

Abstract
Engineering mechanically-robust bioadhesive hydrogels that can withstand large strains may open new opportunities for the sutureless sealing of highly stretchable tissues. While typical chemical modifications of hydrogels, such as increasing the functional group density of crosslinkable moieties and blending them with other polymers or nanomaterials have resulted in improved mechanical stiffness, the modified hydrogels have often exhibited increased brittleness resulting in deteriorated sealing capabilities under large strains. Furthermore, highly elastic hydrogels, such as tropoelastin derivatives are highly expensive. Here, gelatin methacryloyl (GelMA) is hybridized with methacrylate-modified alginate (AlgMA) to impart ion-induced reversible crosslinking that can dissipate energy under strain. The hybrid hydrogels provide a photocrosslinkable, injectable, and bioadhesive platform with an excellent toughness that can be tailored using divalent cations, such as calcium. This class of hybrid biopolymers with more than 600% improved toughness compared to GelMA may set the stage for durable, mechanically-resilient, and cost-effective tissue sealants. This strategy to increase the toughness of hydrogels may be extended to other crosslinkable polymers with similarly-reactive moieties.
Keywords: Tough hydrogel, sutureless tissue sealing, gelatin methacryloyl, alginate, bladder injury, anastomosis
1. Introduction
Minimally invasive sealing of injured tissues and organs is of utmost importance in biomedicine.[1–7] The global market for hemostats and tissue sealants was more than $4.6 billion in 2017, which is expected to grow beyond $10 billion by 2027.[8] The integration of sealant biomaterials with tissues undergoing extensive daily activities, such as stretching and contraction, is an unmet clinical need that requires stretchable and biocompatible material platforms. Various tissue injuries, such as skin and muscle laceration and burn,[9–11] cardiac trauma,[12] lung puncture,[13–18] liver bleeding,[19] cornea,[20,21] and teeth and gum injuries[22–26] as well the anastomosis of tubular tissues[27–30] have been treated with tissue adhesive biomaterials to leverage a regenerative microenvironment while providing a physical support.
Organs that naturally undergo dynamic shape changes, such as lung, heart, and bladder are extremely challenging to seal using an adhesive biomaterial. A suitable adhesive should adhere well to the wet tissue and adapt to the external mechanical stimuli that are often cyclic. Despite significant advances in developing elastic hydrogels, the majority of bioadhesive hydrogels undergo rupture due to brittleness and a fracture energy in the order of 10 J m−2, which is almost two orders of magnitude lower than that of soft tissues, [31–37] To address this challenge, some efforts have been devoted to design stretchable bioadhesives based on crosslinkable biomaterials.
Tough hydrogels that can dissipate energy and undergo plastic deformation have been engineered using a double/multi-network hydrogels. [38–43] As an example, polyacrylamide has been mixed with alginate to increase the toughness of pristine polymer [31,44] via the synergistic contribution of ionically-crosslinkable alginate with covalently crosslinked polyacrylamide. The physical crosslinking of alginate inside the chemically-crosslinked polyacrylamide enabled the hydrogels to stretch beyond 20 times their initial length and provided a fracture energy of ~9000 J m−2. The improved mechanical properties of the hybrid hydrogels have been attributed to the synergy between two mechanisms: (i) crack bridging by the covalent network and (ii) energy dissipation by the secondary, often physical network permitting reversible bond breakage. The covalent crosslinking supports the physical state of hydrogels while the physically-formed, reversible network dissipates the external energy, protecting the scaffold from permanent mechanical damage. As polyacrylamide-based tough hydrogels do not adhere well to tissues, post-treatments for surface modification is necessary after crosslinking these hydrogels. [44]
Another class of elastic and biodegradable sealants based on chemically-modified tropoelastin (MeTro), a recombinant human protein, has been developed recently.[18] Although these hydrogels had lower mechanical strength and elasticity compared to some of the double-network hydrogels, they exhibited higher tissue adhesion and elasticity compared to the commercially available sealants, such as Evicel®, Coseal®, and Progel®. Standard wound closure experiments showed that MeTro (20% w/v) was able to stretch 2 times its initial length before breaking, providing a tissue adhesive hydrogel that could seal defected sites on elastic tissues, such as lung and blood vessels, in vivo. The extremely high production cost of MeTro has significantly limited its translation for clinical applications. Accordingly, there is currently a significant medical need for bioadhesive, biodegradable, and tough tissue sealants that can be produced at a relatively low cost, preferably from naturally-derived, biodegradable materials.
In this work, we aim to develop a tough, cost-effective, biodegradable, bioadhesive, and injectable hydrogel based on naturally-derived, biocompatible materials. We aim to design a hybrid hydrogel based on gelatin and alginate, two of the most clinically-used biomaterials, functionalized with vinyl groups and evaluate their capability in sealing highly-stretchable tissues. We show how methacrylate-modified alginate (AlgMA) contributes to the formation of an energy dissipating ionic network while providing covalent bonding with gelatin methacryloyl (GelMA), imparting unique mechanical properties to the adhesive hybrid hydrogel. We detail how this platform may pave the way for developing a new class of superior injectable hydrogels with promising tissue adhesion capability and toughness.
2. Results and discussion
The hybrid hydrogels are composed of two widely used, biocompatible biopolymers, gelatin and alginate, both modified with vinyl groups via a facile reaction with methacrylic anhydride. The pendant methacrylic/methacryloyl groups can be activated using a broad range of initiators, such as photo-initiators for the photo-crosslinking of biopolymer solutions. The interactions among the biopolymers and tissues are presented in Figure 1. GelMA and AlgMA can be self-crosslinked or covalently attached to each other via the photo-activated reaction of vinyl groups, forming a mechanically resilient network of hydrophilic biopolymers, which can provide a biocompatible microenvironment. The MA groups on these biopolymers may also react with the amine groups on the tissue to form chemical bonds, fostering tissue adhesion. Hydrogen bonds can also be formed between GelMA and AlgMA at the tissue-biopolymer interface, enforcing the tissue adhesion. Importantly, calcium ions physically crosslink AlgMA to form an egg-box structure via the electrostatic interactions between the α-L-(1–4)-guluronate residues (G blocks) and calcium cations.[45,46] The shear force can reversibly increase the distance between the charged moieties in the egg-box structure, reducing their electrostatic attraction, which can be recovered when the external shear is eliminated.
Figure 1. Schematic of a GelMA-AlgMA hybrid hydrogel undergoing photo/ionic crosslinking and tissue adhesion.
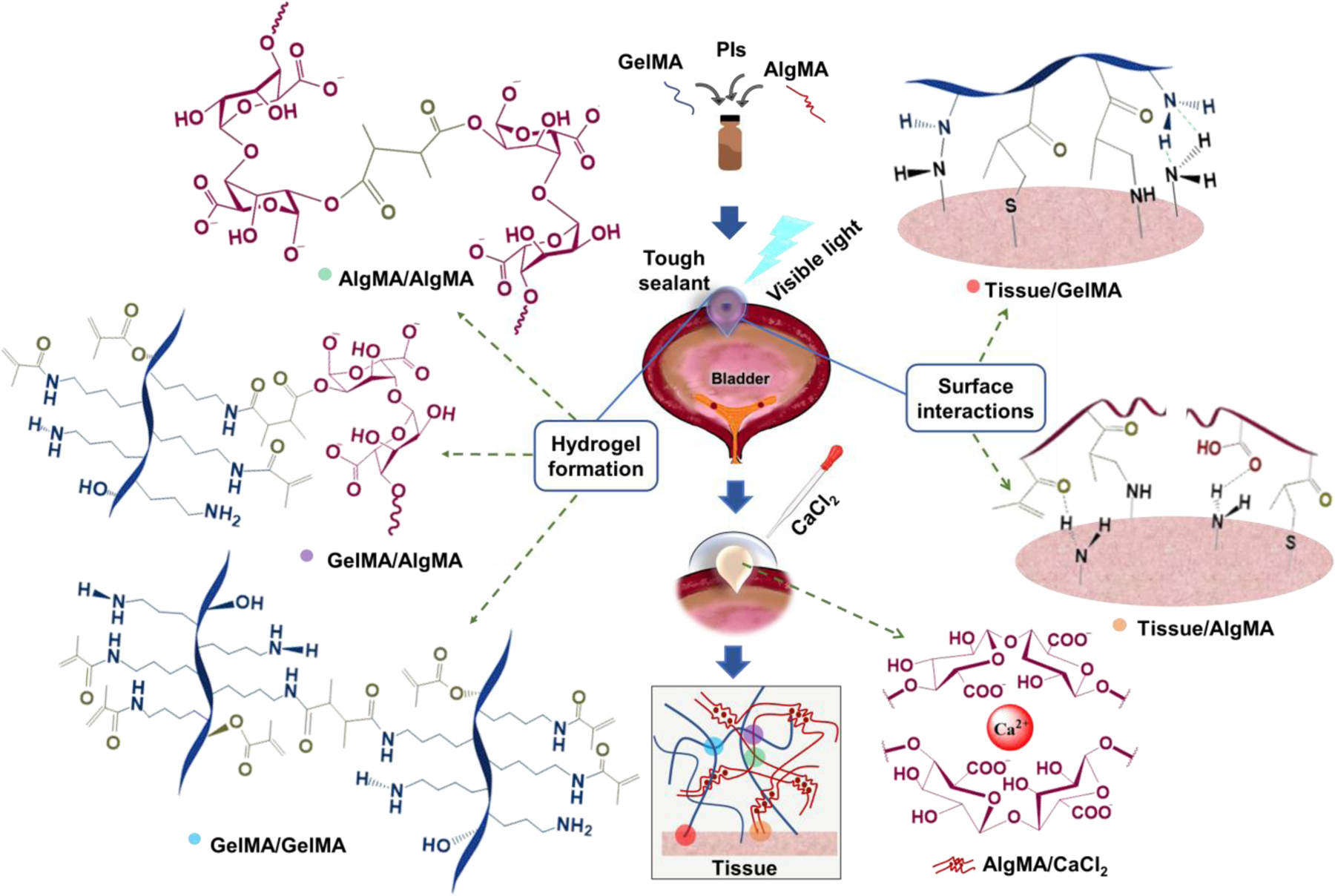
Both AlgMA and GelMA undergo covalent crosslinking through the photo-initiated polymerization of methacrylate/methacryloyl (MA) groups. In AlgMA, the G blocks on the polymer chains form ionic bonds with Ca2+, providing a reversibly crosslinked network. Crosslinking hybrid GelMA/AlgMA hydrogels yields two types of polymer networks intertwined and connected by covalent bonds (via MA groups) supplemented by the Ca2+-mediated physical bonds of AlgMA. GelMA may interact with amine-rich biological tissues through the formation of hydrogen bonds as well as covalent bonding of amine-MA and thiol-MA groups. AlgMA can interact with the tissue via hydrogen bonding, covalent bonding, and/or electrostatic interactions between the carboxylate and amino groups.
