

Abstract
Few human genetic diseases can rely on the availability of as many and as diverse animal models as cystic fibrosis (CF), a multiorgan syndrome caused by functional absence of Cystic Fibrosis Transmembrane Regulator (CFTR). The recent development of highly effective CFTR modulator drug therapies simultaneously highlighted the remarkable clinical improvement achievable with these treatments, the lack of therapeutic alternatives for non-responders, and the need to understand the kinetics of disease upon early life/chronic treatment. These advances have rekindled efforts to leverage animal models to address critical knowledge gaps in CF. This paper provides a concise overview of the areas of interests for therapeutic intervention in the current CF landscape, focusing on the contributions of in vivo models to understand CF pathogenesis, identify therapeutic windows, and develop novel therapies for all CFTR mutations.
Keywords: Cystic Fibrosis, animal models
Introduction
Cystic Fibrosis (CF) is a monogenetic disease caused by absence of functional Cystic Fibrosis Transmembrane Regulator (CFTR), an anion channel that directly regulates chloride and bicarbonate secretion/absorption across epithelia in many organs. Defects in CFTR function may arise from aberrant transcription/translation, folding/degradation, trafficking/stability in the plasma membrane, or gating/conductance. Main manifestations of the disease involve: 1) airway mucus obstruction and inflammation with predisposition to bacterial infections and recurrent exacerbations, leading to progressive tissue damage and respiratory failure; and 2) gastrointestinal, pancreatic and hepatic dysfunctions, which lead to malabsorption and affect intestinal transit, growth, and overall metabolism(1).
CFTR orthologs are expressed in vertebrates, from cartilaginous fish onward (2, 3). This has allowed the development of CF animal models in several species, which have been used to investigate CF pathophysiology and test therapeutic treatments. In order of first appearance in the published literature, CF researchers have generated animal models in the following species: mouse(4, 5), pig(6–10), ferret(11, 12), rat(13–17), zebrafish(18, 19), sheep(20, 21), drosophila(22), and rabbit(23, 24). Other non CFTR-directed animal models have been instrumental to study particular aspects of CF pathophysiology, e.g., defective airway mucus clearance (Scnn1b-Tg mice(25–27), elastase-treated sheep(28–31) or mice(32, 33)) or to simplify complex system biology, e.g., mucosal surface immunology (Xenopous laevis tad pole(34, 35)). Far from being redundant, this variety reflects an effort to provide the CF research community with multiple tools to study CF pathophysiology or therapeutic options, and to overcome the shortcomings inevitably associated with any individual animal model in terms of organ-specific phenotypes, cost, and tractability, as extensively reviewed in the recent past(36–40).
The advent of highly effective modulator drug therapies (HEMT), small molecules that aid mutant CFRT intracellular trafficking and support its function once it reaches the plasma membrane, has transformed health outcomes and quality of life for a large fraction (~90%) of people with CF (PwCF)(41–43). These highly beneficial therapies are projected to further improve longevity of PwCF and their stark success has poised the scientific community to address the lack of treatment for the 10% of PwCF who do not respond to HEMT and to explore the long-term implications and new challenges arising from a longer life with CF. Notably, none of the currently available HEMT directly stemmed from animal model research. So, how relevant are CF animal models in the HEMT era and can they be harnessed to address emerging CF research priorities? The ongoing PROMISE study(44), a large multidisciplinary study focused on the impact of triple combination therapy (elexacaftor/tezacaftor/ivacaftor) on the US population age 6 and older, highlighted specific “areas of interest” as emerging research priority in CF (i.e., clinical change, mucus biology, microbiology, inflammation and host response, gastroenterology, endocrinology, liver disease, nasal airway epithelial cell function, and pediatric inclusion). These priorities are also reflected in the Cystic Fibrosis Foundation drug development pipeline (https://apps.cff.org/trials/pipeline) where the primary therapeutic outcomes involve: restore CFTR function; enhance mucociliary clearance; develop novel anti-inflammatory and anti-infective strategies; and support nutritional/gastrointestinal health. Arguably, animal models provide the unique opportunity to study how CFTR dysfunction affects multiple organ systems, how disease phenotypes relate to each other, and how they are influenced by non-CFTR-related modifiers, e.g., genetic background(45), microbiota(46), sex(47), age(48), and environmental exposures(49). The purpose of this brief review is to summarize the ongoing contribution of CF animal models’ research to the aforementioned priorities, with reference to recently published papers.
Restoration of CFTR function
Pharmacologic correction of defective CFTR function is achievable provided that 1) the mutant CFTR protein is expressed in the target organ, and 2) the mutant CFTR is responsive to the modulation. About 7% of PwCF carry severe mutations (nonsense/missense, splice-site, frameshifts, insertions/deletions) that lead to complete absence of CFTR protein, and are thus non-responsive to HEMT. The challenging goal of introducing functional CFTR in affected tissues using gene therapy approaches has been heavily pursued using CF animal models(50, 51). Wild-type animals have been used to evaluate safety and efficiency of different vectors or delivery methods (e.g., adeno-associated vector in ferret(52), lentiviral vectors in marmoset(53), microspray administration in neonatal pigs(54)). Mutant CFTR-based models have been used to evaluate restoration of CFTR activity in vivo. Functional readout varied depending on the model used and include normalization of pulmonary function parameters in CFTRtm1Unc KO mouse treated with chemically modified hCFTR mRNA(55), increase in basal nasal potential difference towards WT levels in 510X CF rat treated with a tagged CFTR lentiviral vector(56), and restoration of transepithelial Cl− current in freshly excised airways from gut-corrected CF pig treated with an integrating adenovirus-based vector expressing hCFTR (57). Models recapitulating key features of CF lung disease, i.e., airway mucus obstruction and inflammation, have been used to study barriers to vectors’ delivery(58) and stability(59) in inflamed and obstructed airways. Proof-of-concept studies in mice(60) also supported the feasibility of in vivo cell-based therapy, where genetic correction is achieved ex vivo in suitable regenerative cells which are then transplanted to the affected organ(61).
Due to their relatively low maintenance cost, fast life cycle, and amenability to complex gene editing manipulation as compared to larger models (e.g., pigs, ferret, sheep, rabbits), rodents have been the species of choice to generate screening tools for read-through pharmacologic therapy against nonsense/missense mutations. The G542X stop mutation has been introduced in mouse(62) and rat(15). This mutation has also been introduced in sheep with the goal of using this neonatal lethal model to test in utero therapies (21). In an effort to provide screening platforms with the closest DNA and protein sequence similarity to hCFTR, “humanized” CF mice have been generated by transgenic expression of hCFTR on a mCFTR KO background (63). In this model, multiple copies of hCFTR were integrated in a single insertion site, making further mutant models based on this strain suitable to test pharmacologic interventions (e.g., hCFTR G551D, F508del, G542X, W1282X, 3849+10kb C>T), but complicating the application of gene editing approaches. This limitation has been recently overcome by using exon replacement strategies that allow for tailored substitution of the endogenous mouse sequence with the human one, obtaining a hybrid CFTR (exon 11: WT, F508del; exon 12: WT, G542X, and R553X, Hodges, C. et al, Abstract 662 North American Cystic Fibrosis Conference, Virtual event, November 2021, Journal of Cystic Fibrosis Vol. 20, Supplement S314).
