M + ACN + Na + 2H]+3; mass calcd for [C25H30N4O3S2]: 498.1759; found, 498.9551.
2-[2-Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(3-methoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (16)
White amorphous solid; yield: 80%; mp 122–124 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.24–1.83) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (7.30–7.69) m, overlapped peaks, 8H, Ar–H, (4.05) s, 2H, −NCH2CO–, (8.53) s, 1H, −CONHN–, (4.79) s, 2H, −COCH2S–, (5.31) s, 1H, CH-cyclic, −SCHN–, (3.73) s, 3H, CH3O–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0(−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar), 110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 166.4 (−NCH2CO–Aliphatic), 175.4 (−CO-cyclic), 35.6 (−COCH2S−), 82.0 (−SCHN−), 140.6 (C–Ar), 148.3(CH3O–CAr),129.6 (CH–Ar), 122.3 (2CH–Ar), 110.2 (CH–Ar), 53.1 (OCH3–Ar).
HR-MS (ESI) (m/z): 542.7368 [M + 2Na – H]+; mass calcd for [C25H30N4O3S2], 498.1759; found, 498.0087.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-methoxyphenyl)-4-oxothiazolidin-3-yl]acetamide (17)
Yellow amorphous solid; yield: 80%; mp 110–112 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.83) broad singlet, 3H, −CH2CH3, (1.25–1.62) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25), broad singlet, 2H, −S–CH2–CH2–, (7.11–7.50) m, overlapped peaks, 8H, Ar–H, (4.65) s, 2H, −NCH2CO–, (10.52) s, 1H, −CONHN–, (4.96) s, 2H, 2H, −COCH2S–, (5.22) s, 1H, CH-cyclic, −SCHN−), Ar–H), (3.66) s, 3H, CH3O–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2(−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 174.4 (−CO-cyclic), 35.6 (−COCH2S−), 82.0 (−SCHN−), 140.6 (C–Ar), 147.3 (CH3O–Ar),129.6 (CH–Ar), 122.3 (2CH–Ar), 110.2 (CH–Ar), 53.1 (OCH3–Ar).
HR-MS (ESI) (m/z): 574.9204 [M + 2K – H]+; mass calcd for [C25H30N4O3S2]: 498.1759; found, 498.0113.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(2-hydroxyphenyl)-4-oxothiazolidin-3-yl]acetamide (18)
White amorphous solid; yield: 68%; mp 111–113 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.81) broad singlet, 3H, −CH2CH3, (1.25–1.65) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.48) m, overlapped peaks, 8H, Ar–H, (4.55) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.88) s, 2H, 2H, −COCH2S–, (5.07) s, 1H, CH-cyclic, −SCHN−), 8.44 s (1H, OH–Ar.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 117.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.4 (−CO-cyclic), 33.6 (−COCH2S−), 80.0 (−SCHN−), 140.6 (C–Ar), 144.3 (HO–CAr),129.6 (CH–Ar), 128.8 (2CH–Ar), 115.2 (CH–Ar).
HR-MS (ESI) (m/z): 532.7020 [M + ACN + Li]+; mass calcd for [C24H28N4O3S2]: 484.1608; found, 484.7345.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(5-methylfuran-2-yl)-4-oxothiazolidin-3-yl]acetamide (19)
Yellow amorphous solid; yield: 70%; mp 124–126 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.25–1.64) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.17) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.50) m, overlapped peaks, 6H, Ar–H, (4.05) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.94) s, 2H, 2H, −COCH2S–, (5.08) s, 1H, CH-cyclic, −SCHN–, (1.64) s, 3H, CH3–furan.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 34.7 (−CH2S−), 150.6 (−SNCN−), 138.9 (−NCCH−), 135.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar), 110.0 (CH–Ar), 40.03 (−NCH2CO–Aliphatic), 166.0 (−NCH2CO–Aliphatic), 175.0 (−CO-cyclic), 35.6 (−COCH2S−), 82.0 (−SCHN−), 139.7 (2O–CHAr), 107.4 (2CH–Ar), 17.3 (CH3–Ar).
