

Abstract
The strain 68–1 rhesus cytomegalovirus (RhCMV) based vaccine for simian immunodeficiency virus (SIV) can stringently protect rhesus macaques (RMs) from SIV challenge by arresting viral replication early in primary infection. This vaccine elicits unconventional SIV-specific CD8+ T cells that recognize epitopes presented by major histocompatibility complex (MHC)-II and MHC-E instead of MHC-Ia. Although RhCMV/SIV vaccines based on strains that only elicit MHC-II and/or MHC-Ia restricted CD8+ T cells do not protect against SIV, it remains unclear whether MHC-E-restricted T cells are directly responsible for protection and whether these responses can be separated from the MHC-II-restricted component. Using host microRNA (miR)-mediated vector tropism restriction, we show that the priming of MHC-II and MHC-E epitope-targeted responses depended on vector infection of different, non-overlapping cell types in rhesus macaques (RMs). Selective inhibition of RhCMV infection in myeloid cells with miR-142-mediated tropism restriction eliminated MHC-E epitope-targeted CD8+ T cell priming, yielding an exclusively MHC-II epitope-targeted response. Inhibition with the endothelial cell-selective miR-126 eliminated MHC-II epitope-targeted CD8+ T cell priming, yielding an exclusively MHC-E epitope-targeted response. Dual miR-142+miR-126-mediated tropism restriction reverted CD8+ T cell responses back to conventional MHC-Ia epitope targeting. Although the magnitude and differentiation state of these CD8+ T cell responses were generally similar, only the vectors programmed to elicit MHC-E-restricted CD8+ T cell responses provided protection against SIV challenge, directly demonstrating the essential role of these responses in RhCMV/SIV vaccine efficacy.
One Sentence Summary:
RhCMV/SIV vector tropism modulation differentially programs CD8+ T cell response MHC restriction and vaccine efficacy
INTRODUCTION
SIV vaccines based on strain 68–1 RhCMV vectors demonstrate a pattern of efficacy in which 50–60% of vaccinated RMs exhibit early, complete replication arrest and subsequent clearance of initially established infection with highly pathogenic SIVmac239 (1–4). This “control and clear” efficacy has not been observed with any other vaccine modality, suggesting that it arises from specific characteristics of 68–1 RhCMV/SIV vector-elicited immune responses. 68–1 RhCMV/SIV vectors elicit and indefinitely maintain robust SIV insert-specific T cell responses that, in contrast to other vaccine modalities, include a very high proportion of circulating and tissue-based effector-memory CD8+ T cells (TEM) capable of mediating early, effective interception of nascent SIV infection without anamnestic expansion (1–3, 5, 6). In addition, CD8+ T cells elicited by 68–1 RhCMV/SIV vectors target epitopes presented by MHC-II or MHC-E, not MHC-Ia, and include recognition of universal epitopes termed supertopes (7, 8).
While the elicitation of high frequency TEM-biased CD8+ T cell responses is a characteristic of all tested primate CMVs, including circulating (wildtype) strains (4, 9), the unconventional CD8+ T cell epitope targeting manifested by strain 68–1 elicited CD8+ T cells is not (4, 7, 8). This unusual CD8+ T cell priming results from chance genetic changes in the 68–1 RhCMV genome, specifically the deletion or functional inactivation of 8 viral genes in two non-contiguous regions: Rh157.5/Rh157.4 and Rh158-161, orthologous to HCMV UL128-130 and UL146-147, respectively (4). Differential repair of these genes in 68–1 RhCMV leads to either of two distinct CD8+ T cell response types: 1) MHC-Ia-restricted-only, or 2) a mix of MHC-Ia- and MHC-II-restricted CD8+ T cells (table S1). Thus, these 8 gene products either inhibit MHC-E-restricted CD8+ T cell priming or both MHC-E- and MHC-II-restricted CD8+ T cell priming, revealing that an intrinsic capacity of RhCMV to prime an unconventionally restricted CD8+ T cell response is redundantly inhibited in the wildtype virus by their combined activity (4).
The priming of MHC-E-restricted CD8+ T cells requires MHC-E upregulation via the VL9 peptide ligand contained within the RhCMV Rh67 gene product (UL40 in HCMV) (10). Deletion or functional mutation of this viral VL9 sequence results in RhCMV/SIV vectors that exclusively elicit MHC-II-restricted CD8+ T cells. The requirement for Rh67 in MHC-E-restricted CD8+ T cell priming by 68–1 RhCMV/SIV vectors also suggests that this priming requires direct Ag presentation from vector-infected cells. The RhCMV/SIV vectors that manifest MHC-II- and/or MHC-Ia-restricted CD8+ T cell responses do not mediate protection against SIV challenge (table S1), despite the demonstration that their magnitude and phenotype is similar to that of 68–1 RhCMV-elicited responses (4). These findings implicate, but do not directly demonstrate, MHC-E-restricted CD8+ T cell responses as the adaptive immune mediator of SIV replication arrest.
These data highlight the importance of understanding the cell types and mechanisms underlying MHC-E-restricted CD8+ T cell response priming in RMs to better define the requirements for eliciting these efficacy-associated responses in humans. Given the apparent role of direct Ag presentation in MHC-E-restricted CD8+ T cell priming, we hypothesized that CMV infection of specific cell types might determine the elicitation of these responses. Primate CMVs take advantage of diverse cell niches for specific viral activities, including viral amplification (fibroblasts and related mesenchymal cells), viral dissemination (myeloid-derived cells such as monocyte/macrophages and dendritic cells), latency/persistence (myeloid cells and endothelial cells), and viral shedding/transmission (epithelial cells) (11–13). Since these targeted cell types likely differ in their ability to support T cell priming via direct antigen presentation and/or response amplification after viral infection, we sought to determine whether engineering tropism restriction into RhCMV/SIV vectors using cell type-specific host miRs as developed by Barnes et al. (14) might differentially modify CD8+ T cell epitope targeting characteristics.
The 68–1-derived vector 68–1.2 is repaired for both Rh157.5 and Rh157.4 and thus elicits MHC-Ia-restricted T cells (table S1). Incorporation of miR-142 target sequences into essential genes of this vector (Rh108 and Rh156/IE2, orthologs to HCMV UL79 and UL122, respectively) selectively blocks the vector’s ability to infect miR-142-expressing myeloid-derived cells in vitro (4). This tropism restriction dramatically changes the epitope targeting of 68–1.2 vector-elicited CD8+ T cells from 100% conventional MHC-Ia-restricted epitopes to 88% MHC-II-restricted and only 12% MHC-Ia-restricted epitopes (4). Productive infection of non-myeloid derived cells thus seems to favor RhCMV vector priming of MHC-II-restricted CD8+ T cells. However, this observation did not identify the required cell type for this priming or suggest the extent to which tropism affects the priming of MHC-E-restricted CD8+ T cell responses.
Here, we used the miR-restriction approach to limit the viral tropism of 68–1 RhCMV to myeloid cells (miR-142), epithelial cells (miR-205) or endothelial cells (miR-126). This approach revealed that the priming of the MHC-E or MHC-II epitope-targeted CD8+ T cells is dependent on infection of myeloid cells or endothelial cells, respectively. Moreover, tropism-restriction by both miR-142 and miR-126 reverted CD8+ T cell priming back to conventional MHC-Ia-restriction. Epithelial cell restriction had no effect on CD8+ T cell response programming but did abrogate vector shedding in the urine. We further demonstrate that of the tropism-restricted vectors, only the miR-126-restricted RhCMV/SIV vectors expressing either conventional SIV inserts or a supertope-focused SIV insert provided replication arrest-type protection against SIV challenge. These data indicate the critical involvement of RhCMV’s broad viral tropism in unconventional CD8+ T cell response priming, and directly demonstrate the dependence of 68–1 RhCMV/SIV efficacy on MHC-E-restricted CD8+ T cell responses using a tropism-programmed “MHC-E-only” CD8+ T cell vaccine.
RESULTS
Non-fibroblast infection by RhCMV in the absence of a functional pentameric complex
One of the two key genetic changes in 68–1 RhCMV that endows this virus the ability to elicit unconventionally restricted CD8+ T cell responses is the absence of functional Rh157.5 and Rh157.4 expression (4). The products of these two genes contribute 2 of 5 components of the pentameric receptor complex, which is a secondary CMV entry receptor needed for efficient in vitro infection of non-fibroblast cell types, including monocyte/macrophages, endothelial cells, and epithelial cells (11, 15, 16). However, previous work has suggested that 68–1 RhCMV can infect macrophages in vivo, despite the absence of the pentameric receptor complex (17). To confirm this finding and extend it to endothelial cells, we used multiplex RNAscope and immunofluorescence analysis on spleen tissues of 6 RhCMV seronegative RMs obtained 14 days after infection with either genetically full-length (wildtype-like) RhCMV (pentameric complex-intact; n = 3) or the same virus with a 68–1-like Rh157.5 + Rh157.4- and Rh158–161-deleted genetic configuration (pentameric complex-null; n = 3) (18). Although the attenuated Rh157.5 + Rh157.4- and Rh158–161-deleted RhCMV showed considerably less overall spread compared to full length RhCMV, this vector was able to infect macrophages and endothelial cells at the same proportion of overall infected cells as the full-length virus (fig. S1). Thus, pentameric complex-null RhCMV can productively infect both myeloid-derived and endothelial cells in vivo, leaving open the possibility that either or both cell types might contribute to the priming of unconventional CD8+ T cell responses by the 68–1 RhCMV/SIV vector.
Effect of myeloid, endothelial and epithelial tropism restriction on 68–1 RhCMV/SIV vector elicitation of unconventional CD8+ T cell responses
As a first step in determining whether miR-mediated tropism restriction can alter the priming of unconventional CD8+ T cell responses by 68–1 RhCMV, we constructed and validated a 68–1 RhCMV/SIVgag vector with the same miR-142-restricting elements previously used in the 68–1.2 vector backbone (4) (fig. S2). After vaccinating RMs with this construct, flow cytometric intracellular cytokine staining (ICS) assay was used to measure the vector-elicited CD8+ T cell response to 1) a consecutive overlapping SIV Gag 15mer peptide mix (overall response), 2) individual MHC-II and MHC-E SIV Gag supertope peptides, and 3) each individual peptide in the overall SIV Gag 15mer mix, with the MHC restriction of these individual peptide responses determined by MHC-specific blocking analysis, as previously described (4, 8). In contrast to the responses elicited by 68–1 RhCMV/SIVgag (4, 8), the robust SIV Gag-specific responses induced by miR-142-restricted 68–1 RhCMV/SIVgag were entirely MHC-II-restricted (Fig. 1A,B; table S2), indicating that the miR-142-mediated modulation of 68–1 vector tropism (inhibition of myeloid cell infection) completely abrogated MHC-E-restricted CD8+ T cell priming. This immunotype mimics that of Rh67-deleted 68–1 RhCMV/SIV vectors that lack intracellular MHC-E transport due to loss of virus-provided VL9 peptide ligand (10), and together these observations strongly suggest that priming of the 68–1 RhCMV/SIV vector-elicited MHC-E-restricted CD8+ T cell response depends on myeloid cell infection, most likely due to direct Ag presentation by 68–1 RhCMV vector-infected myeloid-derived cells.
Figure 1. Differential CD8+ T cell immunogenicity of single miR-restricted RhCMV/SIV vectors.
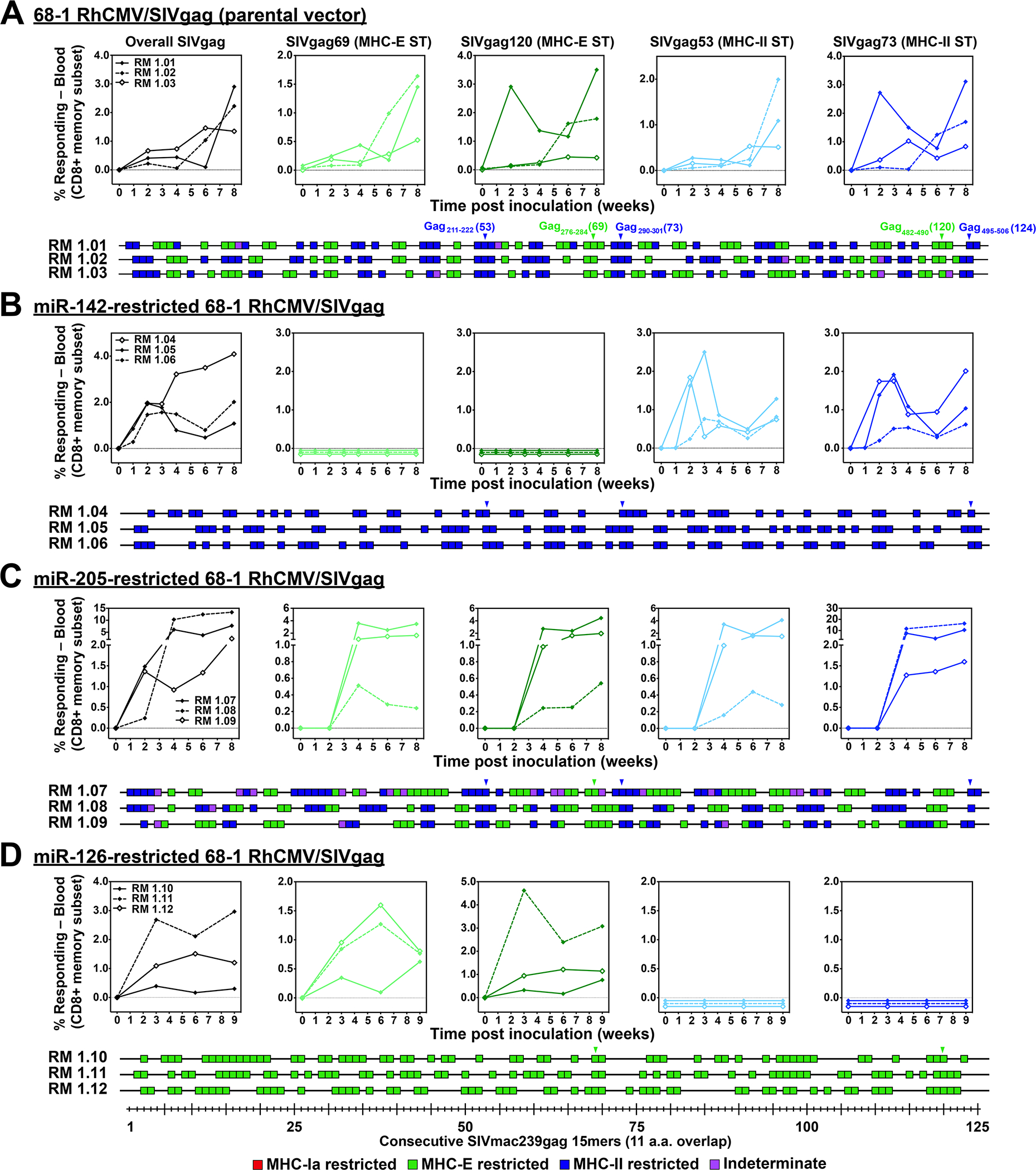
(A-D) Comparison of SIV Gag-specific CD8+ T cell responses elicited by the parental 68–1 RhCMV/SIVgag vector (A) with those of 68–1 RhCMV/SIVgag vectors with miR-142-, miR-205-, and miR-126-mediated tropism restriction (B-D). Peripheral blood CD8+ T cells from 3 representative RMs inoculated with each of these vectors were assessed by flow cytometric ICS assay (TNF-α and/or IFN-γ readout) for responses to 1) a mixture of 125 consecutive 15mer peptides comprising the overall SIV Gag protein sequence (black; top left panels), 2) individual MHC-E (green, top middle panels) and MHC-II-restricted (blue, top right panels) SIV Gag supertopes (STs; MHC-E: Gag69 = Gag276–284, Gag120 = Gag482–490 and MHC-II: Gag53 = Gag211–222, Gag73 = Gag290–301), and 3) each of 125 consecutive 15mer SIV Gag peptides with any above threshold (≥ 0.05% after background subtraction) responses indicated by a box (lower panels). Boxes are colored to reflect MHC restriction based on the ability to inhibit the response with the MHC-E blocking peptide VL9, the MHC-II blocking mAb G46.6, and/or the pan-MHC-I blocking mAb W6/32 (see Methods). Overall analysis of 15mer peptide responses in all RMs vaccinated with these miR-restricted 68–1 RhCMV/SIV vectors are shown in table S2, with estimated epitope densities shown in table S3.
