

Conspectus

Bridged and fused rings are commonly found in biologically important molecules. Current tactics to construct these ring systems are primarily based on stepwise ring formation (i.e., making one ring first followed by making another) and cycloaddition reactions (e.g., Diels–Alder reaction). To seek a complementary and perhaps more unified ring-forming approach, a deconstructive strategy based on C–C bond activation of cyclic ketones has been conceived. The named “cut-and-sew” reaction uses cyclic ketones with a tethered unsaturated moiety as substrates, which involves oxidative addition of a transition metal into the ketone C–C bond followed by intramolecular insertion of the unsaturated unit. This strategy has proved successful to access diverse ring scaffolds that are nontrivial to construct otherwise.
This Account offers a concise summary of our laboratory’s systematic efforts in developing transition metal-catalyzed cut-and-sew reactions for the synthesis of bridged and fused rings over the past 10 years. In particular, we will focus on the reactions using readily available benzocyclobutenones and cyclobutanones. To date, the scope of the cut-and-sew reactions has been greatly expanded. First, diverse unsaturated moieties can serve as suitable coupling partners, such as alkenyl, alkynyl, allenyl, carbonyl, and iminyl groups. Second, a variety of reaction modes have been uncovered. In this account, (4 + 2), (4 + 2 – 1), and (4 + 1) cycloadditions that lead to a range of bridged or fused scaffolds will be summarized. Third, enantioselective transformations have been realized to efficiently construct chiral scaffolds, which are enabled by two strategies: enantio-determining migratory insertion and desymmetrization of cyclobutanones. Fourth, the synthetic applications have been demonstrated in streamlined total syntheses of a number of complex natural products. Compared to conventional synthetic logics, the cut-and-sew reaction allows the development of new bond-disconnecting strategies. Thus, the syntheses of (−)-cycloclavine, (−)-thebainone A, penicibilaenes, and the proposed cycloinumakiol are discussed in more detail.
In addition to the narrative of the development of the cut-and-sew chemistry, this Account also aims to provide core guiding foundations and inspirations toward broader deconstructive synthetic applications through C–C bond cleavage. It is anticipated that more classes of cyclic compounds could serve as the substrates beyond benzocyclobutenones and cyclobutanones, and more diverse unsaturated moieties could be coupled. It can also be envisaged that more innovative utilization of this cut-and-sew strategy in complex organic syntheses will be revealed in the near future.
Key References
Xu, T.; Dong, G. Rhodium-Catalyzed Regioselective Carboacylation of Olefins: A C–C Bond Activation Approach for Accessing Fused-Ring Systems. Angew. Chem., Int. Ed. 2012, 51, 7567–7571.1The concept of the cut-and-sew transformations to construct fused rings is illustrated.
Ko, H.; Dong, G. Cooperative Activation of Cyclobutanones and Olefins Leads to Bridged Ring Systems by a Catalytic [4 + 2] Coupling. Nat. Chem.2014, 6, 739–744.2The challenge of using cyclobutanones in the cut-and-sew reaction is addressed by adding a temporary directing group.
Deng, L.; Chen, M.; Dong, G. Concise Synthesis of (−)-Cycloclavine and (−)-5-epi-Cycloclavine via Asymmetric C–C Activation. J. Am. Chem. Soc.2018, 140, 9652–9658.3The potential of the cut-and-sew reaction is demonstrated in the rapid and enantioselective construction of multiple fused rings in indole alkaloids.
Xue, Y.; Dong, G. Total Synthesis of Penicibilaenes via C–C Activation-Enabled Skeleton Deconstruction and Desaturation Relay-Mediated C–H Functionalization. J. Am. Chem. Soc.2021, 143, 8272–8277.4The “C–C/C–H” two-stage strategy is conceived and demonstrated in concise total synthesis.
1. Introduction
With an increasing demand to escape from flatland,5 synthetic methods that can efficiently generate novel, complex, sp3-rich structures, such as bridged and fused rings, become more and more attractive to medicinal chemists. Conventionally, bridged and fused rings were often prepared via either stepwise ring formation or cycloaddition. The stepwise approaches involve making one ring first and then another; thus, they generally require multistep operations, along with some functional group manipulations and protecting group usages. On the other hand, cycloaddition reactions, such as the Diels–Alder reaction, have been highly powerful in constructing various ring systems and successfully demonstrated in numerous elegant total syntheses.6 Typically, different classes of substrates are required in order to access different ring systems, and certain ring structures, such as those relying on forming “anti-Bredt” intermediates, are more challenging to construct with cycloaddition-based strategies.
Transition metal (TM)-catalyzed carbon–carbon (C–C) bond activation has emerged as a useful tool for devising unusual bond-disconnecting strategies.7 To seek a complementary ring-forming approach, a deconstructive strategy based on C–C bond activation of readily available cyclic ketones has been conceived (Scheme 1A). This named “cut-and-sew” strategy8 uses cyclic ketones with a tethered unsaturated moiety as substrates. It starts with the oxidative addition of a TM into the ketone C–C bond (the “cut” step) to give a reactive metallacycle, followed by intramolecular migratory insertion of the unsaturated unit and reductive elimination to furnish the ring (the “sew” step). It has been hypothesized that, by changing the ring sizes of the cyclic ketones, the length of the linkers, and different unsaturated coupling partners, diverse bridged and fused ring scaffolds would be constructed by this unified method. In addition, considering that the carbonyl moiety in ketones can be extruded under certain conditions during the C–C activation processes, the decarbonylative cut-and-sew reactions (with CO deletion) can also be realized with ketone-based substrates. Moreover, apart from the normal 2π insertion, TM-catalyzed one-carbon ring expansions have also been achieved to generate intriguing products.
Scheme 1. Cut-and-Sew Reactions of Benzocyclobutenones and Cyclobutanones.

