

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) nucleocapsid protein is an essential structural component of mature virions, encapsulating the genomic RNA and modulating RNA transcription and replication. Several of its activities might be associated with the protein's ability to undergo liquid–liquid phase separation. NSARS‐CoV‐2 contains an intrinsically disordered region at its N‐terminus (NTE) that can be phosphorylated and is affected by mutations found in human COVID‐19 infections, including in the Omicron variant of concern. Here, we show that NTE deletion decreases the range of RNA concentrations that can induce phase separation of NSARS‐CoV‐2. In addition, deletion of the prion‐like NTE allows NSARS‐CoV‐2 droplets to retain their liquid‐like nature during incubation. We further demonstrate that RNA‐binding engages multiple parts of the NTE and changes NTE's structural properties. The results form the foundation to characterize the impact of N‐terminal mutations and post‐translational modifications on the molecular properties of the SARS‐CoV‐2 nucleocapsid protein.
Statement
The nucleocapsid protein of SARS‐CoV‐2 plays an important role in both genome packaging and viral replication upon host infection. Replication has been associated with RNA‐induced liquid–liquid phase separation of the nucleocapsid protein. We present insights into the role of the N‐terminal part of the nucleocapsid protein in the protein's RNA‐mediated liquid–liquid phase separation.
Keywords: liquid–liquid phase separation, NMR, nucleocapsid protein, RNA, SARS‐CoV‐2
1. INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has caused an outbreak of a pandemic. The SARS‐CoV‐2 lipid membrane envelopes a positive‐sense, single‐stranded 30 kb large RNA genome. The genome is encapsulated by the nucleocapsid protein (NSARS‐CoV‐2). NSARS‐CoV‐2 is highly abundant and in addition to its structural role, it might play an important role in SARS‐CoV‐2 replication via RNA‐driven liquid–liquid phase separation (LLPS). 1 , 2 , 3 , 4 , 5 , 6 From a therapeutic view, targeting NSARS‐CoV‐2 could be useful to improve the efficacy of currently available vaccines. 7 , 8
NSARS‐CoV‐2 consists of two structured domains flanked by intrinsically disordered regions (Figure 1a). The structured C‐terminal domain is responsible for NSARS‐CoV‐2 dimerization and oligomerization. 11 The flexible linker between the two folded domains includes a serine‐arginine‐rich region (SR‐region), which mediates interaction between the nucleocapsid protein and various protein and RNA partners. 6 , 12 , 13 Due to the high flexibility of the SR‐rich linker region, the N‐terminal folded domains are largely decoupled from the C‐terminal dimer, 2 , 14 , 15 giving the N‐terminal domains enough flexibility to engage in RNA binding. Key to RNA‐binding is a finger‐like basic patch in the N‐terminal domain. 16 In addition, essentially all parts of the nucleocapsid protein have been shown to bind RNA or be involved in binding other partners. 16
FIGURE 1.
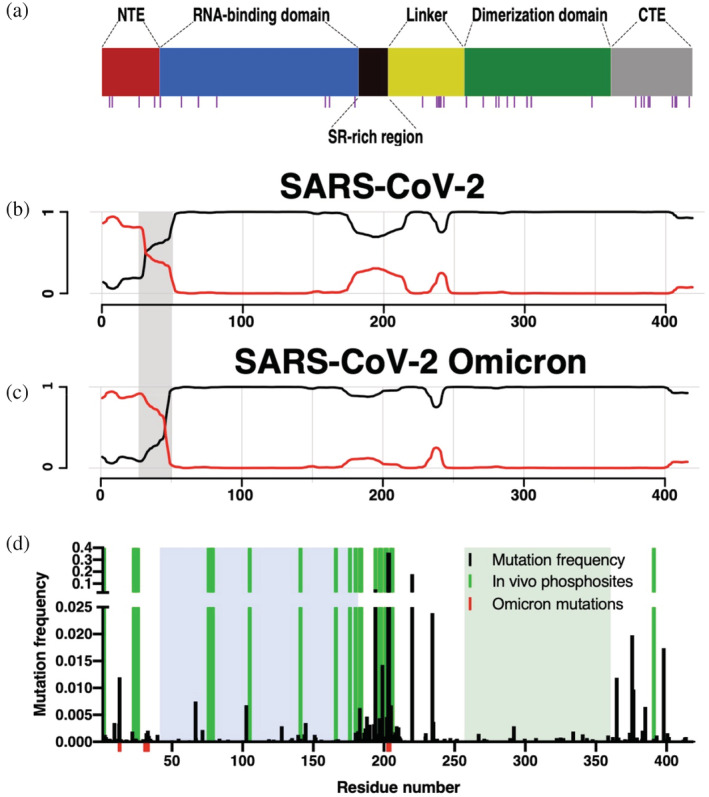
Sequence analysis of the nucleocapsid protein from the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) coronavirus. (a) Domain organization of NSARS‐CoV‐2 with glutamine residues highlighted by magenta bars underneath. (b and c) Prediction of prion‐like domains in the nucleocapsid protein of SARS‐CoV‐2 (b) and the Omicron variant (c) using the PLAAC algorithm. 9 , 10 Background and prion‐like domains are indicated by black and red lines, respectively. The gray rectangle highlights the region with the strongest differences. (d) NSARS‐CoV‐2 mutation and phosphorylation analysis. Amino acid frequency of mutations using 219,909 sequences from the China National Center for Bioinformatics. Mutations present in the Omicron variant of SARS‐CoV‐2 are shown in red. Phosphorylation sites are highlighted in green. Folded domains are highlighted by blue and green boxes.
Most structural biology‐focused research has so far concentrated on the two folded domains of NSARS‐CoV‐2, which are conserved between different coronaviruses. 17 , 18 Less attention on the other hand has been paid to the disordered regions of NSARS‐CoV‐2, despite their suggested roles for modulating NSARS‐CoV‐2 activity. For example, the N‐terminal intrinsically disordered tail (NTE) has been suggested to be responsible for the recruitment of NSARS‐CoV‐2 into stress granules. 19 To close this gap, we characterize here NTE's contribution to NSARS‐CoV‐2 LLPS and binding to RNA, and provide insights into its molecular properties using a combination of phase separation assays, NMR spectroscopy and molecular dynamics (MD) simulations.