The 1HNMR spectra of pre- and post-crosslinked hydrogels are shown in Figure S1 (Supporting Information). The degree of methacrylation for GelMA and AlgMA were ~ 64% and 19%, respectively. Peaks in the shaded area reflects the methacrylation of parent components, gelatin and alginate. The low degree of conjugation for alginate can be attributed to its structural complexity compared to gelatin. For GelMA and AlgMA, ~ 53% and 49% crosslinking of MA groups were observed, respectively, when visible light was exposed to the hydrogels for 4 min. In the 1HNMR spectra, the normalized intensity of vinyl hydrogen related to the acrylic groups decreased upon photo-induced crosslinking. The degree of crosslinking was calculated by integrating the double bond proton peaks (vinyl) considering methyl peaks as reference.[47,48] Same procedure was followed to calculate the percentage of crosslinking for the hybrid hydrogel, which was found to be approximately 65%.
To develop a stretchable, tough sealant derived from two abundant biopolymers, we combined GelMA and AlgMA at a fixed GelMA concentration (20 % w/v). The GelMA concentration was chosen based on our previous studies, establishing the optimum concentration of GelMA to achieve the best sealing performance while providing an injectable pre-gel solution.[20] We studied the effect of AlgMA concentration on the physical properties of hybrid hydrogels. Figure 2a presents the visual difference between GelMA hydrogel and the hybrid GelMA-AlgMA hydrogel under tensile stress. Only 40% of strain is able to break the GelMA hydrogel, seen in the middle panel; however, adding only 2% AlgMA to GelMA, photocrosslinking followed by ion-mediated physical crosslinking of hybrid hydrogel increase the failure strain from 40% to 80%. Examples of tensile strain-stress curves for the hybrid hydrogels are shown in Figure 2b. The tensile stress linearly increases as the strain increases until the hydrogel is broken. The tensile strain at break (Figure 2c) shows that increasing the AlgMA concentration up to 2–3% (w/v) increases the ultimate strain (stretchability) of hybrid hydrogels by 100%. Further increasing AlgMA concentration decreases the stretchability, possibly as a result of increased brittleness. The increased brittleness may be a result of increased chemical crosslinking density, decreasing the length of polymer chains between crosslinking nodes. The shortened chain size results in the decreased fracture energy.[31]
Figure 2. Mechanical and rheological properties of hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA.
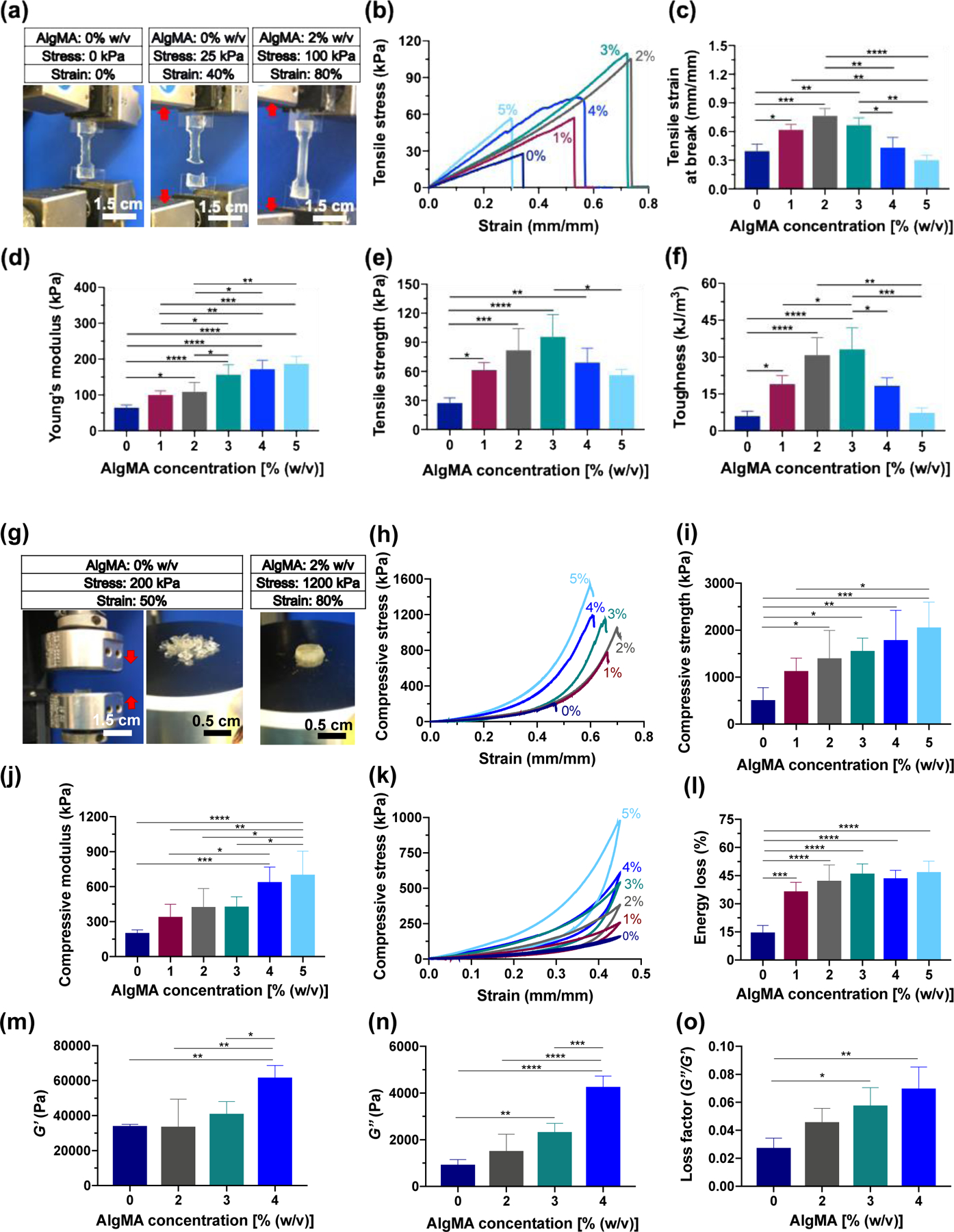
(a) Images of the hybrid hydrogels containing 0% and 2% (w/v) AlgMA undergoing stretching. (b) Representative tensile stress-strain curves, (c) tensile strain at break, (d) Young’s modulus, (e) tensile strength, and (f) toughness for the hybrid hydrogels containing varying concentrations of AlgMA. (g) Images of the hybrid hydrogels undergoing compression, and (h) representative compressive stress-strain curves, (i) compressive strength, (j) compressive modulus, (k) cyclic compressive stress-strain curves, and (l) energy loss for the hybrid hydrogels. The (m) storage modulus, (n) loss modulus, and (o) loss factor at angular frequency = 1 rad s−1 and strain = 0.1% for the hybrid hydrogels. Data are reported as the mean values of at least 5 experiments ± their standard deviation. The statistical analysis was done according to the methods explained in “Statistical analysis” section. Asterisks show the results that are statically significant with p-values < 0.05 (*), 0.01 (**), 0.001 (***), or 0.0001 (****).
The tensile (Young’s) modulus, shown in Figure 2d, continually increases by increasing the AlgMA concentration as a result of increased crosslinking density. At 3% AlgMA, the Young’s modulus is ~ 3 times the modulus in the absence of AlgMA. The tensile strength, i.e., the tensile stress at break, has a similar behavior to ultimate tensile strain. Figure 2e presents the tensile strength of hybrid hydrogels containing varying concentrations of AlgMA. Increasing the AlgMA concentration increases the tensile strength by a factor of 3 at an AlgMA concentration of 2–3%, whereas high AlgMa concentrations (>3%) significantly decreases the tensile strength. Figure 2f shows the toughness of the hybrid hydrogels, obtained from the area under the tensile strain-stress curves. Interestingly, the toughness significantly increases by increasing the AlgMA concentration to 2–3%. At this concentration of AlgMA, the toughness of hybrid hydrogels is more than 300% of the GelMA hydrogel. Increasing the AlgMA concentration further than 3%, significantly decreases the toughness of the hybrid hydrogels. At ≥5% AlgMA, the hybrid hydrogels have toughness very similar to that of an AlgMA-free hydrogel, which is due to the increased brittleness and weakened stretchability of hydrogels.