HR-MS (ESI) (m/z): 543.7320 [M + CH3OH + K]+; mass calcd for [C23H28N4O3S2]; 472.1608; found, 472.7389.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-isopropylphenyl)-4-oxothiazolidin-3-yl] Acetamide (20)
Orange color amorphous solid; yield: 70%; mp 161–163 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.26–1.60) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.23) broad singlet, −S–CH2–CH2–, (7.09–7.51) m, overlapped peaks, 8H, Ar–H, (3.84) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.66) s, 2H, 2H, −COCH2S–, (4.89) s, 1H, CH-cyclic, −SCHN–, (3.23) m, 1H, CH3–CH–CH3, (1.68) dd, J = 15.9 Hz, 8.5 Hz, 6H, −CH–(CH3)2.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 34.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar),110.0 (CH–Ar), 40.09 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.4 (−CO-cyclic), 32.6 (−COCH2S−), 62.0 (−SCHN−), 136.7 (C–Ar), 107.4(C–Ar), 128.4 (2CH–Ar), 126.0 (2CH–Ar), 30.7 (CH–Ar), 23.2 (2CH3).
HR-MS (ESI) (m/z): 543.7332 [M + CH3OH + H]+; mass calcd for [C27H34N4O2S2], 510.2123; found, 510.6997.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[4-oxo-2-(thiophen-2-yl)-thiazolidin-3-yl]acetamide (21)
Yellow amorphous solid; yield: 65%; mp 112–114 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.84) broad singlet, 3H, −CH2CH3, (1.22–1.64) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.19) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.67) m, overlapped peaks, 7H, Ar–H, (4.55) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.95) s, 2H, 2H, −COCH2S–, 2H, −COCH2S–, (5.08) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 39.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 176.4 (−CO-cyclic), 37.6 (−COCH2S−), 75.0 (−SCHN−), 139.4 (SC–Ar), 127.0 (2CH–Ar), 125.5 (SCH–Ar).
HR-MS (ESI) (m/z): 552.8290 [M + 2K]+2; mass calcd for [C22H26N4O2S3], 474.1217; found, 474.9027.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(naphthalen-2-yl)-4-oxothiazolidin-3-yl]acetamide (22)
White amorphous solid; yield: 78%; mp 108–110 °C.
1H NMR [DMSO-d6, 400 MHz, δ (ppm)]: δ(0.83) broad singlet, 3H, −CH2CH3, (1.27–1.54) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (7.07–7.48) m, overlapped peaks, 11H, Ar–H, (4.08) s, 2H, −NCH2CO–, (8.45) s, 1H, −CONHN–, (3.86) s, 2H, −COCH2S–, (4.64) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.9 (−CO-cyclic), 37.6 (−COCH2S−), 79.0 (−SCHN−), 135.2 (C–Ar), 132.3 (2C–Ar), 129.4 (2CH–Ar), 127.0 (2CH–Ar), 125.1 (2CH–Ar).
HR-MS (ESI) (m/z): 524.9692 [M + Li]+; mass calcd for [C28H30N4O2S2]: 518.1810; found, 518.0282.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl]-N-[2-(4-fluorophenyl)-4-oxothiazolidin-3-yl]acetamide (23)
Light yellow amorphous solid; yield: 75%; mp 120–122 °C.
1H NMR [DMSO-d6, 400 Hz, δ (ppm)]: δ(0.82) broad singlet, 3H, −CH2CH3, (1.26–1.65) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.25) broad singlet, 2H, −S–CH2–CH2–, (7.55–7.50) m, overlapped peaks, 8H, Ar–H, (4.02) s, 2H, −NCH2CO–, (8.41) s, 1H, −CONHN–, (3.66) s, 2H, 2H, −COCH2S–, (5.08) s, 1H, CH-cyclic, −SCHN–.
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 40.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar), 110.0 (CH–Ar), 39.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.6 (−CO-cyclic), 39.6 (−COCH2S−), 67.9. (−SCHN−), 137.3 (C–Ar), 132.7 (F–CAr), 130.1 (2CH–Ar), 128.3 (2CH–Ar).
HR-MS (ESI) (m/z): 517.1544 [M + Na + Li + H]3+; mass calcd for [C24H27FN4O2S2]: 486.1559; found, 486.1168.
2-[2-(Hexylthio)-1H-benzo[d]imidazole-1-yl)-N-[4-oxo-2-(p-tolyl)thiazolidin-3-yl]acetamide (24)
Yellow amorphous solid; yield: 70%; mp 109–111 °C.