We next determined whether MHC-II-restricted CD8+ T cell responses required infection of other non-fibroblast cell types using the same cell type-selective miR-mediated tropism restriction approach. We selected two additional miRs for this analysis: miR-126 and miR-205, which are most strongly expressed by endothelial cells (vascular and lymphatic) and epithelial cells, respectively (19–23). We constructed 68–1 RhCMV/SIVgag containing targeting sites for these miRs using the same design as the miR-142-restricted vectors described above and validated the ability of cells expressing these miRs to inhibit productive infection by these vectors (figs. S2, S3). miR-205-restriction of the 68–1 vector had no effect on the epitope targeting of elicited SIV Gag-specific CD8+ T cells relative to responses induced by the parental 68–1 RhCMV/SIVgag vector (Fig. 1C). However, this miR-205-restriction did have a notable in vivo effect: in contrast to the parental 68–1 and miR-142-restricted 68–1 vectors, the miR-205-restricted 68–1 vector was not shed in the urine of vaccinated RMs (24) (fig. S4), a finding that is in keeping with a requirement for productive epithelial cell infection for viral shedding in urine. Unlike the miR-205 restriction, miR-126 restriction of 68–1 RhCMV/SIV had no effect on urine shedding (fig. S4), but this miR-126 restriction manifested a dramatic effect on the epitope targeting of elicited SIV Gag-specific CD8+ T cells, completely abrogating priming of MHC-II-restricted CD8+ T cells (Fig. 1D; table S2). This resulted in vector-elicited responses that were exclusively MHC-E-restricted, including the universal MHC-E-presented supertopes. It should be noted that with both the miR-142- and miR-126-restricted vectors, the first detectable supertope-specific responses at 2–3 weeks post vaccination were type-specific, exclusively either MHC-E- or MHC-II-restricted (Figs. 1B,D; top panels) indicating that these differential response types arise at the time of initial Ag presentation and/or early amplification of these responses, and not from differential selection at later timepoints. Thus, MHC-E- and MHC-II-restricted CD8+ T cell priming requires 68–1 RhCMV/SIV vector infection of two non-overlapping cell types defined by differential miR expression: miR-142-expressing myeloid cells for MHC-E-restricted responses and miR-126-expressing cells, likely endothelial cells, for MHC-II-restricted responses, with no discernable involvement by miR-205-expressing epithelial cells.
Effect of multiple miR-mediated tropism restrictions on 68–1 RhCMV/SIV vector elicitation of unconventional CD8+ T cell responses
We next determined the effects of multiple miR-mediated tropism restrictions in the same vector; 1) miR-142 + miR-126, 2) miR-205 + miR-126, and 3) miR-205 + miR-142 + miR-126 (fig. S5). When used in combination, miR-205 restriction of 68–1 RhCMV/SIV vectors had no detectable influence on CD8+ T cell priming but was able inhibit vector shedding in urine (Fig. 2; table S2; fig. S4). However, the combination of miR-142- + miR-126- restriction (± miR-205) resulted in vectors that had lost the ability to elicit both MHC-E-restricted and MHC-II-restricted CD8+ T cells, reverting the vector-elicited CD8+ T cell responses back to the wildtype immune configuration of entirely conventional MHC-Ia-restriction (Fig. 2; table S2). These data suggest that MHC-Ia-restricted antigen presentation is the default priming pathway when the two unconventional antigen presentation pathways are absent. The minimal estimated breadth of SIV epitopes recognized by CD8+ T cells (see Supplemental Methods) elicited by the miR-142-, miR-126-, and miR-142 + miR-126-programmed 68–1 RhCMV vectors expressing the same full length SIV inserts was similar for all response types, averaging between 6.0 and 6.9 recognized epitopes per 100 amino acids of Gag, 5’-Pol and Rev sequence (table S3).
Figure 2. Differential CD8+ T cell immunogenicity of multi-miR-restricted RhCMV/SIV vectors.
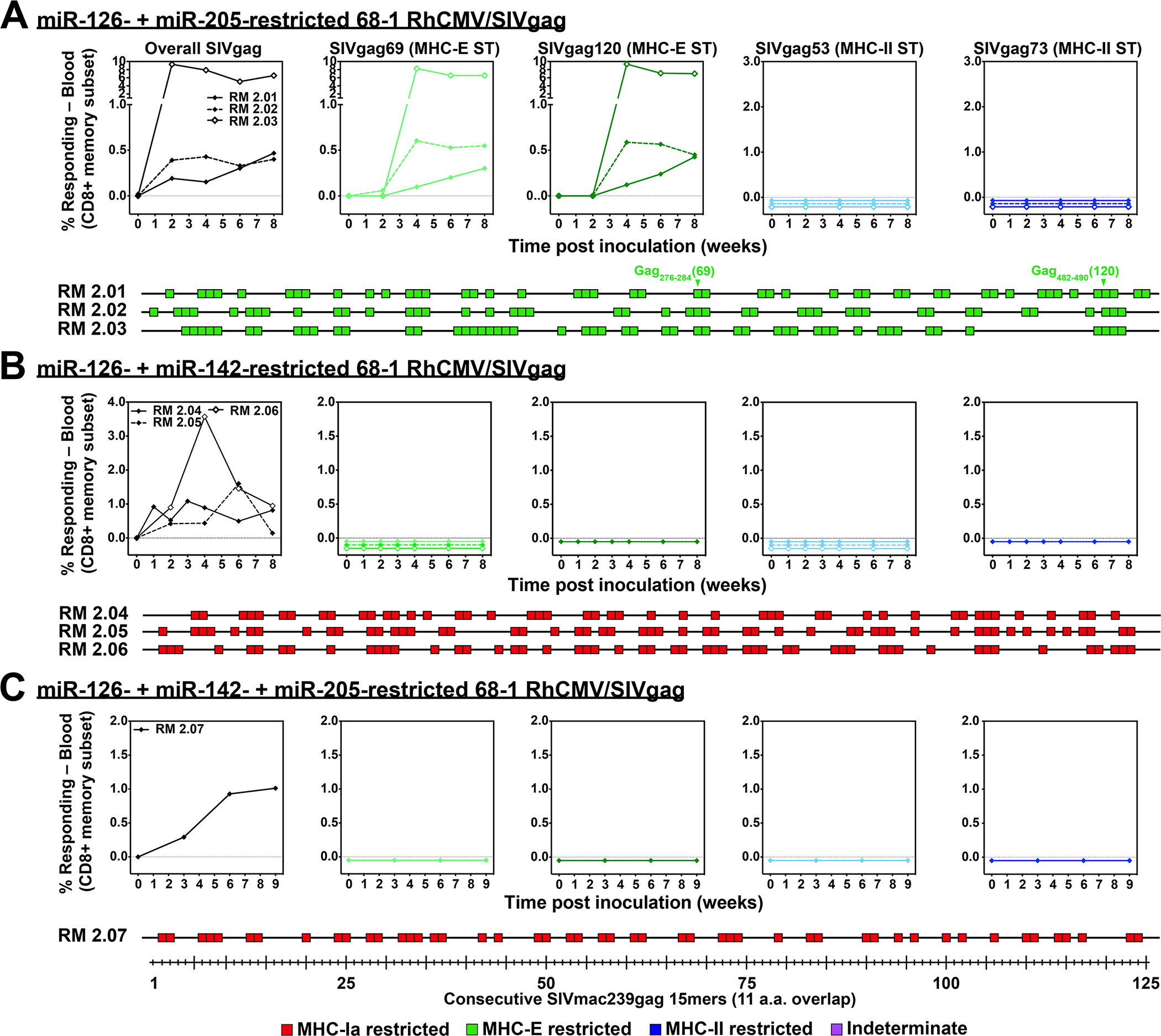
(A-C) Analysis of SIV Gag-specific CD8+ T cell responses elicited by 68–1 RhCMV/SIVgag vectors with multiple miR-mediated tropism restrictions: miR-126 + miR-205 (n = 3 RMs), miR-126 + miR-142 (n = 3 RMs), and miR-126 + miR-142 + miR-205 (n = 1 RM). Analysis was performed as described in Fig. 1. Overall analysis of 15mer peptide responses in all RMs vaccinated with these miR-restricted 68–1 RhCMV/SIV vectors are shown in table S2, with estimated epitope densities shown in table S3.
Design of miR-restricted 68–1 RhCMV/SIV vectors with universal SIV-specific CD8+ T cell responses
A unique characteristic of the unconventionally restricted CD8+ T cell responses elicited by Rh157.5 + Rh157.4- and Rh158–161-deleted RhCMV/SIV vectors is the existence of supertopes – MHC genotype-independent epitopes recognized by vaccine-elicited CD8+ T cells in all RMs [fig. S6; (4, 7, 8)]. The ability to use miR-restriction to independently program these responses to MHC-E-only or MHC-II-only restriction suggested the potential to construct a universal supertope-targeted vaccine in which elicited CD8+ T cell responses are identical with respect to MHC restriction type and epitopes targeted across all vaccinated individuals. To determine the feasibility of this concept, we engineered miR-142-restricted and miR-126-restricted 68–1 RhCMV vectors to express an SIV insert comprised of a linear arrangement of 15mer sequences encoding the MHC-II-restricted or MHC-E-restricted supertopes and, to facilitate supertope processing, the adjacent 4 amino acids on either one or both sides of the supertope 15mer (fig. S7). For these “supertope-focused” vector inserts, we included all the MHC-E or MHC-II supertopes previously identified in the SIV Gag, Retanef and 5’-Pol sequences that are used in our full length SIV insert-expressing vectors (fig. S6). Despite the considerable reduction in total encoded SIV sequence (79.4% and 83.1% reduced from corresponding full length SIV inserts for MHC-II and MHC-E supertope-focused inserts, respectively) and unnatural arrangement of the peptide segments in these inserts, these single supertope-focused miR-restricted 68–1 vectors showed robust overall CD8+ T cell responses to SIV Gag, Rev/Tat/Nef, and 5’-Pol that were not significantly different from the responses to the same SIV proteins generated by RhCMV/SIV vector sets with the conventional full length inserts (Fig. 3A,B). As anticipated, all RMs vaccinated with the supertope-focused insert containing vectors, like those vaccinated with the corresponding full length insert expressing vectors, manifested responses to the appropriate supertopes, and many of these RMs differentially manifested responses to adjacent non-supertope epitopes (subtopes) that were also expressed in the supertope-focused insert (Fig. 3C–F).
Figure 3. Analysis of RhCMV vectors with supertope-focused SIV inserts.
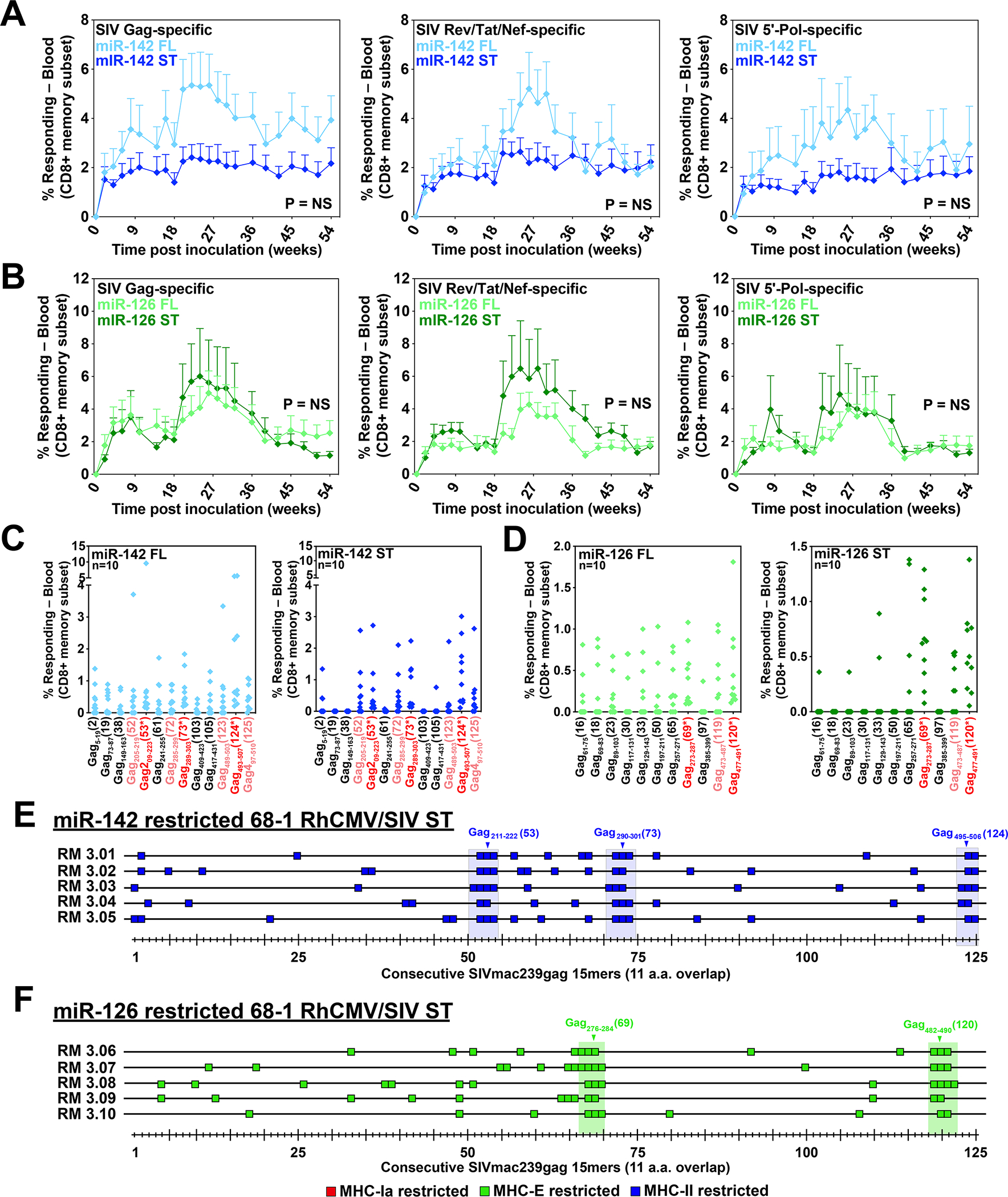
(A,B) Comparison of overall SIV Gag, Rev/Tat/Nef, and 5’-Pol CD8+ T cell responses elicited by a set of three miR-142-restricted (A) or miR-126-restricted (B) 68–1 RhCMV/SIV vectors individually expressing full length Gag, Retanef and 5’-Pol inserts versus the same responses elicited by a single miR-142-restricted or miR-126-restricted 68–1 RhCMV/SIV vector expressing either the MHC-II supertope- or the MHC-E supertope-focused insert (see fig. S7). Responses were determined by flow cytometric ICS assay using overlapping 15mer peptide mixes comprising the overall Gag, Rev/Tat/Nef, and 5’-Pol sequences, as described in Fig. 1. Two-sided Wilcoxon P-values are shown, comparing the area-under-the-curve (AUC) of response to full-length vs. supertope inserts adjusted for multiple comparisons. (C,D) Analysis of the same samples shown in panels A and B at the single 15mer epitope level, comparing responses to supertope peptides (red), adjacent non-supertope peptides in the supertope-focused insert (pink), and selected non-contiguous commonly recognized peptides (black) that are not present in the supertope-focused inserts for the both the miR-142-restricted (MHC-II-only) and miR-126-restricted (MHC-E-only) 68–1 RhCMV/SIV vectors. (E,F) Full analysis of CD8+ T cell responses to overlapping SIV Gag 15mers following vaccination with either the miR-142-restricted (E) or miR-126-restricted (F) 68–1 RhCMV/SIV vectors expressing the MHC-II- and MHC-E-supertope-focused inserts, respectively, in 5 representative RMs per vector, as described in Fig. 1. The blue and green shaded rectangles represent the 15mer peptides that are completely (15/15 amino acids) or mostly (11/15 amino acids) encoded in the MHC-II and MHC-E supertope-focused inserts, with the core supertope 15mers shown by arrowheads. Additional analyses are shown in fig. S8. ST – supertope; FL – full length.