Another important property of bioadhesive materials is their resistance against compression. A suitable bioadhesive must resist against compressive stress and should not break. The stress may originate from daily activities, such as running, or unwanted events, such as falling down o hitting an object. Figure 2g presents the images of the GelMA hydrogel and a hybrid GelMA-AlgMA hydrogel post compression. The GelMA hydrogel undergoes severe mechanical damage at only 50% strain, whereas the hybrid hydrogel including 2% AlgMA withstands 3 times higher compressive stress and holds its integrity at more than 80% strain. Such a remarkable improvement in the mechanical properties of GelMA may be of interest in a broad range of biomedical applications, particularly for developing regenerative, tough cell-laden scaffolds. Figure 2h presents examples of compressive strain-stress curves for the hybrid hydrogels. As can be seen in this figure, at a given strain, the compressive stress of a hybrid hydrogel increases as the AlgMA concentration increases. Figure 2i presents the compressive strength, defined as the maximum stress that the hydrogels can withstand before breaking, versus AlgMA concentration. Increasing the AlgMA concentration continually increases the compressive strength. Similarly, the compressive modulus of hybrid hydrogels increases when the AlgMA concentration increases (Figure 2j). Increased stiffness of hybrid hydrogels is mainly a result of covalent binding among GelMA and AlgMA biopolymers. Figure 2k shows the cyclic compression of hybrid hydrogels. Increasing the AlgMA concentration increases the compressive hysteresis of hydrogels as a result of unzipping the Ca2+-crosslinked G blocks of AlgMA followed by the re-establishing of physical bonds in the egg-box structure. The energy loss, shown in Figure 2l, obtained from the area inside the compressive hysteresis curve increases from ~ 15% for the GelMA hydrogel to ~ 45% when AlgMA concentration is ~2–3% w/v.
Figure 2m–o present the rheological properties of hybrid hydrogels comprising varying concentrations of AlgMA. The storage modulus (G’, Figure 2m) and loss modulus (G’’, Figure 2n) of hybrid hydrogels, measured through small angle oscillatory rheology at oscillatory frequency ~ 1 rad s−1 and strain ~ 0.1% continually increase by increasing the AlgMA concentration. The effect of AlgMA is more pronounced on the loss moduli, possibly because of the improved energy dissipation as a result of reversible disruption of Ca2+-mediated physical bonds between G blocks. The loss factor or damping factor (Figure 2o), defined as the ratio of G’’ to G’ shows that while the hybrid hydrogels have a solid-like behavior, i.e., loss factor << 1, increasing the AlgMA concentration continually increases the loss factor as a result of increased energy dissipation at a low oscillatory strain. The viscoelastic moduli versus oscillatory shear strain (at angular frequency ~ 1 rad s−1) and versus angular frequency (at oscillatory shear strain ~ 0.1%) for the hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA are shown in Figure S2 (Supporting Information). Note that the improvement in the mechanical properties of hybrid hydrogels is not the result of increase solid content (Figure S3, Supporting Information).
The physical properties of hybrid hydrogels, particularly swelling and degradation are important in evaluating their potential for sealing tissues and supporting the tissue regeneration process. A high swelling ratio post crosslinking may result in inevitable stress imposed to the tissue-hydrogel interface, resulting in the delamination of the adhered gel. Figure 3a shows the swelling kinetics of hybrid hydrogels versus incubation time in DPBS at 37°C. Regardless of the AlgMA concentration, the hybrid hydrogels reach their equilibrium swelling in about 4 h post crosslinking. Figure 3b presents the equilibrium swelling ratio of hydrogels after 4 h incubation in DPBS. Increasing the AlgMA concentration increases the swelling ratio from ~ 5% for AlgMA-free GelMA to ~ 20% when the AlgMA concentration is about 3%. The degradation of hybrid hydrogels in the presence of collagenase, one of the main enzymes responsible for degrading collagen in vivo, was evaluated. Figure 3c presents the kinetics of enzyme-mediated degradation of hybrid hydrogels within 5 weeks post crosslinking. In the presence of collagenase (1.25 U/mL), all the hydrogels undergo degradation over time. At low AlgMA concentrations (≤ 2%), in 1 week, more than 40% of the hydrogels degrade and by the 5th week, less than 20% of them were remained. Figure 3d shows the degradation of hydrogels after 5 weeks of incubation in the collagenase solution. Increasing the AlgMA concentration continually decreased the degradation rate of hydrogels from ~ 90% in the absence of AlgMA to ~ 40% when 4–5% AlgMA was present. When the AlgMA concentration is less than 2%, there is no significant change in the degradation of hybrid hydrogels compared to the degradation of GelMA.
Figure 3. Physical properties of hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA.
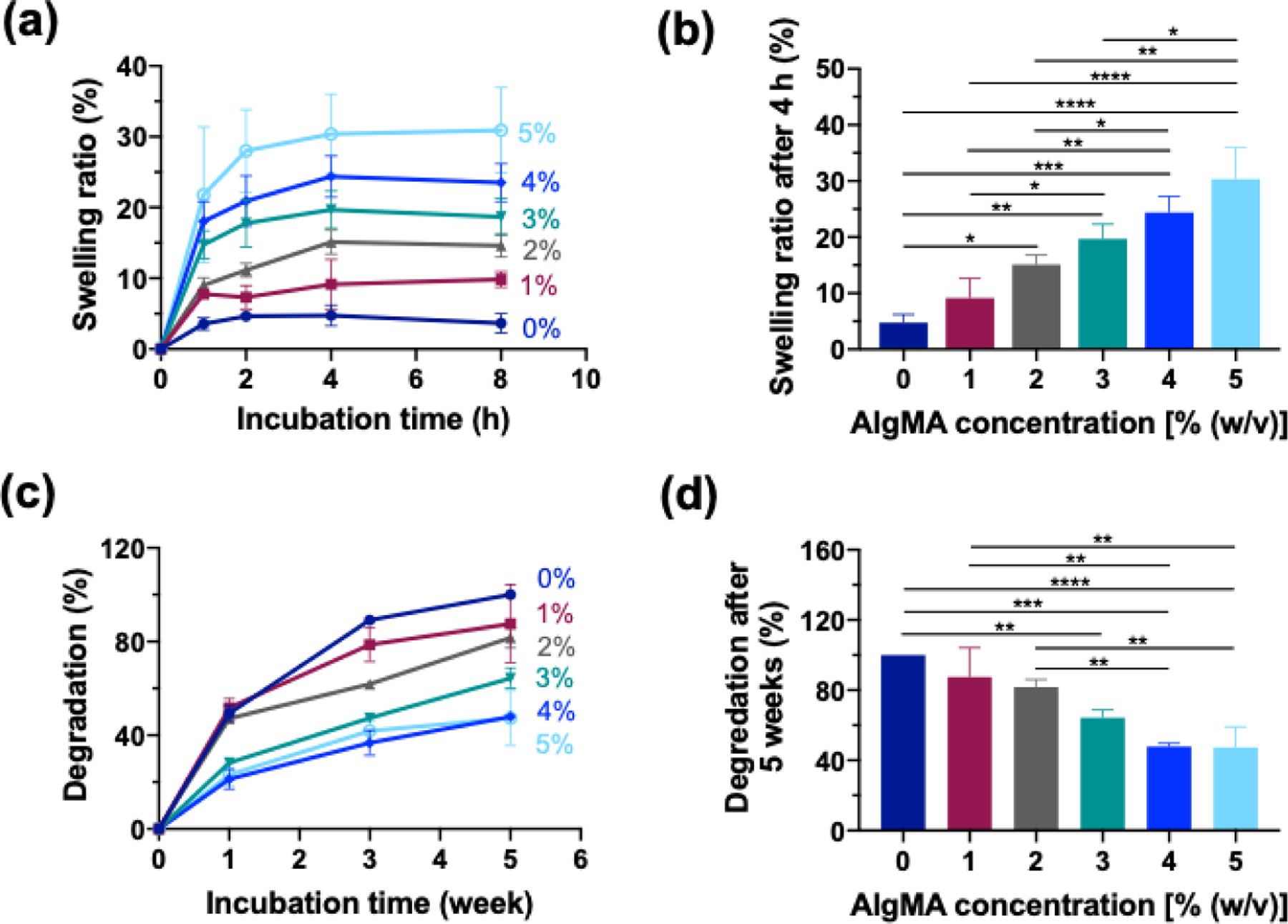
(a) Time-dependent swelling ratio of hydrogels immersed in DPBS at 37 °C. (b) The swelling ratio of the hydrogels after 4 h incubation in DPBS. (c) Time-dependent degradation of the hydrogels immersed in DPBS containing collagenase (1.25 U mL−1) at 37 °C. (d) The degradation of hydrogels after 5 weeks of incubation in DPBS containing collagenase (1.25 U mL−1) at 37 °C. Data are reported as the mean values of at least 5 experiments ± their standard deviation. The statistical analysis was done according to the methods explained in “Statistical analysis” section. Asterisks show the results that are statically significant with p-values < 0.05 (*), 0.01 (**), 0.001 (***), or 0.0001 (****).