Among CF animal models, only ferret(11), pig(6), sheep(21), and rabbit(23, 64) CFTR is naturally responsive to modulators of hCFTR, likely due to a higher degree of sequence and structural homology as compared to other species. Pharmacologic rescue of CFTR in animal models can be used to query critical issues that would be intractable in human subjects, such as effective therapeutic windows, spatio-temporal patterns of disease development, and effect of treatment withdrawal. A remarkable example for these studies featured in-utero and postnatal treatment of ferrets carrying the G551D mutation with the potentiator ivacaftor (VX-770)(12).Resulting data support the efficacy of early intervention to prevent perinatal manifestation of the disease (pancreas, intestine, growth), and highlights how therapeutic regimens need to be maintained to prevent reoccurrence of disease (lung, pancreas). An important corollary of this study is that using the outbred ferret model carrying a defined genetic mutation (G551D) it was possible to appreciate the contribution of disease modifier genes in the manifestation of disease and response to therapy (G551D homozygous ferret with pancreatic insufficiency regardless of VX770 treatment). G551D humanized rats have also been used to test the effect of ivacaftor on age-dependent airway mucus abnormalities(65) and inflammation(66). Notably, accumulating evidence suggest that CFTR function is required for proper organ development, including cartilaginous airways of CF pigs (67), rats (13), and mice (68), or nasal sinuses in CF pigs(69). Congenital absence of vas deferens has been documented in CF ferrets (12), rats(70), rabbits(23), and sheep(21), but not in CF mice, whereas CF pigs exhibit high penetrance of vas deferens and epididymal atresia(71), suggesting species- and organ-specific requirements. These results, along with the increasing availability of different animal models susceptible to pharmacological correction of CFTR, suggest that a range of CFTR function-dependent phenotypes can be exploited to study the developmental components of CF disease.
Mucus biology and mucociliary clearance
Luminal obstruction with inspissated mucous secretions characterize the pathologic presentation of CF in several organs (upper and lower respiratory tract, gastrointestinal tract, pancreas, liver, gallbladder, female and male reproductive tract) and has been directly linked to absence of functional CFTR-mediated Cl− and HCO3− secretion. Understanding of the specific mechanisms and tissue/molecular components involved in the generation and failed clearance of these thick secretions is an active area of investigation (72–75). Abnormalities in airway mucus have been detected in bronchoalveolar lavage from young children with CF, were associated with inflammation, and preceded bacterial infection (76), suggesting a causal role in the pathogenesis of obstructive CF lung disease. This central role has been substantiated by studies in CF animal models, including pigs(7, 77), ferret(78), and mouse(27). The general components of the mucociliary clearance system (mucus producing, secretory, and ciliated cells) are present in all CF animal models, although relevant anatomic and cell composition differences must be considered in comparative studies. For examples, airway submucosal glands are associated with cartilaginous airways throughout human airways, with a similar distribution observed in pigs and ferrets. In contrast, submucosal glands are localized in the trachea in rats, they are further restricted to the most proximal portion of the trachea in mice, and are completely absent in rabbits(79). These differences have been exploited to investigate the contribution of submucosal gland vs. superficial epithelia in the pathogenesis and progression of CF lung disease, and evidences so far suggest that the presence of submucosal glands is critical to develop spontaneous and chronic obstructive lung disease (pig and ferret), whereas failure to clear secretions from the smaller conducting airways might be responsible for creating early, heterogeneously distributed “pathologic niches” in the deeper lung(23, 80). Of note, scRNAseq technologies have recently been used to define the cellular and molecular landscape of surface epithelium and submucosal glands in CF vs. WT pigs at birth(10, 81), establishing a critical, initial timepoint for a much-anticipated time course analysis.
In larger CF animal models, lung disease progression and efficacy of therapeutic interventions can be studied longitudinally using imaging modalities similar to humans, like computer tomography (e.g., ferret (78, 82), pig(77)), whereas other techniques had to be devised to study mucociliary clearance in species with smaller lung volumes. Given the many variables that affect mucociliary clearance in vivo, recent efforts have been focused on developing measurements that can be made on intact tissue or in situ to avoid perturbation of the environment. In particular, intact tracheas have been studied with microoptical coherence tomography (μOCT), which allows measurement of thickness of secretions, ciliary beat frequency, mucociliary transport, and submucosal inflammation (14, 83, 84), or fluorescence particle tracking (85). Although sophisticated methods to quantitatively assess obstructive lung disease in the lower airways of smaller species are under development (86–88), the vast majority of studies relies on histological and morphometric assessment of airway mucus burden and inflammation. Specifically, morphometric analyses have been used to quantify airway mucus burden in Scnn1b-Tg mice treated with mucus-mobilizing agents(89–91), elastase-treated WT mice(32), or CF rats challenged with P.aeruginosa embedded in agar beads (Birket, S. et al. Abstract 72 North American Cystic Fibrosis Conference, Denver, October 2018, Pediatric Pulmonology Vol. 53, Supplement 2).
Infection
Although defective airway mucus clearance likely plays a major role in initiating CF lung disease, chronic bacterial airway infections are the leading cause of morbidity and mortality in PwCF. Spontaneous, chronic airway bacterial infections with a range of pathogens including Staphylococcus, Streptococcus, and Enterococcus have been reported in CF ferrets(11, 92) and CF pigs(7, 93). Overall, rodent models do not exhibit chronic airway bacterial infection, with the only two notable exceptions being 1) congenic C57BL/6 Cftrtm1kth mice, in which a proportion of mice (1/3) exhibit colonization by Bordetella pseudohinzii associated with decreased respiratory rates (94), and 2) Scnn1b-Tg mice which are susceptible to spontaneous colonization by oropharyngeal microflora in the early post-natal period (5–10 post-natal days), but become resistant as they age (27). These models allow study of factors facilitating the establishment and the evolution of airway bacterial infection from the perspective of both the host and the pathogens, as well as pharmacological interventions. Several models of experimentally-induced bacterial infection have also been developed to test specific contributions of host and pathogen factors, with an important distinction between “chronic” vs. “acute” protocols(95). In the absence of a CF-like muco-obstructed and likely hypoxic milieu, i.e., in WT animals, chronic (i.e., lasting more than 1–2 weeks) infections are often obtained by embedding the bacteria in polymer beads which provide a protective microenvironment against mucociliary and immune clearance. Although not without limitations, these models have been critical to demonstrate the contributions of host genetic background(45), to benchmark current therapies (96), and to test novel anti-infective treatments, e.g., phage therapy(97). Acute anti-infectives screening has been performed using aerosolized bacteria models(98), and CF zebrafish embryos(99). Viral infections have been linked to pulmonary exacerbations and progressive decline in lung function, but respiratory viruses routinely cultured during periods of exacerbation in PwCF (i.e., influenza, respiratory syncytial virus, and respiratory virus(100)) have not been systematically studied in CF animal models, likely due to differences in viral tropism across species. Non-tuberculous mycobacteria (NTM) are emerging pathogens for PwCF and are found in ~ 10% of the patient population(101). Pharmacologic screening of anti-NTM compounds or studies investigating the interaction between NTM and the immune system can be performed in non-mammalian models (zebrafish, X. laevis tad pole(35)), in mice deficient in innate or adaptive immune mediators (e.g., GM-CFS KO, SCID, and NOD) or transiently immunosuppressed by chronic corticosteroid treatment(102). Integration of these approaches in animal models recapitulating specific features of CF lung pathology could provide further insights for the detection and treatment of old and emerging microbial threats for PwCF.
