

Abstract
The emergence of novel proteins, beyond these that can be readily made by duplication and recombination of preexisting domains, is elusive. De novo emergence from random sequences is unlikely because the vast majority of random chains would not even fold, let alone function. An alternative explanation is that novel proteins emerge by duplication and fusion of pre‐existing polypeptide segments. In this case, traces of such ancient events may remain within contemporary proteins in the form of reused segments. Together with the late Dan Tawfik, we detected such similar segments, far shorter than intact protein domains, which are found in different environments. The detection of these, “bridging themes,” was based on a unique search strategy, where in addition to searching for similarity of shared fragments, so‐called “themes,” we also explicitly searched for cases in which the sequence segments before and after the theme are dissimilar (both in sequence and structure). Here, using a similar strategy, we further expanded the search and discovered almost 500 additional “bridging themes,” linking domains that are often from ancient folds. The themes, of 20 residues or more (average 53), do not retain their structure despite sharing 37% sequence identity on average. Indeed, conformation flexibility may confer an evolutionary advantage, in that it fits in multiple environments. We elaborate on two interesting themes, shared between Rossmann/Trefoil‐Plexin‐like domains and a β‐propeller‐like domain.
For a Broad Audience
A fundamental question in molecular evolution is how protein domains emerged. Similar segments shared between domains of seemingly distinct origins, may offer clues, as these may be remnants of the evolutionary process through which these domains emerged. However, finding such cases is difficult. Here, we expand the set of such cases which we curated previously, adding segments shared between domains that are considered ancient.
Keywords: ancestral peptides, bridging themes, protein emergence, protein evolutionary patterns, protein space
1. INTRODUCTION
It is commonly accepted that proteins evolve by duplication, mix, and match of autonomously folded domains. 1 But how have the domains themselves emerged? It is tempting to speculate that by analogy, domains also evolve by duplication, mix, and match of smaller protein segments. 2 Indeed, starting with the pioneering discovery of duplication in Ferredoxin by Eck and Dayhoff, 3 evidence to this end accumulate. 2 , 4 , 5 , 6 , 7 , 8 , 9 , 10 Having a repertoire of such ancestral peptides and the domains where they are found furthers our understanding of protein evolution.
One strategy for finding ancestral peptides is based on internal repeats within a domain. 11 A significant advantage of searching for repeats in the same domain is that the search space is very restricted. Indeed, Ferredoxin, the abovementioned first example of such a repeat was identified already in 1966. 3 One example of studied repeat proteins is the β‐propellers, the largest family of tandem repeat proteins. This fold was investigated to explore the peptide‐to‐domain hypothesis. The repeat unit in β‐propellers is a blade, or four anti‐parallel β‐strands, and there are propellers with 4–12 blades. Chandhuri et al. 12 analyzed propeller sequences and showed that they were likely amplified from single blades. In further support that a single blade may have been the ancestral peptide, Tawfik and co‐workers identified a single blade based on ancestral reconstruction that they showed experimentally can multimerize into a five‐bladed propeller. 13 The β‐propeller fold can also show remarkable structural plasticity. 14 For an overview of studies of the evolution of β‐propellers see. 15
Cases where the repeated segment is in domains of seemingly unrelated evolutionary origin are more challenging to find. These so‐called “bridging themes” are of particular interest from an evolutionary standpoint. 9 There may be various explanations for these shared themes: (a) ancient molecular fossils of segments that evolved from a common ancestor of the two domains, 16 (b) a segment copied from one domain and grafted to another, followed by divergence of the two, 17 or (c) the result of convergent evolution toward a shared function. Had we found two identical sequence segments of even a few dozen residues, and as sampling of a specific sequence is an extremely low‐probability event, convergent evolution would not be a likely explanation. 2 In real data, the sequences of the two segments diverged and are merely similar. Thus, we use probabilistic models 18 , 19 to evaluate if there is a likely evolutionary relationship between the two segments. Previous studies searched for such cases and found instances where a similar segment is found in different evolutionary lineages. For example, using Evolutionary Classification of Domains (ECOD) X‐groups annotations 20 to identify different lineages, the set Alva et al. curated (including those identified before them) 10 has 286 such pairs. 9 In collaboration with Tawfik, we found 525 such cases. 9 Within our set, was also a (possibly) ancient shared ancestral segment of the P‐loops and the Rossmanns, 16 just like Tawfik's prediction that it will probably be found. 21
Computational search efforts for bridging themes resulted in different sets. Alva et al. expanded previously known cases, and the ones in their set generally do not overlap with the ones we found (only three X‐group pairs were found in both). Indeed, when a search is successful, the (statistically significant) similar segment within overall different domains is the proof, demonstrating a relationship between the two domains. However, when a search fails, it does not necessarily rule out the existence of relationships between the domains searched, rather, it may be due to the failure of the search procedure. Searching relies on accurate, fast, and sensitive sequence aligners, such as HHSearch 18 and HMMER. 19 Both align a query protein sequence (modeled as a hidden Markov model [HMM]) to a database of many to identify statistically significant cases and align the query to the targets found. The two aligners differ from one another: HHSearch is an HMM‐HMM aligner and searches in a database of HMMs, while HMMER is an HMM‐sequence aligner, and searches within a database of sequences. Our previous search 9 relied on HHSearch, and used themes, 7 which are protein segments that are reused in protein space, as “baits.” 22 We considered all cases where a theme was matched to domains from different X‐groups as candidates for our set. When we aligned a “bait” HMMs to ECOD HMMs, the segments matched to the “bait” could have differed from the segments in the ECOD domain sequences, because the HMM might include insertions and deletions. Hence, we had to identify the segment in the ECOD domain that corresponds to the HMM segment. In contrast, when we use HMMER—an HMM‐sequence aligner, it directly identifies the segments in the ECOD domain sequences.