The adhesive properties of hybrid hydrogels were assessed using standard tissue adhesion protocols. Figure 4a presents the in vitro burst pressure setup, comprising two custom-built compartments sandwiching a collagen sheet. The collagen sheet was perforated to mimic a wound, and the pre-gel solution was pipetted onto it, followed by exposure to visible light to form a bioadhesive gel, sealing the artificial hole. The capability of the hydrogels in sealing the hole was evaluated by introducing air and measuring the pressure until the sealant is damaged and/or detached. Figure 4b presents the representative pressure profiles versus time obtained from the burst pressure experiments. As the air was introduced under the sealed hole, the pressure linearly increased until the sealant was damaged, resulting in an abrupt pressure drop. The pressure drop was whether a result of sealant mechanical failure or delamination, depending on the composition of hybrid hydrogel. The AlgMA-free sealant mainly failed due to the mechanical damage, whereas when 3% of AlgMA was added to GelMA, the mode of failure was delamination. Adding a low concentration of AlgMA to GelMA improved the mechanical properties of the hydrogels. Figure 4c shows the burst pressure of hybrid hydrogels versus the concentration of AlgMA. Increasing AlgMA up to 2–3%, continually increases the burst pressure. The hybrid hydrogels including 3% AlgMA had approximately 2.5 times higher burst pressure than the AlgMA-free gel. Importantly, increasing the AlgMA concentration beyond 3%, adversely affected the burst pressure and deteriorated the performance of the hybrid gels in sealing the hole, because excessive increase in the AlgMA content of hybrid hydrogels increased brittleness and decreased tissue adhesion.
Figure 4. In vitro sealing properties of hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA.
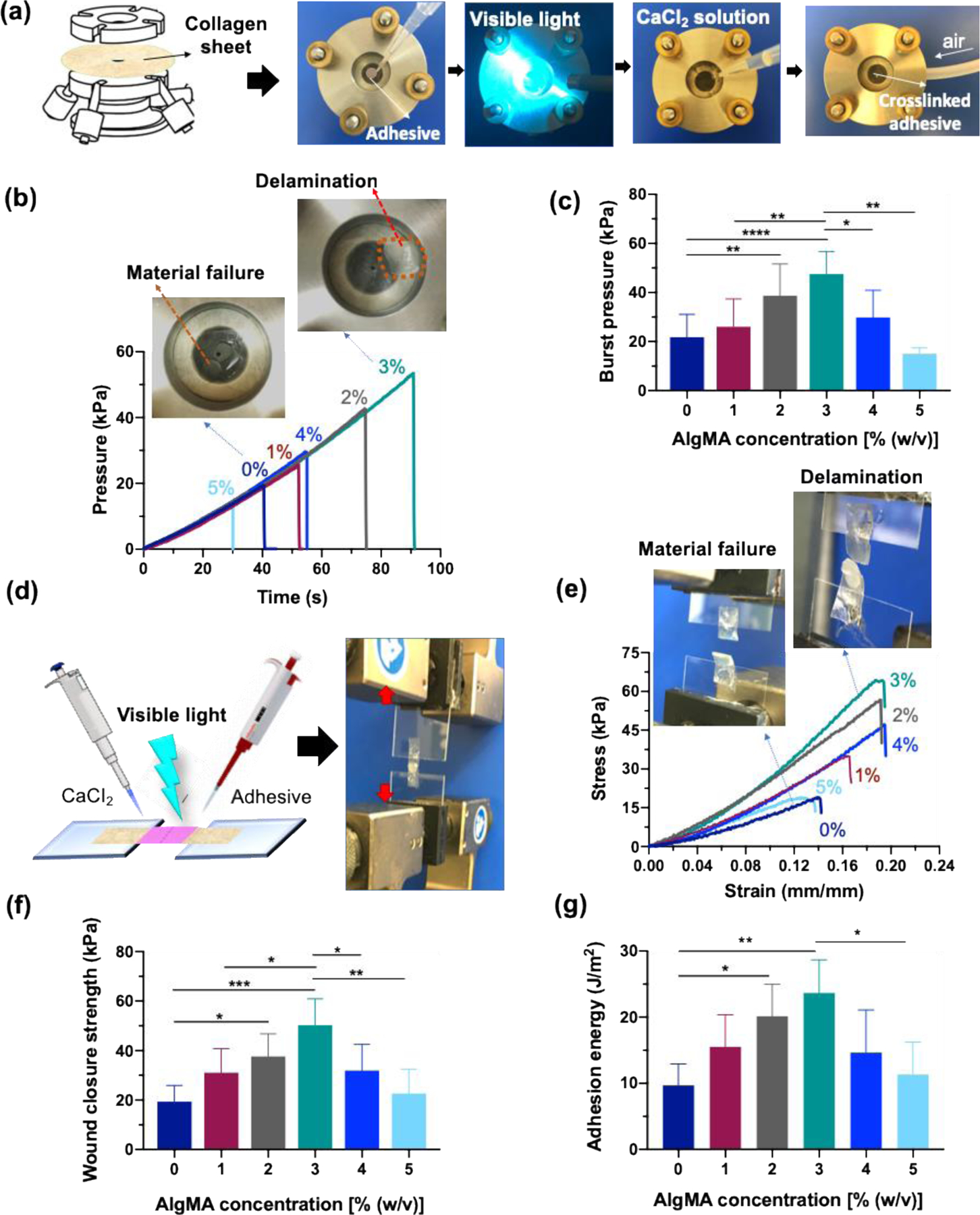
(a) Images showing the burst pressure assessment of double-network hybrid hydrogels prepared via successive photochemical and ion-mediate crosslinking, (b) representative pressure-time curves obtained from the burst pressure tests, and (c) the burst pressure values of hybrid hydrogels containing varying AlgMA concentrations. (d) Wound closure assessment setup, (e) representative stress-strain curves from wound closure experiments, (f) wound closure strength, (g) and adhesion energy of hybrid hydrogels. Data are reported as the mean values of at least 5 experiments ± their standard deviation. The statistical analysis was done according to the methods explained in “Statistical analysis” section. Asterisks show the results that are statically significant with p-values less than 0.05 (*), 0.01 (**), 0.001 (***), or 0.0001 (****).
Figure 4d presents the schematic of wound closure experiments, conducted by performing tensile tests on torn collagen sheet, which were adhered using the sealant hydrogels. The representative tensile strain-stress curves are shown in Figure 4e. Similar to the burst pressure experiments, in the absence of AlgMA or when the concentration of this biopolymer is lower than 3%, the sealant typically ruptures (bulk material) indicating a mechanical failure; however, at 2–3% AlgMA, the failure is mainly due to the hydrogel detachment from the porcine skin, i.e., interfacial delamination. The wound closure strength, defined as the maximum stress that the sealant can tolerate before failure, is shown in Figure 4f. The effect of AlgMA on the wound closure strength of hybrid hydrogels is similar to the burst pressure. There exists an optimum AlgMA concentration at which the wound closure strength is maximized, which is typically in the range of 2–3%, resulting in more than 250% improvement in the wound closure strength. The adhesion energy of the hybrid hydrogels to bond torn pieces of collagen sheets was evaluated using the same setup that was used for the wound closure experiment (Figure 4d). Consistently, the maximum adhesion energy was obtained for hydrogels containing 2–3% AlgMA, which was ~ 150% higher than that for the pure GelMA (Figure 4g).
Overall, the burst pressure for the GelMA-AlgMA (2–3%) hydrogel was more than 300 % higher than that reported for a highly elastic bioadhesive, MeTro gel, while its toughness and stretchability were comparable to MeTro.[18] Given the extremely high production cost of MeTro, the hybrid GelMA-AlgMA sealants demonstrated a high capacity for translation into clinical applications. The burst pressure obtained for GelMA-AlgMA (2–3%) hydrogel was ~ 900% higher than that for the commercially available sealants, Evicel®, Coseal®, and Progel®.[18] The adhesion energy for GelMA-AlgMA hydrogels was comparable to those for fibrin glue and polyethylene glycol (PEG)-based adhesives, but significantly lower than the synthetic bioadhesives, such as cyanoacrylates.[44] However, the high mechanical resilience of GelMA-AlgMA hydrogels combined with their biocompatibility and biodegradability render this class of hybrid hydrogels a superior candidate for sealing highly dynamic tissues.
To simulate the sealing of stretchable organs, we conducted ex vivo sealing experiments on porcine bladder and ureter. Figure 5a shows an ex vivo porcine bladder (5, ai) undergoing manual perforation (5, aii), followed by the deposition of hybrid pre-gel solutions (5, aiii), visible-light mediated crosslinking (5, aiv), CaCl2 treatment (5, av), and filling with DPBS (5, avi). The pressure that the sealed bladder withstands is presented in Figure 5b. As can be seen in this figure, the composition of hybrid hydrogels has a direct effect on the burst pressure. The AlgMA-free sealant provides a burst pressure of ~ 2 kPa, which increases to >5 kPa by increasing the AlgMA concentration to 2%. Further increase in the AlgMA concentration to 5% significantly decreases the burst pressure to a value comparable with the AlgMA-free hydrogel. The effect of AlgMA on the ex vivo burst pressure is similar to that on the tensile strength, toughness, and wound closure strength.
Figure 5. Ex vivo sealing capability of hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA.