2. RESULTS
2.1. Sequence properties of the N‐terminal intrinsically disordered region of NSARS‐CoV ‐2
The ability of proteins to undergo LLPS might be connected to sequence properties found in prion‐like proteins. 20 We applied the PLAAC 9 , 10 algorithm to identify amino acid sequences with prion‐like composition within the NSARS‐CoV‐2 sequence. The analysis reveals pronounced prion‐like properties in the disordered N‐terminal tail and SR‐linker region of NSARS‐CoV‐2 (Figure 1b), in agreement with previous analyses. 5 , 21 Closer inspection shows that the N‐terminal part of NSARS‐CoV‐2 contains several glutamine residues, known to favor prion‐like sequence properties.
An important property of viruses is their rapid adaptation through mutations. To determine if the NTE of NSARS‐CoV‐2 not only has prion‐like, potentially LLPS‐promoting properties but also harbors mutations, we analyzed 219,909 NSARS‐CoV‐2 sequences. We find that most mutations are localized in the intrinsically disordered regions, particularly in the SR‐rich linker region (Figure 1d). Mutations in the SR‐region were suggested to increase the spread of the SARS‐CoV‐2 virus. 22 Notably, the currently predominant SARS‐CoV‐2 variant B.1.1.529 (Omicron; Figure 1c) shares some mutations in the SR‐region with previously characterized variants (R203K, G204R). In addition, the Omicron variant contains a P13L mutation and a Δ31ERS33 deletion which enhances the prion‐like properties of the NTE (Figure 1d). Further support for an important role of the NTE in the life cycle of NSARS‐CoV‐2 comes from the observation that, besides the SR‐region, it can be phosphorylated in SARS‐CoV‐2 infected cells (Figure 1d). 23 , 24
2.2. Influence of the NTE on LLPS of NSARS‐CoV ‐2
Motivated by the above sequence analysis we investigated the regulatory effect of the NTE on the LLPS of NSARS‐CoV‐2. While some studies probed the importance of the NTE for LLPS and biomolecular condensation of NSARS‐CoV‐2, 5 , 19 , 25 its precise role remains unclear. One study suggested that the NTE contributes to the recruitment—potentially through LLPS—into stress granules. 19 Other studies, however, showed that multiple regions of NSARS‐CoV‐2 are important for RNA‐induced LLPS. 5 , 25 To gain additional insights into the contribution of the NTE to LLPS of NSARS‐CoV‐2, we recombinantly prepared two constructs: the 419‐residue full‐length NSARS‐CoV‐2 protein and a 379‐residue construct with deleted NTE (ΔNTESARS‐CoV‐2). The negative charge of polyU is expected to be compensated by the positive charge of the N protein in order to achieve optimal LLPS. Thus, the deletion of the NTE, which reduces the predicted net charge of the protein at pH 7.5 from +25 for the full‐length protein to +20 for ΔNTESARS‐CoV‐2 (assuming a charge of +1 for positive residues and −1 for negative residues), is expected to affect LLPS.
Using the two constructs, we analyzed the effect of the NTE deletion on LLPS for different protein and RNA concentrations. We measured sample turbidities at 350 nm of both full‐length NSARS‐CoV‐2 and ΔNTESARS‐CoV‐2 at protein concentrations up to 50 μM, and in the presence of 0–2 μM polyU (800 kDa), which serves as a simple substitute for viral RNA (Figure 2b,c). The obtained phase diagram of the full‐length protein is in agreement with our previously published data (Figure 2a). 6 In the case of the truncated protein, we also observed maximum turbidity in conditions close to charge neutralization (Figure 2b). However, when we compare in detail the dependence of turbidity on RNA concentration for 50 μM of protein, a clear difference in LLPS behavior is observed between the full‐length and the N‐terminally truncated protein: only the full‐length protein undergoes LLPS in the presence of 1 μM polyU (Figure 2c). To exclude the possibility that our observations are limited to a single experimental condition, we repeated the turbidity measurements of 30 μM ΔNTESARS‐CoV‐2 in 20 mM sodium phosphate buffer at pH 6, 7.5, and 8 and in 20 mM Tris buffer at pH 9, which yielded comparable profiles (Figure S1). In agreement with the turbidity measurements, abundant protein‐dense droplets were seen by differential interference contrast (DIC) and fluorescence microscopy for both proteins at 0.5 μM polyU, but only for the full‐length protein at 1 μM polyU (Figure 2d).
FIGURE 2.
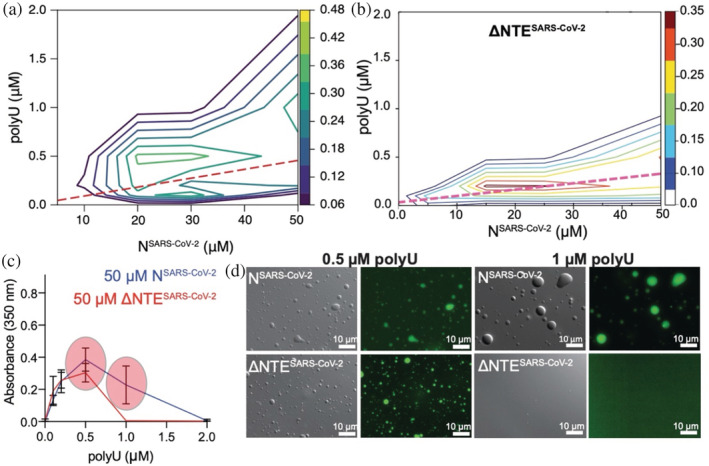
The N‐terminal disordered region of the nucleocapsid protein of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) promotes liquid–liquid phase separation. (a) Phase diagram of polyU‐induced phase separation of NSARS‐CoV‐2 in 20 mM phosphate buffer, pH 7.5, monitored by solution turbidity at 350 nm. Three independent measurements were averaged. The dashed line marks NSARS‐CoV‐2/polyU‐concentrations at which charge neutralization occurs, when we assume a charge of −1 per phosphate group. Figure adapted from Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS‐CoV‐2 phase separates into RNA‐rich polymerase‐containing condensates. Nat Commun. 2020;11:6041. (Figure licensed under: http://creativecommons.org/licenses/by/4.0/.) (b) Phase diagram of ΔNTESARS‐CoV‐2 at protein concentration up to 50 μM and in the presence of increasing polyU RNA concentrations measured as turbidity at 350 nm. Conditions of charge compensation are marked by the dashed magenta line. (c) Absorbance at 350 nm of 50 μM full‐length NSARS‐CoV‐2 or of 50 μM ΔNTESARS‐CoV‐2 in the presence of 0.1, 0.2, 0.5, 1, 2 μM polyU RNA. Red circles highlight differences in the LLPS behavior between the two constructs. (d) Differential interference contrast/fluorescence microscopy images displaying the differences in the phase separation behavior between full‐length NSARS‐CoV‐2 and ΔNTESARS‐CoV‐2.
