

Abstract
Objectives
To determine whether SARS‐CoV‐2 can trigger complement activation, the pathways that are involved and the functional significance of the resultant effect.
Methods
SARS‐CoV‐2 was inoculated into a human lepirudin‐anticoagulated whole blood model, which contains a full repertoire of complement factors and leukocytes that express complement receptors. Complement activation was determined by measuring C5a production with an ELISA, and pretreatment with specific inhibitors was used to identify the pathways involved. The functional significance of this was then assessed by measuring markers of C5a signalling including leukocyte C5aR1 internalisation and CD11b upregulation with flow cytometry.
Results
SARS‐CoV‐2 inoculation in this whole blood model caused progressive C5a production over 24 h, which was significantly reduced by inhibitors for factor B, C3, C5 and heparan sulfate. However, this phenomenon could not be replicated in cell‐free plasma, highlighting the requirement for cell surface interactions with heparan sulfate. Functional analysis of this phenomenon revealed that C5aR1 signalling and CD11b upregulation in granulocytes and monocytes was delayed and only occurred after 24 h.
Conclusion
SARS‐CoV‐2 is a noncanonical alternative pathway activator that progressively triggers complement activation through interactions with heparan sulfate.
Keywords: alternative pathway, complement, coronavirus, heparan sulfate, leukocyte activation, SARS‐CoV‐2
The mechanisms by which SARS‐CoV‐2 activates complement remain unclear, and so here, we utilised an ex vivo human whole blood model to interrogate the pathways and functional responses involved. SARS‐CoV‐2 inoculation in blood caused progressive C5a production over 24 h, which was blocked entirely by inhibitors for factor B, C3, C5 and heparan sulfate. This study therefore provides direct mechanistic evidence for SARS‐CoV‐2 driving complement activation and the requirement for cell surfaces and heparan sulfate.

Introduction
COVID‐19 is a highly contagious respiratory infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). In the last 2 years, this disease has affected over 560 million individuals and caused over 6.3 million deaths. 1 Thus, unprecedented efforts have been put towards vaccine and drug development, but with the possibility of new variants and the inevitability of future pandemics, a fundamental understanding of severe COVID‐19 is still needed. In this context, SARS‐CoV‐2 replicates in an unchecked manner and evades the immune system by exploiting several inborn and acquired weaknesses. 2 , 3 At a critical mass, these virions are believed to trigger a hyperinflammatory response that causes acute respiratory distress syndrome (ARDS). 4 Emerging evidence suggests that the complement system plays a key role in this process. 5 , 6 , 7 Indeed, complement activation has been correlated with disease severity 8 and small case studies have shown that complement inhibition can be effective in critical patients, prompting at least six anticomplement drugs to be taken to clinical trials. 5 However, whilst in vitro studies have demonstrated that specific viral proteins can activate complement in isolated systems, the relationship between the live SARS‐CoV‐2 virus and complement activation remains incompletely understood.
Complement‐mediated disease in COVID‐19 appears to be confined to severely ill patients who are unable to bring the virus under immunological control. In these patients, SARS‐CoV‐2 exploits defects in the interferon system and replicates in an unchecked manner, which at a critical mass, is thought to drive a form of complement‐mediated hyperinflammation. 3 Indeed, evidence of complement activation has been correlated with disease severity and includes serum C5a and C5b‐9 concentrations, 8 leukocyte CD11b expression, 9 which can be due to C5aR1 activation, 10 and postmortem immunochemistry. 7 These features occur on the background of airway complement synthesis 11 and are particularly prominent in individuals who are genetically prone to C5 cleavage, 12 who have elevated mannose‐binding protein levels 13 or who have reduced CD55 expression. 14 Additional investigations suggest that complement activation in severe COVID‐19 can occur through the classical, lectin and alternative pathways. 15 , 16 Thus, complement appears to be a key driver of severe COVID‐19.
Moreover, molecular investigations have provided some insight into the underlying mechanisms that drive complement activation in COVID‐19. Indeed, an initial study utilising the SARS‐CoV‐2 S protein in a specialised functional assay suggests that the virus may activate the alternative pathway by binding to cell surface heparan sulfate, which disinhibits factor H‐mediated complement suppression. 17 In this study, human serum pretreated with recombinant SARS‐CoV‐2 S protein caused complement deposition and cytotoxicity in complement inhibitor‐deficient cells. However, SARS‐CoV‐2 S protein did not trigger complement activation without such cells or after heparan sulfate or factor H supplementation. In addition, a more recent study found that the SARS‐CoV‐2 S and N proteins can activate the lectin pathway via MASP‐2. 18 Thus, early molecular studies using viral proteins suggest that SARS‐CoV‐2 can directly activate the complement system, but conclusive evidence for this with live virus is still outstanding.