On the basis of the rich prior knowledge of C–C activation,7,9 our group has been exploring the TM-catalyzed cut-and-sew reactions and their applications in complex molecule synthesis since 2012.1 In the past 10 years, benzocyclobutenones and cyclobutanones have been the two main classes of substrates we have studied. To date, several different types of cut-and-sew reactions with benzocyclobutenones and cyclobutanones have been developed in our laboratory, including (a) (4 + 2) or (4 + 2 – 1) cycloaddition between benzocyclobutenones and 2π units to construct [m.n.0] fused rings; (b) (4 + 1) cycloaddition between benzocyclobutenones and stryenes to construct 2-indanones; (c) (4 + 2) cycloaddition between α-branched cyclobutanones and 2π units to construct [m.n.0] fused rings; and (d) (4 + 2), (4 + 2 – 1), or (4 + 1) cycloaddition between β-branched cyclobutanones and 2π units to construct [m.n.1] bridged rings. To illustrate how these ring-forming methods can facilitate syntheses of complex molecules, these C–C activation methods have been applied to a number of concise total syntheses (Scheme 1B).3,4,10 In this Account, we first summarize our development of these catalytic cut-and-sew methods with benzocyclobutenones and cyclobutanones, followed by discussions of streamlined syntheses of (−)-cycloclavine, (−)-thebainone A, penicibilaenes, and the proposed cycloinumakiol, as representative examples enabled by such a deconstructive strategy.
2. Development of the Cut-and-Sew Methods
2.1. (4 + 2) Cut-and-Sew Reactions of Benzocyclobutenones
Benzocyclobutenones are a common class of four-membered ring ketones.11,12 They can be easily accessed by diverse methods, including [2 + 2] cycloaddition with benzynes,12b,12c,12e,12g intramolecular nucleophilic addition,12a transition metal-catalyzed intramolecular C–H functionalization,12d,12f and photoinduced cyclization.12h Driven by strain release (their ring strain is higher than saturated cyclobutanones),13 benzocyclobutenones are excellent substrates for transition-metal-mediated C–C bond activation. Inspired by Liebeskind et al.’s seminal organometallic studies,9a,9c we reported the first Rh-catalyzed intramolecular cut-and-sew reaction between benzocyclobutenones and alkenyl groups in 2012 (Scheme 2A).1 Note that the corresponding substrates based on parent cyclobutenones or those fused to nonbenzene aromatics are more difficult to prepare. The cut-and-sew reaction exhibited good functional group tolerance and worked for monosubstituted, 1,1- and 1,2-disubstituted, and trisubstituted alkenyl groups. Thus, it provides rapid access to benzo-fused tricyclic and tetracyclic rings. Later, detailed computational and experimental mechanistic studies showed that the reaction goes through a “rhodium migration” mechanism (Scheme 2B).14 The most favorable reaction path involves oxidative addition into the C(alkyl)–C(carbonyl) bond to generate intermediate 12, followed by decarbonylation and CO-reinsertion to deliver rhodacycle 14. Subsequent 2π-insertion and C–C reductive elimination provide the cut-and-sew product 11. The enantioselective version of the reaction was also reported in 2012, in which DTBM-segphos was found to be a superior ligand to give up to 99% e.e. (Scheme 3).15
Scheme 2. Rhodium-Catalyzed Intramolecular (4 + 2) Reactions between Benzocyclobutenones and Alkenyl Groups.

Scheme 3. Rhodium-Catalyzed Asymmetric Cut-and-Sew Reactions between Benzocyclobutenones and Alkenyl Groups.

Besides alkenyl groups, alkynyl groups were also found to be a suitable coupling partner in the cut-and-sew reaction with benzocyclobutenones (Scheme 4).16 With the C5-unsubstituted or monosubstituted substrates, the resulting (4 + 2) products underwent simultaneous aromatization to form 2-naphthols. In 2018, the same type of reactions was found to be catalyzed by an inexpensive cobalt complex (Scheme 5).17 Comparing to the corresponding Rh catalysis, the cobalt condition not only gives higher yields for some substrates but also allows C8-disubstituted benzocyclobutenones to couple, which were unreactive under the Rh conditions. A combined experimental and computational study shows that the reaction first forms a tetrahedral dicobalt–alkyne complex, which then undergoes oxidative addition into the C1–C2 bond with Co(0), followed by 2π insertion and reductive elimination.
Scheme 4. Rhodium-Catalyzed (4 + 2) Reactions between Benzocyclobutenones and Alkynyl Groups.

Scheme 5. Cobalt-Catalyzed (4 + 2) Reactions between Benzocyclobutenones and Alkynyl Groups.

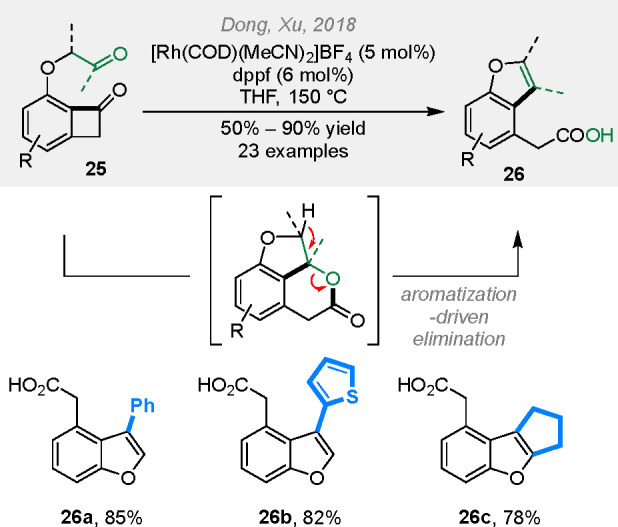
Apart from alkenyl and alkynyl groups, more polar carbon–heteroatom double bonds can also serve as the coupling partners in the cut-and-sew reactions. In 2016, we reported an asymmetric cut-and-sew reaction between benzocyclobutenones and oxime ethers (Scheme 6).18 The combination of two chiral ligands delivered both high yield and high enantioselectivity of the chiral lactam products. The N-OMe group can be easily removed to give free lactams. Analogously, ketones and aldehydes can also serve as the 2π coupling partners (Scheme 7).19 In this case, after generation of the initial (4 + 2) cycloaddition product, the lactone ring is spontaneously opened via elimination to form a more stable benzofuran product (26).
Scheme 6. Rhodium-Catalyzed Asymmetric (4 + 2) Reactions between Benzocyclobutenones and Oxime Ethers.

Scheme 7. Rhodium-Catalyzed Cut-and-Sew Reactions between Benzocyclobutenones and Ketones/Aldehydes.
2.2. (4 + 2) Cut-and-Sew Reactions with Cyclobutanones
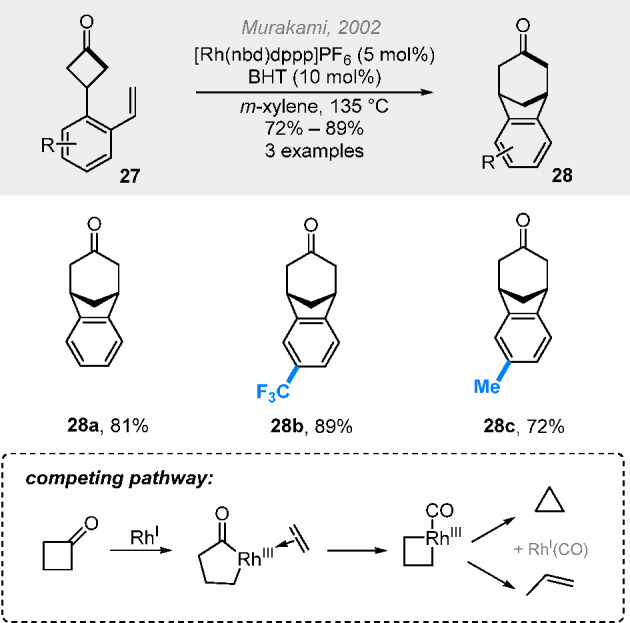
The use of saturated cyclobutanones provides opportunities to access various aliphatic ring systems. For example, β-branched cyclobutanones can undergo (4 + 2) cut-and-sew reactions to generate bridge bicycles. The seminal work by Murakami et al. demonstrated the synthesis of benzo-fused [3.2.1] bridged rings (Scheme 8),9f though the scope is limited to the benzene linker due to the competing decarbonylation associated with the C–C activation of cyclobutanones. To address this challenge, in 2014 we found that the use of 2-aminopyridines20 as an additive can protect the cyclobutanone carbonyl from decarbonylation through the in situ imine formation and accelerate the reaction via acting as a temporary directing group (Scheme 9).2 Diverse [3.3.1] and [3.2.1] bridged rings with various linkers, such as nitrogen- or malonate-based ones, were constructed in good yields. These structures are challenging to access via the traditional type II intramolecular Diels–Alder (IMDA) reaction. Initial success with the enantioselective version of the reaction was also achieved (30b).
Scheme 8. Rhodium-Catalyzed Intramolecular (4 + 2) Reactions with Benzene Linkers.
Scheme 9. Temporary Directing Group-Enabled Cut-and-Sew Reactions between Cyclobutanones and Alkenyl Groups.