For RMs vaccinated with both the miR-142-restricted and miR-126-restricted supertope-focused vectors, initial screening of non-supertope peptides revealed recognition of some SIV Gag peptides that were not included in the supertope-focused vector inserts (Fig. 3C,D). This prompted a more thorough analysis using consecutive 15mer peptides that confirmed reactivity of these supertope-focused insert generated CD8+ T cell responses with some SIV peptide sequences that were not expressed in the vaccine vector (Fig. 3E–F, fig. S8). Of note, these responses were not present prior to vaccination and were identified in samples from different post-vaccination time points. This finding strongly suggests that the CD8+ T cell responses elicited by the supertope-focused inserts expressing RhCMV vectors can be cross-reactive with non-expressed epitopes in the full length SIV sequences, a highly unexpected finding since extensive prior analysis of 68–1 RhCMV vectors expressing conventional protein-based inserts have never shown cross-reactivity to unrelated, non-vector expressed Ags (1, 5, 24, 25)(fig. S9). Cross-reactive MHC-II peptides were quite diverse and considerably different across different RMs. In contrast, cross-reactive MHC-E peptides were more often shared across RMs, although each RM had a unique overall pattern. There were no clear sequence or structural similarities between these cross-reactive peptides or between these cross-reactive peptides and the various supertopes (fig. S6; table S4, S5). This apparent cross-reactivity resulted in average minimal estimated SIV epitope recognition densities outside the supertope-focused inserts of 2.9 and 2.3 epitopes per 100 amino acids of SIV protein sequence for MHC-II-restricted and MHC-E restricted responses elicited by the miR-142-restricted and miR-126-restrected supertope-focused vectors, respectively (table S3). Because of this, the overall estimated minimal epitope density of CD8+ T cell responses elicited by the MHC-II and MHC-E supertope-focused expressing vectors (supertope and non-supertope regions combined) was only reduced by an average of 36% and 42% respectively, relative to the corresponding full length insert-expressing MHC-II-only and MHC-E-only vectors, respectively (table S3).
Relative efficiency of SIV-infected cell recognition by MHC-E-, MHC-II-, MHC-Ia-restricted CD8+ T cells elicited by the different tropism restricted 68–1 RhCMV/SIV vectors
The unconventionally restricted SIV-specific CD8+ T cells elicited by the parental 68–1 RhCMV/SIV vectors specifically respond to autologous SIV-infected CD4+ T cells in vitro in both an MHC-II- and MHC-E-restricted manner (7, 8). Since in vivo function of these responses probably requires SIV-infected cell recognition, we compared the ability of CD8+ T cell responses elicited by the differentially miR-programmed vectors – MHC-II-only, MHC-E-only, and MHC-Ia-only restriction (the former two also including the vectors expressing supertope-focused inserts) – to recognize and respond to autologous SIV-infected CD4+ T cells by ICS assay (fig. S10). Full length SIV insert-elicited CD8+ T cells of all three MHC-restriction types showed specific SIV-infected cell recognition with a similar distribution of response frequencies (Fig. 4A). In addition, MHC-E-restricted and MHC-II-restricted responses elicited by supertope-focused insert expressing vectors showed SIV-infected cell recognition equivalent to the corresponding responses to full length SIV inserts. The MHC-E-only and MHC-II-only restriction types of CD8+ T cell responses elicited by miR-126- and miR-142-restricted 68–1 RhCMV/SIV vectors were validated by VL9 peptide- and anti-MHC-II antibody-mediated blocking of infected cell recognition, respectively (Fig. 4B–F). The SIV-specific CD8+ T cell responses elicited by miR-126- + miR-142-restricted 68–1 RhCMV/SIV vector were blocked by a pan-anti-MHC-I mAb, but not by either of the specific MHC-E or MHC-II inhibitors, consistent with MHC-Ia restriction. Taken together, these data both confirm the MHC-E-only, MHC-II-only, and MHC-Ia-only restriction patterns of CD8+ T cell responses elicited by the different tropism-modified RhCMV vectors and demonstrate that these response types are not significantly different in their ability to mediate in vitro SIV-infected cell recognition.
Figure 4. Comparison of SIV-infected cell recognition by miR-restricted RhCMV/SIV vector-elicited CD8+ T cells.
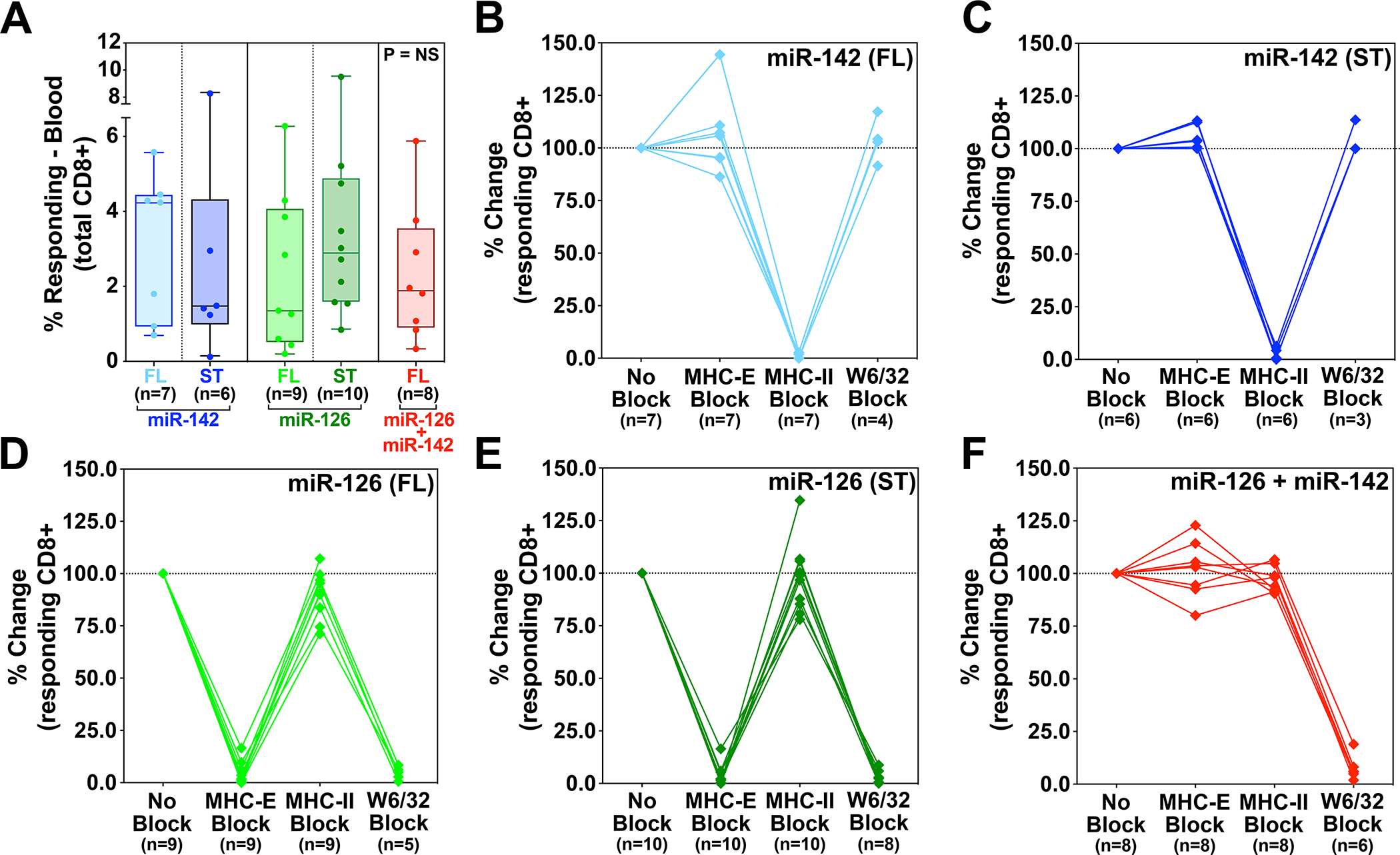
Analysis of autologous SIV-infected CD4+ T cell recognition by CD8β+ T cells isolated from RMs vaccinated twice with the miR-142-, miR-126-, or miR-142 + miR-126-restricted 68–1 RhCMV/SIV vectors, the former 2 vector types including independent analysis of both 3 vector sets expressing full length SIV Gag, Retanef, and 5’-Pol inserts and single supertope-focused vectors, whereas the miR-142 + miR-126-restricted RhCMV vector backbone was only studied with full length inserts. Plateau phase responses (>45 weeks after initial immunization) were defined by TNFα and/or IFN-γ production following peripheral blood-derived CD8β+ T cell incubation with autologous SIVmac239-infected versus mock-infected CD4+ T cells, with the MHC-restriction of the SIV-infected cell recognition determined by blocking analysis (see Fig. S10). (A) Analysis of the net SIV-infected cell-induced CD8+ T cell response frequency (subtracting mock-infected background) for RMs in each vaccine group. Kruskal-Wallis P-values comparing responses between all groups, and between groups with full length and supertope groups pooled, are shown in panel A. (B-F) Demonstration of the ability of anti-MHC-II mAb (MHC-II block), the MHC-E binding peptide VL9 (MHC-E block), or the pan-anti-MHC-I mAb (MHC-Ia and MHC-E block) to differentially inhibit SIV-infected cell recognition by CD8+ T cells in the different vaccine groups, with responses in the presence of blocking agents normalized to the unblocked response. Note that pan MHC-I blocking was not performed on all RMs due to cell number limitations. ST – supertope; FL – full length.
T cell immunogenicity analysis of differentially tropism-programmed 68–1 RhCMV/SIV vectors
The ability to use miR-mediated tropism restriction to program 68–1 RhCMV/SIV vectors to elicit MHC-E-only, MHC-II-only, and MHC-Ia-only CD8+ T cell responses provides the opportunity to: 1) determine the impact of vector tropism-restriction and CD8+ T cell epitope restriction type on quantitative and/or qualitative aspects of long-term response immunogenicity, 2) directly assess the linkage between MHC-E-restricted responses and efficacy, 3) determine whether vectors eliciting a uniform MHC-E-restricted response type manifest improved efficacy over the parental 68–1, and 4) explore, in the setting of single response-type vectors, the extent to which quantitative and/or qualitative aspects of SIV-specific immunogenicity influence protection. In addition, for MHC-E-only and MHC-II-only vectors, we sought to compare the immunogenicity and efficacy of 3 vector sets with vectors individually expressing standard (full length) SIV Gag, Retanef, and 5’Pol inserts vs. single vectors with the (MHC-II or MHC-E) supertope-focused inserts. To address these objectives, we performed a vaccine challenge study to compare the T cell immunogenicity and efficacy of miR-programmed 68–1 RhCMV/SIV vaccines that elicit CD8+ T cell response of the three MHC targeting response types: MHC-II-only (miR-142-restricted; full length and supertope-focused inserts, Groups 1 and 2), MHC-E-only (miR-126-restricted; full length and supertope-focused inserts, Groups 3 and 4), and MHC-Ia-only (miR-126- + miR-142-restricted; full length inserts only, Group 5). Each of these five groups included n = 12–14 cycling female RMs, and an additional 2 cohorts (n = 9 each) of cycling female RMs vaccinated with the parental 68–1 RhCMV/SIV vector set (full length inserts only, Group 6) or left unvaccinated (Group 7) were included as bridging and challenge controls, respectively (Fig. 5A).
Figure 5. Magnitude and durability of miR-restricted RhCMV/SIV vector-elicited T cell responses.