In this paper, we used this strategy, thereby detecting a large new set of bridging themes. This set is complementary to the previously known cases, which collectively represent a repertoire of over thousand bridging themes, many of which link ancient architectures such as P‐loop, Rossmann, Ferredoxin, and TIM‐barrel. Tawfik predicted that β‐propellers, which appear to be of singular lineage, would be evolutionary linked to other architectures (private communication). As he anticipated, such links exists, and are found in our new set of bridging themes. Interestingly, it connects to a Rossmann‐like domain and a Trefoil/Plexin like domain. We elaborate on these cases here.
2. RESULTS
2.1. General properties of the bridging theme dataset
We rely on the ECOD 20 to identify cases where the two domains are considered to be of different evolutionary lineages. In ECOD, the X‐group level is inclusive (more so than in the analog level of the other domain classifications SCOP and CATH), grouping together architecturally similar domains, even if more evidence is needed to deduce a common evolutionary origin. 20 Hence, we deduce that domains in different X‐groups are viewed as of independent evolutionary lineages.
Using HMMER to align our theme HMMs to ECOD 20 domains, we identified 491 pairs of domains that are from different ECOD X‐groups and nonetheless share a segment of at least 20 residues that are similar to each other, that is, bridging themes. The pairs of HMMER bridging themes span 115 ECOD X‐groups from all 20 structural classes, with 108 pairs of X‐groups. Figure 1 shows an overview network of the links among X‐groups, and the data is available for download and browsing as a Cytoscape 23 session in the Supporting Information S1. Compared to the previous bridging themes datasets 9 : 43 of the 115 X‐groups and 19 pairs of X‐groups, are in both sets. We also included a Cytoscape/Cytostruct 24 session with the 491 pairs of domains organized in networks by their X‐groups. Clicking on the edges opens a PyMOL 25 session with the two domains superimposed and the shared segment colored, or a BioEdit 26 session to see the sequence alignment.
FIGURE 1.
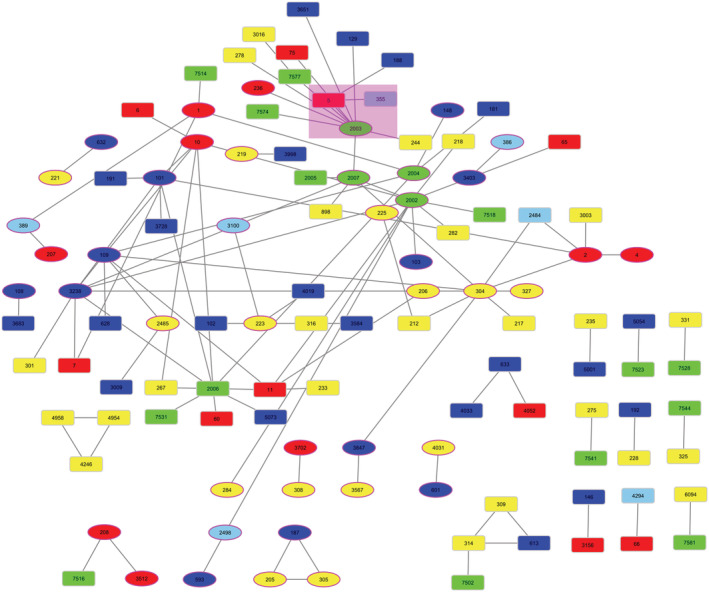
An overview network of the links identified between different Evolutionary Classification of Domains X‐groups. The nodes are the 115 X‐groups found in this bridging themes set, colored according to their architecture: all‐α in blue, all‐β in red, α/β in green, and α + β in yellow. The nodes representing X‐groups that are in the previous bridging theme set are shown as ellipses. Edges connect X‐groups for which we found a bridging theme shared between their domains. The nodes representing Rossmann‐like, β‐propeller, Trefoil/Plexin‐like X‐groups, and bridging themes detailed in the main text are highlighted with a purple background.
Figure 2 shows histograms of properties of the shared segments in the domain pairs of our dataset: the number of aligned residues, the root mean square deviation (RMSD) (calculated for the 469 pairs with all residues present in the PDB file), and the percent sequence identify and similarity. The number of residues was filtered to be above 20, and averages 53, the RMSD averages 12.2 Å, and the percent sequence identify/similarity average 36.7%/70.3%. These values are comparable to their counterparts in the previous 2021 molecular biology & evolution (MBE) bridging themes dataset. 9 The only significant difference is that structure was conserved in about half of the bridging themes in the previous set, and it is less conserved in the current bridging themes set (Figure 2b).
FIGURE 2.