In 2014, the Cramer group reported the highly enantioselective (4 + 2) cut-and-sew reactions based on the benzene-tethered cyclobutanones using alkenyl and carbonyl groups as the coupling partners (Scheme 10).21 In these reactions, DTBM-segphos again proved to be the optional chiral ligand (vide supra, Scheme 3). In 2020, an enantioselective intramolecular cut-and-sew reaction between cyclobutanones and alkynyl groups was disclosed by us (Scheme 11).22 Excellent enantioselectivity was achieved using a cationic Rh-DTBM-segphos catalyst. Interestingly, the in situ generated anti-Bredt bridged rings can be stabilized by rhodium coordination, according to our DFT calculations, which then gives the olefin migration product through hydride transfer. The E alkene was found to be the thermodynamically favored product; in contrast, the kinetically favored Z product predominated with the oxygen-linked substrates (e.g., 40c) when running the reaction at room temperature.
Scheme 10. Rhodium-Catalyzed Cut-and-Sew Reactions between Cyclobutanones and Alkenyl and Carbonyl Groups.

Scheme 11. Rhodium-Catalyzed (4 + 2) Cut-and-Sew Reactions between Cyclobutanones and Alkynyl Groups.

[Rh(C2H4)2Cl]2 (5 mol %), AgSbF6 (10 mol %), (R)-DTBM-segphos (12 mol %), 1,4-dioxane, room temperature.
In addition to the β-branched cyclobutanones, cyclobutanones with an unsaturated unit tethered to the α-position can lead to fused ring formation. In 2018, the cut-and-sew reaction between α-branched cyclobutanones and alkynyl groups was realized to construct [4.3.0] fused enones (Scheme 12).23 Electron-deficient [Rh(CO)2Cl]2 and less bulky, electron-rich PMe2Ph were found to be the optimal catalyst. Both carbon- and nitrogen-based linkers can be adopted; alkyl, aryl and silyl-substituted alkynyl groups are all suitable as coupling partners. Similar to the cut-and-sew reaction mechanism with benzocyclobutenones, it was proposed that the rhodium(I) catalyst first inserts into the less sterically hindered C–C bond in cyclobutanone 43 via oxidative addition to give complex 45, followed by decarbonylation and CO-reinsertion to generate rhodacycle 47. The sequential migratory insertion and reductive elimination finally deliver the fused-ring product 44. Later, the combination of a cationic rhodium complex and DTBM-segphos ligand allowed an efficient kinetic resolution of α-branched cyclobutanones 49 via the selective fused-ring formation (Scheme 13).24 The reaction proceeded at room temperature with high s factors achieved. Under this condition, terminal alkenyl groups can also be coupled. The interesting role of DTBM-segphos in promoting favorable ligand-substrate dispersion interactions has been revealed by the DFT study.
Scheme 12. Rhodium-Catalyzed (4 + 2) Reactions between α-Branched Cyclobutanones and Alkynyl Groups.

Scheme 13. Kinetic Resolution of α-Branched Cyclobutanones via Fused-Ring Formation.

2.3. (4 + 2 – 1) Cut-and-Sew Reactions of Benzocyclobutenones and Cyclobutanones
Considering that the carbonyl moiety in the substrates can be removed by decarbonylation, (4 + 2 – 1)-type transformations, namely, “decarbonylative cut-and-sew” reactions, have also been developed. This provides an unusual strategy to access bridged- or fused-ring scaffolds without bearing a ketone moiety. In 2014, the (4 + 2 – 1) reaction between benzocyclobutenones and alkynyl groups was discovered to give various fused indene products (Scheme 14A).16 The CO extrusion was promoted when running the reaction under reflux in xylene. It was proposed (Scheme 14B) that, after oxidation addition of rhodium(I) into the C1–C8 bond, decarbonylation and CO dissociation take place instead of reinsertion (vide supra, Scheme 2B).
Scheme 14. Rhodium-Catalyzed (4 + 2 – 1) Cut-and-Sew Reactions between Benzocyclobutenones and Alkynyl Groups.

The decarbonylative cut-and-sew reaction with cyclobutanones is more challenging, as it involves a difficult C(sp3)–C(sp3) reductive elimination. In 2016, we found that the use of a monodentate bulky Buchwald ligand enabled a smooth (4 + 2 – 1) cycloaddition between saturated cyclobutanones and alkenyl groups (Scheme 15).25 The bulkiness of the ligand not only promotes CO extrusion but also prevents the coordination of more than one phosphine ligand, thus resulting in coordinative unsaturation at the metal center for olefin coordination. Despite the high reaction temperature required for the reaction, we effectively obtained a range of cyclopentane-bridged rings that are otherwise challenging to prepare.
Scheme 15. Rhodium-Catalyzed (4 + 2 – 1) Reactions between Cyclobutanones and Alkenyl Groups.

2.4. (4 + 1) Cut-and-Sew Reactions of Benzocyclobutenones and Cyclobutanones
During our exploration of an intermolecular cut-and-sew reaction between benzocyclobutenones and styrenes, unexpected (4 + 1) cycloaddition products were obtained as the major products (Scheme 16A),26 in which the terminal carbon of styrenes inserted into the benzocyclobutenone C1–C2 bonds. The use of a “ligandless” cationic rhodium catalyst and a 2-aminopyridine additive were critical for this selectivity. While the double substitution at the C8 position of benzocyclobutenones is needed to prevent substrate decomposition, a range of multisubstituted 2-indanones was efficiently constructed by this method with good functional group tolerance. Further computational studies suggest that, rather than direct C–C reductive elimination, a competing β-H elimination takes place after C–C activation and 2π insertion, and the subsequent hydride reinsertion and reductive elimination lead to the five-membered ring formation (Scheme 16B).
Scheme 16. Rhodium-Catalyzed (4 + 1) Cut-and-Sew Reactions between Benzocyclobutenones and Styrenes.

Ratio of the (4 + 1) versus (4 + 2) products.
In 2015, an intramolecular (4 + 1) cut-and-sew cycloaddition was discovered when using allene-tethered cyclobutanones as the substrates (Scheme 17A),27 which provides rapid access to [4.2.1] and [3.2.1] bicyclic structures. This method shows a good substrate scope on both the cyclobutanone and allene parts. On the basis of the deuterium labeling study, the reaction was proposed to start from the oxidation addition of Rh(I) into the α-C–C bond of cyclobutanone 66 to give intermediate 68, in which the allene moiety coordinates to the rhodium center to prevent decarbonylation (Scheme 17B). The following migratory insertion of the acyl group into the allene central carbon generates allyl-rhodium complex 69, which then undergoes β-H elimination to give diene 70. At this stage, either C–H or C–C migratory insertion followed by reductive elimination can generate the bridged product. In addition, a highly enantioselective version of this reaction was realized using a TADDOL-derived phosphoramidite ligand (Scheme 17C).
Scheme 17. Rhodium-Catalyzed (4 + 1) Cut-and-Sew Reactions between Cyclobutanones and Allenyl Groups.