1H NMR [DMSO-d6, 400 Hz, δ (ppm)]: δ(0.92) broad singlet, 3H, −CH2CH3, (1.25–1.63) m, overlapped peaks, 8H, −CH2–CH2–(CH2)3–, (3.55) broad singlet, 2H, −S–CH2–CH2–, (7.10–7.57) m, overlapped peaks, 8H, Ar–H, (4.10) s, 2H, −NCH2CO–, (11.27) s, 1H, −CONHN–, (3.80) s, 2H, −COCH2S–, (4.90) s, 1H, CH-cyclic, −SCHN–, 1.63 s (3H, CH3–Ar).
13C NMR [DMSO-d6, 100 MHz, δ (ppm)]: 13.9 (−CH3), 22.0 (−CH2−), 27.2 (−CH2−), 29.0 (−CH2−), 31.0 (−CH2−), 37.7 (−CH2S−), 152.6 (−SNCN−), 138.9 (−NCCH−), 134.2 (−NCCH−), 123.0 (2CH–Ar), 115.9 (CH–Ar),110.0 (CH–Ar), 38.9 (−NCH2CO–Aliphatic), 164.4 (−NCH2CO–Aliphatic), 175.9 (−CO-cyclic), 38.6 (−COCH2S−), 67.9 (−SCHN−), 132.6 (C–Ar), 132.2 (CH3C–Ar), 129.0 (2CH–Ar), 128.6 (2CH–Ar), 23.6 (CH3–Ar).
HR-MS (ESI) (m/z): 489.3202 [M + Li]+; mass calcd for [C25H30N4O2S2]: 482.1559; found, 482.2792.
α-Glucosidase Inhibitory Activity
The α-glucosidase inhibition potential of the target compounds 5–24 was evaluated with reference to acarbose.30 The enzyme (0.2 U/mL) taken in phosphate buffer (50 mM, pH. 6.8) was incubated with various concentrations of the compounds at 37 °C for 15 min. Then, p-nitrophenyl-α-d-glucopyranoside (0.7 mM) was added as a substrate. The change in absorbance at 400 nm was monitored up to 30 min by a multiplate spectrophotometer and the compound was replaced by DMSO (7.5% final) as control. All the reactions were performed in triplicate and the percent inhibition was calculated by the following formula
Molecular Docking Studies
The structures of compounds were first drawn in ChemDraw and then transformed to 3D structures and minimized using Chimera. Docking studies of the synthesized compounds were carried out using Autodock Vina in USCF Chimera 1.15 as a docking platform, which is one of the computationally fastest and accurate software used for docking. It is a platform for interactive visualization and evaluation of molecular structures and other related data. Docking simulations were performed on homology modeled α-glucosidase as the receptor protein. The enzyme was first loaded in the working environment and prepared by carrying out the steps; the retrieval of the target enzyme to the working environment, protein structure visualization by USCF chimera, preparation of receptor enzyme and ligand for docking, and final docking of the protein and the ligands as Mol2 files using Autodock Vina in chimera. After docking simulations, the highest-ranked poses were generated for each ligand, and finally, the interpretation and analysis of the docking results were accomplished. Discovery Studio visualizer software was used to display the different interactions between the ligands and the active site residues of the target enzyme.
Conclusions
This research work represents a synthetic approach where 2-mercaptobenzimidazole was alkylated with a 6-carbon alkyl group and the 1H position of the compound was extensively used for modification to see the effect on the biological activities. A total of 20 new derivatives were synthesized and characterized by physicospectral means.
The α-glucosidase inhibition profile of the new derivatives represents good to moderate inhibitory potential as compared to the standard drug acarbose.
The molecular docking studies of the synthesized compounds showed good correlations with the experimental findings. The binding modes of these compounds and their interaction with active site residues revealed them as possible antidiabetic leads.
The degree of activity and docking studies displayed by the novel innovative structural hybrids of benzimidazole and thiazolidinone rings make these compounds new active leads and promising candidates for the development of novel antidiabetics.
Acknowledgments
This work was funded by the Higher Education Commission of Pakistan (HEC) under National Research Project for University (NRPU), no: 20-2892 titled as “Synthesis and characterization of benzimidazole derivatives with potential pharmacological significance”.
The authors declare no competing financial interest.