Inflammation
Chronic inflammation is a hallmark of the multi-organ CF syndrome and it is likely due a combination of recurrent challenges, hyperactive yet inefficient immune responses, and delayed resolution (103). Intrinsic, i.e., due to abnormal CFTR function within the cell, and extrinsic, i.e., due to CFTR-dependent alterations in the cellular milieu, abnormalities have been described for CF neutrophils(104) and macrophages(105). CF pigs and ferrets exhibit spontaneous inflammation in the respiratory and gastrointestinal tracts, and inflammatory markers have been used to track the development and progression of specific organ disease. As these models are amenable to HEMT treatment and might exhibit different levels of organ-specific therapeutic success, it is plausible that they could be harnessed to study the interconnection and developmental dynamics of CF inflammatory responses. In parallel, studies in species considered less exemplary of human CF pathology, like rodents, have highlighted the potential for intrinsic, CFTR-dependent drivers of inflammation. For example, CF mice exhibit an inflammatory imbalance both at baseline (in serum) and after chronic Pseudomonas aeruginosa or lipopolysaccharide (LPS) challenge (in the lung) (106–108). Bone marrow transplant experiments have suggested that cell-based supportive therapy with hematopoietic and mesenchymal stem cells could be beneficial to enhance CF immune regulation (109). Of note, a recent report suggests that the muco-obstructed microenvironment associated with CF lung disease promotes epigenetic reprogramming of resident airway macrophages, shifting their transcriptional profile and activity towards a state that is both hypofunctional and hyperinflammatory(110). These findings share some features with recent single cell RNA sequencing (scRNAseq) analyses of CF vs. healthy control sputa that identified dysregulation in pathways associated with phagocytosis and immune cell regulation in CF macrophages and monocytes populations (111). Finally, humanized G551D rats have been used to test whether pharmacologic correction with ivacaftor would restore the hyperinflammatory lung phenotype present in 6 months-old hG551D rats, both at baseline and after LPS challenge, a surrogate for pulmonary exacerbations. A 7-day treatment with Ivacaftor effectively reduced inflammatory markers in bronchoalveolar lavage from naïve hG551D rats, failed to fully revert the effects of LPS stimulation, suggesting that short-term correction is insufficient to regulate the hyperinflammatory response to a stronger stimulus(66).
Gastrointestinal, pancreatic, and liver disease
Although the respiratory pathology is the primary cause of concern for PwCF, dysfunctions in the gastrointestinal (GI) tract, pancreas, and liver significantly increase the disease burden and affect quality of life. Human CF GI disease is characterized by an ~20% incidence of meconium ileus (MI) at birth (112). In adults, gastroesophageal reflex disease (GERD) affects up to 80% of PwCF), distal intestinal obstruction syndrome (DIOS) is commonly diagnosed, and small intestine bacterial overgrowth (SIBO) is found in 30–50% of PwCF (113). Moreover, PwCF suffer from high incidence of esophageal, gastric, liver, gallbladder, and colon cancer, likely linked to chronic inflammation and tissue damage. Absence of CFTR-mediated bicarbonate and fluid transport in pancreatic ducts causes premature acid-activation of the digestive enzymes secreted by acinar cells and impairs flow of these enzymes into the duodenum, causing malabsorption and concomitant pancreatic inflammation and fatty replacement. These pathologic changes begin before birth and affect 85% of PwCF, and the “collateral damage” in the endocrine pancreas leads to CF-related diabetes in 50% of PwCF, who present with worse lung disease, poorer nutritional status, and increased mortality. Similarly, absence of functional CFTR in the cholangiocytes lining the hepatic biliary ducts leads to cholestasis, microgallbladder, inflammation, and progressive liver fibrosis, another leading source of morbidity and mortality. All CF animal models present gastrointestinal abnormalities of different severities (114, 115) likely due to species-specific physiology as well as CFTR and “vicariate” ion channels’ expression patterns. In CF mice, rats, and rabbits the most noticeable GI phenotype is distal intestinal/ proximal colon obstruction - analogous to DIOS – which is particularly severe after weaning. These species are typically spared from neonatal MI, pancreatic, and hepatic complications (13, 16, 17, 23, 65, 66, 116), except for congenic C57BL/6J Cftrtm1Unc weaned on a liquid diet and aged to up to 2 years, that present with focal hepato-biliary lesions (48, 117). Administration of osmotic laxatives has been used to improve survival and DIOS symptoms in CF mice and other CF animal models, often in conjunction with targeted dietary modifications (23, 93, 118–120). Genetic “gut-correction”, i.e., gut-targeted, transgenic expression of functional CFTR, has also been used to overcome GI pathology in mice, ferrets, and pigs, although stunted growth (121), pancreas-related (9, 11) or metabolic abnormalities (122) still affect these gut-corrected models. GI disease in CF pigs more substantially mirrors human CF, including MI (100% penetrance, requiring surgery at birth), DIOS, diverticulosis, intestinal atresia/stenosis, gastric ulcerations (prophylactically treated with proton pump-inhibitors), alterations in intestinal muscle motility, severe pancreatic pathology detected at birth that requires life-long pancreatic enzymes replacement therapy (PERT), focal biliary cirrhosis, and micro-gallbladder (6, 8, 123, 124). Similar to CF pigs, CF ferrets have been instrumental in illuminating aspects of CF extra-pulmonary pathophysiology, and windows of opportunity for treatment. In CF ferrets, MI is slightly less penetrant than pigs (75%) and gradually transitions to DIOS; mild exocrine pancreas disease is present at birth but within a month progresses towards severe degeneration which requires PERT and causes bouts of inflammation and remodeling leading to glucose intolerance and CF-related diabetes (125). Studies with G551D ferrets suggest that perinatal DIOS and pancreatic degeneration can be prevented by in utero administration of ivacaftor. Conversely, postnatal ivacaftor withdrawal leads to resurgence of these lesions(12). The recently developed CF sheep(20, 21) also exhibit the full range of human CF-like intestinal, pancreatic, and hepatic complications, and the severity of these lesions likely contribute to the dismal survival of this model (live-born lambs are considered non-viable due to severe MI). Although rabbit express CFTR in the pancreatic ducts and CF rabbit exhibit altered lipid metabolism, histologic analyses revealed only mild focal lesions which appear to be dependent on husbandry and overall health status(23).