3. Application in Total Synthesis
The [m.n.0] fused rings and [m.n.1] bridged rings obtained from the cut-and-sew reactions are often found in complex bioactive molecules; thus, a number of total syntheses based on such a deconstructive C–C activation strategy have been achieved.28 In this section, the synthetic efforts from our group are summarized.
3.1. Total Synthesis of Cycloinumakiol (Proposed Structure)
Isolated from extracts of Podocarpus latifolius, cycloinumakiol (1) exhibits a distinct proposed chemical structure from other natural products in the tricyclic inumakiol family.29 In particular, it shows an unusual tetracyclic skeleton featuring a dihydrofuran ring and a quaternary carbon center. From a retrosynthetic aspect (Scheme 18A),10a we envisioned that the isopropyl group on the phenyl ring could be introduced by late-stage arene functionalization, and the tetracyclic core could be constructed via the cut-and-sew reaction with a cyclohexene-tethered benzocyclobutenone. In a forward manner, the benzocyclobutenone substrate (74) was efficiently prepared from the Mitsunobu reaction between phenol 75 and alcohol 76. To enable a highly challenging insertion of a sterically hindered trisubstituted alkenyl group, an electron-deficient [Rh(CO)2Cl]2/P(C6F5)3 catalyst was found to be optimal for this cut-and-sew reaction. With tetracycle 73 in hand, the isopropyl group was installed by a site-selective bromination and a one-pot Suzuki coupling/hydrogenation. After some end-game manipulations, the first total synthesis of the proposed structure of cycloinumakiol (1) was completed in nine steps from compounds 75 and 76, whose structure was unambiguously determined by X-ray crystallography. This effort also let us elucidate the actual structure of cycloinumakiol, which was reassigned to 19-hydroxytotarol.
Scheme 18. Total Synthesis of Cycloinumakiol (Proposed Structure).

3.2. Asymmetric Total Synthesis of (−)-Cycloclavine
Isolated from the seeds of Ipomoea hildebrandtii by Hofmann and co-workers in 196930 and later from Aspergillus japonicas in 1982,31 cycloclavine (2) is a unique member in the ergot alkaloid family because it processes a penta-cyclic core with a unique [3.1.0] structural motif. The sterically congested cyclopropane ring and three contiguous chiral centers including two adjacent quaternary carbons constitute additional challenges for the asymmetric total synthesis of cycloclavine. We envisioned a late-stage reductive amination tactic to construct the pyrrolidine D ring, a rhodium-catalyzed cyclopropanation to form the E ring, and an asymmetric cut-and-sew reaction to build the 6–6–5 fused A/B/C core structure (Scheme 19A).3 In a forward manner, the cut-and-sew precursor benzocyclobutenone 82a was prepared in high yield from commercially available diphenol 83 in three steps (Scheme 19B). After detailed condition optimizations, the combination of cationic rhodium [Rh(COD)2]BF4 and DTBM-segphos was most efficient, giving the desired ketone 81a in 95% yield and 97.5% e.e. This condition also appears to be quite general to access other nitrogen-containing tri- and tetracycles in high yields and excellent enantioselectivity (Scheme 19C). After the cut-and-sew step, the diazo-transfer followed by a Rh-catalyzed diastereoselective cyclopropanation32 of 2-methylallyl chloride 85 delivered the desired cyclopropane product 80 with good diastereoselectivity. In the end game, the SN2-substitution with azide, Boc deprotection and indoline oxidation gave indole 86. A one-pot aza-Wittig/reduction/reductive amination delivered (−)-cycloclavine (2) in 78% yield and >20:1 diastereoselectivity. In summary, this C–C activation-based approach accomplished the asymmetric total synthesis of (−)-cycloclavine in only 10 steps with 30% overall yield.
Scheme 19. Enantioselective Total Synthesis of (−)-Cycloclavine.

3.3. Asymmetric Total Synthesis of (−)-Thebainone A
Morphine (5) and its congeners are among the oldest and most studied alkaloid natural products, many of which have potent neurological and immunological activity.33 They generally possess a poly bridged/fused ring system, a quaternary center, a basic tertiary amine moiety and a 1,2,3,4-tetrasubstituted arene. As a unique member in the morphine-family alkaloids, thebainone A (7) contains an enone moiety in the C ring, which has served as a precursor to synthesize morphine (5) and codeine (6).34 However, the asymmetric synthesis of thebainone A was not reported previously. We devised a deconstructive strategy to synthesize (−)-thebainone A, in which the nitrogen-containing D ring is constructed in the end from an ether precursor (87a), and the fused A/B/C ring core structure, along with a quaternary carbon center, is assembled via an asymmetric cut-and-sew reaction (Scheme 20A).10d
Scheme 20. Deconstructive Asymmetric Total Synthesis of (−)-Thebainone A.

In a forward manner, the cut-and-sew precursor 88a was prepared in four steps from compounds 89 and 90 (Scheme 20B). The challenges of the key C–C activation-enabled (4 + 2) cycloaddition with compound 88a were associated with the presence of an acid-sensitive ketal, a trisubstituted olefin coupling partner, and a relatively long linker. Ultimately, the use of [Rh(COD)2]NTf2 and DTBM-segphos as the catalyst combination afforded high yield and excellent e.e. with 1,2-difluorobenzene as the solvent. This condition can also be used to construct other tetracycles with different olefin coupling partners, linkers, and functional groups (Scheme 20C). With ketone 87a in hand, the cyclic ether C–O bond was selectively cleaved by BBr3, and alkyl bromide 91 was accessed in four steps. Subsequently, ketone protection, SN2 amination, and Ac deprotection were achieved in one pot. The following dehydration with Martin’s sulfurane gave amine 92. The piperidine D ring was constructed by a radical-mediated hydroamination of the alkenyl group through reduction of the tosylamide moiety. Finally, selective deprotection of the middle methyl ether by NaSEt gave ketone 93, followed by desaturation by Stahl’s protocol,35 and furnished the first enantioselective total synthesis of (−)-thebainone A (7). It is worthy to note that intermediate 93 is also a known precursor to morphine (5) and codeine (6).34,36
3.4. Total Syntheses of Penicibilaenes A and B
Isolated from marine fungus Penicillium bilaiae MA-267, penicibilaenes A (8) and B (9) are two sesquiterpenes showing selective and potent activity against plant pathogenic fungus Colletotrichum gloeosporioides.37 They possess an intriguing tricyclo[6.3.1.01,5]dodecane skeleton, which is constituted by [3.3.1]-bridged and [4.3.0]-fused junctions. They also contain six chiral centers with five being contiguous and one all-carbon quaternary stereocenter. Inspired by the biomimetic “two-phase” approach for terpene synthesis,38 a “C–C/C–H” two-stage strategy was proposed to prepare penicibilaenes A and B (Scheme 21A).4 In the “C–H” stage, substituents and additional stereocenters are introduced to the core skeleton via sequential desaturation/β-functionalization of ketones, while in the “C–C” stage the carbon scaffold of the natural products is constructed by the cut-and-sew reaction.
Scheme 21. Total Syntheses of Penicibilaenes A and B.