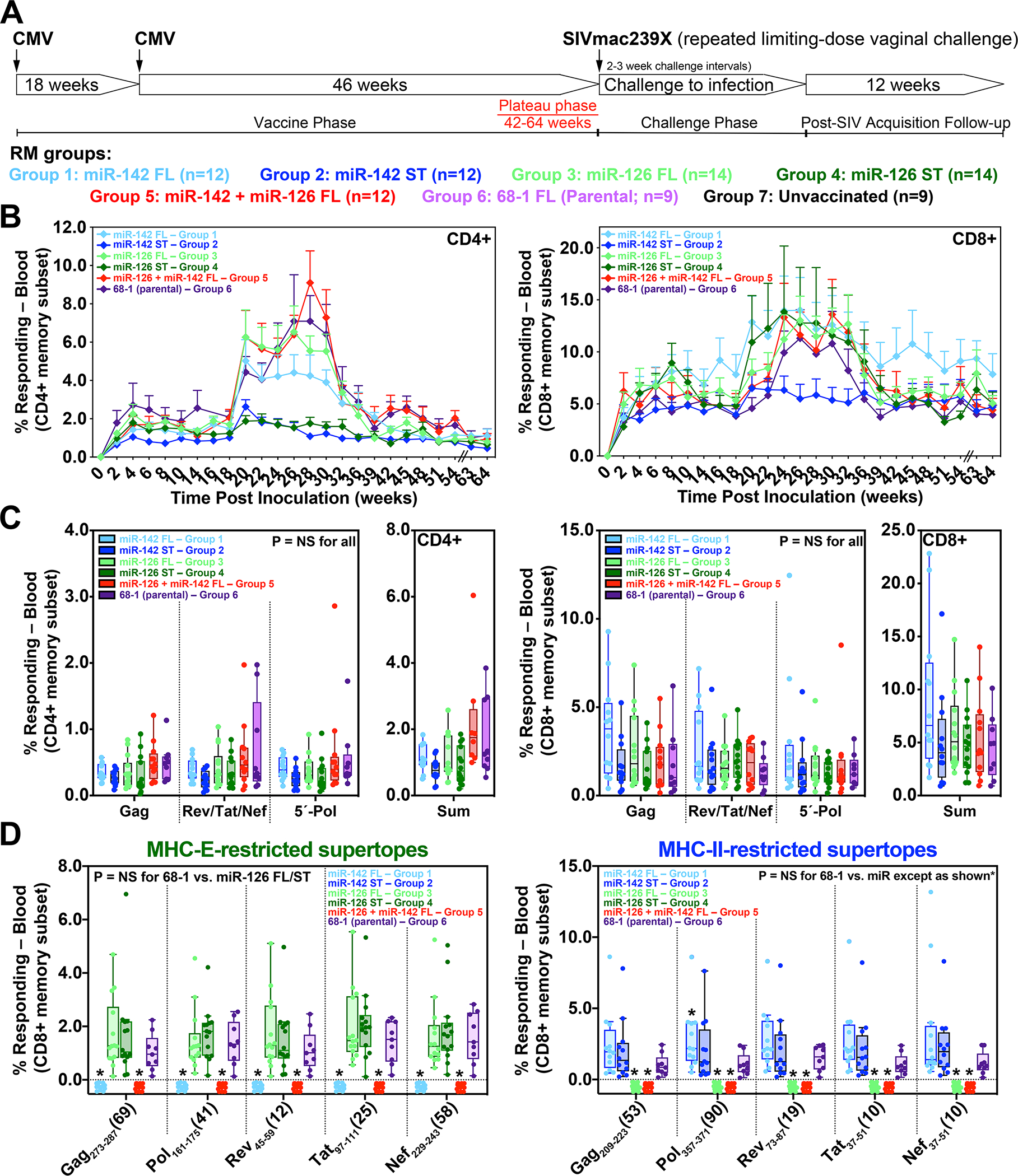
(A) Protocol for the comparison of the immunogenicity and efficacy of differentially miR-restricted 68–1 RhCMV/SIV vaccine in cycling female RMs, including vector sets composed of 3 vectors individually expressing full length SIV Gag, Retanef, and 5’-Pol inserts, and individual vectors expressing MHC-II and MHC-E supertope-focused inserts. (B,C) Longitudinal and plateau-phase analysis of the vaccine-elicited SIV Gag-, Rev/Tat/Nef-, and 5’-Pol-specific CD4+ and CD8+ T cell responses in peripheral blood of RMs vaccinated with the designated vaccines. In B, the background-subtracted frequencies of cells producing TNF-α and/or IFN-γ by flow cytometric ICS assay to overlapping 15mer peptide mixes comprising each of the SIV inserts within the memory CD4+ or CD8+ T cell subsets were summed for overall responses with the figure showing the mean (+ SEM) of these overall responses at each time point. In C, boxplots compare the individual and summed SIV insert-specific CD4+ and CD8+ T cell response frequencies between the vaccine groups during the vaccine phase plateau (each data point is the mean of response frequencies in all samples from weeks 42–64 post-first vaccination). (D) Plateau phase analysis of the vaccine-elicited CD8+ T cell responses to the designated MHC-E- and MHC-II-restricted SIV supertopes in peripheral blood of the indicated vaccine groups by ICS assay. Note that responses elicited by supertope-focused vectors were only analyzed for type-matched responses (e.g., when the indicated epitopic peptides were present in the insert). Wilcoxon rank sum testing (adjusted for multiple comparisons) was used to compare all response parameters shown in panels C and D for the parental 68–1 vaccine to all other vaccines (which are individually designated by the color code shown in panel A). Significant P-values (< 0.05) are designated by *. ST – supertope; FL – full length.
Longitudinal analysis of total SIV-specific CD4+ and CD8+ T cell responses in blood from the RM cohorts vaccinated with the miR-programmed, full length insert-expressing RhCMV/SIV vaccines revealed response profiles that were broadly similar to that of the parent full length insert-expressing 68–1 RhCMV/SIV vaccine (Fig. 5B). Given the design intent of the small, supertope-focused inserts, the miR-142- and miR-126-restricted, supertope-focused vaccines showed reduced total SIV-specific CD4+ T cell responses, primarily after the boost vaccine dose (Fig. 5B). A limited boost response was also observed for the MHC-II-restricted SIV-specific CD8+ T cell response elicited by the miR-142-restricted supertope-only vector, in contrast to a robust boost response in MHC-E-restricted SIV-specific CD8+ T cell responses to the miR-126 restricted supertope-only vector (Fig. 5B). However, since post-boost responses in blood largely return to pre-boost level over the longer term, the end-of-vaccine phase magnitudes of the overall SIV-specific CD4+ and CD8+ T cell responses elicited by all the tested vaccines were not significantly different (Fig. 5C). Similarly, the MHC-II- and MHC-E-restricted supertope-specific CD8+ T cell responses elicited by miR-142- and miR-126-restricted RhCMV/SIV vectors (both full length and supertope-focused inserts), respectively, were similar in magnitude to the corresponding responses elicited by the parental 68–1 RhCMV/SIV vectors (Fig. 5D).
Epitope restriction analysis of responses from 6 RMs in each group confirmed the expected MHC restriction patterns for all miR-restricted RhCMV/SIV vaccines (fig. S8A). For all vaccine groups, the plateau phase SIV Gag-specific CD4+ and CD8+ T cells manifested the characteristic effector-memory bias of RhCMV vector-elicited T cell responses (Fig. 6A) with cytokine synthesis profiles commensurate with this bias (for CD4s and CD8s, expression of TNF-α and MIP-1β alone and TNF-α in combination with IFN-γ, and additionally for CD8s, TNF with IFN-γ and MIP-1β, but not IL-2; Fig. 6B). The proportion of these major cytokine synthesis subsets differed slightly, but significantly, in the responses elicited by the miR-126-restricted RhCMV/SIV vectors relative to the 68–1 parental vector, particularly for the CD8+ T cell responses. The miR-126-restricted RhCMV/SIV vector elicited CD8+ T cell responses showed an increased proportion of cells producing TNF-α in combination with IFN-γ coupled with reduced proportions of cells expressing these 2 cytokines plus MIP-1β and cells producing MIP-1β alone (Fig. 6B), a difference attributable to a 38% reduction in overall MIP-1β-producing cells (95% CI: 8.1–58.5) among the SIV-responding CD8+ T cells in this vaccine group.
Figure 6. Functional differentiation of miR-restricted RhCMV/SIV vector-elicited T cell responses.
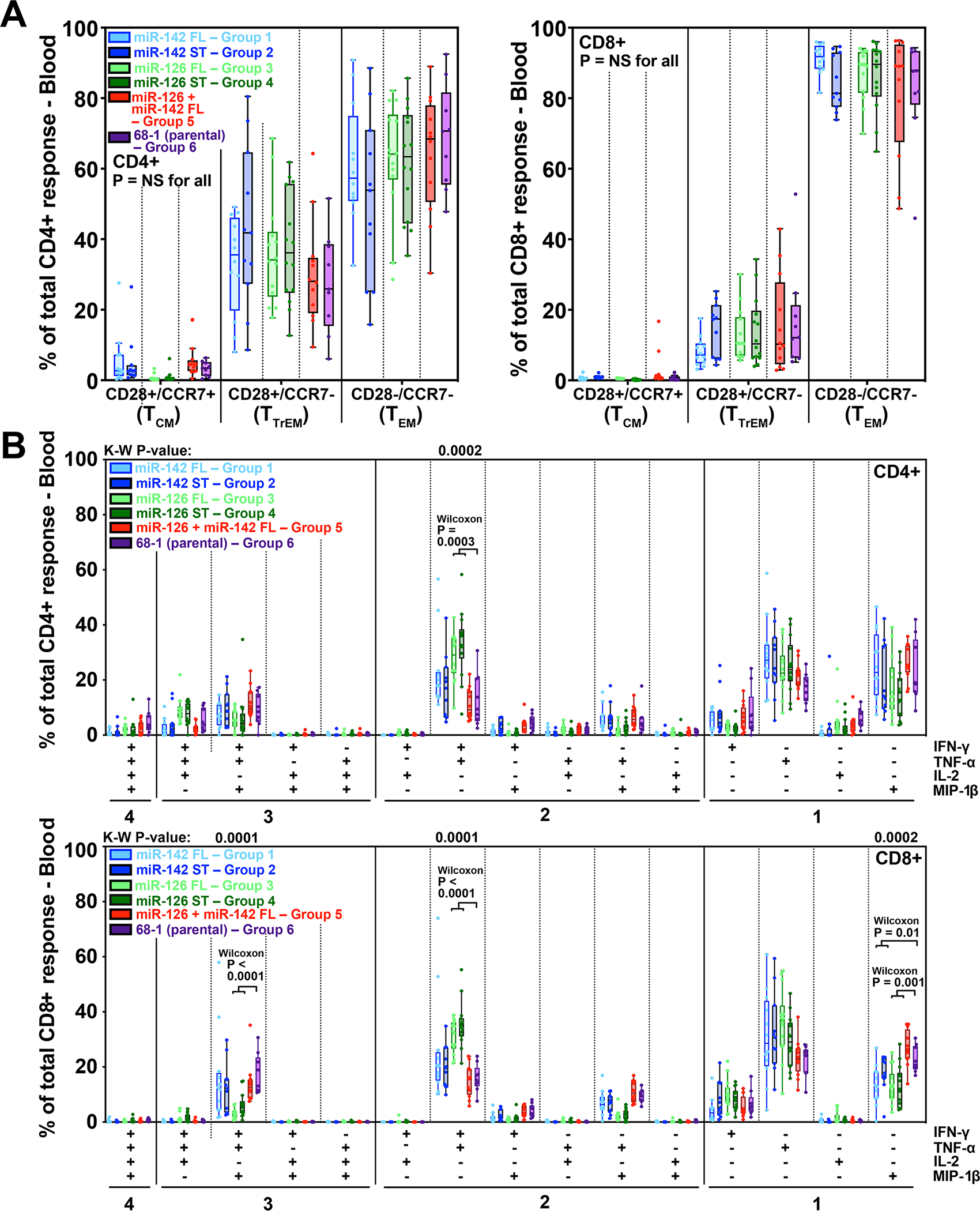
(A) Boxplots compare the memory differentiation phenotype of the vaccine-elicited CD4+ and CD8+ memory T cells in peripheral blood of the same RM cohorts reported in Fig. 5 responding to overall SIV Gag 15mer peptide mix with TNFα and/or IFN-γ production during the post-vaccination plateau phase. Memory differentiation state was based on CD28 and CCR7 expression, delineating central memory (TCM), transitional effector-memory (TTrEM), and effector-memory (TEM), as designated. Kruskal-Wallis P-values comparing response parameters between all treatment groups are shown (adjusted for multiple comparisons). (B) Boxplots compare the frequency of vaccine-elicited CD4+ and CD8+ memory T cells in peripheral blood responding to the overall SIV Gag 15mer peptide mix with TNF-α, IFN-γ, IL-2, and MIP-1β production, alone and in all combinations, in the same samples as panel A. Kruskal-Wallis (K-W) P-values comparing response parameters between all treatment groups are shown where significant (adjusted for multiple comparisons across all fifteen response categories, but only shown for the top four: IFN-γ+/TNF-α+/MIP-1β+, IFN-γ+/TNF-α+, TNF-α+, and MIP-1β+). Where K-W p-values are shown, unadjusted post-hoc Wilcoxon P-values are also shown where significant, comparing miR-142 and miR-126 treatment groups (with supertope-focused and full length insert groups pooled) to 68–1. ST – supertope; FL – full length.
Efficacy analysis of differentially tropism-programmed 68–1 RhCMV/SIV vectors
Starting 65 weeks after first vaccination, all vaccinated RM cohorts (Groups 1–6) and an unvaccinated control RM cohort (Group 7) were subjected to repeated, limiting dose, intra-vaginal SIVmac239 challenge (Fig. 5A), with challenge repeated every 2–3 weeks until take of SIV infection as demonstrated by the onset of measurable plasma viremia and/or de novo induction of SIV Vif- and Env-specific T cell responses. SIV Vif and Env were not present in the vaccine inserts and neither of these responses were detectable prior to challenge in any of the study RMs, including RMs vaccinated with supertope-focused vaccine inserts (Fig. 7; figs. S11, S12). Thus, the post-challenge development of such responses reflects SIV infection, as previously described (1–5). Once the take of SIV infection was documented, RMs with sustained viremia were designated as non-protected, whereas RMs with transient or no viremia were designated protected, with replication arrest of fully competent SIV in these protected RMs confirmed by adoptive transfer of cells, and consequently, SIV infection to naive RMs (2–4). Of the 82 RMs challenged across all groups, all but 4 (1, 2 and 1 RMs in Groups 1, 2 and 6, respectively) demonstrated SIV-infection take (as judged by the above criteria) over the course of the repeated challenge protocol and could be designated as either non-protected or protected for efficacy analysis (Fig. 7; figs. S11, S12; table S6). All unvaccinated control RMs manifested typical SIVmac239 primary infection (Fig. 7A). In contrast, 7 of 8 of RMs vaccinated with the parental 68–1 RhCMV/SIV vector set manifested protection (Fig. 7G), a higher-than-expected frequency in this small cohort than previously observed (1–4). Of the miR-restricted vectors, only the miR-126-restricted (MHC-E-only) vectors were associated with post-infection replication arrest, which was observed in 15 of 28 RMs, including 7 of 14 RMs given vectors with full length SIV inserts and 8 of 14 RMs given the vector with supertope-focused inserts (Figs. 7E,F). All RMs vaccinated with miR-142-restricted vectors (MHC-II-only; both full length and supertope-focused SIV inserts; n = 20) and with the miR-142- + miR-126-restricted vectors (MHC-Ia-only; full length SIV inserts; n = 12) showed progressive SIV infection (Fig. 7B–D), with plateau phase plasma viral loads (log mean levels at weeks 6–10 post-infection) indistinguishable from the unvaccinated controls (fig. S13). Although in previous efficacy assessments of the parental 68–1 RhCMV/SIV vaccine, vaccinated, non-protected RMs showed no difference in plateau phase plasma viremia (1–3), it is notable that in this study, the non-protected RMs given the miR-126-restricted 68–1 RhCMV/SIV vectors expressing full length SIV inserts, manifested a modest (0.58 log), but significant, reduction of log mean viral loads (fig. S13), suggesting that these responses might also have limited conventional anti-viral activity in the absence of successful replication arrest.
Figure 7. Efficacy of miR-restricted RhCMV/SIV vectors.