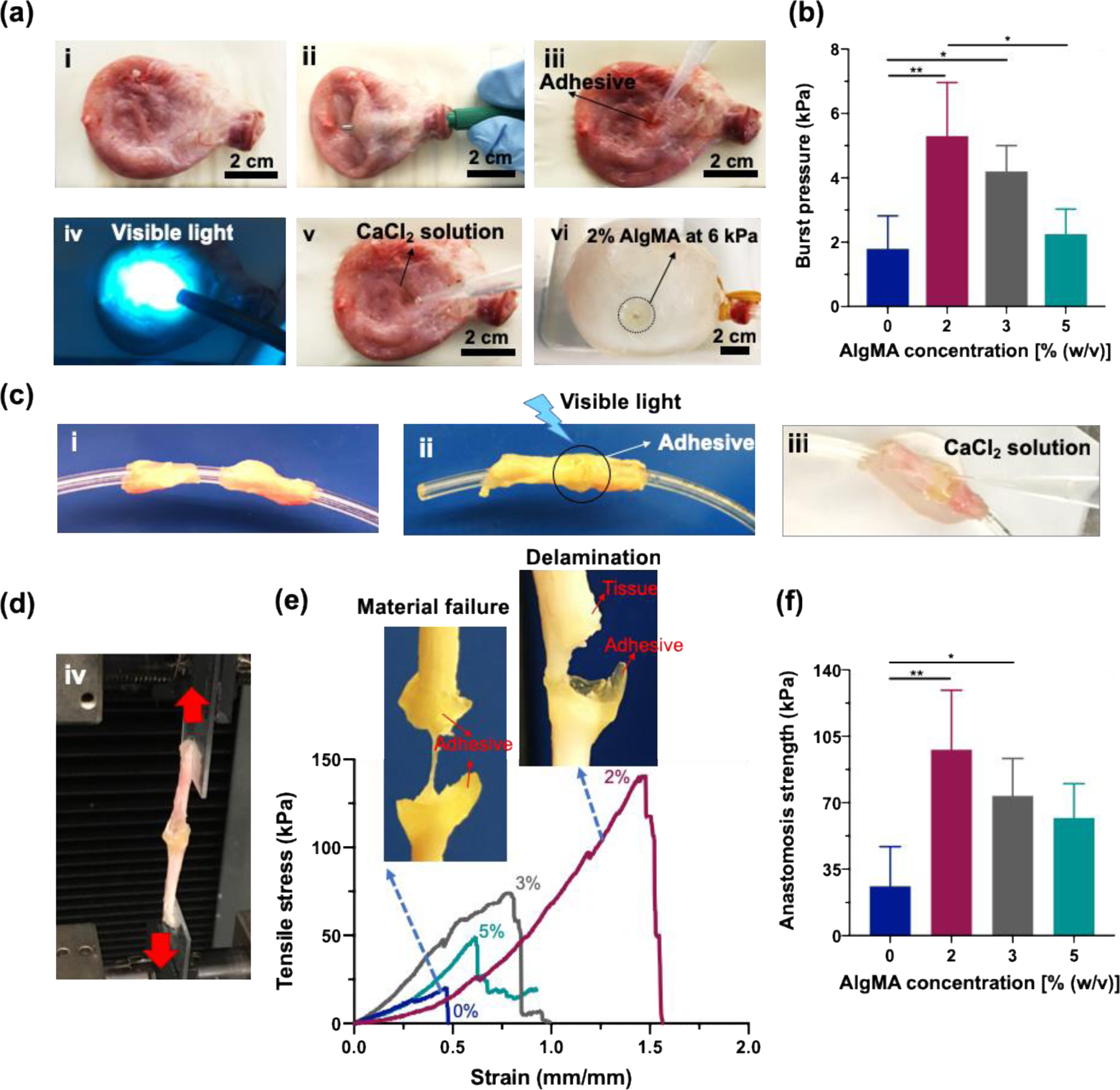
(a) Porcine bladder incision model: images show (a, i) a healthy porcine bladder, (a, ii) a superficial wound created in the bladder before sealing, (a, iii) the wound covered with the hydrogel, (a, iv) the subsequent crosslinking of hydrogel with visible light and (a, v) with a CaCl2 solution, and (a, vi) the sealed bladder filled with water at pressure ~ 6 kPa. (b) Burst pressure of bioadhesive hybrid sealants at varying AlgMA concentrations. (c) Porcine ureter anastomosis model: (c, i-iii) images illustrating the method used for sealing a fully torn porcine ureter, followed by (d) stretching the tissue to test the wound closure capability of the bioadhesive. (e) Representative tensile stress-strain curves and some examples of bioadhesive failure modes during the anastomosis tensile tests. (f) Anastomosis strength of hybrid sealants at varying AlgMA concentrations. Data are reported as the mean values of at least 5 (b) and 4 (f and g) experiments ± their standard deviation. The statistical analysis was done according to the methods explained in “Statistical analysis” section. Asterisks show the results that are statically significant with p-values < 0.05 (*) or 0.01 (**).
We have also evaluated the capability of hybrid hydrogels in connecting two pieces of torn tissues, such as ureter. While conventional ureter anastomosis techniques, such as suturing, is challenging, the hybrid hydrogels can be readily used to bring two pieces of cut ureter (Figure 5, ci) together and adhere them via photocrosslinking (Figure 5, cii) followed by Ca2+-mediated physical bonding (Figure 5, ciii). The anastomosed ureters were examined via the tensile tests (Figure 5d), and the tensile stress versus strain curves were obtained for the hybrid hydrogels containing varying amounts of AlgMA (Figure 5e). In the absence of AlgMA or when the AlgMA concentration is lower than 2%, the sealant ruptures under tensile stress due to the poor mechanical properties, e.g., brittleness. At an optimum concentration of AlgMA (2%), the sealant does not undergo mechanical damages, and at a tensile strength >6 times higher than that of the AlgMA-free sealant, the hybrid sealant detaches from the tissue. The mode of failure attests to the significant improvement in the mechanical properties of GelMA hydrogel when a small amount of AlgMA is added to it, originated from the physically-crosslinked AlgMA, enhancing the hydrogel toughness. Figure 5f presents the adhesion failure stress versus AlgMA concentration. While the failure stress for AlgMA-free sealants is ~ 20 kPa, the failure of hybrid GelMA-AlgMA (2%) sealants occurs at 90 kPa, a four-fold increase in the resilience against tensile failure, which decreases to ~ 60 kPa at 5% AlgMA. The failure energy (Figure 5g) measured based on the area under the tensile stress-strain curves shows that while the AlgMA-free gel has failure energy <10 J m−3, the hybrid hydrogel with the optimum AlgMA concentration (2%) needs ~ 60 J m−3 to detach from the ureter. The improvement in the sealing properties of hybrid hydrogels is not due to the increased solid content (Figure S3, Supporting Information).
The cytotoxicity of novel composite tissue sealants was assessed using the 2D culture of NIH/3T3 fibroblasts on the hydrogels. Figure 6a–f presents the fluorescence images of live (green) and dead (red) cells cultured for 3 and 7 days on composite hydrogels containing 0%, 2%, and 5% AlgMA. Compared to the control (0% AlgMA), the addition of AlgMA to the GelMA hydrogels does not have any significant effect on the cell viability. Fibroblast cells successfully adhere to the composite hydrogels, proliferate, and remain viable throughout the culture period. Figure 6g shows the cell viability, quantified by analyzing live/dead images using ImageJ. The cell viability remains >90% for composite hydrogels containing as high has 5% AlgMA, which is similar to that of the control hydrogel. The metabolic activity of cells seeded on the composite hydrogels in days 1, 3, and 7 is presented in Figure 6h. In each day, there is no significant difference between the metabolic activity of cells adhered onto the hydrogels containing 0–5% AlgMA. In days 3 and 5, the metabolic activity of cells increases by a factor of >2 and >4 compared to day 1, respectively, which is independent of the hydrogel composition. Accordingly, not only the composite hydrogels are not cytotoxic, but also they support cell adhesion and growth, two important phenomena during wound healing processes.
Figure 6. In vitro cytotoxicity assessment of hybrid hydrogels composed of GelMA (20% w/v) and varying concentrations of AlgMA.
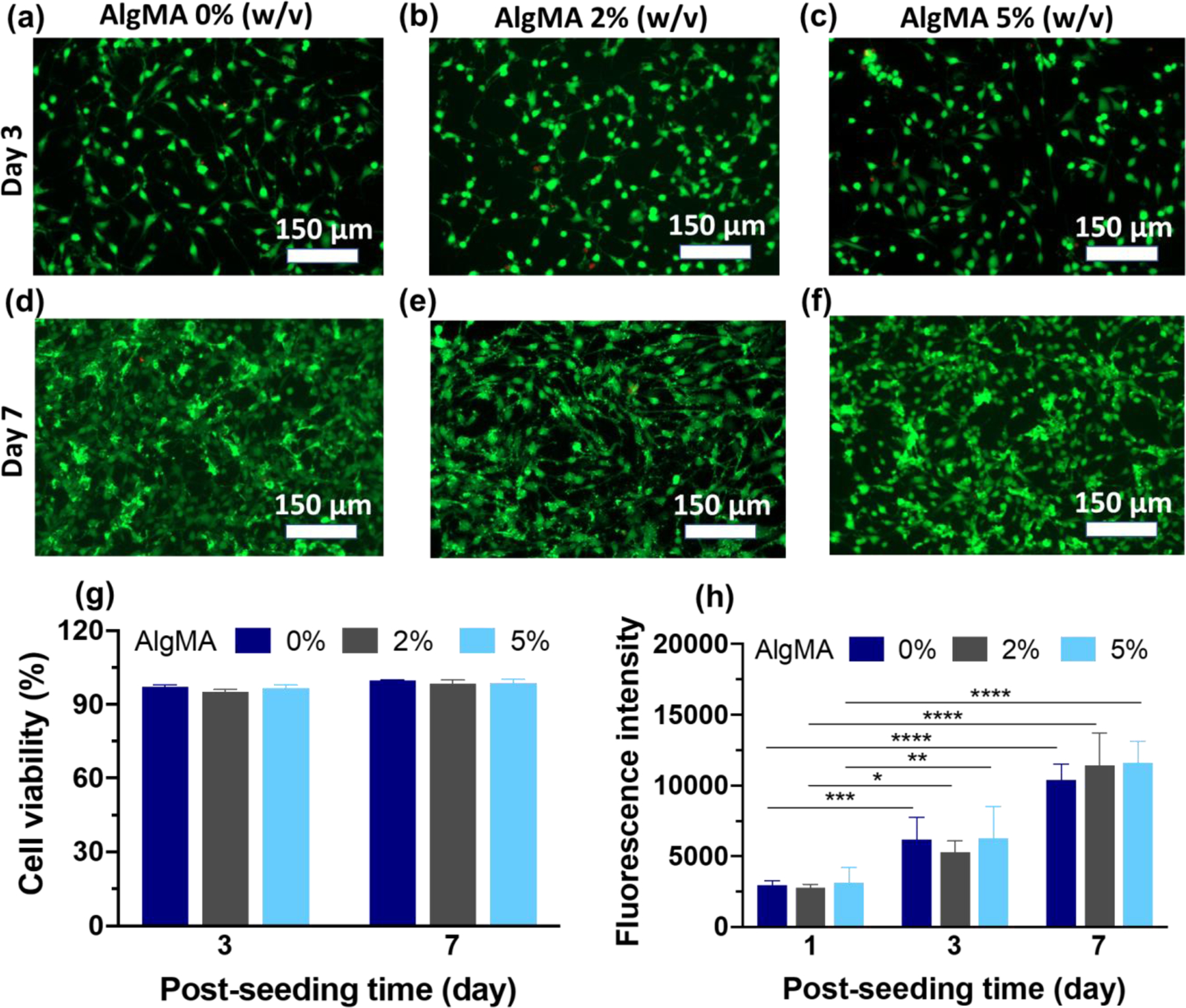
(a-f) Fluorescence images of live and dead cells stained in green and red colors, respectively, after culturing them on the composite hydrogels, (g) viability of fibroblast cells cultured on composite hydrogels, and (h) metabolic activity of fibroblast cells represented by the fluorescence intensity of resazurin converted to fluorescent resorufin. Data are reported as the mean values of at least n = 5 (g) and 4 (h) replicates their standard deviation. The statistical analysis was done according to the methods explained in “Statistical analysis” section. Asterisks show the results that are statically significant with p-values less than 0.05 (*), 0.01 (**), 0.001 (***), or 0.0001 (****).