Recent attention has been given to the influence of the CF gut microbiota on CF multi-organ pathology and susceptibility to colorectal cancer (126, 127). Bacterial overgrowth has been detected in the small intestine of neonatal and adult CF mice(128, 129) and in CF ferrets (92). Alterations in stool microbiota have been detected in young CF rabbits, although it is not clear if these were due to selective administration of osmotic laxative only to the CF rabbits (130). Of note, germ-free CF mice still develop DIOS, and conversely, the presence of inspissated mucosal secretions actively shapes the gut microbiota composition after experimental re-colonization of these mice(46).
Non-vertebrate CF models are also emerging as tools to study aspects of CF GI pathophysiology. Drosophila lacking the evolutionary conserved miR-263 fail to regulate ENaC function in the gut resulting in dehydration of the intestinal surface, enterocytes activation, and gut bacteria overgrowth (131), a phenotype similar to that of Drosophila lacking the CFTR ortholog CG5789(22). Finally, the kinetics and cell types involved in pancreatic degeneration have been studied in the imaging-friendly CF zebrafish(18).
Conclusions:
The breadth and depth of research efforts using CF animal models clearly indicates that their usefulness is continuously expanding, benefiting from current innovations in analytical and gene-editing technologies. On a concluding note, comparison of scRNAseq data across species suggests that main pathways involved in lung development and repair are conserved across species and can be queried through computational analyses regardless of differences in cell composition and species-specific gene expression(132), providing further confidence in the possibility to translate findings generated in animal models to human diseases.
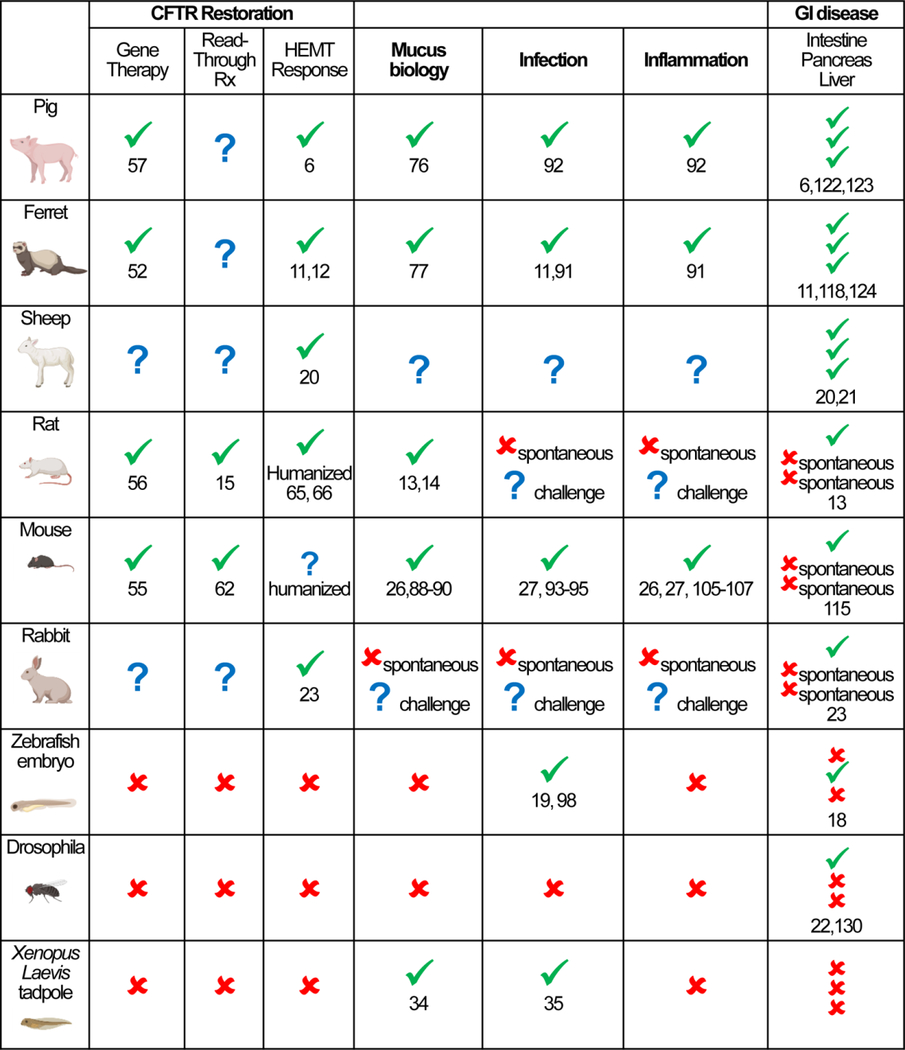
Table 1.
Visual synopsis of CF animal models discussed in this review as referenced to current CF research priorities (CFTR restoration, mucus biology, infection, inflammation, and GI disease). Green check mark indicates documented use through published reports (selected references are provided), red cross mark indicates untested or unsuitable application (for example, absence of spontaneous phenotype), blue question mark indicates applications under development or restricted to a particular variant of the animal model (for example, humanized variant).
|
Acknowledgements:
ALB and BRG are supported by National Institutes of Health grants 1 R01 HL150541-01 to ALB; P01 HL108808, R01 HL136961, UH3 HL123645, and P30 DK065888 to Richard C. Boucher; Cystic Fibrosis Foundation grants GRUBB17XX0 and GRUBB21I0 to BRG, BRAGON21I0 subcontract to ALB, and BOUCHE19R0 to Richard C. Boucher. None of these funding sources had direct involvement in the writing of this report or in the decision to submit it for publication.