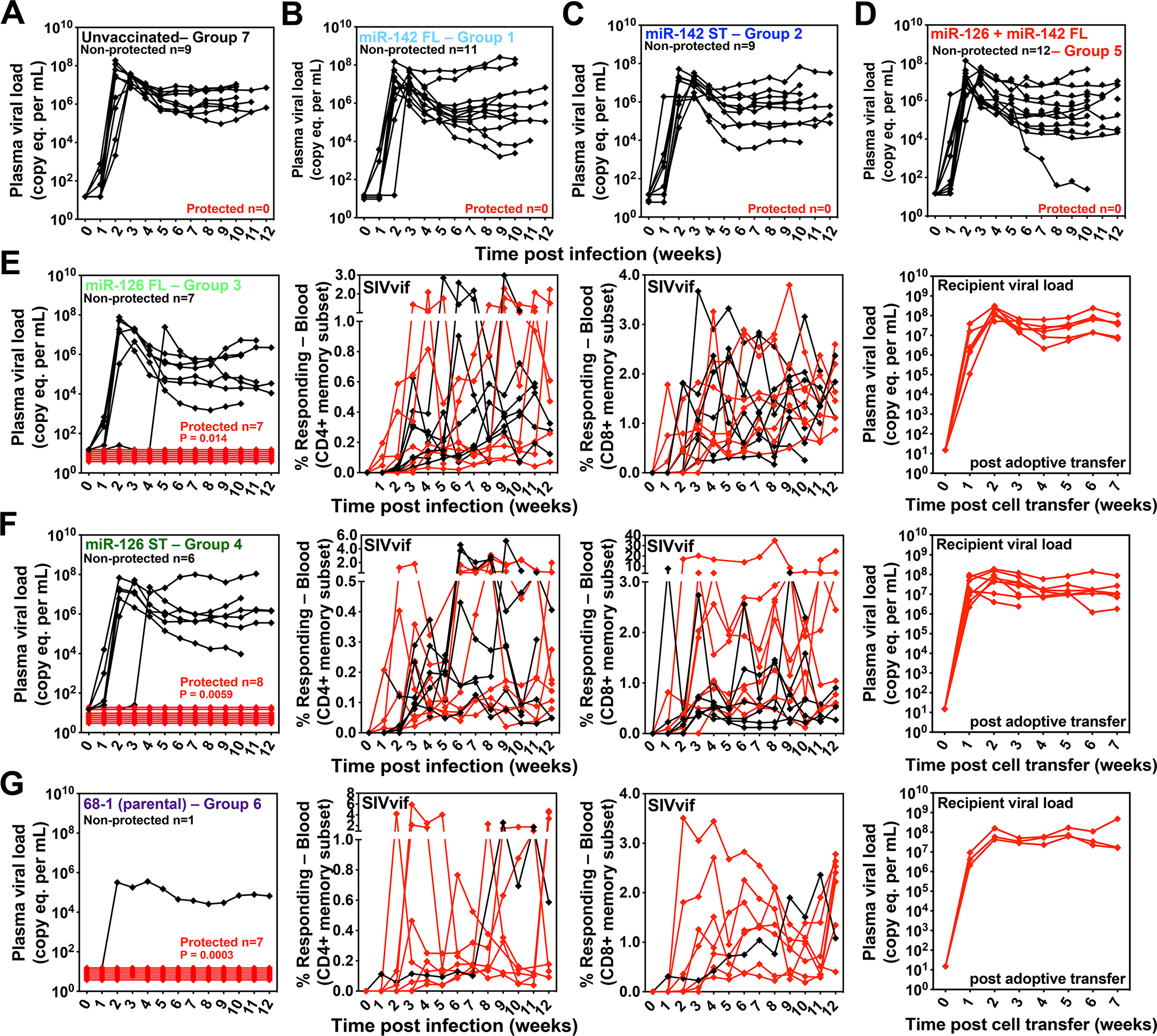
(A-G) Assessment of the outcome of SIV infection after repeated, limiting dose intra-vaginal SIVmac239 challenge of the designated vaccine groups (see Fig. 5A) by longitudinal analysis of plasma viral load (A-D) and/or de novo development of SIV Vif-specific CD4+ and CD8+ T cell responses (E-G). RMs were SIV challenged until the onset of sustained viremia and/or above-threshold SIV Vif-specific T cell responses, with the SIV dose administered 2 or 3 weeks prior to the initial response detection considered the infecting challenge (week 0). The overall n in each panel reflects the total number of RMs with such documented take of SIV infection during the challenge period. RMs with sustained viremia were considered non-protected (black); RMs with no or transient viremia but demonstrating sustained above-threshold SIV Vif-specific T cell responses were considered protected (red). Controlled SIV infection was confirmed in all 15 protected miR-126-restricted 68–1 RhCMV/SIV vector-protected RMs (both full length and supertope-focused inserts) and 3 of 7 parental 68–1 RhCMV/SIV vector-protected RMs by demonstration of sustained plasma SIV viremia in SIV-naïve recipient RMs after adoptive transfer of marrow and/or peripheral lymph node cells from the protected RMs, collected from between day 28 and 56 post-SIV infection (see table S6). Binomial exact P-values are shown where the proportion of protected RMs in a vaccine group differs significantly from the unvaccinated group. ST – supertope; FL – full length.
To confirm the ability of miR-126-restricted 68–1 RhCMV/SIV (MHC-E-only) vectors to mediate replication arrest efficacy regardless of SIV challenge route, we assembled a separate cohort of RMs for intra-rectal SIV challenge that had been vaccinated twice with either the miR-126-restricted 3-vector set expressing full length SIV inserts (n = 9) or the single miR-126-restricted vector expressing the MHC-E supertope-focused SIV insert (n =8). These RMs manifested plateau-phase SIV-specific CD4+ and CD8+ T cell responses of similar magnitude and character as the above-described intravaginally challenged RM cohorts given the same vectors, including exclusively MHC-E-restricted SIV-specific CD8+ T cell responses (Fig. 8A–D; fig. S8B). All 17 of these RMs manifested take of SIVmac239 infection via intrarectal administration with 11 (65%) manifesting typical replication arrest efficacy, including 6 of 9 RMs (67%) given vector with full length inserts and 5 of 8 RMs (63%) given vectors with the MHC-E supertope-focused insert (Fig. 8E,F; Table S6). Across both the intra-vaginally and intra-rectally challenged RM cohorts that were vaccinated with the MHC-E-only miR-126-restricted 68–1 RhCMV/SIV vaccines, there was no correlation between protection and the magnitude of the exclusively MHC-E-restricted CD8+ T cell responses in blood (fig. S14), indicating that our failure to detect such correlation in previous analyses of RMs vaccinated with the parental 68–1 RhCMV/SIV vector sets (2, 3) was not due to our inability to distinguish the presumptively non-protective MHC-II-restricted CD8+ T cell responses from the presumptively protective MHC-E-restricted CD8+ T cell responses. Putting the efficacy results of this study in the context of our previously published results with the parental 68–1 RhCMV/SIV vectors reveals an overall rate of replication arrest of 59% for both intra-rectal and intra-vaginal challenge, with no difference in replication arrest efficacy between the parental 68–1 and MHC-E-only miR-126-restricted 68–1 vectors, and among the latter vectors, no difference between full length and supertope-focused SIV inserts (Fig. 8G).
Figure 8. Efficacy validation of miR-126-restricted (MHC-E-only) RhCMV/SIV vectors.
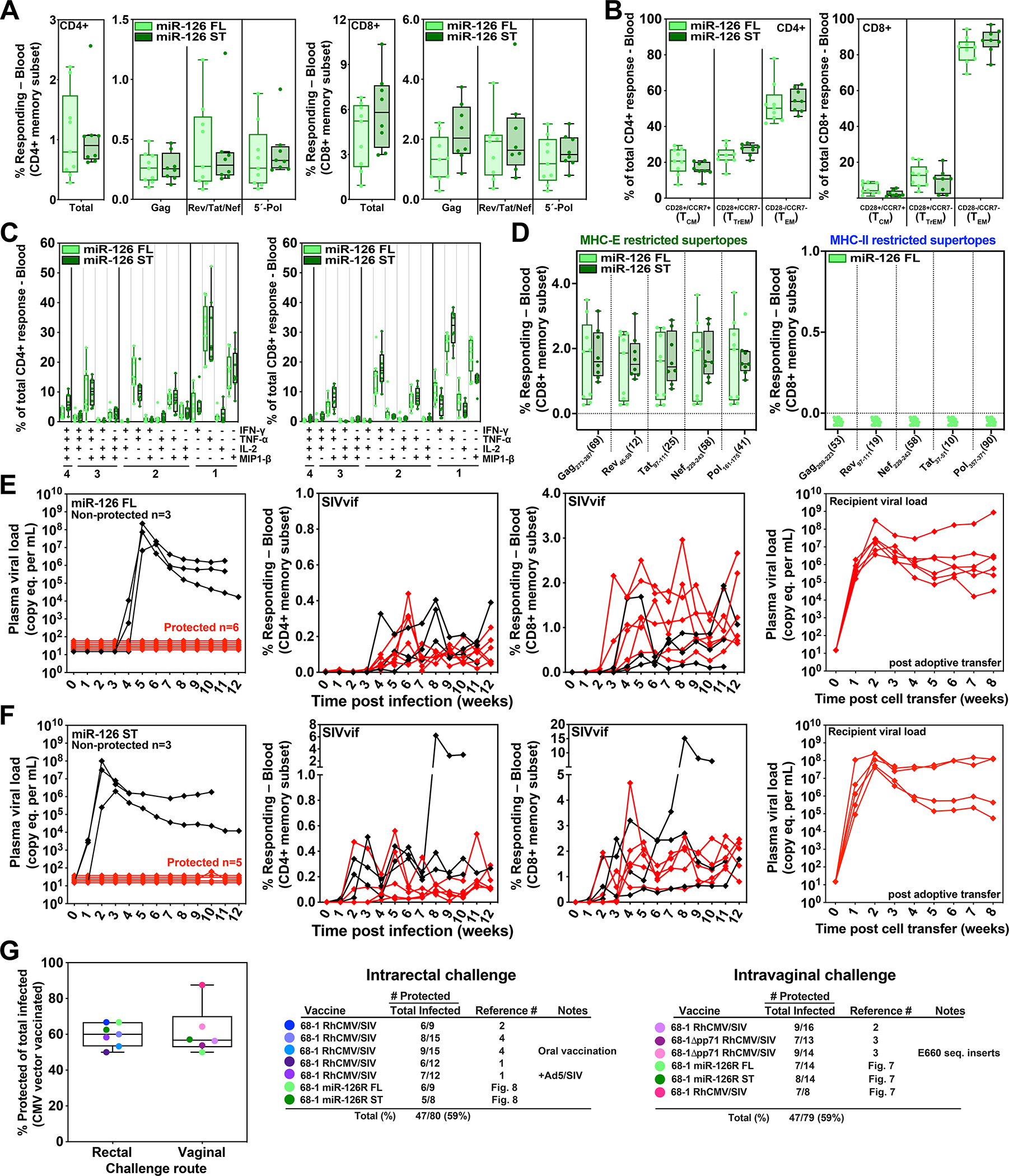
To confirm the efficacy of miR-126-restricted 68–1 RhCMV/SIV vectors after intra-rectal SIVmac239 challenge, we assembled a cohort of 17 RMs – including 9 and 8 RMs that were twice-vaccinated with the set of 3 full length insert expressing vectors or the single supertope-focused vector, respectively – for immunogenicity and efficacy analysis. (A-D) The plateau phase immunogenicity of these vectors, including response magnitude, memory phenotype and cytokine synthesis function, are shown, analyzed as described in Figs. 5 and 6. (E, F). RMs were subjected to repeated limiting dose intrarectal SIVmac239 challenge with outcome determined as described in Fig. 7. Controlled SIV infection was confirmed in all 11 miR-126-restricted 68–1 RhCMV/SIV vector-protected RMs by demonstration of sustained plasma SIV viremia in SIV-naïve recipient RMs after adoptive transfer (right panels) of cells collected from the protected RMs between day 28 and 56 post-SIV infection (see table S6). (G) Comparison summary of the efficacy of miR-126-restricted 68–1 RhCMV/SIV vectors from this study with all published cohorts of RMs vaccinated with non-miR-restricted 68–1 RhCMV/SIV-based vaccines, separated by challenge route. ST – supertope; FL – full length.
DISCUSSION
The finding that 68–1 RhCMV vectors elicit CD8+ T cell responses that exclusively target epitopes presented by either MHC-II or MHC-E demonstrates that virus-induced CD8+ T cell responses are much more complex and flexible than previously thought. This raised fundamental questions regarding the viral and host mechanisms that provide unconventionally restricted CD8+ T cell response types in the absence of any detectable conventional (MHC-Ia-restricted) responses. Since these mechanisms appear to be integral to the ability of 68–1 RhCMV/SIV vaccine-induced immune responses to mediate SIV replication arrest with subsequent viral clearance, their elucidation represents a possible path to an effective HIV vaccine. Here, we first demonstrated that the ability of 68–1 RhCMV/SIV vectors to elicit MHC-E- and MHC-II-restricted CD8+ T cell responses depends on vector infection of two non-overlapping, non-fibroblast cell types that were experimentally defined by cell type-specific miR expression. Secondly, we found that only the vaccines programmed for MHC-E-restricted CD8+ T cell responses were able to mediate replication arrest efficacy despite the observation that, with minor exceptions, all the response types manifested similar long-term response magnitudes, SIV-infected cell recognition capabilities, and functional phenotypes.
The inhibition of 68–1 RhCMV/SIV-elicited MHC-E-restricted CD8+ T cell responses by the myeloid cell-selective miR-142 reflects a requirement for vector infection of macrophages, dendritic cells, and/or their myeloid precursors for MHC-E-restricted CD8+ T cell priming, as these are the RhCMV-targeted cell types that predominantly express this miR (26–28). Given the requirement for MHC-E upregulation mediated by the VL9 peptide within the Rh67 gene product for MHC-E-restricted CD8+ T cell response generation, the most likely explanation for the response programming effect of miR-142 is that inhibition of myeloid cell infection prevents direct MHC-E-restricted Ag presentation by one or more myeloid cell types that are uniquely able to mediate this function. Although the basis of this need for myeloid cell Ag presentation remains to be determined, it is noteworthy that MHC-E expression is relatively high on monocyte/macrophages (increasing with maturation) and that this MHC-E is targeted to autophagic/lysosomal vesicles where it would encounter antigenic peptides that could be exchanged for VL9 (29, 30). In addition, the apparent requirement for myeloid-derived cell infection in this system parallels that of another MHC-E-restricted CD8+ T cell response-inducing organism, Mycobacterium tuberculosis (Mtb) (31), which infects monocyte/macrophages, and is thought to mediate MHC-E-restricted CD8+ T cell priming because of enrichment and peptide loading of MHC-E in the Mtb phagosome with subsequent recycling to the cell surface (32). However, since myeloid cells are thought to play a key role in mediating CMV dissemination (12, 13, 33, 34), it is possible that the miR-142 response programming effect is mediated by a requirement for migratory myeloid cells to disseminate infection or to transport infection to a specialized Ag-presenting cell niche. However, this possibility seems very unlikely given our previous observation that pp71-deleted 68–1 RhCMV/SIV vectors are able to efficiently generate MHC-E-restricted CD8+ T cell responses despite being highly spread-deficient in vivo [~1000-fold attenuated relative to parental 68–1; (3, 25)].
These observations suggest a preliminary model for the mechanisms underlying MHC-E-restricted CD8+ T cell priming and its dependence on myeloid-derived professional Ag presenting cells. However, given that classical MHC-II Ag processing/presentation to CD4+ T cells is mediated by exogenous Ag uptake by professional myeloid-derived Ag presenting cells, particularly dendritic cells (35), it is much more surprising that priming of the 68–1 RhCMV vector-elicited MHC-II-restricted CD8+ T cells has a tropism requirement at all, let alone a strict tropism requirement for a distinct non-myeloid cell type. Yet, our data indicate that this MHC-II-restricted CD8+ T cell priming requires productive vector infection of miR-126-expressing cells and not miR-142- or miR-205-expressing target cells. This requirement for productive vector infection of a specific cell type strongly implies that MHC-II-restricted CD8+ T cell priming by 68–1 RhCMV is, like the MHC-E-restricted CD8+ T cell priming discussed above, mediated by direct Ag presentation from vector-infected cells, just different types of vector-infected cells. Such intracellular Ag processing for direct MHC-II-peptide presentation occurs via both cytosolic and autophagosome processing (36, 37), providing plausible mechanisms for this direct MHC-II-mediated Ag presentation. In terms of the miR-126-expressing cell type mediating this direct presentation, the most likely candidates are one or more endothelial cell types as this miR is a prototypical endothelial cell-selective miR that is highly expressed by both vascular and lymphatic endothelial cells, and play a key role in the development and function of these conduits (19–22). MiR-126 can also be expressed by plasmacytoid dendritic cells and hematopoietic stem cells (38, 39), but plasmacytoid dendritic cells are not thought to be targeted by CMV (40), and both these cell types are reported to co-express miR-142 (26, 38), which, given the lack of miR-142 inhibition of MHC-II-restricted CD8+ T cell response priming, makes their primary involvement in this process unlikely. In contrast, as shown here, 68–1 RhCMV readily infects both vascular and lymphatic endothelial cells in vivo, and endothelial cells are not known be enriched for miR-142 expression (27). Moreover, endothelial cells express MHC-II in situ or readily upregulate MHC-II with inflammation and have been shown to be active participants in engaging adaptive T cell responses (41, 42). What is still unclear is why RhCMV vector-mediated MHC-II-restricted CD8+ T cell priming would be restricted to this cell type, as intrinsic MHC-II processing occurs in many cell types, including professional Ag presenting cells and non-Ag presenting cells (36). It is possible that aspects of CMV infection in endothelial cells that differ from other viral target cells contribute an essential element of this priming mechanism (43–45).