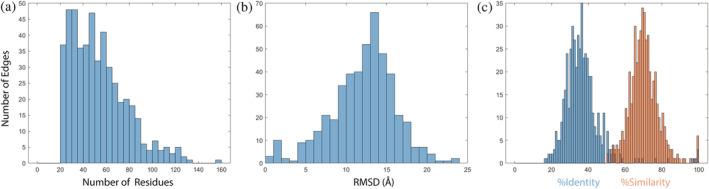
The cumulative properties of the bridging themes in our data set. (a) Length distribution around the average of 53 residues (restricted to more than 20 residues by design). (b) Distribution of RMSD between the Cα‐s of aligned residues in the two variants of the shared theme, after optimal superpositioning. The RMSD was calculated for the 469 pairs with all residues found in the PDB files. The average RMSD is 12.2 Å. (c) Distribution of sequence identity (in blue, averaging 36.7%) and similarity (in orange, averaging 70.3%) among aligned residues in the two variants of the shared themes
One may wonder whether the conformational flexibility of the bridging themes is due to variations of the bridging theme sequences versus due to the sequence contexts before and after the bridging theme. To address this question, we compared two proxy measures: the predicted and true secondary structures of the variations. Namely, using an MSA‐free method, 27 we predicted the secondary structures of bridging themes. MSA‐free methods avoid the interference of evolutionary information. Comparing the difference of predicted secondary elements (Predict_dSS) with the difference of the real ones (True_dSS) of the two variations of a bridging theme, we find a moderate correlation (r pearson = .49, p < 10−3, Figure S2). As RMSD is a more detailed attribute to measure the structural variability, we also computed RMSD of two variations of each bridging theme. Figure S3 shows that when comparing True_dRMSD versus Predict_dSS, the correlation between the two is poor (r pearson = −.28, p < 10−3). This holds true even though the correlation between True_dRMSD and True_dSS is not as poor (r pearson = .43, p < 10−3). Thus, using existing computational tools, we can only conclude that the difference in the secondary structures of the variations of the themes is due both to inherent difference of the bridging sequences themselves (as evidenced by the predicted secondary structure) and the context sequences. Put otherwise, the moderate correlation, suggests that the contexts of the bridging theme influence the conformation of similar segments, but that this impact, is relatively weak, reducing the average fraction of residue pairs sharing identical secondary structure conformations from 0.50 to 0.45.
To investigate the timeline of the evolutionary events involved with these bridging themes, we compared the ages of the folds of the domains in our datasets. We used the age estimates by Edwards et al. 28 which are at the fold‐level of the SCOP classification (Table 1, see Section 4 for details). More than 80% of the domains in our set are estimated to be ancient (age >0.8, on a 0–1 scale with 0 being new‐born and 1 the oldest). We found only a few bridging themes in relatively young domains (age <0.4) in both the new and previous MBE 2021 datasets: in this set only the domains e1nezA2 and e1t7vA2 from one X‐group—233, MHC antigen recognition domain (age <0.4), with variations in e2rgsA2 and e2wngA3 from X‐group 11 Immunoglobulin like beta‐sandwich. That a shared sub‐domain segment can be found not only in very ancient evolutionary lineages, but also in more recent ones, suggests that the evolutionary events that gave rise to these could have also happened more recently. Table 2 details the age difference between the two ECOD domains in our datasets. In both datasets, most of the age gaps of evolutionary links are small (age difference <0.2) and typically both domains are relatively old.
TABLE 1.
The distribution of relative age of ECOD domains involved in the new and previous bridging theme dataset
| Age bin | The number/fraction of domains in the dataset of 2021 MBE | The number/fraction of domains in the dataset of this paper |
|---|---|---|
| 0–0.2 | 1 (0.2%) | 0 (0.0%) |
| 0.2–0.4 | 1 (0.2%) | 2 (0.4%) |
| 0.4–0.6 | 0 (0.0%) | 4 (0.7%) |
| 0.6–0.8 | 141 (30.4%) | 44 (7.9%) |
| 0.8–1.0 | 321 (69.2%) | 504 (91.0%) |
Note: Ancient domains are enriched in both datasets, with only a few domains that are more recent. Age estimation of ECOD domains. Ages are assigned with the dataset from Edwards et al. 28 A relative age of 1.0 and 0.0 represents the oldest and the youngest domain, respectively.
Abbreviation: ECOD, Evolutionary Classification of Domains.
TABLE 2.
The distribution of the age difference of two ECOD domains for each bridging theme
| Age difference bin | The number/fraction of domain pairs in the dataset of 2021 MBE | The number/fraction of domain pairs in the dataset of this paper |
|---|---|---|
| 0–0.2 | 257 (74.5%) | 348 (93.5%) |
| 0.2–0.4 | 76 (22.0%) | 19 (5.1%) |
| 0.4–0.6 | 0 (0.0%) | 3 (0.8%) |
| 0.6–0.8 | 7 (2.0%) | 2 (0.5%) |
| 0.8–1.0 | 5 (1.4%) | 0 (0.0%) |
Note: Most of age differences for each domain pair are small. Age estimation of ECOD domains. Ages are assigned with the dataset from Edwards et al. 28 A relative age of 1.0 and 0.0 represents the oldest and the youngest domain, respectively.
Abbreviation: ECOD, Evolutionary Classification of Domains.