In a forward style, the cut-and-sew precursor 96 was synthesized in three steps from commercially available starting materials 97, 98, and 99 (Scheme 21B). The key C–C activation reaction turned out to be nontrivial, and an important linker effect was found (Scheme 21C). Alkenyl (Lk1), epoxide (Lk2), silyl ether (Lk3), and methyl ether (Lk4) linkers were unsuccessful due to either their instability or lack of rigidity. Eventually, the ester-substituted alkenyl ester linker (Lk5) was most ideal because of its increased rigidity and stability; it also eased the substrate preparation. To address the challenge of coupling with a trisubstituted alkenyl group, 2-amino-3-isopropylpyridine (DG-4) was employed as the temporary carbonyl protecting group and directing group (Scheme 21D). It is also necessary to add zinc triflate to promote the cyclization, though its exact role remains to be uncovered. Ultimately, the desired cut-and-sew product (95) was obtained in useful yield, which then underwent decarboxylation to give tricycle 94.
After constructing the core skeleton of the natural product in the “C–C” stage, the remaining functional groups and stereocenters were installed by taking advantage of a desaturation relay-based strategy involving consecutive ketone α,β-dehydrogenation and β-functionalization. A sequence of Stahl’s oxidation,39 conjugate boration40 and oxidation delivered β-hydroxyl ketone 100. Chelation-controlled methylation41 followed by oxidation with IBX gave a new ketone (101) in high yield and diastereoselectivity. The methyl group at the C2 position was installed through another desaturation42/β-functionalization.43 The rigid half-cage scaffold allowed methyl addition from the less hindered convex face to deliver the desired diastereomer (103). Finally, an alcohol-directed Evans–Saksena reduction44 provided penicibilaene A (8) in 89% yield as a single diastereomer. Acylation of the less bulky secondary alcohol in penicibilaene A (8) delivered penicibilaene B (9). In summary, the first total syntheses of penicibilaenes A (8) and B (9) were accomplished in 13 and 14 steps, respectively, in the longest linear sequence from commercially available starting materials.
4. Summary and Outlook
In this Account, we have summarized our past efforts in the development of cut-and-sew reactions of benzocyclobutenones and cyclobutanones via catalytic C–C activation, as well as their applications in concise total synthesis of complex alkaloids and terpenoids. These methods prove to be useful for the construction of diverse bridged and fused rings that are otherwise more challenging to prepare. The reaction selectivity can be controlled by choices of the catalysts, ligands, and additives. Highly enantioselective cut-and-sew reactions have also been realized. Some of these transformations can even operate at room temperature.
As an outlook, a number of challenges and opportunities still exist for the further development of this type of transformation. First, the substrates so far are mainly restricted to highly strained rings. It would be more attractive if less strained, but readily available five- or six-membered ketones can undergo cut-and-sew reactions to construct medium-sized rings.45 In addition, the unsaturated coupling partners would benefit from a broader scope. For example, the coupling of tetra-substituted alkenyl groups remains to be achieved. Moreover, the type of linkers has large room to expand. To overcome the need of the Thorpe–Ingold effect, more active catalyst systems must be developed. Finally, most of these transformations are catalyzed by rhodium, and it could be an attractive direction to develop more practical and efficient first-row transition metal-catalyzed cut-and-sew reactions.
Acknowledgments
NIGMS (2R01GM109054) and the University of Chicago are acknowledged for research support. We thank Dr. Xin Liu from the University of Chicago for checking the manuscript.
Biographies
Yibin Xue received his B.S. degree in chemistry from Peking University in 2017. He then moved to the University of Chicago to begin his graduate study under the supervision of Professor Guangbin Dong. His research focuses on the total synthesis of natural products through the cut-and-sew reaction and C–C activation of the unstrained C–C bond.
Guangbin Dong received his B.S. degree from Peking University in 2003 and completed his Ph.D. degree in chemistry from Stanford University with Professor Barry M. Trost in 2009. After two years of postdoctoral training with Professor Robert H. Grubbs at California Institution of Technology, he joined the faculty of the University of Texas at Austin in 2011. In 2016, he moved to the University of Chicago as a Professor of Chemistry. His current research interests include transition-metal-catalyzed C–H and C–C bond activation, total synthesis of bioactive natural products, boron chemistry, and the development of new materials.
The authors declare no competing financial interest.
References
- Xu T.; Dong G. Rhodium-Catalyzed Regioselective Carboacylation of Olefins: A C–C Bond Activation Approach for Accessing Fused-Ring Systems. Angew. Chem. Int. Ed 2012, 51, 7567–7571. 10.1002/anie.201202771. [DOI] [PubMed] [Google Scholar]
- Ko H. M.; Dong G. Cooperative Activation of Cyclobutanones and Olefins Leads to Bridged Ring Systems by a Catalytic [4 + 2] Coupling. Nat. Chem. 2014, 6, 739–744. 10.1038/nchem.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Chen M.; Dong G. Concise Synthesis of (−)-Cycloclavine and (−)-5-epi-Cycloclavine via Asymmetric C–C Activation. J. Am. Chem. Soc. 2018, 140, 9652–9658. 10.1021/jacs.8b05549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Dong G. Total Synthesis of Penicibilaenes via C–C Activation-Enabled Skeleton Deconstruction and Desaturation Relay-Mediated C–H Functionalization. J. Am. Chem. Soc. 2021, 143, 8272–8277. 10.1021/jacs.1c04335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; b Lovering F. Escape from Flatland 2: Complexity and Promiscuity. Med. Chem. Comm 2013, 4, 515–519. 10.1039/c2md20347b. [DOI] [Google Scholar]