Due to highly efficient MHC-Ia downregulation in CMV-infected cells (24) and dominant infection of mesenchymal cells that are not professional Ag presenting cells, priming of conventional MHC-Ia-restricted responses by wildtype CMV has been thought to be mediated by cross-priming (46, 47). Since exogenous Ag processing and presentation is almost certainly always occurring in 68–1 RhCMV vector infection, it is not surprising that dual miR-142- and miR-126-mediated restriction of 68–1 RhCMV/SIV, which prevents direct priming of both MHC-E- and MHC-II-restricted CD8+ T cells, results in reversion to a conventional, likely cross-primed MHC-Ia-restricted CD8+ T cell response. However, it is surprising that this constitutive exogenous Ag processing and presentation does not elicit MHC-Ia-restricted CD8+ T cells along-side the MHC-E- and/or MHC-II-restricted CD8+ T cell responses, and that these MHC-Ia-restricted response only appear when the directly primed unconventional responses are inhibited. While limited data in murine models suggests that direct priming might be more potent than cross-priming (48, 49), the complete lack of (cross-primed) MHC-Ia-restricted CD8+ T cell responses in 68–1 RhCMV/SIV vaccinated RMs with unconventional responses is surprising, especially since co-administration of 68–1 + 68–1.2 RhCMV/SIV vectors, the latter programmed for MHC-Ia-restricted responses, results in a mixture of unconventionally and conventionally restricted CD8+ T cell responses (4). These observations suggest that for RhCMV vectors, the directly primed unconventional responses outcompete and/or suppress cross-presented conventional MHC-Ia-restricted priming in local microenvironments, a novel type of immunodominance that adds to the existing complexity of CD8+ T cell immunodominance mechanisms (50, 51). Indeed, because of predictable and distinct T cell responses that arise from the three separate priming mechanisms described here, the RhCMV vector platform offers a useful model to further explore these mechanisms.
Our ability to use miR-mediated tropism modulation of 68–1 RhCMV vectors to individually and exclusively engage each of three distinct CD8+ T cell priming pathways (MHC-Ia, MHC-II, and MHC-E) allowed us to program 68–1 RhCMV/SIV vaccines with respect to each CD8+ T cell epitope targeting response-type, and thereby compare the efficacy of vaccine vectors that primarily differ in CD8+ T cell response targeting. The results were definitive: only the miR-126-restricted vectors, which elicited MHC-E-restricted CD8+ T cell responses, were efficacious, showing the same replication arrest-type efficacy previously characterized for unmodified 68–1 RhCMV/SIV vectors (1–4). Taken together with our previous analysis of gene-modified 68–1 RhCMV/SIV vector efficacy (4, 10), these results conclusively link MHC-E-restricted SIV-specific CD8+ T cell responses with protection against SIV challenge via both intravaginal and intrarectal challenge routes, and strongly imply that these responses mediate the adaptive component of 68–1 RhCMV/SIV vector-mediated protection.
The level of replication arrest-type efficacy observed in RMs vaccinated with the 3-vector set of miR-126-restricted 68–1 RhCMV/SIV expressing full length SIV inserts was maintained in RMs vaccinated with a single miR-126-restricted 68–1 RhCMV/SIV vector expressing an MHC-E supertope-focused insert. Testing the supertope-focused inserts, which for the MHC-E supertope insert was reduced in length from full length inserts by 83%, was intended to assess the ability of supertope-focused responses to mediate both SIV-infected cell recognition and replication arrest-type efficacy. This testing was also intended to inform the corollary questions of whether the remarkable response breadth of full length insert expressing 68–1 RhCMV/SIV vector elicited CD8+ T cells (7, 8) is required for efficient SIV-infected cell recognition and for efficacy, or alternatively, whether the MHC-E supertope-specific responses might comprise a minimal, universal efficacious response. MHC-E supertope-focused vectors did indeed generate responses that were similarly efficient at SIV-infected cell recognition and mediating efficacy as vectors expressing corresponding full length SIV inserts, but our ability to use these data to define a minimal, universal efficacious MHC-E-restricted response was complicated by the unexpected breadth of these responses, including responses to supertope-adjacent, non-supertope epitopes in the insert and cross-reactive recognition of non-supertope epitopes outside the insert. This latter finding was entirely unexpected, as extensive previous analysis has not shown such cross-reactivity to unrelated (non-insert, non-CMV) Ags in RMs vaccinated with 68–1 RhCMV vectors (1, 5, 24, 25). We identified cross-reactivity to single Gag, Rev/Nef/Tat, and Pol peptides during epitope deconvolution analysis, but did not identify cross-reactivity with peptide mixes from the non-vector-expressed SIV Vif or Env proteins in this study.
This cross-reactivity appears to be another unusual feature of the CMV vector platform that results when 68–1 RhCMV vectors selectively express supertope-focused inserts, rather than full length viral protein coding sequences. Our current hypothesis is that the T cell receptors (TCRs) mediating these RhCMV vector-elicited unconventional MHC-E restricted CD8+ T cell responses are intrinsically cross-reactive, with SIV supertope recognition being a prototypical component of this cross-reactivity, such that the extent of cross-reactivity is increased with vectors encoding supertope-focused inserts. Since the observed cross-reactivity is highly diverse with no obvious consistent structural similarity between the targeted cross-reactive peptides, it is possible that the cross-reactivity is not primarily peptide-directed, but rather due to TCR recognition of peptide ligand-induced changes in MHC-E conformation (30), possibly binding in a manner analogous to the atypical docking modes reported for γδ TCR recognition of MR1 (52). Studies to analyze the epitope recognition of cloned TCR from vector-elicited, MHC-E-restricted, SIV supertope-targeted CD8+ T cells have been initiated to address these hypotheses. While the basis of this cross-reactivity and its implications for CMV vector development remain to be resolved in future studies, our finding that RM vaccination with the MHC-E supertope-focused insert-expressing miR-126-restricted 68–1 RhCMV/SIV vector provides >50% replication arrest efficacy after SIV challenge further strengthens the association of MHC-E-restricted CD8+ T cell responses and the replication arrest pattern of vaccine protection, showing for the first time that this protection can be achieved with a single RhCMV/SIV vector, rather than the multi-vector vaccines used in previous studies (1–4).
The binary replication arrest efficacy of 68–1 RhCMV/SIV vaccines is manifested by 59% of vaccinated RMs given either intra-rectal or intra-vaginal SIVmac239 challenge, across multiple studies, with protection not significantly correlated with peak or plateau-phase magnitude of SIV-specific CD8+ T cell responses (1–4). It is notable that the overall (replication arrest-type) efficacy of miR-126-restricted 68–1 RhCMV vectors, across both intra-vaginal and intra-rectal challenge, was not different from that of the parental 68–1 vector, nor was efficacy better correlated with the peak or plateau phase magnitudes of the exclusively MHC-E-restricted SIV-specific CD8+ T cells responses elicited by these vectors. These observations lead to two conclusions: 1) that eliminating MHC-II-restricted responses with miR-126 tropism restriction provides no discernable advantage over the parental 68–1 RhCMV/SIV vaccine with respect to replication arrest efficacy, suggesting that these MHC-II-restricted SIV-specific CD8+ T cell responses are effectively neutral with respect to this type of efficacy, and 2) that a separate component of 68–1 RhCMV/SIV-induced immunity likely contributes to determining efficacy in vaccinees that have MHC-E-restricted SIV-specific CD8+ T cell responses. Indeed, with respect to the latter point, recent analysis of 68–1 RhCMV/SIV vaccine efficacy reveals that in the presence of MHC-E-restricted CD8+ T cell responses, replication arrest protection can be predicted by a sustained whole blood transcriptomic response to vaccination that includes innate and adaptive immune gene pathways that coalesce around a signature of IL-15 signaling (53). Although the physiologic mechanisms by which these signaling pathways contribute to efficacy are unknown, the most straightforward hypothesis is that sustained low-level IL-15 signaling acts on the MHC-E-restricted CD8+ T cells themselves, enabling efficacy by tuning their homing/migration, Ag-receptor triggering thresholds, and/or effector functions (53).
Thus, based on the available evidence to date, the current understanding of the basis of the binary, replication arrest-type efficacy mediated by 68–1 RhCMV/SIV vectors reflects the early effective intercept of SIV infection by high frequency, effector differentiated, innate response-tuned MHC-E-restricted CD8+ T cells – a very different pattern of protection via a very different immune mechanism than any previously reported HIV/SIV vaccine. Indeed, as shown in this paper, the MHC-E-restricted CD8+ T cells associated with replication arrest efficacy are only weakly capable of conventional CD8+ T cell-mediated suppression of ongoing viral replication in vivo (e.g., reduction in plateau phase viral loads). Translation of the more desirable replication arrest-type efficacy to the clinic will almost certainly require that an HCMV/HIV vaccine recapitulate the above-described unconventional immune mechanisms in people. Since effector memory-biased CD8+ T cell responses elicited by efficacious RhCMV vectors, as well as by non-efficacious RhCMV vectors and natural RhCMV infection, are also characteristic of natural HCMV infection, the primary challenge of initial translation efforts will be to equip an HCMV/HIV vector with ability to elicit MHC-E-restricted, HIV-specific CD8+ T cell responses. Natural HCMV infection has been demonstrated to elicit MHC-E-restricted CD8+ T cells to variant UL40-embedded VL9 peptides (54), but not to the broad non-VL9 epitopes/supertopes documented for 68–1 RhCMV. We have, however, previously demonstrated that all of the RhCMV genes shown to be involved in programming MHC-E responses in RhCMV, including inhibitors (Rh157.5/Rh157.4; Rh158–161) and promoters (Rh67) of this priming, can be functionally replaced by their HCMV counterparts (UL128/UL130; UL146/UL147 and UL40) in RhCMV, indicating conservation of these MHC-E-restricted CD8+ T cell priming-related functions in HCMV. This conservation suggests that an HCMV lacking the UL128/UL130 and UL146/UL147 inhibitors, and expressing UL40, will have the biologic potential to mediate the priming of HIV-specific, MHC-E-restricted CD8+ T cells needed for vaccine efficacy, a hypothesis that is currently being tested in a phase I clinical trial (55).
The data presented here suggest that while MHC-E CD8+ T cell responses are required for vaccine efficacy, exclusive MHC-E-restricted CD8+ T cell responses do not enhance efficacy. This suggests that miR-126 restriction-mediation abrogation of MHC-II-restricted CD8+ T cell priming in an orthologous HCMV/HIV vector is dispensable from an efficacy standpoint. However, using miR restriction to prevent productive infection in miR-126-expressing endothelial cells, as well as miR-205-expressing epithelial cells, might enhance vector safety (e.g., reduce potential for vascular and epithelial organ complications) and for the latter, minimize vector shedding in secretions without impacting efficacy. Also, exclusive MHC-E-restricted epitope targeting might be advantageous for other vaccine targets, with the ability to target supertopes in essentially any antigen with miR-126-programmed 68–1-like HCMV vectors, offering the possibility to create MHC-E-restricted, MHC haplotype-independent CD8+ T cell vaccines for cancers and infectious diseases in which the target neoplastic/infected cells express tumor/pathogen-peptide MHC-E complexes (31, 56–59).
MATERIALS AND METHODS
For more details on all methods and materials listed below, please see the supplementary materials.
Study Design.
This study had two major objectives. First, we sought to determine the role of viral tropism in unconventional CD8+ T cell response programming by strain 68–1 RhCMV/SIV vectors, knowing that 1) MHC-II-restricted CD8+ T cell response programming could be induced by tropism-modification of 68–1.2 RhCMV/SIV vectors (4), 2) 68–1 RhCMV/SIV vector induction of MHC-E-restricted CD8+ T cell responses was the result of direct priming (10), and 3) pentamer-null RhCMV vectors were able to infect macrophages and endothelial cells with potential direct Ag-presenting cell function (fig. S1). Our approach to this objective was to construct 68–1 RhCMV/SIV vectors with microRNA-mediated tropism restriction, in which target sites for cell-selective microRNAs were incorporated into the 3’ untranslated region of the essential viral Rh108 and Rh156 genes, thereby inhibiting productive infection in the following specific cell types: myeloid cells using miR-142–3p (miR-142), endothelial cell using miR126–3p (miR-126), and epithelial cells using miR-205–5p (miR-205). RMs were then vaccinated with these vectors and the resulting CD8+ T cell responses were examined for epitope restriction as previously described (4, 8). The number of RMs and peptide responses analyzed for each studied vector are shown in table S1, but it is noteworthy that the MHC restriction status of the responses elicited by miR-restricted vectors was confirmed by analysis of nearly 1,700 peptide responses in 78 different RMs, respectively, all showing consistent patterns of restriction for each vector type. These restricted patterns were then independently confirmed with MHC-specific blocking analysis of vector-elicited CD8+ T cell recognition of autologous SIV-infected CD4+ T cells, allowing us to firmly conclude that 68–1 RhCMV/SIV vector elicited MHC-II-, MHC-E-, and MHC-Ia-restricted CD8+ T cell responses arise from different cell types, and that this priming can be exactly regulated by cell-type specific tropism restriction.
We next sought to compare efficacy of RhCMV/SIV vectors that were differentially programmed via tropism modification for MHC-II-, MHC-E-, and MHC-Ia-restricted CD8+ T cell responses, to test the hypothesis that replication arrest-type efficacy is dependent on MHC-E-restricted CD8+ T cell epitope targeting. Our use of vectors that were genetically identical except for tropism-restriction would allow us to rule out the possibility that non-tropism-related genetic differences might account for any differences in efficacy. For this second objective, we constructed a set of strain 68–1 vectors individually expressing SIVmac239 Gag, Retanef, and 5’-Pol for each distinct immunotype: MHC-II-only (miR-142-restricted), MHC-E-only (miR-126-restricted), and MHC-Ia-only (miR-126 + miR-142-restriced). In addition to the full length insert-expressing vector sets, we also constructed miR-126- and miR-142-restricted (single) vectors expressing supertope-focused inserts consisting of end-to-end juxtaposition of MHC-E and MHC-II supertope regions, respectively, to determine the ability of these supertope-restricted responses to mediate efficacy. We thus used these 5 vaccines – 3 vector sets and 2 supertope-focused single vectors – in an SIVmac239 challenge study with cycling female RMs and repeated limiting dose challenge via an intravaginal route. We used an n = 12 group size for the 2 MHC-II epitope targeting vaccines and 1 MHC-Ia epitope targeting vaccine, and an n = 14 group size for the 2 MHC-E epitope targeting vaccines, with the larger n in the latter groups anticipated to potentially allow more statistical power for elucidating quantitative correlates of protection. In addition, we included positive and negative control groups consisting of unvaccinated RMs or RMs vaccinated with parental (non-miR restricted) 68–1 RhCMV/SIV vector set (n = 9 cycling female RMs for each). These group sizes were projected to allow for thorough comparison of the overall immunogenicity of each vector backbone and to detect per-vaccine-group binary (replication arrest-type) protection levels of 50% at 80% power without multiplicity adjustment. In addition, we confirmed the efficacy of the MHC-E-only (miR-126-restricted) vectors in an independent cohort of 17 RMs challenged with repeated, limiting dose SIVmac239 via an intra-rectal route (n = 9 with a full length insert expressing vector set; n = 8 with a supertope-focused vector). All results from these study RMs are included in the presented data (no data were excluded as outliers). Plasma and cell-associated viral load assays were performed by blinded analysis; however, due to logistical constraints, other staff were not blinded to treatment assignments.
