

Abstract
Brain-derived neurotrophic factor (BDNF) expression increases in the paraventricular nucleus of the hypothalamus (PVN) during hypertensive stimuli including stress and hyperosmolarity, but its role in PVN cardiovascular regulatory mechanisms is unclear. Chronic BDNF overexpression in the PVN has been shown to elevate sympathetic tone and blood pressure in part by modulating central angiotensin (Ang) II mechanisms. However, the cardiovascular effects of short-term increases in PVN levels of BDNF and the mechanisms governing them are unknown. Therefore, we investigated whether acute BDNF microinjections into the PVN of conscious and anesthetized Sprague-Dawley rats induce blood pressure elevations and whether Ang II signaling is involved in these hypertensive responses. In conscious rats, unilateral BDNF (12.5 ng) microinjections into the PVN increased mean arterial pressure (MAP) by 27 ± 1 mm Hg (P < 0.001 vs vehicle), which was significantly attenuated by intracerebroventricular infusion of the Ang II-type-1 receptor (AT1R) antagonist losartan and by ganglionic blockade with intravenous hexamethonium infusion. In anesthetized rats, unilateral PVN microinjection of BDNF increased MAP by 31 ± 4 mm Hg (P < 0.001 vs vehicle), which was prevented by PVN microinjection pretreatments with the high-affinity BDNF receptor TrkB antagonist ANA-12, losartan, the angiotensin converting enzyme inhibitor lisinopril, or by intravenous hexamethonium. Additional experiments in hypothalamic samples including the PVN revealed that BDNF-induced TrkB receptor phosphorylation was prevented by ANA-12 and losartan pretreatments. Collectively, these data indicate that BDNF acting within the PVN acutely raises blood pressure under permissive control of Ang II-AT1R mechanisms and therefore may play an important role in mediating acute pressor responses to hypertensive stimuli.
Keywords: Brain-derived neurotrophic factor, Paraventricular nucleus of the hypothalamus, Angiotensin, TrkB receptors, Blood pressure, Sympathetic nervous system
1. Introduction
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, exerts diverse effects on neuronal structure and function throughout the central nervous system by regulating gene expression, neuronal structure, synaptic development and ion channel function through its high-affinity tyrosine receptor kinase B (TrkB) receptor (Andero et al., 2014). These BDNF-regulated mechanisms in neuroplasticity, memory and learning have been widely studied (Andero et al., 2014; Bekinschtein et al., 2014; Leal et al., 2015; Lu et al., 2014), but recent investigations suggest that BDNF may also be a key regulator of cardiovascular function in medullary and hypothalamic nuclei. For instance, endogenous BDNF tonically regulates blood pressure and efferent sympathetic nerve activity in the nucleus of the solitary tract (NTS) (Clark et al., 2011), and microinjections of exogenous BDNF into the NTS and rostral ventrolateral medulla (RVLM) significantly elevate blood pressure (Clark et al., 2011; Wang and Zhou, 2002).
The paraventricular nucleus of the hypothalamus (PVN) is a major neurohumoral integration site as well as a modulator of sympathetic nervous system activity via parvocellular projections to the NTS, RVLM and intermediolateral spinal column neurons (Dampney et al., 2005). Intriguingly, BDNF mRNA and protein expression in the PVN increase significantly in response to hypertensive stimuli such as stress and hyperosmolarity (Aliaga et al., 2002; Hammack et al., 2009; Smith et al., 1995a), but its role in PVN cardiovascular regulatory mechanisms is unclear. We recently demonstrated that long-term overexpression of BDNF in the PVN via adeno-associated viral vector delivery produces chronic elevations in blood pressure, heart rate and sympathetic activity (Erdos et al., 2015). However, consequences of long-term BDNF upregulation may differ considerably from the effects of short-term increases in BDNF within the PVN like those that occur during acute stress (Smith et al., 1995a).