- For selected reviews, see:; a Nicolaou K. C.; Snyder S. A.; Montagnon T.; Vassilikogiannakis G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed 2002, 41, 1668–1698. . [DOI] [PubMed] [Google Scholar]; b Ylijoki K. E. O.; Stryker J. M. [5 + 2] Cycloaddition Reactions in Organic and Natural Product Synthesis. Chem. Rev. 2013, 113, 2244–2266. 10.1021/cr300087g. [DOI] [PubMed] [Google Scholar]; c Yin Z.; He Y.; Chiu P. Application of (4 + 3) Cycloaddition Strategies in the Synthesis of Natural Products. Chem. Soc. Rev. 2018, 47, 8881–8924. 10.1039/C8CS00532J. [DOI] [PubMed] [Google Scholar]; d Min L.; Liu X.; Li C.-C. Total Synthesis of Natural Products with Bridged Bicyclo[m.n.1] Ring Systems via Type II [5 + 2] Cycloaddition. Acc. Chem. Res. 2020, 53, 703–718. 10.1021/acs.accounts.9b00640. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Murakami M.; Ito Y. Cleavage of Carbon–Carbon Single Bonds by Transition Metals. Top. Organomet. Chem. 1999, 3, 97–129. 10.1007/3-540-68525-1_5. [DOI] [Google Scholar]; b van der Boom M. E.; Milstein D. Cyclometalated Phosphine-Based Pincer Complexes: Mechanistic Insight in Catalysis, Coordination, and Bond Activation. Chem. Rev. 2003, 103, 1759–1792. 10.1021/cr960118r. [DOI] [PubMed] [Google Scholar]; c Jun C.-H. Transition Metal-Catalyzed Carbon–Carbon Bond Activation. Chem. Soc. Rev. 2004, 33, 610–618. 10.1039/B308864M. [DOI] [PubMed] [Google Scholar]; d Tobisu M.; Chatani N. Catalytic Reactions Involving the Cleavage of Carbon–Cyano and Carbon–Carbon Triple Bonds. Chem. Soc. Rev. 2008, 37, 300–307. 10.1039/B702940N. [DOI] [PubMed] [Google Scholar]; e Chen F.; Wang T.; Jiao N. Recent Advances in Transition-Metal-Catalyzed Functionalization of Unstrained Carbon–Carbon Bonds. Chem. Rev. 2014, 114, 8613–8661. 10.1021/cr400628s. [DOI] [PubMed] [Google Scholar]; f Dong G.C–C Bond Activation; Springer-Verlag: Berlin, 2014; Vol. 346. [Google Scholar]; g Cleavage of Carbon–Carbon Single Bonds by Transition Metals; Murakami M., Chatani N., Eds.; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]; h Souillart L.; Cramer N. Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev. 2015, 115, 9410–9464. 10.1021/acs.chemrev.5b00138. [DOI] [PubMed] [Google Scholar]; i Fumagalli G.; Stanton S.; Bower J. F. Recent Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev. 2017, 117, 9404–9432. 10.1021/acs.chemrev.6b00599. [DOI] [PubMed] [Google Scholar]; j Song F.; Gou T.; Wang B.-Q.; Shi Z.-J. Catalytic Activations of Unstrained C–C Bond Involving Organometallic Intermediates. Chem. Soc. Rev. 2018, 47, 7078–7115. 10.1039/C8CS00253C. [DOI] [PubMed] [Google Scholar]; k Deng L.; Dong G. Carbon–Carbon Bond Activation of Ketones. Trends Chem. 2020, 2, 183–198. 10.1016/j.trechm.2019.12.002. [DOI] [Google Scholar]; l Xia Y.; Dong G. Temporary or Removable Directing Groups Enable Activation of Unstrained C–C Bonds. Nat. Rev. Chem. 2020, 4, 600–614. 10.1038/s41570-020-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Murakami M.; Ishida N. Cleavage of Carbon–Carbon σ-Bonds of Four-Membered Rings. Chem. Rev. 2021, 121, 264–299. 10.1021/acs.chemrev.0c00569. [DOI] [PubMed] [Google Scholar]; n Nakao Y. Metal-mediated C–CN Bond Activation in Organic Synthesis. Chem. Rev. 2021, 121, 327–344. 10.1021/acs.chemrev.0c00301. [DOI] [PubMed] [Google Scholar]
- Chen P.-h.; Billett B. A.; Tsukamoto T.; Dong G. “Cut and Sew” Transformations via Transition-Metal-Catalyzed Carbon–Carbon Bond Activation. ACS Catal. 2017, 7, 1340–1360. 10.1021/acscatal.6b03210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal studies of C–C activation of four-membered ketones, see:; a Huffman M. A.; Liebeskind L. S.; Pennington W. T. Synthesis of Metallacyclopentenones by Insertion of Rhodium into Cyclobutenones. Organometallics 1990, 9, 2194–2196. 10.1021/om00158a009. [DOI] [Google Scholar]; b Huffman M. A.; Liebeskind L. S. Nickel(0)-Catalyzed Synthesis of Substituted Phenols from Cyclobutenones and Alkynes. J. Am. Chem. Soc. 1991, 113, 2771–2772. 10.1021/ja00007a072. [DOI] [Google Scholar]; c Huffman M. A.; Liebeskind L. S.; Pennington W. T. Reaction of Cyclobutenones with Low-Valent Metal Reagents to Form η4- and η2-Vinylketene Complexes. Reaction of η4-Vinylketene Complexes with Alkynes to Form Phenols. Organometallics 1992, 11, 255–266. 10.1021/om00037a047. [DOI] [Google Scholar]; d Murakami M.; Amii H.; Ito Y. Selective Activation of Carbon–Carbon Bonds Next to a Carbonyl Group. Nature 1994, 370, 540–541. 10.1038/370540a0. [DOI] [Google Scholar]; e Murakami M.; Amii H.; Shigeto K.; Ito Y. Breaking of the C–C Bond of Cyclobutanones by Rhodium(I) and Its Extension to Catalytic Synthetic Reactions. J. Am. Chem. Soc. 1996, 118, 8285–8290. 10.1021/ja9604525. [DOI] [Google Scholar]; f Murakami M.; Itahashi T.; Ito Y. Catalyzed Intramolecular Olefin Insertion into a Carbon–Carbon Single Bond. J. Am. Chem. Soc. 2002, 124, 13976–13977. 10.1021/ja021062n. [DOI] [PubMed] [Google Scholar]
- a Xu T.; Dong G. Coupling of Sterically Hindered Trisubstituted Olefins and Benzocyclobutenones by C–C Activation: Total Synthesis and Structural Revision of Cycloinumakiol. Angew. Chem. Int. Ed 2014, 53, 10733–10736. 10.1002/anie.201404802. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qiu B.; Li X.-T.; Zhang J.-Y.; Zhan J.-L.; Huang S.-P.; Xu T. Catalytic Enantioselective Synthesis of 3,4-Polyfused Oxindoles with Quaternary All-Carbon Stereocenters: A Rh-Catalyzed C–C Activation Approach. Org. Lett. 2018, 20, 7689–7693. 10.1021/acs.orglett.8b03412. [DOI] [PubMed] [Google Scholar]; c Zhang Y.; Shen S.; Fang H.; Xu T. Total Synthesis of Galanthamine and Lycoramine Featuring an Early-Stage C–C and a Late-Stage Dehydrogenation via C–H Activation. Org. Lett. 2020, 22, 1244–1248. 10.1021/acs.orglett.9b04337. [DOI] [PubMed] [Google Scholar]; d Hou S.-H.; Prichina A. Y.; Dong G. Deconstructive Asymmetric Total Synthesis of Morphine-Family Alkaloid (−)-Thebainone A. Angew. Chem. Int. Ed 2021, 60, 13057–13064. 10.1002/anie.202103553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Flores-Gaspar A.; Martin R. Recent Advances in the Synthesis and Application of Benzocyclobutenones and Related Compounds. Synthesis 2013, 45, 563–580. 