Rhesus macaques.
These experiments used a total of 225 purpose-bred male and female RMs (M. mulatta) of Indian genetic background, including 1) 43 RMs for delineation of SIV Retanef and 5’-Pol supertopes (and confirmation of SIV Gag supertopes), 2) 105 RMs for immunogenicity and/or efficacy analysis of parental and tropism-restricted 68–1 RhCMV/SIV vectors, 3) 9 RMs for unvaccinated controls, 4) 29 RMs as adoptive transfer recipients, and 5) 39 RMs vaccinated with 68–1 RhCMV expressing different inserts for response specificity analysis, the latter including 30 RMs from a previously published study (2). In addition, tissues from 6 previously reported (18) RhCMV seronegative RMs that were vaccinated with a reconstituted (wildtype-like), full length RhCMV with (n = 3) and without (n = 3) the double deletion (ΔRh157.4/.5 + ΔRh158–161) that mimics the key genetic changes in strain 68–1 RhCMV were used for immunohistochemical analysis of RhCMV cell type tropism (tissues obtained from necropsy 14 days post vaccination). RhCMV vectors were routinely dosed at 106-107 infectious units for immunogenicity analysis and 5×106 infectious units per vector for efficacy analysis, all via subcutaneous administration.
RM care and all experimental protocols and procedures were approved by the ONPRC Institutional Animal Care and Use Committee (IACUC). The ONPRC is a Category I facility. The Laboratory Animal Care and Use Program at the ONPRC is fully accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC) and has an approved Assurance (#A3304–01) for the care and use of animals on file with the NIH Office for Protection from Research Risks. The IACUC adheres to national guidelines established in the Animal Welfare Act (7 U.S.C. Sections 2131–2159) and the Guide for the Care and Use of Laboratory Animals (8th Edition) as mandated by the U.S. Public Health Service Policy.
Generation, recovery and characterization of recombinant RhCMV vectors.
SIV insert [SIV Gag, Retanef, 5’-Pol (protease and reverse transcriptase)]-expressing vaccine vectors based on RhCMV 68–1 (GenBank #MT157325) have been described previously (1, 4). Other RhCMV constructs used in this study were based on the partially repaired RhCMV 68–1.2 strain (GenBank #MT157326) and on the reconstructed full length RhCMV clone (GenBank #MT157327) (4, 18). Following the original 68–1 vector design, full length SIV inserts were introduced into the Rh211 open reading frame (ORF) in RhCMV 68–1 and 68–1.2 where transgene expression was driven by an EFIα promoter. Supertope-focused SIV inserts were expressed by replacing the Rh107 ORF. The RhCMV 68–1 and 68–1.2 -miR and -scrambled control vectors expressing SIV antigens were generated using galactokinase- (galK-) mediated bacterial artificial chromosome (BAC) recombination (60). All vectors derived from BACs were reconstituted in primary embryonal rhesus fibroblasts (RFs) via electroporation of purified BAC DNA. Virus titers of all RhCMV stocks were determined by fifty-percent tissue culture infective dose (TCID50) assay on RFs and expression of the SIV immunological marker was confirmed by immunoblot.
To analyze the growth restriction of the 68–1-miRNA vectors, multi-step growth assays were performed on RFs transiently transfected with the appropriate miRNA or a negative control mimic (Horizon Discovery). 12-well plates seeded with RFs were transfected with 20 pmol miRNA mimic/well (Dharmacon) using RNAiMax (Invitrogen) according to the manufacturer’s instructions. 24 h later the cells were infected with 68–1-miRNA vector or the scrambled control virus at a multiplicity of infection of 0.01. Supernatants were collected at the indicated timepoints and titered using a standard plaque assay on RFs. To analyze growth restriction in primary rhesus endothelial cells [maintained in EBM-2 basal medium with EGM-2 SingleQuots™ supplement excluding Heparin (Lonza), as well as 10% FBS and antibiotics] and epithelial cells (maintained in 50% DMEM, 50% DMEM/F12 as well as 1x non-essential amino acids, 10% FBS and antibiotics), 12-well plates seeded with rhesus endothelial cells or epithelial cells were infected with 68–1.2-miR-126 or 68–1.2-miR-205 vectors, respectively, or the scrambled control virus at a multiplicity of infection of 5. Supernatants were collected at the indicated timepoints and titered using standard plaque assay on RFs.
Immunologic assays.
SIV-specific CD4+ and/or CD8+ T cell responses were measured in peripheral blood mononuclear cells (PBMC) by flow cytometric intracellular cytokine analysis, as previously described (1–3).
SIV detection assays.
Plasma SIV RNA levels were determined using an SIV Gag-targeted quantitative RT-PCR format assay, with 6 replicate reactions analyzed per extracted sample for assay thresholds of 15 SIV RNA copies/ml, as previously described (2, 3, 62).
Vector shedding assays (urine cocultures).
Filter-sterilized (0.4 mm) urine was collected from RMs at the designated timepoints post-vaccination and centrifuged at 16,000 g for 1 h at 4°C to concentrate virus for co-culture on rhesus fibroblasts. Cell lysates were prepared after we observed extensive cytopathic effects, or after 42 days of coculture if cytopathic effects were minimal or not observed. The prepared cell lysates were assessed for the presence of RhCMV based on expression of RhCMV or SIV antigens by immunoblotting (24, 25).
RhCMV vector immunofluorescence analysis.
Immunofluorescent phenotyping of RhCMV vector+ cells using RNAscope was performed on FFPE spleen and lymph node sections as previously reported (18). Immunofluorescent phenotyping of RhCMV vector+ cells using RhCMV-specific mAbs was performed on FFPE spleen and lymph node sections similar to our previous report with some modifications (18).
Statistical Analysis.
Boxplots show jittered points and a box from 1st to 3rd quartiles (IQR) and a line at the median, with whiskers extending to the farthest data point within 1.5*IQR above and below the box. For all immune responses, we compared treatment groups with two-sided Wilcoxon rank-sum tests (pairwise comparisons) or the Kruskal-Wallis test (comparisons between ≥ 2 groups). When applicable, we performed post-hoc Wilcoxon rank-sum tests between pairs of treatment groups, without adjustment for multiple comparisons. For longitudinal analyses, we calculated the area under the curve (AUC) or the plateau average value for each RM, as specified in the figure legends. For analysis of T cell responses to specific cytokines, we transformed response data to the log scale and compared treatment groups using an ordinary linear model. To compare time-to-event data, we used the Mantel-Haenszel log-rank test. All P-values for analyses of protection efficacy are based on two-sided exact tests of binomial proportions. In all cases (except where noted), we adjust P-values for multiple comparisons using the using the Holm procedure for family-wise error rate control. All analyses were performed in R v3.6.0 with the package Exact v2.0 (63).
Supplementary Material
Raw data file: quantitative immunologic data for all Figures (Excel spreadsheet)
Raw data file: non-quantitative epitope analysis data (Excel spreadsheet)
Supplemental Materials and Methods
Supplemental Figures
Fig. S1. The pentameric receptor complex is not required for RhCMV vector infection of macrophages or endothelial cells in vivo.
Fig. S2. Effect of individual miR-mediated tropism restrictions on 68–1 RhCMV replication in cell culture.
Fig. S3. miR-126- and miR-205-mediated tropism restriction of 68–1.2 RhCMV in their corresponding cell types.
Fig. S4. Effect of miR-mediated tropism restrictions on 68–1 RhCMV/SIV vector shedding in urine.
Fig. S5. Effect of combination miR-mediated tropism restrictions on 68–1 RhCMV replication in cell culture.
Fig. S6. Documentation of supertopes (universal epitopes) in the SIV Gag, Retanef and 5’-Pol vector inserts.
Fig. S7. Amino acid sequence of MHC-II and MHC-E supertope-focused vector inserts.
Fig. S8. Analysis of MHC restriction patterns of the SIV epitope-specific responses elicited by different miR-restricted 68–1 RhCMV/SIV vaccine vectors used in the challenge studies.
Fig. S9. Analysis of cross-reactivity with non-vaccine expressed Ags after vaccination with 68–1 RhCMV vectors.
Fig. S10. Assay procedure and gating strategy for flow cytometric cytokine production-based SIV-infected cell recognition analysis.
Fig. S11. Induction of SIV Vif-specific T cell responses with take of SIV infection after challenge (RM groups 1, 2, 5, 7)
Fig. S12. Induction of SIV Env-specific T cell responses with take of SIV infection after challenge (RM groups 1–7)
Fig. S13. Comparison of viral set points of 68–1 RhCMV/SIV vaccinated, but unprotected, RMs in each vaccine group relative to unvaccinated controls.
Fig. S14. The magnitude of the SIV-specific, MHC-E-restricted CD8+ T cell response in blood at peak and plateau during the vaccine phase does not predict efficacy.
Supplemental Tables
Table S1. Summary of RhCMV gene products influencing the priming of unconventionally restricted CD8+ T cell responses.
Table S2. Summary of restriction-assigned 15mer peptide responses for all study RMs.
Table S3. Epitope density analysis of differentially tropism-restricted (response programmed) 68–1 RhCMV/SIV vectors.
Table S4. List 15mers with sequences outside the MHC-E supertope-focused insert (≤ 4 amino acid overlap) that are recognized by CD8+ T cell responses elicited by the miR-126-restricted 68–1 RhCMV vectors expressing MHC-E supertope-focused SIV inserts.
Table S5. List 15mers with sequences outside the MHC-II supertope-focused insert (≤ 4 amino acid overlap) that are recognized by CD8+ T cell responses elicited by the miR-142-restricted 68–1 RhCMV vectors expressing MHC-II supertope-focused SIV inserts.
Table S6. Cells used for adoptive transfer analysis of replication competent SIV in protected RMs.
Table 1:
Effect of miR-mediated tropism restriction on 68-1 RhCMV/SIV vector urine shedding, CD8+ T cell response programming and vaccine efficacy
| miR restriction | Vector tropism restriction (cell type) | Urine shedding | MHC targeting of vector-elicited CD8+ T cells | SIV efficacy (replication arrest) |
|---|---|---|---|---|
| none | none | yes | MHC-E and MHC-II | yes |
| miR-142 | myeloid | yes | MHC-II alone | no |
| miR-126 | endothelial | yes | MHC-E alone | yes |
| miR-205 | epithelial | no | MHC-E and MHC-II | not done |
| miR-142 + miR-126 | myeloid + endoth. | not done | MHC-Ia alone | no |
Acknowledgments:
We thank B. Miles, M. Fischer, A. Barber-Axthelm, C. Shriver-Munsch, T. Swanson, A. Sylwester, S. Hagen, E. McDonald, E. Ainslie, A. McNett, and K. Rothstein for technical or administrative assistance; B. Keele for providing SIVmac239 challenge virus; and A. Townsend for figure preparation.
Funding:
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) grants UM1 AI124377 and U19 AI128741 to LJP; the Oregon National Primate Research Center Core grant from the National Institutes of Health, Office of the Director (P51 OD011092); contracts from the National Cancer Institute (#s HHSN261200800001E and 75N1019D00024) to JDL; and the Bill and Melinda Gates Foundation grant OPP1107409.
Footnotes
Competing interests: OHSU, LJP, SGH, JAN, and KF have a substantial financial interest in Vir Biotechnology, Inc., a company that may have a commercial interest in the results of this research and technology. LJP, SGH and KF are also consultants to Vir Biotechnology, Inc. LJP, JAN, KF, SGH and MHH are co-inventors of US patent 10,688,164 “CMV vectors comprising microRNA recognition elements” licensed to Vir Biotechnology, Inc. LJP, JAN, KF, SGH, MHH and DM are co-inventors of US patent 10,532,099 “Cytomegalovirus vectors eliciting T cells restricted by major histocompatibility complex E molecules”, licensed to Vir Biotechnology, Inc. These potential individual and institutional conflicts of interest have been reviewed and managed by OHSU.
Data and materials availability:
All data associated with this study are present in the paper or Supplementary Materials, including raw data files for main figures (tables S7, S8). The computer code used to perform statistical analysis is available at 10.5281/zenodo.6468337.RhCMV/SIV vectors can be obtained through an MTA.