References
- Rasool S. R.; Saadon A. Barbituric acid as a core for some new heterocyclic substituted derivatives. J. Appl. Chem. 2014, 3, 74–81. [Google Scholar]
- Sen J.; Srivastava S. Synthesis and antimicrobial activity of some new 2-[2″, 3″-dinitrophenyl]-3-(substituted aryl)-3, 3a-dihydrobenzimidazo [2, 1-a] pyrazolo [3, 4-d] thiazoles derivatives. Int. J. Theor. Appl. Sci. 2009, 1, 41–47. [Google Scholar]
- Chauhan D.; Hati S.; Priyadarshini R.; Sen S. Transcriptome analysis predicts mode of action of benzimidazole molecules against Staphylococcus aureus UAMS-1. Drug Dev. Res. 2019, 80, 490–503. 10.1002/ddr.21523. [DOI] [PubMed] [Google Scholar]
- Ali M.; Ali S.; Khan M.; Rashid U.; Ahmad M.; Khan A.; Al-Harrasi A.; Ullah F.; Latif A. Synthesis, biological activities, and molecular docking studies of 2-mercaptobenzimidazole based derivatives. Bioorg. Chem. 2018, 80, 472–479. 10.1016/j.bioorg.2018.06.032. [DOI] [PubMed] [Google Scholar]
- Yadav S.; Narasimhan B.; Lim S. M.; Ramasamy K.; Vasudevan M.; Shah S. A. A.; Selvaraj M. Synthesis, characterization, biological evaluation and molecular docking studies of 2-(1H-benzo [d] imidazole-2-ylthio)-N-(substituted 4-oxothiazolidin-3-yl) acetamides. Chem. Cent. J. 2017, 11, 137. 10.1186/s13065-017-0361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandarajagopal K.; Tiwari R. N.; Bothara K.; Sunilson J. A. J.; Dineshkumar C.; Promwichit P. 2-Mercaptobenzimidazole derivatives: synthesis and anticonvulsant activity. Adv. Appl. Sci. Res. 2010, 1, 132–138. [Google Scholar]
- Tahlan S.; Kumar S.; Narasimhan B. Antimicrobial potential of 1 H-benzo [d] imidazole scaffold: a review. BMC Chem. 2019, 13, 18. 10.1186/s13065-019-0521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer H. H.; Ali O. M.; El-Kafaween I. K. Synthesis of some new nucleosides derived from 2-mercapto benzimidazole with expected biological activity. Orient. J. Chem. 2017, 33, 2303–2310. 10.13005/ojc/330519. [DOI] [Google Scholar]
- Alkhafaji A.; Muhi-Eldeen Z.; Al-Kaissi E.; Al-Muhtaseb N.; Arafat T.; Ghattas M. A. Synthesis, structural elucidation, and evaluation of antimicrobial activity of 5-ethoxy-2-mercaptobenzimidazole derivatives. Int. J. Med. Health Res. 2018, 7, 117–127. [Google Scholar]
- Keri R. S.; Hiremathad A.; Budagumpi S.; Nagaraja B. M. Comprehensive review in current developments of benzimidazole-based medicinal chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. 10.1111/cbdd.12462. [DOI] [PubMed] [Google Scholar]
- Nevade S. A.; Lokapure S. G.; Kalyane N. V. Synthesis and pharmacological evaluation of some novel 2-mercapto benzimidazole derivatives. J. Korean Chem. Soc. 2013, 57, 755–760. 10.5012/jkcs.2013.57.6.755. [DOI] [Google Scholar]
- Syunyakov T.; Neznamov G. Evaluation of the therapeutic efficacy and safety of the selective anxiolytic afobazole in generalized anxiety disorder and adjustment disorders: Results of a multicenter randomized comparative study of diazepam. Ter. Arkh. 2016, 88, 73–86. 10.17116/terarkh201688873-86. [DOI] [PubMed] [Google Scholar]
- Aboul-Enein H.; El Rashedy A. Benzimidazole derivatives as antidiabetic agents. Med. Chem. 2015, 5, 318–325. 10.4172/2161-0444.1000280. [DOI] [PubMed] [Google Scholar]