The dataset includes pairs in which one of the domains is a β‐propeller and the other is not. There are three β‐propeller domains in these pairs: e3adeA1 (ECOD classification 5.1.3.16, 6 bladed), e4l1mA1 (ECOD classification 5.1.4.31, 7 bladed), and e3nvqB1(ECOD classification 5.1.4.36, 7 bladed). The other X‐group in the pair of domains are either the α/β Rossmann‐like domains (superfamilies 2003.1.[1,5]), the Trefoil/Plexin domain‐like domain (family 355.1.1.2), or the all‐α nuclear receptor ligand‐binding domain (families 188.1.1.[1,8]). We elaborate on the two former cases.
2.2. Evolutionary link between β‐propeller and Rossmann fold
Figure 3 shows a bridging theme shared between the Rossmann‐like e2yxeA1 (ECOD X‐group 2003) and the β‐propeller‐like e4l1mA1 (ECOD X‐group 5). The sequences of these two variations of the shared theme appear to be homologous (22 aligned residues with 45% identity and 68% similarity). Most identical residues in the alignment are prebiotic amino acids (Gly, Val, Ile, Ala, Thr, Glu). In contrast, the segments before and after the bridging theme are not similar (Figure 3e). Despite the high sequence similarity, the two variations are structurally different (Figure 3a). The theme in e2yxeA1 overlaps with the ancestral β‐α‐β motif described in our joint work with Tawfik, 16 which links two ancient protein families from distinct evolutionarily lineages—Rossmann‐like and P‐loop‐like domains. The variation in e4l1mA1 is a β‐hairpin, corresponding to a part of the blade repeat of the seven‐bladed β‐propeller domain.
FIGURE 3.
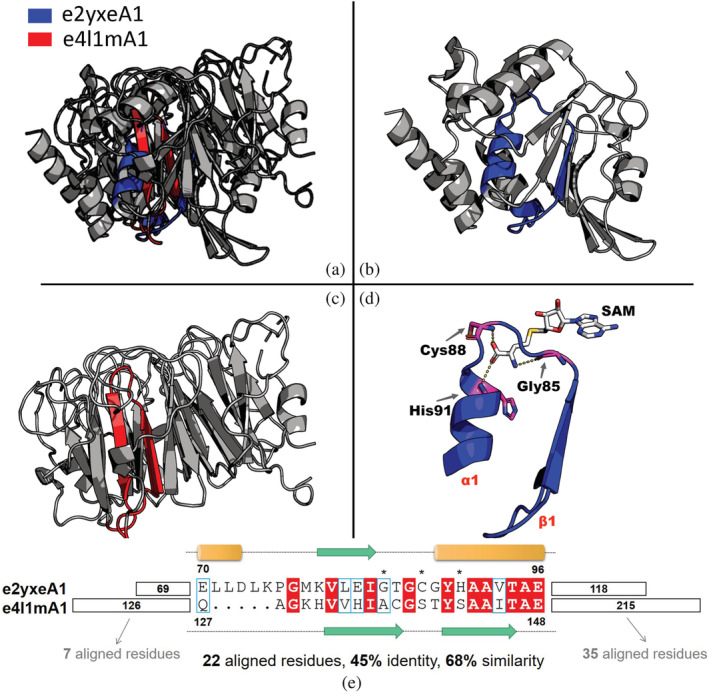
A bridging theme shared between Rossmann domain e2yxeA1 and β‐propeller domain e4l1mA1. (a) The overall structures of e2yxeA1 and e4l1mA1 are superposed based on the theme alignment. The structure of the theme in (b) e2yxeA1 and (c) e4l1mA1 is colored in blue and red, respectively. Both global structures and theme structures differ in these two domains. (d) A close view of the interaction between the theme of e2yxeA1 and the estimated binding mode of the S‐adenosylmethionine (SAM) ligand (see Section 4). Three residues mediating binding are shown as pink sticks: Gly85, Cys88, and His91. (e) The sequence alignment of the bridging theme in these two domains. The residue numbers of the beginnings and ends of the theme variations, taken from Evolutionary Classification of Domains, are indicated. There are 22 aligned residues with 45% identity and 68% similarity. Identical residues are colored in red and similar residues are marked with blue frames. Secondary structure elements are shown as cylinder, arrow and dotted line for α helix, β strand and loop, respectively. Three residues interacting with SAM are marked with asterisks. The local alignment of the segments before and after the bridging theme has only 7 and 35 aligned residues, respectively. All sequence alignments in this work were generated in ESPript 3.0. 29
The variation of this theme in the Rossmann domain includes several residues that mediate S‐adenosylmethionine binding (Figure 3d). A “β‐turn,” which is regarded as a unique feature of Rossmann MTases, can be observed in the Gly‐rich loop region of e2yxeA1, with a motif “TGCG.” As previous studies showed, 30 Cys88 of this motif binds the oxygen atom of the ligand's methionine using a backbone nitrogen atom. His91 located right after this “β‐turn” motif and Gly85 near the tip of α1 interact with the oxygen atom and nitrogen atoms of the methionine moiety, respectively. Interestingly, all residues that mediate these interactions in the theme of e2yxeA1 are not aligned to the theme of e4l1mA1 (Figure 3e). Indeed, to the best of our knowledge, the variation of this theme in e4l1mA is not involved in ligand binding, at least not of nucleotide cofactors.