Glossary
- PwCF
People with CF
- HEMT
highly effective modulator drug therapies
- μOCT
microoptical coherence tomography
- NTM
Non-tuberculous mycobacteria
- scRNAseq
single cell RNA sequencing
- MI
meconium ileus
- GERD
gastroesophageal reflex disease
- DIOS
distal intestinal obstruction syndrome
- SIBO
small intestine bacterial overgrowth
- PERT
pancreatic enzymes replacement therapy
Footnotes
Conflict of interest statement: The authors have no competing interests
References cited and recommended reading:
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, and Bush A. Cystic fibrosis. Nat Rev Dis Primers. 2015;1:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bose SJ, Scott-Ward TS, Cai Z, and Sheppard DN. Exploiting species differences to understand the CFTR Cl- channel. Biochem Soc Trans. 2015;43(5):975–82. [DOI] [PubMed] [Google Scholar]
- 3.Cui G, Hong J, Chung-Davidson YW, Infield D, Xu X, Li J, et al. An Ancient CFTR Ortholog Informs Molecular Evolution in ABC Transporters. Dev Cell. 2019;51(4):421–30 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros. 2011;10 Suppl 2:S152–71. [DOI] [PubMed] [Google Scholar]
- 5.Guilbault C, Saeed Z, Downey GP, and Radzioch D. Cystic fibrosis mouse models. American journal of respiratory cell and molecular biology. 2007;36(1):1–7. [DOI] [PubMed] [Google Scholar]
- 6.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science (New York, NY). 2008;321(5897):1837–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.••.Bouzek DC, Abou Alaiwa MH, Adam RJ, Pezzulo AA, Reznikov LR, Cook DP, et al. Early Lung Disease Exhibits Bacteria-Dependent and -Independent Abnormalities in Cystic Fibrosis Pigs. Am J Respir Crit Care Med. 2021;204(6):692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]; Most recent update on the lung phenotype in CF pigs
- 8.Ostedgaard LS, Meyerholz DK, Chen JH, Pezzulo AA, Karp PH, Rokhlina T, et al. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Science translational medicine. 2011;3(74):74ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123(6):2685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurman AL, Li X, Villacreses R, Yu W, Gong H, Mather SE, et al. A Single-Cell Atlas of Large and Small Airways at Birth in a Porcine Model of Cystic Fibrosis. American journal of respiratory cell and molecular biology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120(9):3149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.••.Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Science translational medicine. 2019;11(485). [DOI] [PMC free article] [PubMed] [Google Scholar]; Proof-of-concept study for in-utero therapy with CFTR modulator
- 13.Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PloS one. 2014;9(3):e91253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birket SE, Davis JM, Fernandez CM, Tuggle KL, Oden AM, Chu KK, et al. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight. 2018;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.•.Sharma J, Abbott J, Klaskala L, Zhao G, Birket SE, and Rowe SM. A Novel G542X CFTR Rat Model of Cystic Fibrosis Is Sensitive to Nonsense Mediated Decay. Front Physiol. 2020;11:611294. [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of the first rat model to study CF non-sense mutations.
- 16.McCarron A, Cmielewski P, Reyne N, McIntyre C, Finnie J, Craig F, et al. Phenotypic Characterization and Comparison of Cystic Fibrosis Rat Models Generated Using CRISPR/Cas9 Gene Editing. Am J Pathol. 2020;190(5):977–93. [DOI] [PubMed] [Google Scholar]
- 17.Dreano E, Bacchetta M, Simonin J, Galmiche L, Usal C, Slimani L, et al. Characterization of two rat models of cystic fibrosis-KO and F508del CFTR-Generated by Crispr-Cas9. Animal Model Exp Med. 2019;2(4):297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navis A, and Bagnat M. Loss of cftr function leads to pancreatic destruction in larval zebrafish. Dev Biol. 2015;399(2):237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cafora M, Deflorian G, Forti F, Ferrari L, Binelli G, Briani F, et al. Phage therapy against Pseudomonas aeruginosa infections in a cystic fibrosis zebrafish model. Sci Rep. 2019;9(1):1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan Z, Perisse IV, Cotton CU, Regouski M, Meng Q, Domb C, et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight. 2018;3(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.•.Viotti Perisse I, Fan Z, Van Wettere A, Liu Y, Leir SH, Keim J, et al. Sheep models of F508del and G542X cystic fibrosis mutations show cellular responses to human therapeutics. FASEB Bioadv. 2021;3(10):841–54. [DOI] [PMC free article] [PubMed] [Google Scholar]; Most recent update on the development of CF sheep
- 22.Kim K, Lane EA, Saftien A, Wang H, Xu Y, Wirtz-Peitz F, et al. Drosophila as a model for studying cystic fibrosis pathophysiology of the gastrointestinal system. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(19):10357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.•.Xu J, Livraghi-Butrico A, Hou X, Rajagopalan C, Zhang J, Song J, et al. Phenotypes of CF rabbits generated by CRISPR/Cas9-mediated disruption of the CFTR gene. JCI Insight. 2021;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed update on the phenotype of CF rabbits
- 24.Yang D, Liang X, Pallas B, Hoenerhoff M, Ren Z, Han R, et al. Production of CFTR-DeltaF508 Rabbits. Front Genet. 2020;11:627666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mall M, Grubb BR, Harkema JR, O’ Neal WK, and Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. D - 9502015. 2004;10(5):487–93. [DOI] [PubMed] [Google Scholar]
- 26.Livraghi-Butrico A, Grubb BR, Kelly EJ, Wilkinson KJ, Yang H, Geiser M, et al. Genetically determined heterogeneity of lung disease in a mouse model of airway mucus obstruction. Physiol Genomics. 2012;44(8):470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.•.Livraghi-Butrico A, Kelly EJ, Klem ER, Dang H, Wolfgang MC, Boucher RC, et al. Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal immunology. 2012;5(4):397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence of lung disease independent of bacterial infection in airway muco-obstructed mice
- 28.Terryah ST, Fellner RC, Ahmad S, Moore PJ, Reidel B, Sesma JI, et al. Evaluation of a SPLUNC1-derived peptide for the treatment of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L192–L205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabater JR, Sackner MA, Adams JA, and Abraham WM. Whole body periodic acceleration in normal and reduced mucociliary clearance of conscious sheep. PloS one. 2019;14(11):e0224764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nickolaus P, Jung B, Sabater J, Constant S, and Gupta A. Preclinical evaluation of the epithelial sodium channel inhibitor BI 1265162 for treatment of cystic fibrosis. ERJ Open Res. 2020;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim MD, Baumlin N, Yoshida M, Polineni D, Salathe SF, David JK, et al. Losartan Rescues Inflammation-related Mucociliary Dysfunction in Relevant Models of Cystic Fibrosis. Am J Respir Crit Care Med. 2020;201(3):313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Blanco JA, Fakih D, Arike L, Rodriguez-Pineiro AM, Martinez-Abad B, Skansebo E, et al. Attached stratified mucus separates bacteria from the epithelial cells in COPD lungs. JCI Insight. 2018;3(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voynow JA, Fischer BM, Malarkey DE, Burch LH, Wong T, Longphre M, et al. Neutrophil elastase induces mucus cell metaplasia in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1293–302. [DOI] [PubMed] [Google Scholar]