- Čačić M.; Molnar M.; Šarkanj B.; Has-Schön E.; Rajković V. Synthesis and antioxidant activity of some new coumarinyl-1, 3-thiazolidine-4-ones. Molecules 2010, 15, 6795–6809. 10.3390/molecules15106795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Saraf S. K. 4-Thiazolidinone–A biologically active scaffold. Eur. J. Med. Chem. 2008, 43, 897–905. 10.1016/j.ejmech.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Singh R.; Sonar P. K.; Saraf S. K. Novel 4-thiazolidinone derivatives as anti-infective agents: synthesis, characterization, and antimicrobial evaluation. Biochem. Res. Int. 2016, 2016, 8086762. 10.1155/2016/8086762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreca M. L.; Chimirri A.; De Luca L.; Monforte A.-M.; Monforte P.; Rao A.; Zappalà M.; Balzarini J.; De Clercq E.; Pannecouque C.; et al. Discovery of 2,3-diaryl-1,3-thiazolidin-4-ones as potent anti-HIV-1 agents. Bioorg. Med. Chem. Lett. 2001, 11, 1793–1796. 10.1016/s0960-894x(01)00304-3. [DOI] [PubMed] [Google Scholar]
- Bonde C. G.; Gaikwad N. J. Synthesis and preliminary evaluation of some pyrazine containing thiazolines and thiazolidinones as antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 2151–2161. 10.1016/j.bmc.2004.02.024. [DOI] [PubMed] [Google Scholar]
- Sigalapalli D. K.; Pooladanda V.; Kadagathur M.; Guggilapu S. D.; Uppu J. L.; Godugu C.; Bathini N. B.; Tangellamudi N. D. Novel chromenyl-based 2-iminothiazolidin-4-one derivatives as tubulin polymerization inhibitors: Design, synthesis, biological evaluation and molecular modelling studies. J. Mol. Struct. 2021, 1225, 128847. 10.1016/j.molstruc.2020.128847. [DOI] [Google Scholar]
- Türe A.; Ergül M.; Ergül M.; Altun A.; Küçükgüzel İ. Design, synthesis, and anticancer activity of novel 4-thiazolidinone-phenylaminopyrimidine hybrids. Mol. Divers. 2021, 25, 1025–1050. 10.1007/s11030-020-10087-1. [DOI] [PubMed] [Google Scholar]
- Almasirad A.; Ghadimi M.; Mirahmadi S.; Ahmadian Kodakan P.; Jahani R.; Nazari M.; Rezaee E.; Azizian H.; Rabizadeh P.; Tabatabai S. A.; et al. Design, synthesis, and preliminary pharmacological evaluation of novel thiazolidinone derivatives as potential benzodiazepine agonists. Mol. Divers. 2022, 26, 769. 10.1007/s11030-021-10182-x. [DOI] [PubMed] [Google Scholar]
- Taha M.; Shah S. A. A.; Afifi M.; Imran S.; Sultan S.; Rahim F.; Khan K. M. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 2018, 77, 586–592. 10.1016/j.bioorg.2018.01.033. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Struct., Funct., Bioinf. 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Colovos C.; Yeates T. O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P.; Künzli M.; Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan S.; Somani R.; Shirodkar P.; Kadam V. Microwave assisted synthesis and antimicrobial activity of some newer mannich bases. Int. J. PharmTech Res. 2009, 1, 811–815. [Google Scholar]
- Javid M. T.; Rahim F.; Taha M.; Rehman H. U.; Nawaz M.; wadood S.; Imran I.; Uddin A.; Mosaddik K. M.; Khan K. M. Synthesis, in vitro α-glucosidase inhibitory potential and molecular docking study of thiadiazole analogs. Bioorg. Chem. 2018, 78, 201–209. 10.1016/j.bioorg.2018.03.022. [DOI] [PubMed] [Google Scholar]
- Elrayess R.; Ghareb N.; Azab M. M.; Said M. M. Synthesis and antimicrobial activities of some novel benzimidazole and benzotriazole derivatives containing β-lactam moiety. Life Sci. J. 2013, 10, 1784–1793. [Google Scholar]
- Eisa H. M.; Barghash A.-e. M.; Badr S. M.; Farahat A. A. Synthesis and antimicrobial activity of certain benzimidazole and fused benzimidazole derivatives. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2010, 49, 1515–1525. [Google Scholar]
- Kashtoh H.; Hussain S.; Khan A.; Saad S. M.; Khan J. A. J.; Khan K. M.; Perveen S.; Choudhary M. I. Oxadiazoles and thiadiazoles: Novel α-glucosidase inhibitors. Bioorg. Med. Chem. 2014, 22, 5454–5465. 10.1016/j.bmc.2014.07.032. [DOI] [PubMed] [Google Scholar]