2.3. Evolutionary link between β‐propeller and Trefoil/Plexin domains
Figure 4 shows another bridging theme that links the Trefoil/Plexin‐like e1olzA3 (ECOD X‐group 355) and the β‐propeller‐like e3nvqB1 (ECOD X‐group 5). The two variations of this bridging theme have clearly emerged from a shared origin (35 aligned residues with 51% identity and 71% similarity, Figure 4b). The structure of this theme is context‐dependent (Figure 4a): the e1olzA3 variation contains two β strands and a short α helix, while the e3nvqB1 variation includes several β strands and disordered loops. The variation of this theme in e1olzA3 is continuous, whereas that of e3nvqB1 is discontinuous and taken from several blades. Functionally, both 1olz and 3nvq belong to vertebrate Semaphorins, a secreted and membrane protein with various functions. Interestingly, the domain e1olzA3 is in the protein chain 1olz, and lies between a β‐propeller domain (e1olzA2) and an IgG‐like domain (e1olzA1). The β‐propeller domain e3nvqB1 performs similar functions to the β‐propeller e1olzA2 that is neighboring on the same chain with the Trefoil/Plexin‐like e1olzA3. However, the analog parts in the β‐propeller on the same chain, do not align well with the sequence of e1olzA3. Although from distinct ECOD X‐groups, e1olzA3 and e3nvqB1 are highly functionally relevant. In Semaphorin, the β‐propeller domain (e3nvqB1) is essential for signaling through receptors, and the PSI domain (e1olzA3), a cysteine‐rich domain existing in Plexins, Semaphorins and Integrins, is responsible for the correct positioning of the binding site of the propeller. 31 Given that the bridging theme observed in these two domains may be a result of gene duplication, it is attractive to further study the evolutionary history of these domains in Semaphorins.
FIGURE 4.
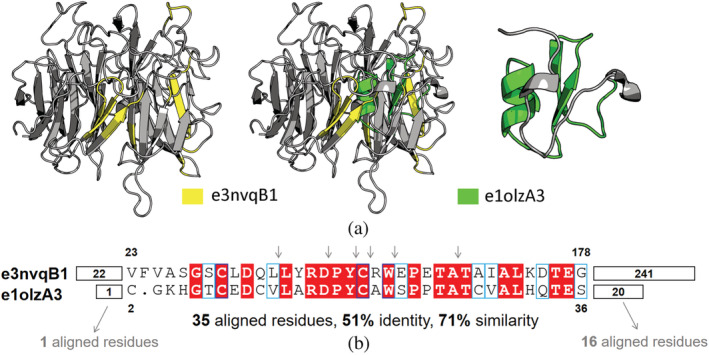
A bridging theme shared between β‐propeller like domain e3nvqB1 and Trefoil/Plexin‐like domain e1olzA3. (a) Center panel: The two domains are optimally superposed according to the sequence alignment of the themes. Side panels: The structures of e3nvqB1 (left) and e1olzA3 (right) with their themes highlighted in yellow and green. Despite the high sequence identity, the structures of the themes in the two domains differ. (b) Sequence alignment of two variations of the bridging theme shows evidence of homology (35 aligned residues with 51% identity and 71% similarity). The residue numbers of the beginnings and ends of the theme variations, taken from Evolutionary Classification of Domains, are indicated. For clarity, insertions of the discontinuous theme in e3nvqB1 are simply marked as arrows. The numbers before and after the bridging theme represent the number of residues in corresponding regions, 1 and 16, respectively.
3. DISCUSSION
Using HMMER (rather than HHSearch), we can directly search for incidences of our theme HMMs within ECOD sequences. This identified 491 domain pairs from 108 pairs of ECOD X‐groups, spanning 115 X‐groups. The lengths of the shared segments and their sequence similarity offers strong support for their homology.
Our new dataset shares 19 pairs of X‐groups with the previous one and adds 89 novel pairs, demonstrating that the search procedures HMMER and HHSearch vary. Indeed, both are state‐of‐the‐art tools for sensitive sequence similarity detection, and that they find different cases implies that one should use both, select the meaningful instances (e.g., based on length of shared segment and its sequence similarity), and collect these to a combined set. Using the same search workflow with different aligners holds promise to find a more comprehensive dataset.
The structure of the bridging themes in our dataset differ, suggesting a flexibility in conformation allowing these segments to fit in diverse contexts. Our comparison of secondary structures with and without their sequence contexts provides a preliminary glimpse into the impact of variations of both bridging theme sequence and context sequences on structures of these similar segment. We used a secondary structure prediction method that does not use an MSA of homologous proteins, to avoid averaging over many samples, yet in doing so we are relying on much less accurate prediction. We see that the secondary structure prediction differences and the true secondary structures differences vary, but not dramatically. On the other hand, even though the true secondary structure differences and the RMSD differences are correlated, the RMSD differences do not correlate with the predicted secondary structure differences. This suggests that the structural differences as measured by RMSD are due to cases where the sequence context is different and also, to cases where the (MSA‐free) secondary structure prediction is inaccurate. The structural plasticity of the bridging theme can be an inherent property of the theme itself, where the sequence variations in it already led to different structures. Alternatively, the structural plasticity may be due to a dramatically different context, leading to different biophysical environments within their respective domains. Deciphering for different cases what is the dominant contributing factor, is a very challenging question, which is well‐beyond the scope of this study.