We previously reported that NSARS‐CoV‐2 droplets become less dynamic over time. 6 We, therefore, repeated the analysis with ΔNTESARS‐CoV‐2 by measuring fluorescence recovery after photobleaching (FRAP) with freshly prepared droplets in presence of charge‐compensating amounts of polyU (assuming a charge of −1 per phosphate group), followed by a second measurement after 1 hour waiting time (Figure 3). In contrast to the full‐length protein, we observed similar profiles for early and late time points of ΔNTESARS‐CoV‐2 droplets, that is, that no change of dynamics over time occurred. Deletion of the NTE thus allows NSARS‐CoV‐2 droplets to retain their liquid‐like nature.
FIGURE 3.
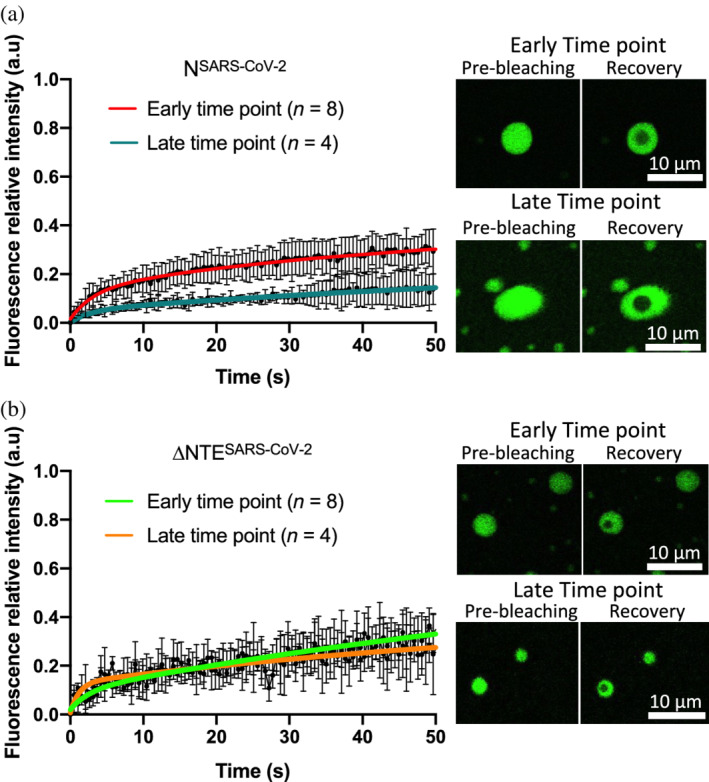
Time‐dependent change in diffusion of phase‐separating NSARS‐CoV‐2. (a) Left panel, fluorescence recovery of 30 μM NSARS‐CoV‐2 droplets formed in presence of 0.3 μM polyU measured at early (red) and late (blue) time points. Error bars represent standard deviations for n = 8 and n = 4 for early and late time points, respectively; right panel, representative micrographs showing pre‐ and post‐bleaching. Scale bars = 10 μm. (b) Left panel, fluorescence recovery of 30 μM ΔNTESARS‐CoV‐2 droplets formed in presence of 0.3 μM polyU measured at early (green) and late (orange) time points. Error bars represent standard deviations for n = 8 and n = 4 for early and late time points, respectively; right panel, representative micrographs showing pre‐ and post‐bleaching. Scale bars = 10 μm
2.3. Structural biases in the NTE of NSARS‐CoV ‐2
To better understand the molecular basis of the influence of the NTE on the RNA‐induced phase separation of NSARS‐CoV‐2, we characterized the structural properties of the NTE using NMR spectroscopy and MD simulations. Based on previous studies, already demonstrating that the NTE is disordered, 5 , 26 , 27 we prepared a synthetic peptide that comprises the NTE of NSARS‐CoV‐2. Focusing on the isolated NTE region has the advantage of decreased NMR signal overlap, which arises from the protein's intrinsically disordered linker and C‐terminal regions (Figure 4a).
FIGURE 4.
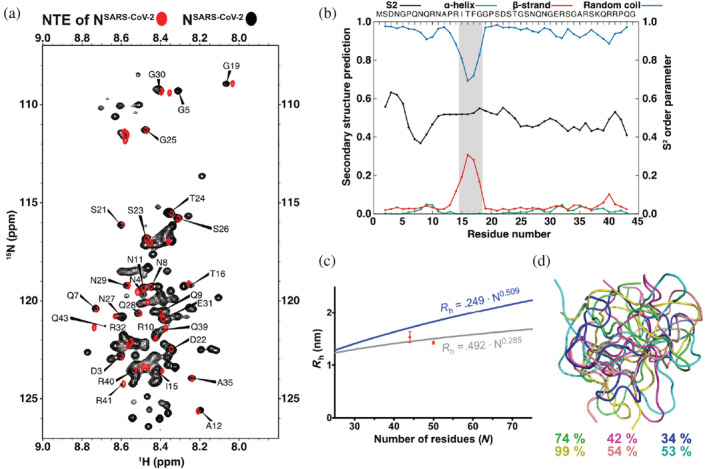
The N‐terminal region of the nucleocapsid protein of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) forms a compact structure with transient β‐structure. (a) Superposition of 1H‐15N HSQC spectra of full‐length NSARS‐CoV‐2 in black and the NTE of NSARS‐CoV‐2 in red measured at 5°C with amino‐acid assignments. (b) Order parameter, S2, and secondary structure prediction from the experimental chemical shifts of the NTE peptide of NSARS‐CoV‐2 using the TALOS+ software. 28 The region with transient β‐structure is highlighted in gray. (c) Hydrodynamic radius of the NTE of NSARS‐CoV‐2 determined by NMR (44 residues) and MD simulations (50 residues). The upper (blue) and lower (light gray) curves indicate the estimated values for an IDP and a globular protein, respectively. 29 (d) Representative structures of the NTE of NSARS‐CoV‐2 obtained from six independent MD simulations. The occupancy of the most populated cluster in each of the six MD simulations is indicated on the side.