10.1055/s-0032-1316850. [DOI] [Google Scholar]; b Chen P.-h.; Dong G. Cyclobutenones and Benzocyclobutenones: Versatile Synthons in Organic Synthesis. Chem.—Eur. J. 2016, 22, 18290–18315. 10.1002/chem.201603382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Aidhen I. S.; Ahuja J. R. A Novel Synthesis of Benzocyclobutenones. Tetrahedron Lett. 1992, 33, 5431–5432. 10.1016/S0040-4039(00)79113-1. [DOI] [Google Scholar]; b Mariet N.; Ibrahim-Ouali M.; Santelli M. First [2 + 2]-Cycloaddition of a 3,4-Didehydropyridine and a Ketene Dialkyl Acetal. Tetrahedron Lett. 2002, 43, 5789–5791. 10.1016/S0040-4039(02)01176-0. [DOI] [Google Scholar]; c Tsujiyama S.; Suzuki K. Preparation of Benzocyclobutenone Derivatives Based on an Efficient Generation of Benzynes. Org. Synth 2007, 84, 272–284. 10.15227/orgsyn.084.0272. [DOI] [Google Scholar]; d Álvarez-Bercedo P.; Flores-Gaspar A.; Correa A.; Martin R. Pd-Catalyzed Intramolecular Acylation of Aryl Bromides via C–H Functionalization: A Highly Efficient Synthesis of Benzocyclobutenones. J. Am. Chem. Soc. 2010, 132, 466–467. 10.1021/ja909811t. [DOI] [PubMed] [Google Scholar]; e Im G-Y. J.; Bronner S. M.; Goetz A. E.; Paton R. S.; Cheong P. H.-Y.; Houk K. N.; Garg N. K. Indolyne Experimental and Computational Studies: Synthetic Applications and Origins of Selectivities of Nucleophilic Additions. J. Am. Chem. Soc. 2010, 132, 17933–17944. 10.1021/ja1086485. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Flores-Gaspar A.; Gutiérrez-Bonet Á.; Martin R. N-Heterocyclic Carbene Dichotomy in Pd-Catalyzed Acylation of Aryl Chlorides via C–H Bond Functionalization. Org. Lett. 2012, 14, 5234–5237. 10.1021/ol3023819. [DOI] [PubMed] [Google Scholar]; g Chen P.-H.; Savage N. A.; Dong G. Concise Synthesis of Functionalized Benzocyclobutenones. Tetrahedron 2014, 70, 4135–4146. 10.1016/j.tet.2014.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Yano T.; Kawasaki T.; Yuhki T.; Ishida N.; Murakami M. Synthetic Approach to Benzocyclobutenones Using Visible Light and a Phosphonate Auxiliary. Org. Lett. 2018, 20, 1224–1227. 10.1021/acs.orglett.8b00160. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhou Q.; Fang H.; Lu P. Dancing on Ropes - Enantioselective Functionalization of Preformed Four-Membered Carbocycles. Chin. J. Chem. 2022, 40, 1346–1358. 10.1002/cjoc.202100879. [DOI] [Google Scholar]
- Lu G.; Fang C.; Xu T.; Dong G.; Liu P. Computational Study of Rh-Catalyzed Carboacylation of Olefins: Ligand-Promoted Rhodacycle Isomerization Enables Regioselective C–C Bond Functionalization of Benzocyclobutenones. J. Am. Chem. Soc. 2015, 137, 8274–8283. 10.1021/jacs.5b04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T.; Ko H. M.; Savage N. A.; Dong G. Highly Enantioselective Rh-Catalyzed Carboacylation of Olefins: Efficient Syntheses of Chiral Poly-Fused Rings. J. Am. Chem. Soc. 2012, 134, 20005–20008. 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]
- Chen P.-h.; Xu T.; Dong G. Divergent Syntheses of Fused β-Naphthol and Indene Scaffolds by Rhodium-Catalyzed Direct and Decarbonylative Alkyne–Benzocyclobutenone Couplings. Angew. Chem. Int. Ed 2014, 53, 1674–1678. 10.1002/anie.201310100. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Li X.; Chen S.; Chen P.-h.; Billett B. A.; Huang Z.; Dong G. Cobalt-Catalyzed Intramolecular Alkyne/Benzocyclobutenone Coupling: C–C Bond Cleavage via a Tetrahedral Dicobalt Intermediate. ACS Catal. 2018, 8, 845–849. 10.1021/acscatal.7b03852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Xu T.; Li H.; Dong G. Enantioselective Rh-Catalyzed Carboacylation of C=N Bonds via C–C Activation of Benzocyclobutenones. J. Am. Chem. Soc. 2016, 138, 369–374. 10.1021/jacs.5b11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.; Zhang Y.; Qiu B.; Wang Y.; Qin Y.; Dong G.; Xu T. Rhodium(I)-Catalyzed Carboacylation/Aromatization Cascade Initiated by Regioselective C–C Activation of Benzocyclobutenones. Angew. Chem. Int. Ed 2018, 57, 2859–2863. 10.1002/anie.201713179. [DOI] [PubMed] [Google Scholar]
- Park Y. J.; Park J.-W.; Jun C.-H. Metal–Organic Cooperative Catalysis in C–H and C–C Bond Activation and Its Concurrent Recovery. Acc. Chem. Res. 2008, 41, 222–234. 10.1021/ar700133y. [DOI] [PubMed] [Google Scholar]
- a Souillart L.; Parker E.; Cramer N. Highly Enantioselective Rhodium(I)-Catalyzed Activation of Enantiotopic Cyclobutanone C–C Bonds. Angew. Chem. Int. Ed 2014, 53, 3001–3005. 10.1002/anie.201311009. [DOI] [PubMed] [Google Scholar]; b Souillart L.; Cramer N. Highly Enantioselective Rhodium(I)-Catalyzed Carbonyl Carboacylations Initiated by C–C Bond Activation. Angew. Chem. Int. Ed 2014, 53, 9640–9644. 10.1002/anie.201405834. [DOI] [PubMed] [Google Scholar]
- Hou S.-H.; Yu X.; Zhang R.; Deng L.; Zhang M.; Prichina A. Y.; Dong G. Enantioselective Type II Cycloaddition of Alkynes via C–C Activation of Cyclobutanones: Rapid and Asymmetric Construction of [3.3.1] Bridged Bicycles. J. Am. Chem. Soc. 2020, 142, 13180–13189. 10.1021/jacs.0c05647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Jin L.; Dong G. Fused-Ring Formation by an Intramolecular “Cut-and-Sew” Reaction between Cyclobutanones and Alkynes. Angew. Chem. Int. Ed 2018, 57, 2702–2706. 10.1002/anie.201712487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Fu Y.; Lee S. Y.; Wang C.; Liu P.; Dong G. Kinetic Resolution via Rh-Catalyzed C–C Activation of Cyclobutanones at Room Temperature. J. Am. Chem. Soc. 2019, 141, 16260–16265. 10.1021/jacs.9b09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Ko H. M.; Dong G. Synthesis of Bridged Cyclopentane Derivatives by Catalytic Decarbonylative Cycloaddition of Cyclobutanones and Olefins. Angew. Chem. Int. Ed 2016, 55, 13867–13871. 10.1002/anie.201608158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi S.; Zhang Z.; Xia Y.; Dong G. Rhodium-Catalyzed (4 + 1) Cycloaddition between Benzocyclobutenones and Styrene-Type Alkenes. Angew. Chem. Int. Ed 2022, 61, e202202703 10.1002/anie.202202703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Dong G. (4 + 1) vs (4 + 2): Catalytic Intramolecular Coupling between Cyclobutanones and Trisubstituted Allenes via C–C Activation. J. Am. Chem. Soc. 2015, 137, 13715–13721. 