The ages of the domains with bridging themes are relatively old. This age distribution hints to the scenario that reuse of protein segments played a crucial role in the emergence and early evolution of ancestral proteins. This discrepancy at different stages of protein evolution may result from the continuous evolution of genetic mechanisms, including the changes of DNA replication, RNA transcription, protein synthesis, and even the drastic shift from “RNA/Peptide World” to “DNA/Protein World.” Additionally, similar segments appear extensively in protein families which share a similar estimated age, while domains with distinct ages more seldom share a bridging theme. Age difference of a bridging theme pair may help to distinguish among evolutionary scenarios behind the shared segment. Pairs with a large age difference, may be perhaps due to a copy‐paste event, rather than a common ancestor. Notice, however, that for our age estimates we relied on data for SCOP folds, which added an extra step of mapping the ECOD domains to their SCOP folds.
We discovered two potential evolutionary links between β‐propeller and other protein folds. The theme linking propellers to an ancient fold, Rossmanns, is particularly captivating. Apart from e2yxeA1 discussed above, 10 other Rossmann domains share a similar bridging theme with the e4l1mA1 propeller. The detected bridging theme of e4l1mA1 in these domain pairs are nearly the same, while the corresponding regions in different Rossmann domains vary slightly, including different secondary elements. However, all bridging themes of these Rossmann domains belong to the above‐mentioned β‐α‐β motif. Nearly all residues that interact with S‐adenosylmethionine in these Rossmann domains do not align to any residue in e4l1mA1. Rather, the respective amino acids correspond to deletions in the propeller. For example, in e3eeyA1 there are two deletions in its bridging theme with e4l1mA1 (Figure S1): one is a four residues loop, connecting helix α1 with strand β1 of the Rossmann domain, and the other is a 19‐residue deletion that includes the β2 strand, where the conserved ribose‐binding Asp is located. Similar insertion/deletion events were observed in the previous bridging theme dataset. There, we proposed that they may have emerged later than the shared sequence. 9
While one cannot differentiate based on shared segment in the β‐propeller and the other domains if this shared segment is a remnant of an ancestral domain, or piece copied from one and grafted into the other, the following argument support that these are pieces that were grafted into the β‐propellers. The structural plasticity of the β‐propellers suggest that they can accommodate insertions. 14 , 15 The Rossmann fold is more ancient than the β‐propeller domain, 32 and in particular we believe Rossmanns, found throughout all kingdoms of life, emerged before this specific β‐propeller, which is a component of the ubiquitination system in eukaryotes. It may have been that this bridging theme migrated from Rossmann domains to β‐propellers, after which the nucleotide binding segments, no longer needed in the β‐propeller, were deleted. In the bridging theme shared between e1olzA3 and e3nvqB1, the two domains appearing in Semaphorins (with one in the same chain as a β‐propeller), also suggest that a piece from one domain in the chain was grafted to another domain in the chain.
These observations suggest two directions for future study: Experiments can be designed to test these scenarios. For instance, considering the nearly continuous bridging theme shared between e2yxeA1 and e4l1mA1 and its short length, it is tempting to combine deep mutation scanning and high‐throughput sequencing to exhaustively characterize structural element composition and biochemical properties of mutants in this 22‐residue‐long region as many as possible, either in a complete structural context or, preferably, short peptide, where possible. Given that we still lack adequate evidence to propose the evolutionary history of these two distinct protein folds from a structural and functional perspective, these experiments can describe a landscape of biophysical and biochemical properties of the shared theme, facilitate our understanding of potential evolutionary pathways among these protein families, and thus help us to distinguish homology and analogy of sequences and structures. On the computational front, one can search for bridging themes shared among domains of different X‐groups (i.e., different evolutionary lineages) that can be found on the same chain. If we find any, these may be viable candidates for the so‐called “copy‐paste” events.
Finally, while our sequence search strategy was very conservative and based on stringent sequence similarity thresholds, we cannot rule out the possibility that at least some of the bridging themes, both the ones currently introduced and the ones from our previous publication, 9 are due to convergent evolution. This is less of a concern when structure and/or function is preserved between the theme variations but might be the case for the putative evolutionary link between Rossmann and β‐propeller, where both changed.
4. METHODS
We generated HMMER HMMs 19 for each of the 12,689 themes 7 in our dataset from the same .sto MSA files that we have previously used to search for bridging themes. 9 Then, we searched the database of all 70%NR ECOD sequences. 20 We then analyzed with a python script the output of these, to identify cases where the same theme was aligned (with an E‐value lower than 0.001) to domains of different X‐groups. This resulted in a set of pairs of domains, where a segment that was aligned to the “bait” theme is marked within each of the domains. In the cases where two domains were identified using more than one “bait” theme (which happens, because our themes overlap each other), we kept only the pair that was assigned the highest HMMER score.