3. Conclusions
Developing injectable, tough, and cost-effective tissue adhesive biomaterials is an unmet clinical need for the minimally invasive sealing of damaged tissues, particularly when sutures or staples are not favorable. We have developed a composite biopolymer made up of GelMA, a widely used biomaterial for tissue engineering and regenerative medicine, and AlgMA that can be readily injected/pipetted into the lesion and adhere to the tissue via visible light-mediated crosslinking. The chemically photocrosslinked hybrid hydrogels undergo physical crosslinking using divalent ions, such as Ca2+, to form a reversible egg-box-like network of G blocks. While the chemically-crosslinked MA groups of GelMA and AlgMA contribute to maintaining the structural integrity of the hybrid hydrogels, the physical crosslinking provides a polymer network that dissipates energy under stress, imparting stretchability and toughness to the hybrid hydrogels. This class of protein-polysaccharide based composite hydrogels are tissue adhesive and provide excellent sealing properties at low concentrations of AlgMA (2–3%), outperforming current elastic tissue adhesives. We speculate that these novel hybrid hydrogels may leverage the sutureless tissue sealing, particularly in challenging organs that undergo severe mechanical stress during daily activities.
4. Materials and methods
4.1. Materials
Type A porcine gelatin, alginic acid sodium salt from brown algae (low viscosity alginate), methacrylic anhydride (MA; purity≥94%), triethanolamine (TEA; purity≥99%), N-vinylcaprolactam (VC; purity≥98%), eosin Y, calcium chloride dihydrate (purity≥99%), dimethyl sulfoxide-d6 (DMSO-d6; purity≥99%), and deuterium oxide (D2O, purity≥99.9%) were all purchased from Sigma-Aldrich. Type II collagenase, Dulbecco’s phosphate-buffered saline (DPBS), Dulbecco’s Modified Eagle Media (DMEM), fetal bovine serum (FBS), and antibiotic-antimycotic (penicillin/streptomycin, P/S) were manufactured by GIBCO/Life Technologies and purchased from Thermo Fisher Scientific.
4.2. Synthesis of gelatin methacryloyl (GelMA) and methacrylate-modified alginate (AlgMA)
GelMA was prepared according to the literature.[49,50] In brief, 10 g of type A gelatin from porcine skin was added to 100 mL of DPBS to make a 10% w/v gelatin solution, which was stirred at 250 rpm and 50 °C to fully dissolve gelatin. The reaction vessel was then covered with aluminum foil, and 8 mL of MA was added dropwise to the gelatin solution and allowed to react at 50 °C for 2 h. The reaction was stopped following a 3-time dilution with DPBS at 50 °C, and the mixture was dialyzed against deionized (DI) water using a 12–14 kDa dialysis tubing (Spectrum™ Spectra/Por™ 4 RC Dialysis Membrane Tubing) for 1 week at 50 °C to remove the impurities and methacrylic acid. The purified solution was lyophilized in a freeze dryer to yield a white foam.
Alginate was modified with methacrylic groups based on a method described in the literature.[51] Briefly, 2.5 g of low viscosity alginate was dissolved in 100 mL of DI water at room temperature to yield a 2.5% w/v alginate solution. The same volume of MA (100 mL) was added to the alginate solution dropwise and stirred at 250 rpm and room temperature. To separate the product (AlgMA) from the solution, the mixture was poured into 500 mL of ethanol and stirred until AlgMA precipitated. The precipitate was then vacuum filtered using a 5 µm filter paper (50, Whatman™), dissolved in DI water (100 mL), and precipitated out for the second time using 500 mL of ethanol. This process was repeated at least three times to remove salts and remaining MA from the precipitate. Finally, the precipitate was collected on a filter paper and dried at room temperature overnight.
4.3. Preparation of GelMA-AlgMA hybrid hydrogels
A GelMA solution (20% w/v) including varying concentrations of AlgMA (0 to 5% w/v) was prepared by dissolving GelMA and AlgMA in 1 mL of DPBS at 37 °C. The visible light photoinitiators, VC, TEA, and eosin Y, were added to the mixture in proportion to the total concentration of methacrylated solid content, i.e., GelMA and AlgMA (see Table S1, Supporting Information). The mixture was then placed at 80 °C in an oven for 10 min, vortexed, and incubated at 37 °C overnight to allow AlgMA to fully dissolve in the DPBS. The pre-gel solutions were covalently crosslinked through visible light irradiation (wavelength = 450–550 nm, intensity ~ 100 mWcm−2, Genzyme FocalSeal LS1000 Xenon Light) for 4 min. For the ionic crosslinking of AlgMA post chemical crosslinking, it was incubated in a CaCl2 solution (concentration = 0.13 times that of AlgMA, Table S1, Supporting Information) at room temperature for at least 5 min.
4.4. Chemical characterization
NMR spectroscopy was used for the chemical characterization of biopolymers and crosslinked hydrogels. 1HNMR spectra was recorded for all samples at room temperature on a 400 MHz Bruker AV400 spectrometer. All spectra were corrected with phase and base line, and the solvent peak was fixed before quantifying the degree of methacrylation. To quantify the degree of methacrylation, GelMA and AlgMA were separately dissolved in D2O at a concentration of 10 mg/mL, and the degree of substitution was estimated based on literature.[47,48] To assess the crosslinking degree of bioadhesives, the hybrid hydrogels containing GelMA 20% w/v and 3% w/v AlgMA were photocrosslinked according to the method explained in section 4.3. The samples were then immersed in DMSO for 7 days at 37 °C and sonicated for 1 h every day to partially dissolve the hydrogels prior to conducting the NMR spectroscopy. The degree of crosslinking of hybrid hydrogels were calculated by integrating the double bond protons (vinyl) δ 5.28 in consideration with the methyl group of acrylate at δ 1.8 as reference peak. For quantitative analyses, at least 3 spectra were analyzed for each hydrogel.
4.5. Mechanical and rheological characterizations
An Instron mechanical tester (5943 Series) was used for the tensile and compression testing of hydrogels. At least 5 samples were tested for each condition, and the results were averaged. To prepare samples for the tensile test, 250 µL of a pre-gel solution was transferred to a dumbbell-shaped polydimethylsiloxane (PDMS) mold (gage length = 10 mm, gage width = 5 mm, gage thickness = 1.5 mm, and fillet radius = 2 mm) and crosslinked with visible light for 4 min. The samples were then removed from the mold and immersed in 3 mL of their corresponding CaCl2 solution for 30 min to ionically crosslink AlgMA. After removing the CaCl2 solution, the samples were blotted using Kimwipe, and the two ends of samples were attached to two pieces of glass slides using Super glue (Gel Control). The glass slides were then gripped with Instron jaws, and the tensile tests were performed at a constant strain rate (10 mm/min). Tensile strength was defined as the maximum stress at the point of failure, elastic (Young’s) modulus was the slope of the stress-strain curve, and toughness was calculated based on the area under the stress-strain curve.
For compression testing, 75 µL of a pre-gel solution was transferred to a cylindrical PDMS mold (diameter = 6 mm and height = 1.5 mm), and a glass coverslip was placed on the mold to flatten the top surface. The samples were then crosslinked with visible light followed by CaCl2 incubation as explained before. The compression tests were performed using Instron at a strain rate of 0.3 mm/min up to the failure point. Compressive modulus was calculated based on the slope of linear stress-strain curve up to strain = 0.2 mm/mm. Cyclic compression tests were performed at the same strain rate up to strain = 0.5 mm/mm, and the energy loss was calculated as
Rheological studies were conducted using a modular compact rheometer (MCR 302, Anton Paar, Graz, Austria). The hydrogel samples (n = 4) with diameter = 8 mm and height = 1.5 mm were prepared following the same method used for the compression tests. The samples were then placed onto a sandblasted flat plate and confined with an 8 mm diameter plate to perform the rheological tests using the rheometer. Mineral oil (M5904 from Sigma-Aldrich) was applied around the plate to prevent water evaporation and maintain the hydrogel moist. Shear strain sweeps were conducted at 37 °C, strain = 0–100% and angular frequency = 1 rad s−1, and frequency sweeps were performed at angular frequency = 0–100 rad s−1 and strain = 0.1%. The storage modulus (G’), loss modulus (G’’), and loss factor (G’’/G’) were reported at angular frequency = 1 rad s−1and strain = 0.1%.