Therefore, here we inoculated SARS‐CoV‐2 virus into a lepirudin‐anticoagulated human blood model and used ELISAs and flow cytometry to measure complement activation and functionality, respectively. Using this model, we show that SARS‐CoV‐2 activates the alternative complement pathway, requiring interactions with cell surfaces and heparan sulfate, and in doing so causes delayed leukocyte activation through C5a‐C5aR1 signalling.
Results
SARS‐CoV‐2 activates the alternative complement pathway in whole blood, requiring interactions with heparan sulfate
We first investigated whether SARS‐CoV‐2 could activate complement by inoculating the virus at a multiplicity of infection (MOI) 0.1 and 1.0 in a lepirudin‐anticoagulated human blood model and subsequently measuring plasma C5a with an ELISA. This model simulates a naïve and complete complement system and contains all soluble and membrane‐bound complement factors, receptors and inhibitors, thereby enabling it to detect all pathways of complement activation and their functional consequences 19 Indeed, SARS‐CoV‐2‐inoculated whole blood at MOI 1.0 showed an increase in C5a of 5–10 ng mL−1 and 30–50 ng mL−1 above that in the mock control at 30 min and 24 h, respectively (Figure 1a). This occurred on the background of the de novo complement activation permitted by lepirudin, as reflected in the increased baseline observed at 24 h following mock infection. By contrast, this did not occur in isolated plasma (Figure 1b), which suggests that a cellular component is required for appropriate SARS‐CoV‐2‐mediated complement activation. Likewise, mock‐treated plasma had higher C5a levels than mock‐treated whole blood, as the former lacks cell surface complement receptors (e.g. C5aR1, CR1, CR3 and CR4), which sequestrate activated complement fragments.
Figure 1.
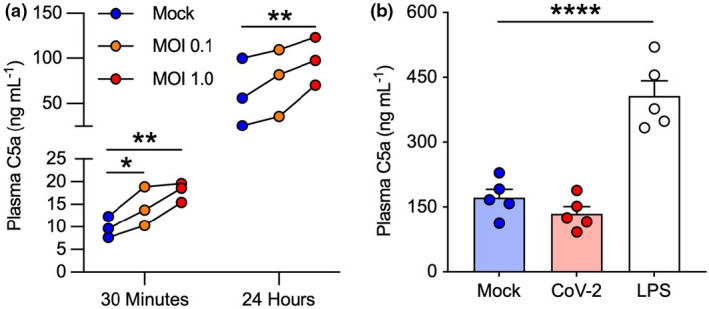
SARS‐CoV‐2 activates complement in whole blood but not in plasma. C5a levels were assessed with an ELISA in SARS‐CoV‐2‐inoculated lepirudin‐anticoagulated (a) human blood at 30 min and 24 h (n = 3, three technical replicates) and (b) plasma at 24 h (n = 5, two technical replicates). Different donors were used in a and b. Columns and whiskers depict mean values and SEM, respectively. MOI, multiplicity of infection; CoV‐2, SARS‐CoV‐2 MOI 1.0; *P < 0.05, **P < 0.01, ****P < 0.0001, using a one‐way ANOVA.
To further delineate the pathways involved, coronavirus‐inoculated whole blood was also pretreated with complement and heparan sulfate inhibitors based on previous studies. 17 Indeed, specific blockade of the alternative pathway through factor B inhibition with LNP023, significantly attenuated complement activation (Figure 2a). Moreover, inhibition of heparan sulfate using either EGCG or the synthetic heparan sulfate mimetic PG545 significantly reduced activation of complement in this model (Figure 2b). As expected, global complement inhibition at the level of C3 (compstatin) or C5 (eculizumab) also significantly blocked SARS‐CoV‐2 mediated C5a generation (Figure 2c and d). Thus, given that the C3, C5 and heparan sulfate inhibitors are membrane impermeable, these findings suggest that SARS‐CoV‐2 activates the alternative complement pathway through cell surface‐dependent and heparan sulfate‐dependent mechanisms.
Figure 2.