Our goal was to filter a meaningful representative set of domain pairs. In each pair of ECOD F‐groups with a different X‐group classification, we kept the representative pair with the highest combined alignment score. The combined alignment score is calculated as the optimal global alignment score of the similar segments (global_segment) minus the average of the optimal local alignment scores of the segments before and after the similar one (local_before + local_after)/2. This selects the pair with the most similar aligned segment, flanked by the most dissimilar segments before and after. Because we noticed that there were many pairs where the aligned segment covers one of the domains almost entirely, we added the condition that the total number of residues in the segments before and after in each of the domains must be at least 20. Finally, similar to previous methods, 9 we calculated a p‐value measure for the significance of the alignment score with respect to scores of alignments of random segments (drawn from the same distribution). For this, we estimated the parameters of the extreme value distribution from the scores of the alignments between the first segment, and 1,000 randomly chosen segments drawn from a multinomial distribution estimated from the second segment. Finally, we filtered the set to keep only pairs where the aligned segments are more than 20 residues, and the p‐value is lower than .001. We also calculated for the alignments: its length (number of residues), percent sequence similarity/identity, and in cases all the aligned residues are in the PDB file, the RMSD of Cα atoms of the aligned residues after optimal superpositioning.
We relied on the age estimates by Edwards et al., 28 where the age of each fold in the SCOP hierarchy are inferred by conducting maximum parsimony methods based on the NCBI common taxonomy tree. To map between ECOD and SCOP entities, we used the following procedure: For each ECOD domain, we searched with HHSearch the sequence against the SCOP95 database with default settings. The SCOP Fold of the best hit was assigned to the ECOD domain, and then the relative age of this SCOP Fold was taken from the Edwards et al.'s dataset to be the estimated age of that ECOD domain. If the HHSearch probability of the SCOP domain hit for an ECOD domain is lower than 90%, we could not assign this ECOD domain an age.
We considered a three states description of secondary structures (helix, strand and loop). To avoid the interference by homology, we used SPIDER3‐Single algorithm to predict secondary structures of each bridging theme sequence without the contexts before and after it. 27 Unlike most of secondary structure prediction algorithms based on HMMs or PSSMs to include more evolutionary features, the advanced SPIDER3‐Single is implemented in deep whole‐sequence learning and require only one sequence as the input for the neural network. For each bridging theme pair, we predicted two variations of the shared segment alone and compared their secondary structures. The difference of two variations in secondary elements was simply computed as the fraction of residue pairs that share the same secondary structure state in two variations. STRIDE 33 was used to assign secondary structures to these bridging themes from structures.
ECOD domain e2yxeA1 is an apo structure without its ligand. Therefore, we estimated the position of S‐adenosylmethionine via superposing e2yxeA1 with the ligand‐bound e3lbfA1, a domain from the same F‐group. Both e2yxeA1 and e3lbfA1 have a canonical “β‐turn,” which mediate ligand binding. The loop containing the “β‐turn” and the S‐adenosylmethionine of e3lbfA1 (residue 84–90, IGTGCGY) was aligned to the corresponding loop of e2yxeA1 (residue 82–88, IGTGSGY). The loops from both domains share a highly similar structure (Cα RMSD of 0.2 Å). We utilized the ligand from e3lbfA1 as the estimated ligand bound by e2yxeA1 after superposition and generated Figure 3d.
AUTHOR CONTRIBUTIONS
Kaiyu Qiu: Data curation (supporting); investigation (supporting); writing – review and editing (supporting); Rachel Kolodny: Conceptualisation; investigation; writing (original draft; review editing), software, data curation; Nir Ben‐Tal: Conceptualisation, investigation, writing (original draft; review editing).
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
Danny was our dear colleague and friend, and he is deeply missed. We thought about him a lot while working on this paper, debating what would his opinion be on this and that. Surely, his insight would have better deciphered the evolutionary clues that we collected here. This study was supported by ISF grants 450/16 and 1764/21 and VW foundation grant 94747. Nir Ben‐Tal's research was supported in part by the Abraham E. Kazan Chair in Structural Biology, Tel Aviv University.
Qiu K, Ben‐Tal N, Kolodny R. Similar protein segments shared between domains of different evolutionary lineages. Protein Science. 2022;31(9):e4407. 10.1002/pro.4407
Review Editor: John Kuriyan
Funding information Israel Science Foundation, Grant/Award Numbers: 1764/21, 450/16; VW foundation, Grant/Award Number: 94747
Contributor Information
Nir Ben‐Tal, Email: bental@tauex.tau.ac.il.
Rachel Kolodny, Email: trachel@cs.haifa.ac.il.
DATA AVAILABILITY STATEMENT
Data available in article Supporting Information S1.