- 34.Dubaissi E. A ‘tad’ of hope in the fight against airway disease. Biochem Soc Trans. 2020;48(5):2347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez A, Shoen C, Cynamon M, Dimitrakopoulou D, Paiola M, Pavelka MS Jr., et al. Developing Tadpole Xenopus laevis as a Comparative Animal Model to Study Mycobacterium abscessus Pathogenicity. Int J Mol Sci. 2021;22(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keiser NW, and Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med. 2011;17(6):478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semaniakou A, Croll RP, and Chappe V. Animal Models in the Pathophysiology of Cystic Fibrosis. Front Pharmacol. 2018;9:1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.••.McCarron A, Parsons D, and Donnelley M. Animal and Cell Culture Models for Cystic Fibrosis: Which Model Is Right for Your Application? Am J Pathol. 2021;191(2):228–42. [DOI] [PubMed] [Google Scholar]; Most recent, comprehensive review of CF animal models
- 39.McCarron A, Donnelley M, and Parsons D. Airway disease phenotypes in animal models of cystic fibrosis. Respir Res. 2018;19(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, et al. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. 2018;17(2S):S28–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet (London, England). 2019;394(10212):1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med. 2019;381(19):1809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middleton PG, and Taylor-Cousar JL. Development of elexacaftor - tezacaftor - ivacaftor: Highly effective CFTR modulation for the majority of people with Cystic Fibrosis. Expert review of respiratory medicine. 2021;15(6):723–35. [DOI] [PubMed] [Google Scholar]
- 44.••.Nichols DP, Donaldson SH, Frederick CA, Freedman SD, Gelfond D, Hoffman LR, et al. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J Cyst Fibros. 2021;20(2):205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of critical research needs for PwCF in the HEMT era
- 45.Lore NI, Sipione B, He G, Strug LJ, Atamni HJ, Dorman A, et al. Collaborative Cross Mice Yield Genetic Modifiers for Pseudomonas aeruginosa Infection in Human Lung Disease. mBio. 2020;11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meeker SM, Mears KS, Sangwan N, Brittnacher MJ, Weiss EJ, Treuting PM, et al. CFTR dysregulation drives active selection of the gut microbiome. PLoS Pathog. 2020;16(1):e1008251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pashuck TD, Franz SE, Altman MK, Wasserfall CH, Atkinson MA, Wronski TJ, et al. Murine model for cystic fibrosis bone disease demonstrates osteopenia and sex-related differences in bone formation. Pediatr Res. 2009;65(3):311–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durie PR, Kent G, Phillips MJ, and Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164(4):1481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geiser M, Stoeger T, Casaulta M, Chen S, Semmler-Behnke M, Bolle I, et al. Biokinetics of nanoparticles and susceptibility to particulate exposure in a murine model of cystic fibrosis. Part Fibre Toxicol. 2014;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.•.Tang Y, Yan Z, and Engelhardt JF. Viral Vectors, Animal Models, and Cellular Targets for Gene Therapy of Cystic Fibrosis Lung Disease. Hum Gene Ther. 2020;31(9–10):524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent, comprehensive review of CF animal models for gene therapy
- 51.Ensinck M, Mottais A, Detry C, Leal T, and Carlon MS. On the Corner of Models and Cure: Gene Editing in Cystic Fibrosis. Front Pharmacol. 2021;12:662110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Y, Yan Z, Lin S, Huntemann ED, Feng Z, Park SY, et al. Repeat Dosing of AAV2.5T to Ferret Lungs Elicits an Antibody Response That Diminishes Transduction in an Age-Dependent Manner. Mol Ther Methods Clin Dev. 2020;19:186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farrow N, Cmielewski P, Delhove J, Rout-Pitt N, Vaughan L, Kuchel T, et al. Towards Human Translation of Lentiviral Airway Gene Delivery for Cystic Fibrosis: A One-Month CFTR and Reporter Gene Study in Marmosets. Hum Gene Ther. 2021;32(15–16):806–16. [DOI] [PubMed] [Google Scholar]
- 54.Cooney AL, and Sinn PL. Intratracheal aerosolization of viral vectors to newborn pig airways. Biotechniques. 2020;68(5):235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haque A, Dewerth A, Antony JS, Riethmuller J, Schweizer GR, Weinmann P, et al. Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis. Sci Rep. 2018;8(1):16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reyne N, Cmielewski P, McCarron A, Delhove J, Parsons D, and Donnelley M. Single-Dose Lentiviral Mediated Gene Therapy Recovers CFTR Function in Cystic Fibrosis Knockout Rats. Front Pharmacol. 2021;12:682299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cooney AL, Thornell IM, Singh BK, Shah VS, Stoltz DA, McCray PB Jr., et al. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. American journal of respiratory cell and molecular biology. 2019;61(6):747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim N, Duncan GA, Hanes J, and Suk JS. Barriers to inhaled gene therapy of obstructive lung diseases: A review. J Control Release. 2016;240:465–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brinks V, Lipinska K, de Jager M, Beumer W, Button B, Livraghi-Butrico A, et al. The Cystic Fibrosis-Like Airway Surface Layer Is not a Significant Barrier for Delivery of Eluforsen to Airway Epithelial Cells. J Aerosol Med Pulm Drug Deliv. 2019;32(5):303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrow N, Cmielewski P, Donnelley M, Rout-Pitt N, Moodley Y, Bertoncello I, et al. Epithelial disruption: a new paradigm enabling human airway stem cell transplantation. Stem Cell Res Ther. 2018;9(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allan KM, Farrow N, Donnelley M, Jaffe A, and Waters SA. Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front Pharmacol. 2021;12:639475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McHugh DR, Steele MS, Valerio DM, Miron A, Mann RJ, LePage DF, et al. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PloS one. 2018;13(6):e0199573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gawenis LR, Hodges CA, McHugh DR, Valerio DM, Miron A, Cotton CU, et al. A BAC Transgene Expressing Human CFTR under Control of Its Regulatory Elements Rescues Cftr Knockout Mice. Sci Rep. 2019;9(1):11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cho DY, Zhang S, Skinner DF, Lim DJ, Banks C, Grayson JW, et al. Ivacaftor restores delayed mucociliary transport caused by Pseudomonas aeruginosa-induced acquired cystic fibrosis transmembrane conductance regulator dysfunction in rabbit nasal epithelia. Int Forum Allergy Rhinol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Birket SE, Davis JM, Fernandez-Petty CM, Henderson AG, Oden AM, Tang L, et al. Ivacaftor Reverses Airway Mucus Abnormalities in a Rat Model Harboring a Humanized G551D-CFTR. Am J Respir Crit Care Med. 2020;202(9):1271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Green M, Lindgren N, Henderson A, Keith JD, Oden AM, and Birket SE. Ivacaftor partially corrects airway inflammation in a humanized G551D rat. Am J Physiol Lung Cell Mol Physiol. 