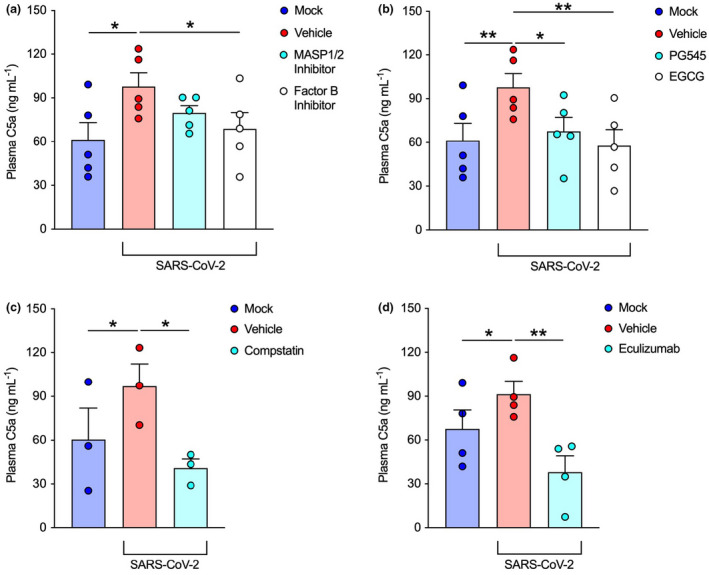
SARS‐CoV‐2 activates the alternative pathway through interactions with heparan sulfate. Plasma C5a levels were assessed with an ELISA in SARS‐CoV‐2‐inoculated lepirudin‐anticoagulated human blood pretreated with various inhibitors and mimetics. These included inhibitors for (a) MASP1/2 (SFMI‐1 10 μm; Lectin Complement Pathway inhibitor) and factor B (LNP023 10 μm; Alternative Complement Pathway inhibitor) (n = 5, two technical replicates); (b) heparan sulfate (EGCG 100 μm) and a mimetic for heparan sulfate (PG545 100 μg mL−1) (n = 5, two technical replicates); (c) C3 (Compstatin 20 μm; global Complement Pathway inhibitor) (n = 3, three technical replicates) and (d) C5 (Eculizumab 100 μg mL−1; terminal Complement Pathway inhibitor) (n = 4, two technical replicates). SARS‐CoV‐2 was inoculated at MOI 1.0 for 24 h. Columns and whiskers depict mean values and SEM, respectively. MOI, multiplicity of infection; *P < 0.05, **P < 0.01, using a one‐way ANOVA.
Flow cytometry optimization and incidental findings on monocyte activation
Next, we sought to determine whether SARS‐CoV‐2‐mediated complement activation was sufficient to induce a functional response. As C5a stimulation of myeloid cells causes C5aR1 internalisation and CD11b upregulation at the cell surface, 10 we inoculated the whole blood model with SARS‐CoV‐2 (MOI 0.1 and 1.0) for 3 and 24 h and used flow cytometry to measure C5aR1 and CD11b surface expression on neutrophils, eosinophils and monocytes. To do so, we trialled a flow cytometry panel under inoculation conditions and found the gating properties to be slightly altered (Supplementary figure 1) compared to fresh blood. Indeed, whilst granulocyte markers were unaffected, monocyte markers including CD16 and HLA‐DR were upregulated in a dose‐dependent manner (Supplementary figure 2a), which suggests that other immune phenomena were present in this assay. Thus, pan‐monocytes without subset differentiation were examined in this study.
SARS‐CoV‐2 causes delayed complement‐mediated leukocyte activation
Given that immune stimulators induce rapid changes in myeloid cell activation, we first tested whether SARS‐CoV‐2 could cause complement‐mediated leukocyte activation after 3 h in the human blood model. However, at this time point, flow cytometry revealed few alterations in cell activation markers in neutrophils, eosinophils and monocytes (data not shown). We therefore extended the period of SARS‐CoV‐2 incubation to 24 h and detected significant C5aR1 internalisation and CD11b upregulation on neutrophils, eosinophils and monocytes (Figure 3). Interestingly, at 3 h post‐inoculation, CD11b upregulation without concomitant C5aR1 internalisation was noted in neutrophils, which suggests that these cells can react acutely to SARS‐CoV‐2 through receptors independent of complement (Supplementary figure 2b).
Figure 3.