REFERENCES
- 1. Kessel A, Ben‐Tal N. Introduction to proteins: Structure, function, and motion. New York: Chapman and Hall/CRC, 2018. [Google Scholar]
- 2. Lupas AN, Ponting CP, Russell RB. On the evolution of protein folds: Are similar motifs in different protein folds the result of convergence, insertion, or relics of an ancient peptide world? J Struct Biol. 2001;134(2–3):191–203. [DOI] [PubMed] [Google Scholar]
- 3. Eck RV, Dayhoff MO. Evolution of the structure of ferredoxin based on living relics of primitive amino acid sequences. Science. 1966;152(3720):363–366. [DOI] [PubMed] [Google Scholar]
- 4. Friedberg I, Godzik A. Connecting the protein structure universe by using sparse recurring fragments. Structure. 2005;13(8):1213–1224. [DOI] [PubMed] [Google Scholar]
- 5. Goncearenco A, Berezovsky IN. Computational reconstruction of primordial prototypes of elementary functional loops in modern proteins. Bioinformatics. 2011;27(17):2368–2375. [DOI] [PubMed] [Google Scholar]
- 6. Alva V, Remmert M, Biegert A, Lupas AN, Söding J. A galaxy of folds. Protein Sci. 2010;19(1):124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nepomnyachiy S, Ben‐Tal N, Kolodny R. Complex evolutionary footprints revealed in an analysis of reused protein segments of diverse lengths. Proc Natl Acad Sci. 2017;114(44):11703–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nepomnyachiy S, Ben‐Tal N, Kolodny R. Global view of the protein universe. Proc Natl Acad Sci USA. 2014;111(32):11691–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kolodny R, Nepomnyachiy S, Tawfik DS, Ben‐Tal N. Bridging themes: Short protein segments found in different architectures. Mol Biol Evol. 2021;38(6):2191–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alva V, Söding J, Lupas AN. A vocabulary of ancient peptides at the origin of folded proteins. Elife. 2015;4:e09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alva V, Lupas AN. From ancestral peptides to designed proteins. Curr Opin Struct Biol. 2018;48:103–109. [DOI] [PubMed] [Google Scholar]
- 12. Chaudhuri I, Söding J, Lupas AN. Evolution of the β‐propeller fold. Proteins. 2008;71(2):795–803. [DOI] [PubMed] [Google Scholar]
- 13. Smock RG, Yadid I, Dym O, Clarke J, Tawfik DS. De novo evolutionary emergence of a symmetrical protein is shaped by folding constraints. Cell. 2016;164(3):476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Afanasieva E, Chaudhuri I, Martin J, et al. Structural diversity of oligomeric β‐propellers with different numbers of identical blades. Elife. 2019;8:e49853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mylemans B, Voet ARD, Tame JRH. The taming of the screw: The natural and artificial development of β‐propeller proteins. Curr Opin Struct Biol. 2021;68:48–54. [DOI] [PubMed] [Google Scholar]
- 16. Longo LM, Jabłońska J, Vyas P, et al. On the emergence of P‐Loop NTPase and Rossmann enzymes from a Beta‐Alpha‐Beta ancestral fragment. Elife. 2020;9:e64415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Longo LM, Kolodny R, McGlynn SE. Evidence for the emergence of β‐trefoils by ‘Peptide Budding’ from an IgG‐like β‐sandwich. PLoS Comput Biol. 2022;18(2):e1009833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soding J. Protein homology detection by HMM‐HMM comparison. Bioinformatics. 2005;21(7):951–960. [DOI] [PubMed] [Google Scholar]
- 19. Finn RD, Clements J, Eddy SR. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011;39(suppl_2):W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng H, Schaeffer RD, Liao Y, et al. ECOD: An evolutionary classification of protein domains. PLoS Comput Biol. 2014;10(12):e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Romero Romero ML, Rabin A, Tawfik DS. Functional proteins from short peptides: Dayhoff's hypothesis turns 50. Angew Chem Int Ed. 2016;55(52):15966–15971. [DOI] [PubMed] [Google Scholar]
- 22. Kolodny R. Searching protein space for ancient sub‐domain segments. Curr Opin Struct Biol. 2021;68:105–112. [DOI] [PubMed] [Google Scholar]
- 23. Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nepomnyachiy S, Ben‐Tal N, Kolodny R. CyToStruct: Augmenting the network visualization of cytoscape with the power of molecular viewers. Structure. 2015;23(5):941–948. [DOI] [PubMed] [Google Scholar]
- 25. Schrodinger, LLC . The PyMOL Molecular Graphics System, Version 1.3r1. 2010.
- 26. Hall TA. BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Paper presented at: Nucleic acids symposium series. 1999.
- 27. Heffernan R, Paliwal K, Lyons J, Singh J, Yang Y, Zhou Y. Single‐sequence‐based prediction of protein secondary structures and solvent accessibility by deep whole‐sequence learning. J Comput Chem. 2018;39(26):2210–2216. [DOI] [PubMed] [Google Scholar]
- 28. Edwards H, Abeln S, Deane CM. Exploring fold space preferences of new‐born and ancient protein superfamilies. PLoS Comput Biol. 2013;9(11):e1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chouhan BPS, Maimaiti S, Gade M, Laurino P. Rossmann‐fold methyltransferases: Taking a “β‐Turn” around their cofactor, S‐adenosylmethionine. Biochemistry. 2018;58(3):166–170. [DOI] [PubMed] [Google Scholar]
- 31. Liu H, Juo ZS, Shim AH‐R, et al. Structural basis of semaphorin‐plexin recognition and viral mimicry from Sema7A and A39R complexes with PlexinC1. Cell. 2010;142(5):749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang M, Caetano‐Anollés G. The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world. Structure. 2009;17(1):66–78. [DOI] [PubMed] [Google Scholar]
- 33. Frishman D, Argos P. Knowledge‐based protein secondary structure assignment. Proteins. 1995;23(4):566–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
Data available in article Supporting Information S1.