REFERENCES AND NOTES
- 1.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M Jr., Lifson JD, Picker LJ, Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473, 523–527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen SG, Piatak M Jr., Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ, Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen SG, Marshall EE, Malouli D, Ventura AB, Hughes CM, Ainslie E, Ford JC, Morrow D, Gilbride RM, Bae JY, Legasse AW, Oswald K, Shoemaker R, Berkemeier B, Bosche WJ, Hull M, Womack J, Shao J, Edlefsen PT, Reed JS, Burwitz BJ, Sacha JB, Axthelm MK, Fruh K, Lifson JD, Picker LJ, A live-attenuated RhCMV/SIV vaccine shows long-term efficacy against heterologous SIV challenge. Sci Transl Med 11, eaaw2607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malouli D, Hansen SG, Hancock MH, Hughes CM, Ford JC, Gilbride RM, Ventura AB, Morrow D, Randall KT, Taher H, Uebelhoer LS, McArdle MR, Papen CR, Espinosa Trethewy R, Oswald K, Shoemaker R, Berkemeier B, Bosche WJ, Hull M, Greene JM, Axthelm MK, Shao J, Edlefsen PT, Grey F, Nelson JA, Lifson JD, Streblow D, Sacha JB, Fruh K, Picker LJ, Cytomegaloviral determinants of CD8+ T cell programming and RhCMV/SIV vaccine efficacy. Sci Immunol 6, eabg5413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M Jr., Lifson JD, Nelson JA, Jarvis MA, Picker LJ, Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15, 293–299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masopust D, Picker LJ, Hidden memories: frontline memory T cells and early pathogen interception. J Immunol 188, 5811–5817 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Fruh K, Picker LJ, Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340, 1237874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, Reed JS, Gilbride RM, Ainslie E, Morrow DW, Ford JC, Selseth AN, Pathak R, Malouli D, Legasse AW, Axthelm MK, Nelson JA, Gillespie GM, Walters LC, Brackenridge S, Sharpe HR, Lopez CA, Fruh K, Korber BT, McMichael AJ, Gnanakaran S, Sacha JB, Picker LJ, Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science, 351, 714–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ, Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168, 29–43 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Verweij MC, Hansen SG, Iyer R, John N, Malouli D, Morrow D, Scholz I, Womack J, Abdulhaqq S, Gilbride RM, Hughes CM, Ventura AB, Ford JC, Selseth AN, Oswald K, Shoemaker R, Berkemeier B, Bosche WJ, Hull M, Shao J, Sacha JB, Axthelm MK, Edlefsen PT, Lifson JD, Picker LJ, Fruh K, Modulation of MHC-E transport by viral decoy ligands is required for RhCMV/SIV vaccine efficacy. Science 372, eabe9233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerna G, Kabanova A, Lilleri D, Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines (Basel) 7, (3):70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinzger C, Digel M, Jahn G, Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325, 63–83 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Jarvis MA, Nelson JA, Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Curr Opin Microbiol 5, 403–407 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Barnes D, Kunitomi M, Vignuzzi M, Saksela K, Andino R, Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe 4, 239–248 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lilja AE, Shenk T, Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc Natl Acad Sci USA 105, 19950–19955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Shenk T, Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci USA 102, 18153–18158 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assaf BT, Mansfield KG, Westmoreland SV, Kaur A, Oxford KL, Diamond DJ, Barry PA, Patterns of acute rhesus cytomegalovirus (RhCMV) infection predict long-term RhCMV infection. J Virol 86, 6354–6357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taher H, Mahyari E, Kreklywich C, Uebelhoer LS, McArdle MR, Mostrom MJ, Bhusari A, Nekorchuk M, E X, Whitmer T, Scheef EA, Sprehe LM, Roberts DL, Hughes CM, Jackson KA, Selseth AN, Ventura AB, Cleveland-Rubeor HC, Yue Y, Schmidt KA, Shao J, Edlefsen PT, Smedley J, Kowalik TF, Stanton RJ, Axthelm MK, Estes JD, Hansen SG, Kaur A, Barry PA, Bimber BN, Picker LJ, Streblow DN, Fruh K, Malouli D, In vitro and in vivo characterization of a recombinant rhesus cytomegalovirus containing a complete genome. PLoS Pathog 16, e1008666 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN, The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15, 261–271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D, miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15, 272–284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahamud MR, Geng X, Ho YC, Cha B, Kim Y, Ma J, Chen L, Myers G, Camper S, Mustacich D, Witte M, Choi D, Hong YK, Chen H, Varshney G, Engel JD, Wang S, Kim TH, Lim KC, Srinivasan RS, GATA2 controls lymphatic endothelial cell junctional integrity and lymphovenous valve morphogenesis through miR-126. Development 146, dev184218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kontarakis Z, Rossi A, Ramas S, Dellinger MT, Stainier DYR, Mir-126 is a conserved modulator of lymphatic development. Dev Biol 437, 120–130 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Ferrari E, Gandellini P, Unveiling the ups and downs of miR-205 in physiology and cancer: transcriptional and post-transcriptional mechanisms. Cell Death Dis 11, 980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, Axthelm MK, Nelson JA, Jarvis MA, Picker LJ, Fruh K, Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328, 102–106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall EE, Malouli D, Hansen SG, Gilbride RM, Hughes CM, Ventura AB, Ainslie E, Selseth AN, Ford JC, Burke D, Kreklywich CN, Womack J, Legasse AW, Axthelm MK, Kahl C, Streblow D, Edlefsen PT, Picker LJ, Fruh K, Enhancing safety of cytomegalovirus-based vaccine vectors by engaging host intrinsic immunity. Sci Transl Med 11, eaaw2603 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CZ, Li L, Lodish HF, Bartel DP, MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T, A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moller R, Schwarz TM, Noriega VM, Panis M, Sachs D, Tortorella D, tenOever BR, miRNA-mediated targeting of human cytomegalovirus reveals biological host and viral targets of IE2. Proc Natl Acad Sci USA 115, 1069–1074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Camilli G, Cassotta A, Battella S, Palmieri G, Santoni A, Paladini F, Fiorillo MT, Sorrentino R, Regulation and trafficking of the HLA-E molecules during monocyte-macrophage differentiation. J Leukoc Biol 99, 121–130 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Walters LC, Harlos K, Brackenridge S, Rozbesky D, Barrett JR, Jain V, Walter TS, O’Callaghan CA, Borrow P, Toebes M, Hansen SG, Sacha JB, Abdulhaqq S, Greene JM, Fruh K, Marshall E, Picker LJ, Jones EY, McMichael AJ, Gillespie GM, Pathogen-derived HLA-E bound epitopes reveal broad primary anchor pocket tolerability and conformationally malleable peptide binding. Nat Commun 9, 3137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joosten SA, Sullivan LC, Ottenhoff TH, Characteristics of HLA-E Restricted T Cell Responses and Their Role in Infectious Diseases. J Immunol Res 2016, 2695396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grotzke JE, Harriff MJ, Siler AC, Nolt D, Delepine J, Lewinsohn DA, Lewinsohn DM, The Mycobacterium tuberculosis phagosome is a HLA-I processing competent organelle. PLoS Pathog 5, e1000374 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang S, Springer LE, Rao HZ, Espinosa Trethewy RG, Bishop LM, Hancock MH, Grey F, Snyder CM, Hematopoietic cell-mediated dissemination of murine cytomegalovirus is regulated by NK cells and immune evasion. PLoS Pathog 17, e1009255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford LB, Kim JH, Collins-McMillen D, Lee BJ, Landais I, Held C, Nelson JA, Yurochko AD, Caposio P, Human Cytomegalovirus Encodes a Novel FLT3 Receptor Ligand Necessary for Hematopoietic Cell Differentiation and Viral Reactivation. mBio 9, e00682–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neefjes J, Jongsma ML, Paul P, Bakke O, Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11, 823–836 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Schmid D, Pypaert M, Munz C, Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veerappan Ganesan AP, Eisenlohr LC, The elucidation of non-classical MHC class II antigen processing through the study of viral antigens. Curr Opin Virol 22, 71–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agudo J, Ruzo A, Tung N, Salmon H, Leboeuf M, Hashimoto D, Becker C, Garrett-Sinha LA, Baccarini A, Merad M, Brown BD, The miR-126-VEGFR2 axis controls the innate response to pathogen-associated nucleic acids. Nat Immunol 15, 54–62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, Schira G, Amendola M, Quattrini A, Martino S, Orlacchio A, Dick JE, Biffi A, Naldini L, Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Transl Med 2, 58ra84 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Yun TJ, Igarashi S, Zhao H, Perez OA, Pereira MR, Zorn E, Shen Y, Goodrum F, Rahman A, Sims PA, Farber DL, Reizis B, Human plasmacytoid dendritic cells mount a distinct antiviral response to virus-infected cells. Sci Immunol 6, eabc7302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pober JS, Tellides G, Participation of blood vessel cells in human adaptive immune responses. Trends Immunol 33, 49–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santambrogio L, Berendam SJ, Engelhard VH, The Antigen Processing and Presentation Machinery in Lymphatic Endothelial Cells. Front Immunol 10, 1033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Momtaz S, Molina B, Mlera L, Goodrum F, Wilson JM, Cell type-specific biogenesis of novel vesicles containing viral products in human cytomegalovirus infection. J Virol, 95, e02358–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bughio F, Umashankar M, Wilson J, Goodrum F, Human Cytomegalovirus UL135 and UL136 Genes Are Required for Postentry Tropism in Endothelial Cells. J Virol 89, 6536–6550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rue CA, Jarvis MA, Knoche AJ, Meyers HL, DeFilippis VR, Hansen SG, Wagner M, Fruh K, Anders DG, Wong SW, Barry PA, Nelson JA, A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J Virol 78, 12529–12536 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Busche A, Jirmo AC, Welten SP, Zischke J, Noack J, Constabel H, Gatzke AK, Keyser KA, Arens R, Behrens GM, Messerle M, Priming of CD8+ T cells against cytomegalovirus-encoded antigens is dominated by cross-presentation. J Immunol 190, 2767–2777 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Munks MW, Pinto AK, Doom CM, Hill AB, Viral interference with antigen presentation does not alter acute or chronic CD8 T cell immunodominance in murine cytomegalovirus infection. J Immunol 178, 7235–7241 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Xu RH, Remakus S, Ma X, Roscoe F, Sigal LJ, Direct presentation is sufficient for an efficient anti-viral CD8+ T cell response. PLoS Pathog 6, e1000768 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sei JJ, Haskett S, Kaminsky LW, Lin E, Truckenmiller ME, Bellone CJ, Buller RM, Norbury CC, Peptide-MHC-I from Endogenous Antigen Outnumber Those from Exogenous Antigen, Irrespective of APC Phenotype or Activation. PLoS Pathog 11, e1004941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yewdell JW, Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25, 533–543 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Chen W, McCluskey J, Immunodominance and immunodomination: critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv Cancer Res 95, 203–247 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Rice MT, von Borstel A, Chevour P, Awad W, Howson LJ, Littler DR, Gherardin NA, Le Nours J, Giles EM, Berry R, Godfrey DI, Davey MS, Rossjohn J, Gully BS, Recognition of the antigen-presenting molecule MR1 by a Vdelta3+ gammadelta T cell receptor. Proc Natl Acad Sci USA 118, e2110288118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barrenäs F, Hansen SG, Law L, Driscoll C, Green RR, Smith E, Chang J, Golez I, Peng X, Whitmore L, Newhouse D, Hughes CM, Morrow D, Randall K, Selseth AN, Ford JC, Gilbride RM, Oswald K, Shoemaker R, Fast R, Bosche W, Axthelm MK, Fukazawa Y, Pavlakis GN, Fourati S, Sekaly R-P, Lifson JD, Komorowski J, Kosmider E, Edlefsen PT, Picker LJ, Gale M Jr., A sustained vaccine-induced transcriptional signature featuring IL-15 response predicts RhCMV/SIV vector efficacy. PLoS Pathog 17, e1009278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jouand N, Bressollette-Bodin C, Gerard N, Giral M, Guerif P, Rodallec A, Oger R, Parrot T, Allard M, Cesbron-Gautier A, Gervois N, Charreau B, HCMV triggers frequent and persistent UL40-specific unconventional HLA-E-restricted CD8+ T cell responses with potential autologous and allogeneic peptide recognition. PLoS Pathog 14, e1007041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.VIR-1111: A Prototype Human CMV-based Vaccine for Human Immunodeficiency Virus (HIV) in Healthy Volunteers, ClinicalTrials.gov Identifier: NCT04725877, VirBiotechnology; (2021). [Google Scholar]
- 56.Burwitz BJ, Hashiguchi PK, Mansouri M, Meyer C, Gilbride RM, Biswas S, Womack JL, Reed JS, Wu HL, Axthelm MK, Hansen SG, Picker LJ, Fruh K, Sacha JB, MHC E-restricted CD8+ T cells target Hepatitis B virus-infected human hepatocytes. J Immunol 204, 2169–2176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goncalves MA, Le Discorde M, Simoes RT, Rabreau M, Soares EG, Donadi EA, Carosella ED, Classical and non-classical HLA molecules and p16(INK4a) expression in precursors lesions and invasive cervical cancer. Eur J Obstet Gynecol Reprod Biol 141, 70–74 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Jimenez P, Geraghty DE, Garrido F, Analysis of HLA-E expression in human tumors. Immunogenetics 54, 767–775 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Benevolo M, Mottolese M, Tremante E, Rollo F, Diodoro MG, Ercolani C, Sperduti I, Lo Monaco E, Cosimelli M, Giacomini P, High expression of HLA-E in colorectal carcinoma is associated with a favorable prognosis. J Transl Med 9, 184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG, Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33, e36 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen SG, Zak DE, Xu G, Ford JC, Marshall EE, Malouli D, Gilbride RM, Hughes CM, Ventura AB, Ainslie E, Randall KT, Selseth AN, Rundstrom P, Herlache L, Lewis MS, Park H, Planer SL, Turner JM, Fischer M, Armstrong C, Zweig RC, Valvo J, Braun JM, Shankar S, Lu L, Sylwester AW, Legasse AW, Messerle M, Jarvis MA, Amon LM, Aderem A, Alter G, Laddy DJ, Stone M, Bonavia A, Evans TG, Axthelm MK, Fruh K, Edlefsen PT, Picker LJ, Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat Med 24, 130–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansen SG, Piatak M, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ, Addendum: Immune clearance of highly pathogenic SIV infection. Nature 547, 123–124 (2017). [DOI] [PubMed] [Google Scholar]
- 63.R Development Core Team. R, A Language and Environment for Statistical Computing. http://www.R-project.org (Vienna, Austria, 2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw data file: quantitative immunologic data for all Figures (Excel spreadsheet)
Raw data file: non-quantitative epitope analysis data (Excel spreadsheet)
Supplemental Materials and Methods
Supplemental Figures
Fig. S1. The pentameric receptor complex is not required for RhCMV vector infection of macrophages or endothelial cells in vivo.
Fig. S2. Effect of individual miR-mediated tropism restrictions on 68–1 RhCMV replication in cell culture.
Fig. S3. miR-126- and miR-205-mediated tropism restriction of 68–1.2 RhCMV in their corresponding cell types.
Fig. S4. Effect of miR-mediated tropism restrictions on 68–1 RhCMV/SIV vector shedding in urine.
Fig. S5. Effect of combination miR-mediated tropism restrictions on 68–1 RhCMV replication in cell culture.
Fig. S6. Documentation of supertopes (universal epitopes) in the SIV Gag, Retanef and 5’-Pol vector inserts.
Fig. S7. Amino acid sequence of MHC-II and MHC-E supertope-focused vector inserts.
Fig. S8. Analysis of MHC restriction patterns of the SIV epitope-specific responses elicited by different miR-restricted 68–1 RhCMV/SIV vaccine vectors used in the challenge studies.
Fig. S9. Analysis of cross-reactivity with non-vaccine expressed Ags after vaccination with 68–1 RhCMV vectors.
Fig. S10. Assay procedure and gating strategy for flow cytometric cytokine production-based SIV-infected cell recognition analysis.
Fig. S11. Induction of SIV Vif-specific T cell responses with take of SIV infection after challenge (RM groups 1, 2, 5, 7)
Fig. S12. Induction of SIV Env-specific T cell responses with take of SIV infection after challenge (RM groups 1–7)
Fig. S13. Comparison of viral set points of 68–1 RhCMV/SIV vaccinated, but unprotected, RMs in each vaccine group relative to unvaccinated controls.
Fig. S14. The magnitude of the SIV-specific, MHC-E-restricted CD8+ T cell response in blood at peak and plateau during the vaccine phase does not predict efficacy.
Supplemental Tables
Table S1. Summary of RhCMV gene products influencing the priming of unconventionally restricted CD8+ T cell responses.
Table S2. Summary of restriction-assigned 15mer peptide responses for all study RMs.
Table S3. Epitope density analysis of differentially tropism-restricted (response programmed) 68–1 RhCMV/SIV vectors.
Table S4. List 15mers with sequences outside the MHC-E supertope-focused insert (≤ 4 amino acid overlap) that are recognized by CD8+ T cell responses elicited by the miR-126-restricted 68–1 RhCMV vectors expressing MHC-E supertope-focused SIV inserts.
Table S5. List 15mers with sequences outside the MHC-II supertope-focused insert (≤ 4 amino acid overlap) that are recognized by CD8+ T cell responses elicited by the miR-142-restricted 68–1 RhCMV vectors expressing MHC-II supertope-focused SIV inserts.
Table S6. Cells used for adoptive transfer analysis of replication competent SIV in protected RMs.
Data Availability Statement
All data associated with this study are present in the paper or Supplementary Materials, including raw data files for main figures (tables S7, S8). The computer code used to perform statistical analysis is available at 10.5281/zenodo.6468337.RhCMV/SIV vectors can be obtained through an MTA.