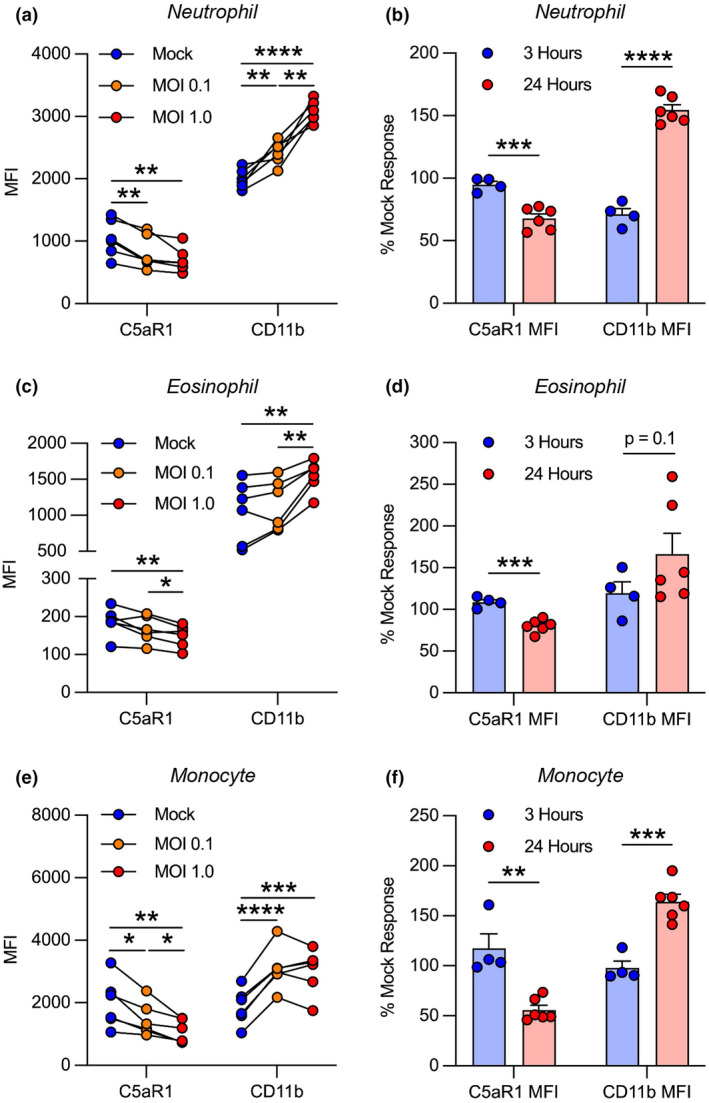
SARS‐CoV‐2 causes delayed complement‐mediated activation in innate leukocytes. SARS‐CoV‐2‐inoculated lepirudin‐anticoagulated human blood was analyzed with flow cytometry. SARS‐CoV‐2 caused dose‐dependent C5aR1 internalisation and CD11b upregulation in (a) neutrophils, (c) eosinophils and (e) monocytes at 24 h postinoculation (n = 6). Comparison of C5aR1 and CD11b responses between SARS‐CoV‐2‐inoculated blood at 3 (n = 4) and 24 h (n = 6) expressed as a percentage change from mock inoculation (b, d, f). Columns and whiskers depict mean values and SEM, respectively. MOI, multiplicity of infection; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using a one‐way ANOVA or t‐test.
Anti‐C5/C5aR1 drugs eculizumab and PMX205 inhibit SARS‐CoV‐2‐induced complement‐mediated inflammation
To investigate the functional role of the terminal complement pathway in SARS‐CoV‐2‐mediated leukocyte activation, we next pretreated the whole blood model with a C5 inhibitor (eculizumab) and a C5aR1 antagonist (PMX205) prior to viral inoculation. After 24 h, both drugs inhibited SARS‐CoV‐2‐dependent C5aR1 internalisation and CD11b upregulation in neutrophils and to a lesser extent in eosinophils (Figure 4a–d). Interestingly, in eosinophils, the inhibitors reversed these effects beyond the mock baseline, which may reflect inhibition of de novo complement activation. By contrast, these drugs were only able to partially inhibit C5aR1 internalisation and did not lower CD11b upregulation in monocytes (Figure 4e and f). This latter finding suggests that complement at the level of C5 is not involved in this process in these cells.
Figure 4.
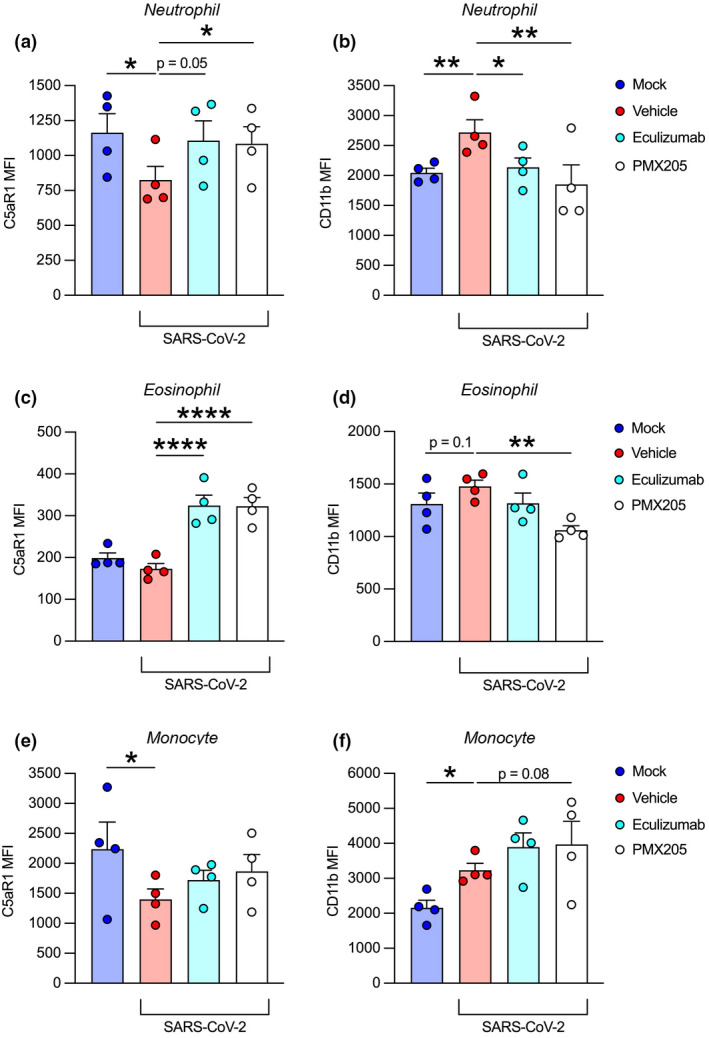
C5/C5aR1 inhibition attenuates SARS‐CoV‐2‐induced leukocyte activation. Anti‐C5/C5aR1 inhibitors eculizumab (100 μg mL−1; C5 inhibitor) and PMX205 (10 μm; C5aR1 antagonist) were administered to lepirudin‐anticoagulated whole blood (n = 4) prior to SARS‐CoV‐2 inoculation at MOI 1.0 for 24 h. Flow cytometry was used to measure activation markers C5aR1 and CD11b in terms of MFI on neutrophils (a, b), eosinophils (c, d) and monocytes (e, f). Columns and whiskers depict mean values and SEM, respectively. MOI, multiplicity of infection; MFI, median fluorescence intensity; *P < 0.05, **P < 0.01, ****P < 0.0001, using a one‐way ANOVA.