4.6. Assessment of swelling and degradation
In order to test the physical characteristics of bioadhesive hydrogels, cylindrical samples (diameter = 8 mm and thickness = 1.5 mm, 5 samples for each condition) were prepared in PDMS molds following the procedure explained in section 4.3. The weight of wet samples after crosslinking was measured (W0), and the samples were allowed to swell in DPBS at 37 °C for 1, 2, 4, and 8 h. At the end of experiments, the samples were removed from DPBS, gently blotted with Kimwipe, and their weight was measured (Wt). The swelling ratio was calculated as
For the degradation assessment, the cylindrical samples were immersed in a DPBS solution containing 10 µg mL−1 of collagenase at 37 °C. The samples were removed from the solution at desired incubation times, rinsed 3 times with DI water, lyophilized, and the dry weight was recorded. The degradation ratio was calculated as
where Wi is the initial dry weigh of samples before immersion in the collagenase solution, and Wd is their dry weight after incubation in the collagenase solution at a given time.
4.7. Assessment of cytocompatibility
Cytocompatibility of the hydrogels was evaluated in 2D culture using a mouse fibroblast cell line (NIH/3T3, ATCC). 3T3 cells were cultured in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic (P/S) and incubated at 37° C and 5% CO2. The media was changed every other day and cells were passaged once they reached the confluency of 90%. To assess the cytotoxicity of the hydrogels, disc-shaped GelMA (20% w/v) samples (diameter = 8 mm and thickness = 1.5 mm, 4 samples for each condition) containing 0, 2, and 5% w/v AlgMA were prepared according to the method explained in section 4.3 and were sterilized with UV light for 2 h. The samples were transferred to 48-well plates, and 100 µL of a cell suspension containing 100,000 cells mL−1 were seeded on top of the hydrogels. The cell viability was assessed on days 3 and 7 of culture using LIVE/DEAD™ viability/cytotoxicity kit (Invitrogen). Cells were visualized on a fluorescence microscope (Axio Observer 5, Zeiss), and ImageJ software (150.i) was used to estimate the number of living (green) and dead (red) cells. Cell viability was determined as the ratio of the living cells to the total number of cells. The metabolic activity of cells was measured on days 1, 3, and 7 of culture using PrestoBlue™ reagent (Invitrogen). For this, PrestoBlue was mixed with the media at the ratio of 1 to 9, and 500 µL of the solution was added onto the hydrogels and incubated at 37° C for 2 h. The fluorescence intensity resulting from the cell activity was measured at excitation and emission wavelengths of 560 nm and 590 nm, respectively, using a fluorescence plate reader (vis Synergy 2, BioTek). The readings were normalized with respect to the control well without any cells and reported based on post-seeding time for each sample.
4.8. In vitro assessment of tissue sealing properties
4.8.1. In vitro burst pressure
In vitro burst pressure tests were performed following the standard protocol for the burst strength of surgical sealants (ASTM F2392–04 [52]). Collagen sheets (Weston 19–0102-W 33 mm Collagen Sausage Casing) were cut into round pieces (diameter = 30 mm) and rinsed three times with DI water to remove glycerin and soften the sheets. A circular defect (diameter = 1 mm) was created in the center of collagen sheets using a biopsy punch, and sheets were immersed in DBPS for 10 min at room temperature prior to use. A collagen sheet was then removed from DPBS, gently blotted with Kimwipe to remove the excess liquid, and placed onto the burst pressure device. The pre-gel solution (20 µL) was applied on the defect and irradiated with visible light for 4 min, followed by 5 min exposure to 1 mL of CaCl2 solution (see Table S1, Supporting Information). The burst pressure testing system was connected to a syringe pump (Aaddin-1000HP, WPI Inc.), and air was blown under the sealed collagen sheet at a constant rate of 20 mL min−1. Pressure was registered using a sensor (PASCO Wireless Pressure) and monitored by SPARKvue (version 4.0.0.18, PASCO) software. Burst pressure was defined as the maximum pressure at which the adhesive failed due to the material rupture or delamination from the collagen sheet. At least 5 samples were tested for each condition, and the burst pressure values were averaged.
4.8.2. In vitro wound closure test
Wound closure strengths of the hydrogels were examined following the standard test method for the wound closure strength of tissue adhesives and sealants, ASTM F2458–05, [53] with some modifications. In brief, a collagen sheet was cut into rectangular pieces of (5×20 mm2) and stored in DPBS at room temperature for 10 min prior to the experiments. To prepare the samples, the collagen sheet was blotted with Kimwipe to remove the excess liquid, and each end of the sheet was glued to two pieces of glass slides, which were 6 mm apart using Super glue. An incision was made in the center of collagen sheet using a razor blade, and 20 µL of the pre-gel solution was applied onto the incision to cover the area (5×6 mm2). Subsequently, the adhesive was crosslinked with the visible light and CaCl2 as explained previously. The glass slides were then transferred to the Instron mechanical tester, and special attention was paid to avoid distortion and unnecessary mechanical stress. The sample was then stretched at the constant rate of 1 mm min−1, and the wound closure strength of sealant materials was determined at the point of rupture, and the adhesion energy was calculated based on a method reported for measuring adhesion energy between stretchable materials.[54] In brief, the total energy used for hydrogel debonding and deformation was calculated based on the area under the stress-strain curves. The energy that was consumed for the hydrogel deformation at the site of defect was estimated based on the tensile stress-stain behavior of hydrogels shown in Fig. 2b. The adhesion energy was calculated by subtracting the deformation energy from total energy, which was normalized based on the area of hydrogel that was detached from the collagen sheet under tensile stress. At least 5 replicates were tested for each condition, and the results were averaged.
4.9. Ex vivo sealing tests
For the ex vivo experiments, the tissues were obtained from discarded animals provided by UCLA animal facility. The tissues were stored not more than 24 h in the refrigerator at 4 °C prior to the experiments.
4.9.1. Ex vivo burst pressure
The pressure required to rupture/detach the bioadhesives from the ex vivo porcine bladder model were measured using at least 5 hydrogels per condition. A circular defect (diameter = 3) mm was made on the bladder using a biopsy punch (Figure 5aii), and 100 µL of the pre-gel solution was applied onto the defect, irradiated with visible light for 4 min, and incubation in 1 mL of CaCl2 solution for 10 min. The bladder was gradually filled with DI water at a constant rate of 20 mL min−1 using a peristaltic pump (BT100–1l, Longer Pump), and the pressure was monitored using the same setup explained in section 4.8. The burst pressure was defined as the maximum pressure at which water started leaking from the sealed defect, resulting in dropped or plateaued pressure.
4.9.2. Ex vivo wound closure
The ex vivo wound closure efficacy of hydrogels (n = 4) was investigated using a porcine ureter anastomosis model. The ureter was cut into 30 mm-long pieces and a plastic tube with a diameter of 1 mm was inserted in the ureter. An incision was made at the center of ureter using a razor blade, and 100 µL of the bioadhesive was applied all around the incision, which was irradiated with visible light for 4 min. CalCl2 solution (1 mL) was subsequently applied onto the adhesive and incubated for 5 min. The tube was removed, and the two ends of the ureter were fixed onto two pieces of glass slides using Super glue. The glass slides were then placed onto the Instron mechanical tester and stretched at a rate of 1 mm min−1. The anastomosis strength was defined as the maximum stress at which the sealant ruptured or detached from the tissue.
4.10. Statistical analysis
The data are presented as mean values of at least 3 replicates ± their standard deviation. Statistical analysis was performed using one-way analysis of variance (ANOVA) and followed by Tukey’s multiple comparison test using on GraphPad Prism software (version 8.2.1). Statically significant differences were identified as * (p<0.05), ** (p<0.01), ***(p<0.001), or ****(p<0.0001).
Supplementary Material
Acknowledgements
M.T. acknowledges financial support from Fonds de la recherche en santé du Québec. R.T. would like to acknowledge the financial support from the Scientific and Technological Research Council of Turkey (TUBITAK) 2214-A International Doctorate Research Fellowship Program (App. No: 1059B141700084) and from Istanbul University Cerrahpasa, Engineering Faculty, Chemistry Department. A.K. would like to acknowledge funding from the National Institutes of Health (1R01EB023052-01A1, 1R01HL140618-01). A.S. would like to acknowledge the postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR) and the startup fund from the Pennsylvania State University.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Maryam Tavafoghi, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA..
Amir Sheikhi, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; Department of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA..
Rumeysa Tutar, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; Department of Chemistry, Faculty of Engineering, Istanbul University Cerrahpasa, Avcılar-Istanbul, Turkey..
Jamileh Jahangiry, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA..
Avijit Baidya, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA..
Reihaneh Haghniaz, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA..
Ali Khademhosseini, Department of Bioengineering, University of California, Los Angeles, 410 Westwood Plaza, Los Angeles, CA 90095, USA.; Center for Minimally Invasive Therapeutics (C-MIT), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; California NanoSystems Institute (CNSI), University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, CA 90095, USA.; Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, 5531 Boelter Hall, Los Angeles, CA 90095, USA.; Department of Radiological Sciences, David Geffen School of Medicine, University of California, Los Angeles, 10833 Le Conte Ave, Los Angeles, CA 90095, USA.; Jonsson Comprehensive Cancer Centre, University of California, Los Angeles, 10833 Le Conte Ave, Los Angeles, CA 90024, USA..