Figure 4a shows a superposition of the natural abundance 1H‐15N spectrum of the NTE peptide with that of full‐length NSARS‐CoV‐2 at 5°C. At this low temperature, only residues from the disordered regions of NSARS‐CoV‐2 are observable, while the folded N‐ and C‐terminal domains are broadened beyond detection (Figure 4a). Overall, more cross‐peaks are seen in the spectrum of full‐length NSARS‐CoV‐2, as expected for highly dynamic IDP regions of the NTE, the SR‐rich linker region and the C‐terminal tail. In addition, good agreement is present between many of the cross‐peaks of the isolated NTE (red cross‐peaks) and the corresponding cross‐peaks of the 1H‐15N spectrum of full‐length NSARS‐CoV‐2 (black cross‐peaks in Figure 4a).
We then obtained the sequence‐specific assignment of the NTE peptide using a combination of two‐dimensional NOESY and TOCSY spectra and mapped it onto the 1H‐15N HSQC spectrum (Figure 4a). The analysis showed that deviations in peak positions between the isolated NTE and the full‐length protein occur mainly at NTE's termini. Notably, there is little signal overlap between the chemical shifts of a peptide corresponding to the SR‐rich linker region and the cross‐peaks in the 1H‐15N HSQC of full‐length NSARS‐CoV‐2 (Figure S2). In contrast to the NTE region, the SR‐rich linker region likely has different conformational properties in the isolated state or interacts with one of the folded domains.
Next, we analyzed the presence of transient secondary structure in the NTE using the assigned NMR chemical shifts (Figure 4b). We find that residues 13–19 display a propensity to transiently populate extended/β‐structure‐like conformations. The propensity of residues 13–19 for extended/β‐structure‐like conformations is in agreement with chemical shifts reported for NSARS‐CoV‐2 constructs comprising the NTE as well as the N‐terminal folded domain and the linker region, 26 , 27 while a previously reported MD simulation suggested the presence of a transient α‐helix between residues 31 and 38 but did not identify any β‐structure in the NTE. 2 The secondary structure analysis based on chemical shifts did not provide any evidence for α‐helical structure between residues 31 and 38 suggesting that its population might be very small or require additional binding partners for stabilization. In addition, we observed a pronounced compaction of the NTE in the MD simulation (Figure 4c; Figure S3). The hydrodynamic radius of the isolated NTE corresponds to the value expected for a globular structure of the same sequence length, in good agreement with experimentally determined hydrodynamic radius values (Figure 4c; Figure S4). Previously, MD simulations of the NTE (residues 1–49) reported a radius of gyration of ~18 Å. Comparison with our hydrodynamic radius is however difficult, because the ratio between radius of gyration and hydrodynamic radius strongly depends on the overall shape and compaction of IDPs. 30
2.4. RNA interacts with NTE
The above‐described influence of the NTE on RNA‐induced phase separation of NSARS‐CoV‐2 (Figure 2), suggests that the NTE directly binds to RNA. To gain insight into a potential direct interaction between the NTE and RNA, we performed an NMR titration of full‐length NSARS‐CoV‐2 with increasing concentrations of polyU. In agreement with the previously determined LLPS properties, 1 , 3 , 5 , 6 the NMR sample became highly turbid when we added 0.5 μM polyU to 50 μM NSARS‐CoV‐2, and upon further increase to 1.5 μM polyU the sample turned transparent again. In parallel to the changes in sample turbidity, many cross‐peaks disappeared from the 1H‐15N HSQC of NSARS‐CoV‐2 (Figure S5), but partially reappeared at high RNA concentration (Figure 5a).
FIGURE 5.
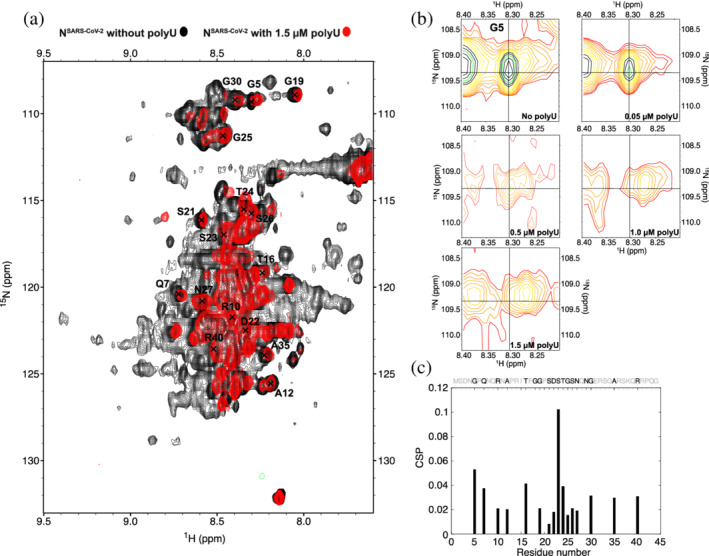
RNA‐interaction of the N‐terminal intrinsically disordered region of NSARS‐CoV‐2. (a) Superposition of 1H‐15N HSQC spectra of full‐length NSARS‐CoV‐2 without polyU RNA and in the presence of 1.5 μM polyU. Spectra were measured at 5°C. (b) 1H‐15N HSQC spectra of the NSARS‐CoV‐2‐residue G5 in the presence of 0, 0.05, 0.5, 1.0, and 1.5 μM polyU RNA. (c) Chemical shift perturbation of the NTE upon interaction with 1.5 μM polyU RNA (NTE sequence shown above; unassigned/overlapped residues are displayed in light gray)
The disappearance of the NMR signals at conditions of LLPS could arise from cluster formation between NSARS‐CoV‐2/RNA. Considering that we confirmed droplet formation at the same protein/RNA concentrations by microscopy, we reason that the effect predominantly arises from increased relaxation rates in the protein when it is concentrated into the viscous phase of NSARS‐CoV‐2/RNA droplets. 31 Additional 1H‐13C XL‐ALSOFAST HSQC experiments further showed that even for the more flexible methyl groups, RNA‐induced LLPS of NSARS‐CoV‐2 causes severe line broadening (Figure S6).