Discussion
Results support clinical findings of complement in COVID‐19
Overall, the complement activation observed in this study is consistent with the current clinical and molecular understanding of COVID‐19. Foremostly, in vivo complement profiling in severe COVID‐19 reveals activation of the alternative pathway 16 and in vitro studies have shown that recombinant viral S protein can activate this same pathway through interactions with heparan sulfate. 17 However, in contrast to our whole blood model, complement activation during COVID‐19 is probably a multifactorial phenomenon driven by multiple complement pathways, 7 , 16 , 18 nonspecific DAMP release, 5 genetic susceptibility to complement activation, 12 , 13 and local complement synthesis. 11 But in this regard, our participants were seronegative for anti‐SARS‐CoV‐2 IgG, which suggests that the classical pathway was not activated in these experiments (Supplementary figure 3). Additionally, given that C5a‐mediated immune activation typically occurs within 60 min, 10 the delayed response requiring 24 h in our model is consistent with the gradual progression 4 and upregulation of CD11b on leukocytes in severe cases. 9 Thus, these results strongly support the emerging paradigm of heparan sulfate‐ and alternative complement pathway‐mediated disease in severe COVID‐19.
Complement activation appears to be localised to organs that support replication
When placed in the context of in vivo viral titres, our results also suggest that SARS‐CoV‐2‐mediated complement activation is localised to organs that support replication and have high viral loads. On average COVID‐19 patients have median viral titres of ~106 RNA copies mL−1 in their airways; as determined from nasopharyngeal swaps, sputum and bronchoalveolar lavage fluid; and ~103 RNA copies mL−1 in their serum, in which the former is at least 10 times higher in severe cases than that in mild cases. 20 , 21 By comparison, in this study whole blood inoculated with SARS‐CoV‐2 at MOI 0.1 and 1.0 was exposed to virion concentrations of ~3.5–5.5 × 105 and ~3.5–5.5 × 106 FFU mL−1, respectively. Thus, complement activation in severe COVID‐19 is most likely localised to tissues that support replication (e.g. lung parenchyma) with changes in plasma complement occurring as a secondary phenomenon. This implies that anticomplement drugs in severe COVID‐19 require significant tissue distribution, which may have limited the efficacy of the parenteral drugs that have so far been tested in clinical trials. In this sense, SARS‐CoV‐2 can be viewed as an organ‐based activator of complement that poses unique challenges to drug development.
Complement and heparan sulfate in severe COVID‐19
Furthermore, complement activation through heparan sulfate appears to be an inadvertent phenomenon. Indeed, this polysaccharide post‐translational modification promotes specific protein–protein interactions, which in this case concentrates factor H on host cells to prevent excessive complement activation. 22 Therefore, whilst SARS‐CoV‐2 principally uses heparan sulfate to dock with angiotensin‐converting enzyme 2 to enable infection, 23 it also secondarily activates complement by causing factor H disinhibition. In our whole blood model, this was most likely mediated by CD44v3 on lymphocytes but in vivo is probably the result of interactions with a range of membrane‐bound (e.g. syndecan 1–4) and extracellular matrix proteoglycans (e.g. perlecan). 24 Interestingly, S protein–heparan sulfate interactions can also disrupt antithrombin and heparin cofactor II activity, and thus, heparan sulfate warrants further investigation as a multifaceted drug target in severe COVID‐19. 25
Concluding remarks
COVID‐19 continues to cause more deaths than any other pandemic in living memory. Mounting evidence suggests that complement plays a key role in severe disease, and here, we show that in blood, SARS‐CoV‐2 interacts with cell surface heparan sulfate and activates the alternative pathway, which ultimately drives innate leukocyte activation through C5a‐C5aR1 signalling. In doing so, these findings support the use of targeted anti‐complement treatments in severe COVID‐19.