2021;320(6):L1093–L100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meyerholz DK, Stoltz DA, Gansemer ND, Ernst SE, Cook DP, Strub MD, et al. Lack of cystic fibrosis transmembrane conductance regulator disrupts fetal airway development in pigs. Lab Invest. 2018;98(6):825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bonvin E, Le Rouzic P, Bernaudin JF, Cottart CH, Vandebrouck C, Crie A, et al. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulator-deficient mice. The Journal of physiology. 2008;586(13):3231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang EH, Pezzulo AA, Meyerholz DK, Potash AE, Wallen TJ, Reznikov LR, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122(9):1898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plyler ZE, Birket SE, Schultz BD, Hong JS, Rowe SM, Petty CF, et al. Non-obstructive vas deferens and epididymis loss in cystic fibrosis rats. Mech Dev. 2019;155:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pierucci-Alves F, Akoyev V, Stewart JC 3rd, Wang LH, Janardhan KS, and Schultz BD. Swine models of cystic fibrosis reveal male reproductive tract phenotype at birth. Biol Reprod. 2011;85(3):442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boucher RC. Muco-Obstructive Lung Diseases. N Engl J Med. 2019;380(20):1941–53. [DOI] [PubMed] [Google Scholar]
- 73.Wine JJ, Hansson GC, Konig P, Joo NS, Ermund A, and Pieper M. Progress in understanding mucus abnormalities in cystic fibrosis airways. J Cyst Fibros. 2018;17(2S):S35–S9. [DOI] [PubMed] [Google Scholar]
- 74.Hansson GC. Mucus and mucins in diseases of the intestinal and respiratory tracts. J Intern Med. 2019;285(5):479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morrison CB, Markovetz MR, and Ehre C. Mucus, mucins, and cystic fibrosis. Pediatr Pulmonol. 2019;54 Suppl 3:S84–S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.••.Esther CR Jr., Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Science translational medicine. 2019;11(486). [DOI] [PMC free article] [PubMed] [Google Scholar]; Pathogenesis of lung disease in children with CF
- 77.Xie Y, Ostedgaard L, Abou Alaiwa MH, Lu L, Fischer AJ, and Stoltz DA. Mucociliary Transport in Healthy and Cystic Fibrosis Pig Airways. Annals of the American Thoracic Society. 2018;15(Suppl 3):S171–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.••.Rosen BH, Evans TIA, Moll SR, Gray JS, Liang B, Sun X, et al. Infection Is Not Required for Mucoinflammatory Lung Disease in CFTR-Knockout Ferrets. Am J Respir Crit Care Med. 2018;197(10):1308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence of lung disease independent of bacterial infection in CF ferrets
- 79.Choi HK, Finkbeiner WE, and Widdicombe JH. A comparative study of mammalian tracheal mucous glands. J Anat. 2000;197 Pt 3:361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tiddens HA, and Rosenow T. What did we learn from two decades of chest computed tomography in cystic fibrosis? Pediatr Radiol. 2014;44(12):1490–5. [DOI] [PubMed] [Google Scholar]
- 81.Yu W, Moninger TO, Thurman AL, Xie Y, Jain A, Zarei K, et al. Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs. Proceedings of the National Academy of Sciences of the United States of America. 2022;119(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernandez-Petty CM, Hughes GW, Bowers HL, Watson JD, Rosen BH, Townsend SM, et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight. 2019;4(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.••.Pieper M, Schulz-Hildebrandt H, Mall MA, Huttmann G, and Konig P. Intravital microscopic optical coherence tomography imaging to assess mucus-mobilizing interventions for muco-obstructive lung disease in mice. Am J Physiol Lung Cell Mol Physiol. 2020;318(3):L518–L24. [DOI] [PMC free article] [PubMed] [Google Scholar]; Translational application of OCT imaging in mice
- 84.Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, et al. Defective innate immunity and hyperinflammation in newborn cystic fibrosis transmembrane conductance regulator-knockout ferret lungs. American journal of respiratory cell and molecular biology. 2015;52(6):683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rogers TD, Ostrowski LE, Livraghi-Butrico A, Button B, and Grubb BR. Mucociliary Clearance in Mice Measured by Tracking Trans-tracheal Fluorescence of Nasally Aerosolized Beads. Sci Rep. 2018;8(1):14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wielputz MO, Eichinger M, Zhou Z, Leotta K, Hirtz S, Bartling SH, et al. In vivo monitoring of cystic fibrosis-like lung disease in mice by volumetric computed tomography. Eur Respir J. 2011;38(5):1060–70. [DOI] [PubMed] [Google Scholar]
- 87.Stahr CS, Samarage CR, Donnelley M, Farrow N, Morgan KS, Zosky G, et al. Quantification of heterogeneity in lung disease with image-based pulmonary function testing. Sci Rep. 2016;6:29438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murrie RP, Werdiger F, Donnelley M, Lin YW, Carnibella RP, Samarage CR, et al. Real-time in vivo imaging of regional lung function in a mouse model of cystic fibrosis on a laboratory X-ray source. Sci Rep. 2020;10(1):447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, et al. An Improved Inhaled Mucolytic to Treat Airway Muco-Obstructive Diseases. Am J Respir Crit Care Med. 2019;199(2):171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Graeber SY, Zhou-Suckow Z, Schatterny J, Hirtz S, Boucher RC, and Mall MA. Hypertonic saline is effective in the prevention and treatment of mucus obstruction, but not airway inflammation, in mice with chronic obstructive lung disease. American journal of respiratory cell and molecular biology. 2013;49(3):410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou Z, Treis D, Schubert SC, Harm M, Schatterny J, Hirtz S, et al. Preventive but not late amiloride therapy reduces morbidity and mortality of lung disease in betaENaC-overexpressing mice. Am J Respir Crit Care Med. 2008;178(12):1245–56. [DOI] [PubMed] [Google Scholar]
- 92.Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TI, et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. American journal of respiratory cell and molecular biology. 2014;50(3):502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Science translational medicine. 2010;2(29):29ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Darrah R, Bonfield T, LiPuma JJ, Litman P, Hodges CA, Jacono F, et al. Cystic Fibrosis Mice Develop Spontaneous Chronic Bordetella Airway Infections. J Infect Pulm Dis. 2017;3(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kukavica-Ibrulj I, and Levesque RC. Animal models of chronic lung infection with Pseudomonas aeruginosa: useful tools for cystic fibrosis studies. Lab Anim. 2008;42(4):389–412. [DOI] [PubMed] [Google Scholar]