Despite the strong LLPS‐induced broadening of the cross‐peaks in the 1H‐15N HSQC spectra of NSARS‐CoV‐2, we could follow a few selected residues at increasing polyU concentrations. This is illustrated for the cross‐peak of G5 in Figure 5b. In the absence of polyU, a single peak is observed. The peak weakens upon addition of 0.05 μM polyU, that is, at the onset of LLPS (Figure 2b,c). At the conditions of maximum LLPS (0.5 μM polyU, as assessed by turbidity; Figure 2b,c) the cross‐peak appears as doublet (Figure 5b). With further increase of the polyU concentration, the G5 signal at the original position is further broadened such that only the newly generated cross‐peak remains at the highest tested polyU concentration (1.5 μM; Figure 5b). The observations suggest that we detect both an RNA‐free conformation and an RNA‐bound conformation in the NTE. The two conformations are in slow exchange on the NMR chemical shift time scale. In addition, the RNA‐bound conformation of G5 remains sufficiently dynamic in order to be observable in conditions of strong LLPS. Similar observations were made for other cross‐peaks in the NTE (Figure S7).
Further support for a direct interaction between the NTE of NSARS‐CoV‐2 and RNA is accessible at a concentration of 1.5 μM polyU, that is, the high RNA concentration at which no LLPS is present anymore (Figure 2b,c) and from NMR measurements of a protein comprising residue 1–257 of NSARS‐CoV‐2 without and with RNA (Figure S8). Removal of the C‐terminal dimerization domain and the C‐terminal disordered tail decreases the formation of C‐terminal‐mediated oligomerization, which might be modulated by RNA. At the same time, this construct contains two other parts of the protein (the globular RNA‐binding domain and the Ser/Arg‐rich region) that are known to bind to RNA, thus enabling to investigate the binding of RNA to the NTE in the context of known RNA‐binding regions. While the folded N‐terminal domain remains invisible in experiments with both protein constructs, likely due to strongly increased relaxation rates associated to its RNA‐binding, many cross‐peaks of NTE residues are visible (Figure 5a). Careful inspection of selected NTE cross‐peaks, which are not severely affected by signal overlap, reveals chemical shift perturbations and signal broadening across the NTE sequence (Figure 5c; Figure S8).
3. DISCUSSION
NSARS‐CoV‐2 undergoes LLPS with RNA. 1 , 2 , 3 , 5 , 6 The formation of phase‐separated NSARS‐CoV‐2/RNA‐dense condensates might influence viral replication, 6 , 32 drive genome packaging, 3 or modulate the stress‐response. 2 , 33 It is, therefore, important to gain insight into the molecular structural basis of RNA‐induced NSARS‐CoV‐2 phase separation. In the current study, we show that the N‐terminal intrinsically disordered region (NTE) modulates phase separation of NSARS‐CoV‐2 and changes the droplet aging properties through direct binding to RNA. We further provide evidence that RNA‐binding engages multiple parts of the NTE and changes its structural properties.
The N protein is an essential component of positive‐sense coronaviruses, several of which have been identified in humans. While N protein's two folded domains are well conserved among coronaviruses, the intrinsically disordered regions have little sequence conservation. They vary in both amino acid composition and sequence length and thus their physico‐chemical properties. The NTE of NSARS‐CoV‐2 is rich in asparagine and glutamine residues, a typical feature of prion‐like proteins (Figure 1b). There is growing evidence that prion‐like regions play an important role in the development of neurodegenerative diseases through favoring protein aggregation. 34 Notably, the mutations of the Omicron variant increase the content of the prion‐like character in the NTE. We further show that deletion of the NTE weakens the ability of NSARS‐CoV‐2 to form liquid‐like droplets with polyU RNA under conditions of low ionic strength (Figure 2). Rather than abolishing LLPS, however, ΔNTESARS‐CoV‐2 binding saturates at lower RNA concentrations. This can be rationalized on the basis of a lower net charge upon deletion of the NTE and the importance of charge neutralization for RNA‐induced LLPS of NSARS‐CoV‐2. 6 In addition, we show that the droplets of the truncated N protein remain fluid in contrast to the full‐length protein. 6 Our data, thus, demonstrate that the NTE can influence difference aspects of the RNA‐induced LLPS of NSARS‐CoV‐2 in vitro. Experiments in cells are required to understand, how these in vitro properties translate to the formation of NSARS‐CoV‐2‐associated condensates in infected cells. In combination with previously published results indicating that the NTE is important for recruitment of NSARS‐CoV‐2 to stress granules, 19 our work suggests that the NTE can contribute to different LLPS‐associated activities of NSARS‐CoV‐2.
In contrast to the N‐terminal globular domain of NSARS‐CoV‐2, 35 the NTE does not contain a known RNA binding motive. We, however, detected a direct interaction between the NTE and RNA. Electrostatic interactions are likely to be the main driving force for this interaction, in particular with a positively‐charged patch at the C‐terminus of the NTE, which contains three arginine and one lysine residue (Figure 2b). Consistent with an involvement of this positively charged region in RNA‐binding, the cross‐peaks of some of these residues disappeared in the presence of RNA due to intermediate chemical exchange. However, the NMR signal perturbations were not restricted to the C‐terminus of the NTE, but spread over the whole NTE (Figure 5c). In addition, we observed the presence of two distinct conformations for several residues likely corresponding to an RNA‐free and RNA‐bound form. This indicates that RNA binding to the NTE is not restricted to residues capable of electrostatic interactions, in agreement with a recently suggested RNA‐binding mechanism. 36 We further note that the most affected residue of the NTE, S23, is phosphorylated in vivo, 23 , 24 suggesting that phosphorylation of the NTE might influence NSARS‐CoV‐2's ability to phase separate. Consistent with this hypothesis, we previously showed that phosphorylation of the SR‐region of NSARS‐CoV‐2 by the SRSF protein kinase 1 (SRPK1) influences NSARS‐CoV‐2 phase separation. 6 Modulation of phosphorylation pathways thus might influence NSARS‐CoV‐2 condensation in infected cells and serve as a useful entry point for therapeutic intervention against COVID‐19. 37
4. MATERIALS AND METHODS
4.1. Protein preparation
DNA encoding for NSARS‐CoV‐2 (UniProtKB ‐ P0DTC9 obtained from ThermoFisher Scientific, GeneArt) was cloned using BamHI and HindIII restriction enzymes into a pET28 vector, comprising a Z2 solubility tag and a N‐terminal 6× HIS‐Tag for protein purification. BL21(DE3) competent cells were transformed with the cloned plasmids, with kanamycin resistance for colony selection (ThermoFisher Scientific, Invitrogen) and grown in LB until reaching OD600 of 0.7–0.8. Cell cultures were then induced with 0.5 mM IPTG and grown overnight at 37°C. For 15N‐labeled protein the cells were grown in LB until reaching an OD600 of 0.7–0.8, then centrifuged and washed with M9 salts. Cells were subsequently resuspended in minimal M9 medium with 2 g/L of 15NH4Cl as a sole nitrogen source. After 1 h the cell cultures were induced with 0.5 mM IPTG and grown overnight at 37°C. Grown cell cultures were harvested by centrifugation and either used directly or stored at −80°C. Full‐length plasmid was used as a template for PCR to clone DNA of ΔNTESARS‐CoV‐2 and a shorter construct without the dimerization domain (residues 1–257) to new plasmids, which were subsequently expressed and purified using the same protocol as for the full‐length protein.