Methods
Study approval
This research was approved by the University of Queensland Human Research Ethics Committee (2020/HE000559) and all participants gave informed written consent. This research was also approved by the University of Queensland Biosafety Committee (IBC/390B/SCMB/2020, IBC/1301/SCMB/2020, IBC/376B/SBMS/2020 and IBC/447B/SCMB/2021), such that all experiments were performed in a biosafety level 3 facility at the School of Chemistry and Molecular Biosciences at The University of Queensland, Australia. Specifically, all pathogenic materials were handled in a class II biosafety cabinet and all approved personnel used disposal Tychem 2000 coveralls (Dupont, Wilmington, NC, USA; #TC198T YL) and Versaflo‐powered air‐purifying respirators (3M, Saint Paul, MN, USA; #902‐03‐99). Surface disinfection was performed with 70% ethanol while liquid and solid waste underwent autoclave sterilisation (121°C for 45 min).
Participants
Whole blood was drawn from healthy individuals (Supplementary table 1) with a 4.9 mL S‐Monovette (SARSTEDT, Nümbrecht, Germany; #04.1926.001) and anticoagulated with 50 μg mL−1 of Lepirudin (Pharmion, Boulder, CO, USA), which is a direct thrombin inhibitor and an anticoagulant that permits ex vivo complement activation. 10 Türk's solution (Sigma‐Aldrich, Saint Louis, MO, USA; #109277) was used as per manufacturer guidelines to perform total leukocyte counts for MOI calculations.
Cell line and virus
An African green monkey kidney cell line (Vero E6 cells) was cultured in DMEM (Gibco, Waltham, MA, USA) supplemented with 10% heat‐inactivated foetal calf serum (HIFCS) (Bovogen, Melbourne, VIC, Australia) and 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin (P/S). Cells were maintained at 37°C with 5% CO2. The Queensland SARS‐CoV‐2 early isolate QLD02 (GISAID accession number EPI_ISL_407896) was recovered from a patient on 30/01/2020 by the Queensland Health Forensic & Scientific Services. VeroE6 cells were then inoculated with this isolate and an aliquot (passage 2) was provided. Viral stock (passage 3) was then generated through inoculation of VeroE6 cells in DMEM with 2% HIFCS and P/S and stored at −80°C. The viral titre was determined by an immuno‐plaque assay (iPA) as previously described. 26
Whole blood inoculation
For the ELISA experiments, 100 μL of whole blood was inoculated with SARS‐CoV‐2 at an MOI of 0.1 or 1.0, LPS O111:B4 (200 μg mL−1; Sigma‐Aldrich; #L2630‐100MG) or a mock solution (i.e. DMEM with 2% HIFCS and P/S) for 30 min or 24 h at 37°C. Samples were then centrifuged at 2000 g for 10 min at 4°C and plasma was aliquoted and stored at −80°C for downstream analysis. Certain samples were pretreated with the following for 30 min at 37°C: SFMI‐1 (MASP1/2 inhibitor 10 μm; synthesised in house 27 ), LNP023 (factor B inhibitor, 10 μm; AdooQ Bioscience, Irvine, CA, USA; #A18905), compstatin analogue (C3 inhibitor, 20 μm; Wuxi AppTec Ltd, Shanghai, China; #C15031904), eculizumab (C5 inhibitor, 100 μg mL−1; Ichorbio, Wantage, UK; #ICH4005), EGCG (heparan sulfate inhibitor, 100 μm; Sigma‐Aldrich; E4143‐50MG) or pixatimod/PG545 (heparan sulfate mimetic, 100 μg mL−1; synthesised in house 28 ). This assay was repeated with plasma isolated from whole blood after centrifugation at 2000 g for 10 min at room temperature. For the flow cytometry experiments, whole blood was mixed 1:1 with RPMI1640 (Gibco; #42401‐018) and inoculated with SARS‐CoV‐2 at an MOI of 0.1 or 1.0 or a mock solution (as above) and incubated for 3 or 24 h at 37°C with 5% CO2. Certain samples were pretreated with PMX205 (10 μm; synthesised as previously described 29 ) or eculizumab (as above) for 30 min at 37°C with 5% CO2. Due to the limitations of working within a biosafety level 3 facility, we were unable to include inhibitor‐only controls in our experiments.
ELISA
A C5a ELISA (R&D, Minneapolis, MN, USA; #DY2037) was performed on plasma samples from whole blood inoculated with SARS‐CoV‐2 for 30 min and 24 h as per the manufacturer's guidelines.