- 96.Cigana C, Ranucci S, Rossi A, De Fino I, Melessike M, and Bragonzi A. Antibiotic efficacy varies based on the infection model and treatment regimen for Pseudomonas aeruginosa. Eur Respir J. 2020;55(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen F, Cheng X, Li J, Yuan X, Huang X, Lian M, et al. Novel Lytic Phages Protect Cells and Mice against Pseudomonas aeruginosa Infection. J Virol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kirby BD, Al Ahmar R, Withers TR, Valentine ME, Valentovic M, Long TE, et al. Efficacy of Aerosolized Rifaximin versus Tobramycin for Treatment of Pseudomonas aeruginosa Pneumonia in Mice. Antimicrob Agents Chemother. 2019;63(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pont S, and Blanc-Potard AB. Zebrafish Embryo Infection Model to Investigate Pseudomonas aeruginosa Interaction With Innate Immunity and Validate New Therapeutics. Front Cell Infect Microbiol. 2021;11:745851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kiedrowski MR, and Bomberger JM. Viral-Bacterial Co-infections in the Cystic Fibrosis Respiratory Tract. Front Immunol. 2018;9:3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Degiacomi G, Sammartino JC, Chiarelli LR, Riabova O, Makarov V, and Pasca MR. Mycobacterium abscessus, an Emerging and Worrisome Pathogen among Cystic Fibrosis Patients. Int J Mol Sci. 2019;20(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Riva C, Tortoli E, Cugnata F, Sanvito F, Esposito A, Rossi M, et al. A New Model of Chronic Mycobacterium abscessus Lung Infection in Immunocompetent Mice. Int J Mol Sci. 2020;21(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Perrem L, and Ratjen F. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr Pulmonol. 2019;54 Suppl 3:S46–S55. [DOI] [PubMed] [Google Scholar]
- 104.Laucirica DR, Garratt LW, and Kicic A. Progress in Model Systems of Cystic Fibrosis Mucosal Inflammation to Understand Aberrant Neutrophil Activity. Front Immunol. 2020;11:595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bruscia EM, and Bonfield TL. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J Innate Immun. 2016;8(6):550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paroni M, Moalli F, Nebuloni M, Pasqualini F, Bonfield T, Nonis A, et al. Response of CFTR-deficient mice to long-term chronic Pseudomonas aeruginosa infection and PTX3 therapy. J Infect Dis. 2013;208(1):130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Litman PM, Day A, Kelley TJ, and Darrah RJ. Serum inflammatory profiles in cystic fibrosis mice with and without Bordetella pseudohinzii infection. Sci Rep. 2021;11(1):17535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bruscia EM, Zhang PX, Barone C, Scholte BJ, Homer R, Krause DS, et al. Increased susceptibility of Cftr−/− mice to LPS-induced lung remodeling. Am J Physiol Lung Cell Mol Physiol. 2016;310(8):L711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van Heeckeren AM, Sutton MT, Fletcher DR, Hodges CA, Caplan AI, and Bonfield TL. Enhancing Cystic Fibrosis Immune Regulation. Front Pharmacol. 2021;12:573065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hey J, Paulsen M, Toth R, Weichenhan D, Butz S, Schatterny J, et al. Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat Commun. 2021;12(1):6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schupp JC, Khanal S, Gomez JL, Sauler M, Adams TS, Chupp GL, et al. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am J Respir Crit Care Med. 2020;202(10):1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sathe M, and Houwen R. Is meconium ileus associated with worse outcomes in cystic fibrosis? J Cyst Fibros. 2019;18(6):746. [DOI] [PubMed] [Google Scholar]
- 113.S JB, Hachem C, and Abraham JM. Luminal Gastrointestinal Manifestations of Cystic Fibrosis. Curr Gastroenterol Rep. 2021;23(3):4. [DOI] [PubMed] [Google Scholar]
- 114.Olivier AK, Gibson-Corley KN, and Meyerholz DK. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am J Physiol Gastrointest Liver Physiol. 2015;308(6):G459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gibson-Corley KN, and Engelhardt JF. Animal Models and Their Role in Understanding the Pathophysiology of Cystic Fibrosis-Associated Gastrointestinal Lesions. Annu Rev Pathol. 2021;16:51–67. [DOI] [PubMed] [Google Scholar]
- 116.Grubb BR, and Gabriel SE. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. Am J Physiol. 1997;273(2 Pt 1):G258–66. [DOI] [PubMed] [Google Scholar]
- 117.Bodewes FA, van der Wulp MY, Beharry S, Doktorova M, Havinga R, Boverhof R, et al. Altered intestinal bile salt biotransformation in a cystic fibrosis (Cftr−/−) mouse model with hepato-biliary pathology. J Cyst Fibros. 2015;14(4):440–6. [DOI] [PubMed] [Google Scholar]
- 118.Lord R, Fairbourn N, Mylavarapu C, Dbeis A, Bowman T, Chandrashekar A, et al. Consuming Genistein Improves Survival Rates in the Absence of Laxative in DeltaF508-CF Female Mice. Nutrients. 2018;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun X, Olivier AK, Yi Y, Pope CE, Hayden HS, Liang B, et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am J Pathol. 2014;184(5):1309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vitko M, Valerio DM, Rye PD, Onsoyen E, Myrset AH, Dessen A, et al. A novel guluronate oligomer improves intestinal transit and survival in cystic fibrosis mice. J Cyst Fibros. 2016;15(6):745–51. [DOI] [PubMed] [Google Scholar]
- 121.Hodges CA, Grady BR, Mishra K, Cotton CU, and Drumm ML. Cystic fibrosis growth retardation is not correlated with loss of Cftr in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ballard ST, Evans JW, Drag HS, and Schuler M. Pathophysiologic Evaluation of the Transgenic Cftr “Gut-Corrected” Porcine Model of Cystic Fibrosis. Am J Physiol Lung Cell Mol Physiol. 2016;311(4):L779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.•.Zarei K, Meyerholz DK, and Stoltz DA. Early intrahepatic duct defects in a cystic fibrosis porcine model. Physiol Rep. 2021;9(14):e14978. [DOI] [PMC free article] [PubMed] [Google Scholar]; Characterization of liver disease in CF pigs
- 124.Zarei K, Stroik MR, Gansemer ND, Thurman AL, Ostedgaard LS, Ernst SE, et al. Early pathogenesis of cystic fibrosis gallbladder disease in a porcine model. Lab Invest. 2020;100(11):1388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.•.Rotti PG, Xie W, Poudel A, Yi Y, Sun X, Tyler SR, et al. Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. Am J Pathol. 2018;188(4):876–90. [DOI] [PMC free article] [PubMed] [Google Scholar]; Characterization of the kinetics of pancreatic disease in CF ferrets
- 126.Karb DB, and Cummings LC. The Intestinal Microbiome and Cystic Fibrosis Transmembrane Conductance Regulator Modulators: Emerging Themes in the Management of Gastrointestinal Manifestations of Cystic Fibrosis. Curr Gastroenterol Rep. 2021;23(10):17. [DOI] [PubMed] [Google Scholar]
- 127.Scott P, Anderson K, Singhania M, and Cormier R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int J Mol Sci. 2020;21(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Norkina O, Burnett TG, and De Lisle RC. Bacterial overgrowth in the cystic fibrosis transmembrane conductance regulator null mouse small intestine. Infect Immun. 2004;72(10):6040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Canale-Zambrano JC, Auger ML, and Haston CK. Toll-like receptor-4 genotype influences the survival of cystic fibrosis mice. Am J Physiol Gastrointest Liver Physiol. 2010;299(2):G381–90. [DOI] [PubMed] [Google Scholar]
- 130.Liang X, Bouhamdan M, Hou X, Zhang K, Song J, Hao K, et al. Intestinal Dysbiosis in Young Cystic Fibrosis Rabbits. J Pers Med. 2021;11(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim K, Hung RJ, and Perrimon N. miR-263a Regulates ENaC to Maintain Osmotic and Intestinal Stem Cell Homeostasis in Drosophila. Dev Cell. 2017;40(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.••.Raredon MSB, Adams TS, Suhail Y, Schupp JC, Poli S, Neumark N, et al. Single-cell connectomic analysis of adult mammalian lungs. Sci Adv. 2019;5(12):eaaw3851. [DOI] [PMC free article] [PubMed] [Google Scholar]; Novel analytical algorithm to query common pathways of lung development and repair across different mammalian species