The cell pellets were resuspended in lysis buffer (25 mM HEPES, 300 mM NaCl, 1 mM EDTA, pH 8.0) supplemented with CaCl2, MnCl2, MgCl2, lysozyme, DNAse I (SigmaAldrich, Roche, 04536282001), and cOmplete™, EDTA‐free Protease Inhibitor Cocktail (SigmaAldrich, Roche, 5056489001). All purification steps were performed at 4°C to avoid precipitation. The cells were lysed using sonication and the protein solution was cleared from cell debris by ultracentrifugation at 75,000 g at 4°C for 30 min (Optima XPN‐80, Beckman Coulter). The supernatant was filtered with 0.2 μm filter and diluted to reduce the NaCl concentration to 100 mM. The sample was then loaded onto HiTrap SP HP column (Cytiva) and the bound protein was eluted with 1 M NaCl. The flow‐through was loaded onto the column several times in order to remove bound nucleic acid, as indicated by a ratio A260/A280 > 1, until there was no protein left. Collected fractions with A260/A280 > 1 were prone to aggregate, and were therefore kept separately from the fractions with A260/A280 < 1. Both samples were then diluted to 500 mM NaCl concentration and loaded onto HisTrap FF column (Cytiva). The bound protein was eluted with 500 mM imidazole. After overnight dialysis (25 mM TRIS, 170 mM NaCl, 5 mM imidazole, pH 8.0) the His‐Tag was cleaved off by a TEV protease (as confirmed by SDS PAGE gel) and samples were loaded again onto HisTrap FF column. At this point, the protein was still binding to the column. Reasoning that a cut protein bound to the column via electrostatic interactions with a small portion of an uncut protein, the cut protein was eluted with 300 mM NaCl. The bound uncut protein was subsequently eluted by 500 mM imidazole. The last purification step was done using size exclusion chromatography, HiLoad 26/600 Superdex 200 pg (buffer composition: 25 mM TRIS, 300 mM NaCl, pH 8.0). The A260/A280 ratio of 0.55 measured in the final fractions confirmed the absence of nucleic acid contamination. Protein purity was verified using SDS page gel. Samples were then concentrated and exchanged to either NMR buffer (50 mM sodium phosphate, 10% D2O, 0.01% NaN3, pH 6.8) or buffer for phase separation experiments (20 mM sodium phosphate, pH 7.6).
The synthetic NTE peptide (residues 1–44 of NSARS‐CoV‐2) was bought from GenScript. The NTE peptide purity was estimated to be over 98%. The SR peptide (residues 182–197 of NSARS‐CoV‐2) was produced as described previously. 6
4.2. Turbidity measurements
Phase diagrams of full‐length NSARS‐CoV‐2 and ΔNTESARS‐CoV‐2 were determined using a NanoDrop spectrophotometer (ThermoFisher Scientific, Invitrogen). Polyuridylic acid (polyU; molecular weight = 800–1,000 kDa) was purchased from Sigma‐Aldrich (P9528). The same batch was used throughout the measurements. According to dynamic light scattering measurements, this batch is characterized by an average molecular weight of 800 kDa. PolyU (in concentrations from 0 to 2 μM) was added right before the experiments, followed by thoroughly pipetting and measurement of turbidity at 350 nm UV‐Vis. Averaged turbidity values and the error bars were derived from measurements of three independent, freshly prepared samples. Turbidity measurements of 30 μM ΔNTESARS‐CoV‐2 were repeated using 20 mM sodium phosphate buffer at pH 6, 7.5, and 8 and in 20 mM Tris at pH 9.
4.3. DIC/fluorescence microscopy
Full‐length NSARS‐CoV‐2 and ΔNTESARS‐CoV‐2 proteins were labeled using Alexa‐fluor 488™ (green) microscale protein labeling kits (ThermoFisher Scientific, Invitrogen). Small amounts (~0.3 μl) of fluorescently‐labeled NSARS‐COV‐2 were premixed with unlabeled NSARS‐CoV‐2 and diluted to 30–50 μM final concentration with 20 mM sodium phosphate buffer at pH 7.5. 0.3 or 0.5 μM polyU RNA was added to NSARS‐CoV‐2 or ΔNTESARS‐CoV‐2, respectively, achieving approximately charge compensation. 5 μl of the sample were subsequently loaded onto a slide and covered with an 18 mm coverslip. DIC and fluorescent micrographs were acquired on a Leica DM6B microscope with a 63× objective (water immersion) and processed using Fiji software (NIH).
4.4. FRAP measurements
FRAP measurements have been performed on 30 μM full‐length NSARS‐CoV‐2 or ΔNTESARS‐CoV‐2 in presence of 0.3 μM polyU in 20 mM sodium phosphate buffer (NaP), pH 7.5 on a Leica TCS SP8 confocal microscope using a 63× objective (oil immersion) and a 488 argon laser line. Prior measurement, unlabeled FL‐ or 41‐419 NSARS‐CoV‐2 were mixed with Alexa Fluor 488 lysine labeled FL‐ or 41‐419 NSARS‐CoV‐2.
Fluorescence recoveries have been recorded as previously described. 6 Upon addition of polyU, that is, on freshly formed droplets early time points were recorded within a time <15 min, late time points have been acquired after 1 h of sample incubation on the glass.