Flow cytometry
Whole blood inoculated with SARS‐CoV‐2 was blocked with 5 μL of TruStain (Biolegend, San Diego, CA, USA; #422302) for 10 min at room temperature and then stained for granulocyte and monocyte markers, complement receptors and viability for 15 min at room temperature. The specific reagents included CD14‐PerCP‐Cy5.5 (Biolegend; #301824), CD16 APC‐Cy7 (Biolegend; #302018), HLA‐DR BV785 (Biolegend; #307642), CD88 (C5aR1) PE‐Cy7 (Biolegend; #344308), CD45 BV605 (Biolegend; #368524), CD66b FITC (Biolegend; #305104), CD11b AF700 (Biolegend; #301356) and Zombie Aqua (Biolegend; #423102). Samples were then fixed and lysed with 2 mL of BD FACSLyse (BD, Franklin Lakes, NJ, USA; #349202) for 15 min at room temperature and inverted 10 times at the 0‐ and 7.5‐min marks. Lysed samples were then centrifuged at 600 g for 5 min at room temperature. Leukocytes were then resuspended in PBS for flow cytometry acquisition on a BD LSR Fortessa II. Data analysis was performed with FlowJo v10.6.2.
SARS‐CoV‐2 serology testing
Trimeric SARS‐CoV‐2 spike protein was coated at 2 μg mL−1 on an ELISA plate overnight. 30 Plates were blocked for 1 h at room temperature with a blocking buffer (PBS containing 0.05% Tween‐20 and milk sera diluent/blocking solution—Seracare, Milford, CT, USA). Plasma from the whole blood used for the C5a ELISA study, a positive plasma control NIBSC 20/130 and a pre‐COVID‐19 serum‐negative control were serially diluted in blocking buffer and added to the plate for 1 h at 37°C. Plates were washed and probed by goat anti‐human HRP antibody (1:2500) in blocking buffer for 1 h at 37°C. Tetramethylbenzidine substrate solution and sulfuric acid stop solution were then added prior to absorbance analysis. NIBSC 20/130A is a human covalence serum obtained from the National Institute for Biological Standards and Control (URL: https://www.nibsc.org/documents/ifu/20‐130.pdf).
Statistical analysis
Statistical analysis was performed with GraphPad Prism Software v9.0.2 using paired or unpaired one‐way ANOVAs with Mann–Whitney U‐tests or t‐tests.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Martin W Lo: Conceptualization; formal analysis; investigation; methodology; project administration; visualization; writing – original draft. Alberto A Amarilla: Conceptualization; investigation; methodology; writing – review and editing. John D Lee: Validation; writing – review and editing. Eduardo A Albornoz: Project administration; writing – review and editing. Naphak Modhiran: Investigation; writing – review and editing. Richard J Clark: Resources. Vito Ferro: Resources; supervision. Mohit Chhabra: Resources. Alexander A Khromykh: Funding acquisition; project administration; writing – review and editing. Daniel Watterson: Funding acquisition; methodology; project administration; resources; supervision; writing – review and editing. Trent M Woodruff: Conceptualization; formal analysis; funding acquisition; methodology; project administration; resources; supervision; visualization; writing – review and editing.
Supporting information
Supplementary figures 1–3
Supplementary table 1
Acknowledgments
We thank the Queensland Health Forensic and Scientific Services, Queensland Department of Health, for providing the QLD02 SARS‐CoV‐2 isolate. We acknowledge funding support from the National Health and Medical Research Council (APP2009957 to TMW), Queensland Department of State Development, Tourism and Innovation (AQIRF110‐2020‐CV Industry Research Fellowship to JDL), The Australian Infectious Diseases Research Centre (COVID‐19 seed grant to AAK) and the Medical Research Future Fund (APP1202445‐2020 MRFF Novel Coronavirus Vaccine Development Grant).
Contributor Information
Daniel Watterson, Email: d.watterson@uq.edu.au.
Trent M Woodruff, Email: t.woodruff@uq.edu.au.