References
- [1].Kinaci A, Algra A, Heuts S, O’Donnell D, van der Zwan A, van Doormaal T, World Neurosurg 2018, 118, 368. [DOI] [PubMed] [Google Scholar]
- [2].Grunzweig KA, Ascha M, Kumar AR, J. Plast. Reconstr. Aesthetic Surg 2019, 72, 871. [DOI] [PubMed] [Google Scholar]
- [3].Foster LJR, Bioadhesion Biomim. Nat. Appl 2015, 203. [Google Scholar]
- [4].Scognamiglio F, Travan A, Rustighi I, Tarchi P, Palmisano S, Marsich E, Borgogna M, Donati I, de Manzini N, Paoletti S, J. Biomed. Mater. Res. Part B Appl. Biomater 2016, 104, 626. [DOI] [PubMed] [Google Scholar]
- [5].Plat VD, Bootsma BT, van der Wielen N, Straatman J, Schoonmade LJ, van der Peet DL, Daams F, Int. J. Surg 2017, 40, 163. [DOI] [PubMed] [Google Scholar]
- [6].Annabi N, Yue K, Tamayol A, Khademhosseini A, Eur. J. Pharm. Biopharm 2015, 95, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhu W, Chuah YJ, Wang D-A, Acta Biomater 2018, 74, 1. [DOI] [PubMed] [Google Scholar]
- [8].NBC29, “Global Hemostats and Tissue Sealants market to grow at 8.03% of CAGR by 2027,” can be found under https://www.nbc29.com/story/40159960/global-hemostats-and-tissue-sealants-market-to-grow-at-803-of-cagr-by-2027, 2019.
- [9].Han L, Liu K, Wang M, Wang K, Fang L, Chen H, Zhou J, Lu X, Adv. Funct. Mater 2018, 28, 1704195. [Google Scholar]
- [10].Ghobril C, Grinstaff MW, Chem. Soc. Rev 2015, 44, 1820. [DOI] [PubMed] [Google Scholar]
- [11].Chen T, Chen Y, Rehman HU, Chen Z, Yang Z, Wang M, Li H, Liu H, ACS Appl. Mater. Interfaces 2018, 10, 33523. [DOI] [PubMed] [Google Scholar]
- [12].Rousou JA, J. Card. Surg. Incl. Mech. Biol. Support Hear. Lungs 2013, 28, 238. [DOI] [PubMed] [Google Scholar]
- [13].Madaghiele M, Demitri C, Sannino A, Ambrosio L, Burn. trauma 2014, 2, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saxena AK, J. Thorac. Cardiovasc. Surg 2010, 139, 496. [DOI] [PubMed] [Google Scholar]
- [15].Otani Y, Tabata Y, Ikada Y, Ann. Thorac. Surg 1999, 67, 922. [DOI] [PubMed] [Google Scholar]
- [16].Araki M, Tao H, Nakajima N, Sugai H, Sato T, Hyon S-H, Nagayasu T, Nakamura T, J. Thorac. Cardiovasc. Surg 2007, 134, 1241. [DOI] [PubMed] [Google Scholar]
- [17].Kobayashi H, Sekine T, Nakamura T, Shimizu Y, J. Biomed. Mater. Res. An Off. J. Soc. Biomater. Japanese Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater 2001, 58, 658. [DOI] [PubMed] [Google Scholar]
- [18].Annabi N, Zhang Y-N, Assmann A, Sani ES, Cheng G, Lassaletta AD, Vegh A, Dehghani B, Ruiz-Esparza GU, Wang X, Sci. Transl. Med 2017, 9, eaai7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang R, Li J, Chen W, Xu T, Yun S, Xu Z, Xu Z, Sato T, Chi B, Xu H, Adv. Funct. Mater 2017, 27, 1604894. [Google Scholar]
- [20].Sani ES, Kheirkhah A, Rana D, Sun Z, Foulsham W, Sheikhi A, Khademhosseini A, Dana R, Annabi N, Sci. Adv 2019, 5, eaav1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Grinstaff MW, Biomaterials 2007, 28, 5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Monteiro N, Thrivikraman G, Athirasala A, Tahayeri A, França CM, Ferracane JL, Bertassoni LE, Dent. Mater 2018, 34, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ducret M, Montembault A, Josse J, Pasdeloup M, Celle A, Benchrih R, Mallein-Gerin F, Alliot-Licht B, David L, Farges J-C, Dent. Mater 2019, 35, 523. [DOI] [PubMed] [Google Scholar]
- [24].Fukushima KA, Marques MM, Tedesco TK, de Carvalho GL, Gonçalves F, Caballero-Flores H, Morimoto S, Moreira MS, Arch. Oral Biol 2018. [DOI] [PubMed] [Google Scholar]
- [25].Galler KM, Brandl FP, Kirchhof S, Widbiller M, Eidt A, Buchalla W, Göpferich A, Schmalz G, Tissue Eng. Part A 2018, 24, 234. [DOI] [PubMed] [Google Scholar]
- [26].Kargozar S, Milan PB, Baino F, Mozafari M, in Nanoeng. Biomater. Regen. Med, Elsevier, 2019, pp. 13–38. [Google Scholar]
- [27].Vyas KS, Saha SP, Expert Opin. Biol. Ther 2013, 13, 1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hirai K, Tabata Y, Hasegawa S, Sakai Y, Tissue Eng J Regen. Med 2016, 10, E433. [DOI] [PubMed] [Google Scholar]
- [29].Smith DJ, Brat GA, Medina SH, Tong D, Huang Y, Grahammer J, Furtmüller GJ, Oh BC, Nagy-Smith KJ, Walczak P, Nat. Nanotechnol 2016, 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soucy JR, Shirzaei Sani E, Portillo Lara R, Diaz D, Dias F, Weiss AS, Koppes AN, Koppes RA, Annabi N, Tissue Eng. Part A 2018, 24, 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z, Nature 2012, 489, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vakalopoulos KA, Wu Z, Kroese L, Kleinrensink G-J, Jeekel J, Vendamme R, Dodou D, Lange JF, Ann. Surg 2015, 261, 323. [DOI] [PubMed] [Google Scholar]
- [33].Wallace DG, Cruise GM, Rhee WM, Schroeder JA, Prior JJ, Ju J, Maroney M, Duronio J, Ngo MH, Estridge T, et al. , J. Biomed. Mater. Res 2001, 58, 545. [DOI] [PubMed] [Google Scholar]
- [34].Dastjerdi AK, Pagano M, Kaartinen MT, McKee MD, Barthelat F, Acta Biomater 2012, 8, 3349. [DOI] [PubMed] [Google Scholar]
- [35].Haraguchi K, Takehisa T, Adv. Mater 2002, 14, 1120. [Google Scholar]
- [36].Moretti M, Wendt D, Schaefer D, Jakob M, Hunziker EB, Heberer M, Martin I, J. Biomech 2005, 38, 1846. [DOI] [PubMed] [Google Scholar]
- [37].Lake AG, Thomas GJ, Proc. R. Soc. London. Ser. A. Math. Phys. Sci 1967, 300, 108. [Google Scholar]
- [38].Tuncaboylu DC, Sari M, Oppermann W, Okay O, Macromolecules 2011, 44, 4997. [Google Scholar]
- [39].Gong JP, Katsuyama Y, Kurokawa T, Osada Y, Adv. Mater 2003, 15, 1155. [Google Scholar]
- [40].Yu QM, Tanaka Y, Furukawa H, Kurokawa T, Gong JP, Macromolecules 2009, 42, 3852. [Google Scholar]
- [41].Webber RE, Creton C, Brown HR, Gong JP, Macromolecules 2007, 40, 2919. [Google Scholar]
- [42].Henderson KJ, Zhou TC, Otim KJ, Shull KR, Macromolecules 2010, 43, 6193. [Google Scholar]
- [43].Haque MA, Kurokawa T, Kamita G, Gong JP, Macromolecules 2011, 44, 8916. [Google Scholar]
- [44].Li J, Celiz AD, Yang J, Yang Q, Wamala I, Whyte W, Seo BR, V Vasilyev N, Vlassak JJ, Suo Z, et al. , Science (80–. ) 2017, 357, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ahmed S, Alginates: Applications in the Biomedical and Food Industries, John Wiley & Sons, 2019. [Google Scholar]
- [46].Li L, Fang Y, Vreeker R, Appelqvist I, Mendes E, Biomacromolecules 2007, 8, 464. [DOI] [PubMed] [Google Scholar]
- [47].Wang Z, Tian Z, Menard F, Kim K, Biofabrication 2017, 9, 44101. [DOI] [PubMed] [Google Scholar]
- [48].Boddupalli A, Bratlie KM, J. Biomed. Mater. Res. Part A 2018, 106, 2934. [DOI] [PubMed] [Google Scholar]
- [49].Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H, Biomacromolecules 2000, 1, 31. [DOI] [PubMed] [Google Scholar]
- [50].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Coates EE, Riggin CN, Fisher JP, J. Biomed. Mater. Res. - Part A 2013, 7, 1962. [DOI] [PubMed] [Google Scholar]
- [52].ASTM F2392-04(2015), Standard Test Method for Burst Strength of Surgical Sealants, ASTM International, West Conshohocken, PA, Www.Astm.Org, 2015.
- [53].ASTM F2458-05(2015), Standard Test Method for Wound Closure Strength of Tissue Adhesives and Sealants, ASTM International, West Conshohocken, PA, Www.Astm.Org, 2015.
- [54].Tang J, Li J, Vlassak JJ, Suo Z, Soft Matter 2016, 12, 1093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.