Regions of interest (ROIs) of ~4 μm in diameter have been bleached with 50% of laser power, recovery was imaged at low laser intensity (5%). One hundred frames were recorded with one frame per 523 ms. The data have been processed using FIJI software (NIH).
FRAP recovery curves were obtained using standard protocols. Briefly, for each FRAP measurement intensities for pre‐bleaching, bleaching and post‐bleaching ROIs have been measured. A pre‐bleaching ROI corresponded to a selected region in the droplet before bleaching; the bleached ROI corresponded to the bleached area while the reference ROI corresponded to an area which did not experience bleaching. The fluorescence intensity measured for each of the described ROIs was corrected by background subtraction: a region where no fluorescence was detected was used to calculate the background. Thus, the FRAP recovery was calculated as:
The value obtained was then corrected by multiplication with the acquisition bleaching correction factor (ABCF), calculated as:
FRAP curves were normalized according to:
Values were averaged from four recordings for both early and late time points and the resulting FRAP curves ± standard deviation (std) were fitted for the early time points to a mono‐exponential function:
For the late time points, a bi‐exponential function provided the best fitting:
The mobile and immobile fractions were calculated using the parameters a and c derived from each fitting, according to the following equations:
4.5. NMR spectroscopy
NMR experiments were acquired on a 700 MHz Bruker Avance III spectrometer equipped with a triple resonance TCI CryoProbe with Z‐gradient and on an 800 MHz Bruker Neo spectrometer equipped with a triple resonance TCI CryoProbe with Z‐gradient. Experiments were performed at 278 and 298 K. Chemical shifts of the NTE were assigned using a combination of two‐dimensional TOCSY and NOESY NMR spectra and transferred to 1H‐15N and 1H‐13C HSQC to obtain N, Cα and Cβ chemical shifts. Assigned chemical shifts were deposited in BMRB database (BMRB ID: 51504). The hydrodynamic radius of the NTE was determined using NMR pulse field gradient experiments with DSS (concentration ∼1 mM) used as an internal reference. 1‐D 1H spectra were collected employing the standard Bruker pulse program ledbpgppr2s1d. The gradient length was set to 1,300 μs, and the diffusion delays to 40 and 200 ms for DSS and protein, respectively. For each delay, a set of experiments with gradient strength increasing from 5% to 95% was acquired with 5% steps (100% gradient strength corresponding to 53.5 G/cm). For each spectrum 64 scans and 16,384 points with a spectral width of 11,364 Hz were acquired. Signal intensities corresponding to DSS and the NTE amide signals were integrated using TopSpin (Bruker Biospin) and fitted according to Stokes–Einstein relation. 29 Hydrodynamic radius of the NTE was calculated from the diffusion coefficients using the known hydrodynamic radius of DSS (3.43 Å).
Spectra were processed using TopSpin 4.0.6 (Bruker) or NMRPipe 10.6 38 and analyzed using SPARKY (Goddard TD & Kneller DG [2008] SPARKY 3. University of California, San Francisco). Assigned chemical shifts (N, NH, Cα, Cβ, and Hα) were used to predict secondary structures in the NTE using TALOS+. 28
4.6. MD simulations
The starting structure of the N‐terminal fragment of NSARS‐CoV‐2 was built with Flexible Meccano. 39 Subsequently the peptide was equilibrated with 50,000 steps of energy minimization. To further equilibrate the system, 100 ps each of volume (NVT) and pressure (NPT) equilibration were performed. The MD simulations were carried out in GROMACS (version 2018.3) using the AMBER99SB‐ILDN force field and the TIP3P water model at a temperature of 300 K, 1 bar of pressure and with a coupling time (ζT) of 0.1 ps. The mixtures were solvated in water with 150 mM NaCl, ensuring overall charge neutrality. The particle mesh Ewald algorithm was used for calculation of the electrostatic term, with a radius of 16 Å for the grid‐spacing and Fast Fourier Transform. The cut‐off algorithm was applied for the non‐coulombic potential with a radius of 10 Å. The LINCS algorithm was used to contain bonds and angles. MD simulations were performed during 100 ns in 2 fs steps and saving the coordinates of the system every 10 ps. Five repetitions were done for each simulation, using a different structure from the ensemble of conformations generated by Flexible Meccano. The hydrodynamic radius was calculated with the software HullRad (Version 7). 40 To derive the final hydrodynamic radius, the average of the radius of the structures of the last 50 ns of each simulation was taken together with the standard deviation.
Supporting Information S1 includes an analysis of LLPS of the full‐length NSARS‐CoV‐2 at different pH values. In addition, we include NMR data, such as diffusion experiments and spectra of NSARS‐CoV‐2 amide and methyl spectral regions acquired at different concentrations of polyU.
AUTHOR CONTRIBUTIONS
Milan Zachrdla: Formal analysis (equal); investigation (equal); methodology (equal); project administration (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Adriana Savastano: Formal analysis (equal); investigation (equal); methodology (equal); project administration (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Alain Ibanez de Opakua: Formal analysis (equal); investigation (equal); methodology (equal); validation (equal); visualization (equal); writing review and editing (equal). Markus Zweckstetter: Project administration (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal); resources (equal). Maria‐Sol Cima‐Omori: Investigation (equal); resources (equal).
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENT
Markus Zweckstetter was supported by the European Research Council (ERC) under the EU Horizon 2020 research and innovation program (grant agreement No. 787679). Open Access funding enabled and organized by Projekt DEAL.
Zachrdla M, Savastano A, Ibáñez de Opakua A, Cima‐Omori M‐S, Zweckstetter M. Contributions of the N‐terminal intrinsically disordered region of the severe acute respiratory syndrome coronavirus 2 nucleocapsid protein to RNA‐induced phase separation. Protein Science. 2022;31(9):e4409. 10.1002/pro.4409
Milan Zachrdla, Adriana Savastano, and Alain Ibáñez de Opakua were equally contributed to this article.
Review Editor: Carol Post
Funding information H2020 European Research Council, Grant/Award Number: 787679
DATA AVAILABILITY STATEMENT
All data are available from the authors upon reasonable request.