References
- 1. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis 2020; 20: 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taefehshokr N, Taefehshokr S, Hemmat N, Heit B. Covid‐19: perspectives on innate immune evasion. Front Immunol 2020; 11: 580641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hadjadj J, Yatim N, Barnabei L et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 2020; 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berlin DA, Gulick RM, Martinez FJ. Severe Covid‐19. N Engl J Med 2020; 383: 2451–2460. [DOI] [PubMed] [Google Scholar]
- 5. Lo MW, Kemper C, Woodruff TM. COVID‐19: complement, coagulation, and collateral damage. J Immunol 2020; 205: 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woodruff TM, Shukla AK. The complement C5a‐C5aR1 GPCR axis in COVID‐19 therapeutics. Trends Immunol 2020; 41: 965–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Satyam A, Tsokos MG, Brook OR, Hecht JL, Moulton VR, Tsokos GC. Activation of classical and alternative complement pathways in the pathogenesis of lung injury in COVID‐19. Clin Immunol 2021; 226: 108716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma L, Sahu SK, Cano M et al. Increased complement activation is a distinctive feature of severe SARS‐CoV‐2 infection. Sci Immunol 2021; 6: eabh2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta R, Gant VA, Williams B, Enver T. Increased complement receptor‐3 levels in monocytes and granulocytes distinguish COVID‐19 patients with pneumonia from those with mild symptoms. Int J Infect Dis 2020; 99: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herrmann JB, Muenstermann M, Strobel L et al. Complement C5a receptor 1 exacerbates the pathophysiology of N. meningitidis sepsis and is a potential target for disease treatment. mBio 2018; 9: e01755‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan B, Freiwald T, Chauss D et al. SARS‐CoV‐2 drives JAK1/2‐dependent local complement hyperactivation. Sci Immunol 2021; 6: eabg0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Valenti L, Griffini S, Lamorte G et al. Chromosome 3 cluster rs11385942 variant links complement activation with severe COVID‐19. J Autoimmun 2021; 117: 102595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eriksson O, Hultström M, Persson B et al. Mannose‐binding lectin is associated with thrombosis and coagulopathy in critically ill COVID‐19 patients. J Thromb Haemost 2020; 120: 1720–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramlall V, Thangaraj PM, Meydan C et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS‐CoV‐2 infection. Nat Med 2020; 26: 1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarlhelt I, Nielsen SK, Jahn CXH et al. SARS‐CoV‐2 antibodies mediate complement and cellular driven inflammation. Front Immunol 2021; 12: 767981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Defendi F, Leroy C, Epaulard O et al. Complement alternative and mannose‐binding lectin pathway activation is associated with COVID‐19 mortality. Front Immunol 2021; 12: 742446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS‐CoV‐2 spike proteins is blocked by factor D inhibition. Blood 2020; 136: 2080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ali YM, Ferrari M, Lynch NJ et al. Lectin pathway mediates complement activation by SARS‐CoV‐2 proteins. Front Immunol 2021; 12: 714511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mollnes TE, Storm BS, Brekke OL, Nilsson PH, Lambris JD. Application of the C3 inhibitor compstatin in a human whole blood model designed for complement research – 20 years of experience and future perspectives. Semin Immunol 2022; 12: 101604. [DOI] [PubMed] [Google Scholar]
- 20. Zheng S, Fan J, Yu F et al. Viral load dynamics and disease severity in patients infected with SARS‐CoV‐2 in Zhejiang province, China, January‐March 2020: retrospective cohort study. BMJ 2020; 369: m1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsukagoshi H, Shinoda D, Saito M et al. Relationships between viral load and the clinical course of COVID‐19. Viruses 2021; 13: 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loeven MA, Rops AL, Berden JH, Daha MR, Rabelink TJ, van der Vlag J. The role of heparan sulfate as determining pathogenic factor in complement factor H‐associated diseases. Mol Immunol 2015; 63: 203–208. [DOI] [PubMed] [Google Scholar]
- 23. Clausen TM, Sandoval DR, Spliid CB et al. SARS‐CoV‐2 infection depends on cellular heparan sulfate and ACE2. Cell 2020; 183: 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011; 3: a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng Y, Zhao J, Li J et al. SARS‐CoV‐2 spike protein causes blood coagulation and thrombosis by competitive binding to heparan sulfate. Int J Biol Macromol 2021; 193: 1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amarilla AA, Modhiran N, Setoh YX et al. An optimized high‐throughput Immuno‐plaque assay for SARS‐CoV‐2. Front Microbiol 2021; 12: 625136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kocsis A, Kekesi KA, Szasz R et al. Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose‐binding lectin‐associated serine protease (MASP)‐1 and ‐2: significant contribution of MASP‐1 to lectin pathway activation. J Immunol 2010; 185: 4169–4178. [DOI] [PubMed] [Google Scholar]
- 28. Chhabra M, Wimmer N, He QQ, Ferro V. Development of improved synthetic routes to Pixatimod (PG545), a sulfated oligosaccharide‐steroid conjugate. Bioconjug Chem 2021; 32: 2420–2431. [DOI] [PubMed] [Google Scholar]
- 29. Li XX, Lee JD, Massey NL et al. Pharmacological characterisation of small molecule C5aR1 inhibitors in human cells reveals biased activities for signalling and function. Biochem Pharmacol 2020; 180: 114156. [DOI] [PubMed] [Google Scholar]
- 30. Watterson D, Wijesundara DK, Modhiran N et al. Preclinical development of a molecular clamp‐stabilised subunit vaccine for severe acute respiratory syndrome coronavirus 2. Clin Transl Immunology 2021; 10: e1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–3
Supplementary table 1