10.1021/jacs.5b09799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reviews of using C–C activation in total synthesis, see:; a Murakami M.; Ishida N. Potential of Metal-Catalyzed C–C Single Bond Cleavage for Organic Synthesis. J. Am. Chem. Soc. 2016, 138, 13759–13769. 10.1021/jacs.6b01656. [DOI] [PubMed] [Google Scholar]; b Wang B.; Perea M. A.; Sarpong R. Transition Metal-Mediated C–C Single Bond Cleavage: Making the Cut in Total Synthesis. Angew. Chem. Int. Ed 2020, 59, 18898–18919. 10.1002/anie.201915657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K. P.; Ratnayake R.; Colburn N. H.; Wilson J. A.; Henrich C. J.; McMahon J. B.; Beutler J. A. Inhibitors of the Oncogenic Transcription Factor AP-1 from Podocarpus latifolius. J. Nat. Prod 2011, 74, 374–377. 10.1021/np100736y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffacher D.; Niklaus P.; Tscherter H.; Weber H. P.; Hofmann A. Cycloclavin, Ein Neues Alkaloid aus Ipomoea hildebrandtii Vatke—71: Mutterkornalkaloide. Tetrahedron 1969, 25, 5879–5887. 10.1016/S0040-4020(01)83095-7. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Koike M.; Abe M. Isolation of Cycloclavine from the Culture Broth of Aspergillus japonicus SAITO. Agric. Biol. Chem. 1982, 46, 1921–1922. 10.1271/bbb1961.46.1921. [DOI] [Google Scholar]
- Davies H. M. L.; Nagashima T.; Klino J. L. Stereoselectivity of Methyl Aryldiazoacetate Cyclopropanations of 1,1-Diarylethylene. Asymmetric Synthesis of a Cyclopropyl Analogue of Tamoxifen. Org. Lett. 2000, 2, 823–826. 10.1021/ol005563u. [DOI] [PubMed] [Google Scholar]
- Blakemore P. R.; White J. D. Morphine, the Proteus of Organic Molecules. Chem. Commun. 2002, 1159–1168. 10.1039/b111551k. [DOI] [PubMed] [Google Scholar]
- a Gates M.; Tschudi G. The Synthesis of Morphine. J. Am. Chem. Soc. 1952, 74, 1109–1110. 10.1021/ja01124a538. [DOI] [Google Scholar]; b Gates M.; Tschudi G. The Synthesis of Morphine. J. Am. Chem. Soc. 1956, 78, 1380–1393. 10.1021/ja01588a033. [DOI] [Google Scholar]
- Diao T.; Wadzinski T. J.; Stahl S. S. Direct aerobic α,β-Dehydrogenation of Aldehydes and Ketones with a Pd(TFA)2/4,5-Diazafluorenone Catalyst. Chem. Sci. 2012, 3, 887–891. 10.1039/C1SC00724F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Weller D. D.; Rapoport H. A Practical Synthesis of Codeine from Dihydrothebainone. J. Med. Chem. 1976, 19, 1171–1175. 10.1021/jm00232a001. [DOI] [PubMed] [Google Scholar]; b Rice K. C. A Rapid, High-Yield Conversion of Codeine to Morphine. J. Med. Chem. 1977, 20, 164–165. 10.1021/jm00211a036. [DOI] [PubMed] [Google Scholar]; c Rice K. C. Synthetic Opium Alkaloids and Derivatives. A Short Total Synthesis of (±)-Dihydrothebainone, (±)-Dihydrocodeinone, and (±)-Nordihydrocodeinone as an Approach to a Practical Synthesis of Morphine, Codeine, and Congeners. J. Org. Chem. 1980, 45, 3135–3137. 10.1021/jo01303a045. [DOI] [Google Scholar]
- Meng L.-H.; Li X.-M.; Liu Y.; Wang B.-G. Penicibilaenes A and B, Sesquiterpenes with a Tricyclo[6.3.1.01,5]dodecane Skeleton from the Marine Isolate of Penicillium bilaiae MA-267. Org. Lett. 2014, 16, 6052–6055. 10.1021/ol503046u. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Chen K.; Baran P. S. Total Synthesis of Eudesmane Terpenes by Site-Selective C–H Oxidations. Nature 2009, 459, 824–828. 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]; b Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. 14-Step Synthesis of (+)-Ingenol from (+)-3-Carene. Science 2013, 341, 878–882. 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]; c Kanda Y.; Nakamura H.; Umemiya S.; Puthukanoori R. K.; Murthy Appala V. R.; Gaddamanugu G. K.; Paraselli B. R.; Baran P. S. Two-Phase Synthesis of Taxol. J. Am. Chem. Soc. 2020, 142, 10526–10533. 10.1021/jacs.0c03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao T.; Stahl S. S. Synthesis of Cyclic Enones via Direct Palladium-Catalyzed Aerobic Dehydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 14566–14569. 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-s.; Zhugralin A. R.; Hoveyda A. H. Efficient C–B Bond Formation Promoted by N-Heterocyclic Carbenes: Synthesis of Tertiary and Quaternary B-Substituted Carbons through Metal-Free Catalytic Boron Conjugate Additions to Cyclic and Acyclic α,β-Unsaturated Carbonyls. J. Am. Chem. Soc. 2009, 131, 7253–7255. 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovskiy A.; Kopp F.; Knochel P. Soluble Lanthanide Salts (LnCl3·2LiCl) for the Improved Addition of Organomagnesium Reagents to Carbonyl Compounds. Angew. Chem. Int. Ed 2006, 45, 497–500. 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- Mukaiyama T.; Matsuo J.-i.; Kitagawa H. A New and One-Pot Synthesis of α,β-Unsaturated Ketones by Dehydrogenation of Various Ketones with N-tert-Butyl Phenylsulfinimidoyl Chloride. Chem. Lett. 2000, 29, 1250–1251. 10.1246/cl.2000.1250. [DOI] [Google Scholar]
- Cao M.-Y.; Ma B.-J.; Lao Z.-Q.; Wang H.; Wang J.; Liu J.; Xing K.; Huang Y.-H.; Gan K.-J.; Gao W.; Wang H.; Hong X.; Lu H.-H. Optically Active Flavaglines-Inspired Molecules by a Palladium-Catalyzed Decarboxylative Dearomative Asymmetric Allylic Alkylation. J. Am. Chem. Soc. 2020, 142, 12039–12045. 10.1021/jacs.0c05113. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Chapman K. T.; Carreira E. M. Directed Reduction of β-Hydroxy Ketones Employing Tetramethylammonium Triacetoxyborohydride. J. Am. Chem. Soc. 1988, 110, 3560–3578. 10.1021/ja00219a035. [DOI] [Google Scholar]
- a Xia Y.; Ochi S.; Dong G. Two-Carbon Ring Expansion of 1-Indanones via Insertion of Ethylene into Carbon–Carbon Bonds. J. Am. Chem. Soc. 2019, 141, 13038–13042. 10.1021/jacs.9b07445. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang R.; Xia Y.; Dong G. Intermolecular [5 + 2] Annulation between 1-Indanones and Internal Alkynes by Rhodium-Catalyzed C–C Activation. Angew. Chem. Int. Ed 2021, 60, 20476–20482. 10.1002/anie.202106007. [DOI] [PMC free article] [PubMed] [Google Scholar]