REFERENCES
- 1. Chen H, Cui Y, Han X, et al. Liquid–liquid phase separation by SARS‐CoV‐2 nucleocapsid protein and RNA. Cell Res. 2020;30:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cubuk J, Alston JJ, Incicco JJ, et al. The SARS‐CoV‐2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat Commun. 2021;12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iserman C, Roden CA, Boerneke MA, et al. Genomic RNA elements drive phase separation of the SARS‐CoV‐2 nucleocapsid. Mol Cell. 2020;80:1078–1091.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mu J, Xu J, Zhang L, et al. SARS‐CoV‐2‐encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci. 2020;63:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perdikari TM, Murthy AC, Ryan VH, Watters S, Naik MT, Fawzi NL. SARS‐CoV‐2 nucleocapsid protein phase‐separates with RNA and with human hnRNPs. EMBO J. 2020;39:e106478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS‐CoV‐2 phase separates into RNA‐rich polymerase‐containing condensates. Nat Commun. 2020;11:6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burbelo PD, Riedo FX, Morishima C, et al. Sensitivity in detection of antibodies to nucleocapsid and spike proteins of severe acute respiratory syndrome coronavirus 2 in patients with coronavirus disease 2019. J Infect Dis. 2020;222:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dangi T, Class J, Palacio N, Richner JM, Penaloza MacMaster P. Combining spike‐ and nucleocapsid‐based vaccines improves distal control of SARS‐CoV‐2. Cell Rep. 2021;36:109664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lancaster AK, Nutter‐Upham A, Lindquist S, King OD. PLAAC: A web and command‐line application to identify proteins with prion‐like amino acid composition. Bioinformatics. 2014;30:2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lo YS, Lin SY, Wang SM, et al. Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013;587:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nikolakaki E, Giannakouros T. SR/RS motifs as critical determinants of coronavirus life cycle. Front Mol Biosci. 2020;7:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bessa LM, Guseva S, Camacho‐Zarco AR, et al. The intrinsically disordered SARS‐CoV‐2 nucleoprotein in dynamic complex with its viral partner nsp3a. Sci Adv. 2022;8:eabm4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ribeiro‐Filho HV, Jara GE, Batista FAH, et al. Structural dynamics of SARS‐CoV‐2 nucleocapsid protein induced by RNA binding. PLoS Comput Biol. 2022;18:e1010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jack A, Ferro LS, Trnka MJ, et al. SARS‐CoV‐2 nucleocapsid protein forms condensates with viral genomic RNA. PLoS Biol. 2021;19:e3001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang C‐K, Hsu Y‐L, Chang Y‐H, et al. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: Implications for ribonucleocapsid protein packaging. J Virol. 2009;83:2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dinesh DC, Chalupska D, Silhan J, et al. Structural basis of RNA recognition by the SARS‐CoV‐2 nucleocapsid phosphoprotein. PLoS Pathog. 2020;16:e1009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. The SARS coronavirus nucleocapsid protein – Forms and functions. Antiviral Res. 2014;103:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Shi C, Xu Q, Yin H. SARS‐CoV‐2 nucleocapsid protein undergoes liquid–liquid phase separation into stress granules through its N‐terminal intrinsically disordered region. Cell Discov. 2021;7:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sprunger ML, Jackrel ME. Prion‐like proteins in phase separation and their link to disease. Biomolecules. 2021;11:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tetz G, Tetz V. Prion‐like domains in eukaryotic viruses. Sci Rep. 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Syed AM, Taha TY, Tabata T, et al. Rapid assessment of SARS‐CoV‐2 evolved variants using virus‐like particles. Science. 2021;374(6575):1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davidson AD, Williamson MK, Lewis S, et al. Characterisation of the transcriptome and proteome of SARS‐CoV‐2 reveals a cell passage induced in‐frame deletion of the furin‐like cleavage site from the spike glycoprotein. Genome Med. 2020;12:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bouhaddou M, Memon D, Meyer B, et al. The global phosphorylation landscape of SARS‐CoV‐2 infection. Cell. 2020;182:685–712.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carlson CR, Asfaha JB, Ghent CM, et al. Phosphoregulation of phase separation by the SARS‐CoV‐2 N protein suggests a biophysical basis for its dual functions. Mol Cell. 2020;80:1092–1103.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guseva S, Perez LM, Camacho‐Zarco A, et al. 1H, 13C and 15N Backbone chemical shift assignments of the n‐terminal and central intrinsically disordered domains of SARS‐CoV‐2 nucleoprotein. Biomol NMR Assign. 2021;15:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schiavina M, Pontoriero L, Uversky VN, Felli IC, Pierattelli R. The highly flexible disordered regions of the SARS‐CoV‐2 nucleocapsid N protein within the 1–248 residue construct: Sequence‐specific resonance assignments through NMR. Biomol NMR Assign. 2021;15:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marsh JA, Forman‐Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. 2010;98:2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nygaard M, Kragelund BB, Papaleo E, Lindorff‐Larsen K. An efficient method for estimating the hydrodynamic radius of disordered protein conformations. Biophys J. 2017;113:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abyzov A, Blackledge M, Zweckstetter M. Conformational dynamics of intrinsically disordered proteins regulate biomolecular condensate chemistry. Chem Rev. 2022;122:6719–6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cascarina SM, Ross ED. A proposed role for the SARS‐CoV‐2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 2020;34:9832–9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo L, Li Z, Zhao T, et al. SARS‐CoV‐2 nucleocapsid protein phase separates with G3BPs to disassemble stress granules and facilitate viral production. Sci Bull. 2021;66:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scialò C, De Cecco E, Manganotti P, Legname G. Prion and prion‐like protein strains: Deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses. 2019;11:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Forsythe HM, Rodriguez Galvan J, Yu Z, et al. Multivalent binding of the partially disordered SARS‐CoV‐2 nucleocapsid phosphoprotein dimer to RNA. Biophys J. 2021;120:2890–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krainer G, Welsh TJ, Joseph JA, et al. Reentrant liquid condensate phase of proteins is stabilized by hydrophobic and non‐ionic interactions. Nat Commun. 2021;12:1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X, Verma A, Garcia G, et al. Targeting the coronavirus nucleocapsid protein through GSK‐3 inhibition. Proc Natl Acad Sci USA. 2021;118:e2113401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 39. Ozenne V, Bauer F, Salmon L, et al. Flexible‐meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics. 2012;28:1463–1470. [DOI] [PubMed] [Google Scholar]
- 40. Fleming PJ, Fleming KG. HullRad: Fast calculations of folded and disordered protein and nucleic acid hydrodynamic properties. Biophys J. 2018;114:856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
All data are available from the authors upon reasonable request.