Recent findings suggest a complex, bidirectional interaction between BDNF and angiotensin (Ang) II signaling pathways that may involve Ang II-induced upregulation of BDNF expression to mediate changes in ion channel function (Becker et al., 2015), BDNF-induced upregulation of Ang II type-1 receptor (AT1R) expression and central AT1R-dependent elevations in sympathetic tone, blood pressure and heart rate (Erdos et al., 2015). Furthermore, BDNF expression and Ang II-mediated pathways in the PVN are both stimulated by acute stress and hyperosmotic stimuli (Aliaga et al., 2002; Chen and Toney, 2001; Davern et al., 2009; Saavedra et al., 2004; Smith et al., 1995a). Based on these observations, we hypothesize that acute increases in PVN levels of BDNF elicit hypertensive and tachycardic responses that are mediated by activation of the sympathetic nervous system through an interaction with PVN Ang II signaling mechanisms.
To test this hypothesis, we performed a series of microinjection experiments in conscious and anesthetized animals. First, we investigated the effects of acute BDNF infusion into the PVN of conscious rats to evaluate cardiovascular responses free from the compounding effects of anesthesia (Brahim and Thut, 1984) with or without pretreatment by central infusion of the AT1R inhibitor losartan or intravenous infusion of the ganglionic blocker hexamethonium. Then we examined specific Ang II and BDNF interactions within the PVN by performing microinjections in anesthetized rats using double-barreled glass capillaries to allow infusion of inhibitors and BDNF into the same region of the PVN. Finally, we studied potential involvement of AT1Rs in BDNF-induced activation and autophosphorylation of TrkB receptors in freshly isolated hypothalamic brain punches from the area of the PVN. In this study we provide functional and biochemical evidence for a synergistic interaction between BDNF and Ang II-AT1R signaling in the PVN that contributes to acute increases in blood pressure and establish BDNF as a novel pro-hypertensive mediator in the PVN.
2. Materials and methods
All animal housing, handling, surgical and experimental procedures were conducted within an animal care facility accredited by the Association for the Assessment and Accreditation of Laboratory Care International at the University of Vermont, in accordance with the USA Public Health Service Policy on Human Care and Use of Laboratory Animals and the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Experiments were performed in male Sprague-Dawley (SD) rats obtained from Charles River (Saint-Constant, QC, Canada) at 7–9 weeks of age. Rats were housed individually with a 12-hour light/dark cycle (lights on at 6:00 am), with free access to food (standard chow) and water. Surgeries were performed under continuous isoflurane anesthesia (5% induction; 2–3% maintenance) delivered in oxygen. Depth of anesthesia was assured by close monitoring of blood pressure, heart rate, and lack of eye blink and hind paw pinch reflex responses. Survival surgeries were conducted using aseptic technique and carprofen (5 mg/kg/day s.c.) was administered for post-surgical analgesia at the beginning of surgery and for two days during post-surgical recovery. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Vermont.
2.1. Experimental design and surgical procedures
2.1.1. Microinjections in conscious rats
These experiments were designed to test the effect of acute BDNF injections into the PVN on blood pressure and heart rate in conscious animals, and to investigate the involvement of central AT1Rs and the autonomic nervous system. Under isoflurane anesthesia, intracranial guide cannulae (PlasticsOne, Roanoke, VA) targeting the right PVN (26-ga diameter; inserted 1.8 mm posterior to bregma; 1.7 mm lateral; tilted 10° laterally toward the midline and lowered to 7 mm beneath the surface of the skull) and the left lateral ventricle (22-ga diameter; inserted 0.8 mm posterior to bregma; 0.9 mm lateral and lowered to 3 mm beneath the surface of the skull) were surgically implanted and secured with stainless steel screws and dental cement. A dummy cannula was fastened into each guide. After a 5-day recovery period, rats were surgically outfitted with femoral artery (all groups) and vein (hexamethonium + BDNF group only, see below) catheters under isoflurane anesthesia 24–48 h prior to testing. Catheters were threaded out of a 0.5-cm incision in the interscapular region and sutured into place.
On the day of the experiment, the femoral artery catheter was connected to a pressure transducer (MEMSCAP, Inc., Durham, NC) using an extension catheter, while the femoral vein catheter was connected to a 1-mL syringe. The dummy cannulae were carefully removed from the PVN and intracerebroventricular (icv) guides, and injectors extending 1 mm below the guide cannulae were inserted and secured. Animals were then allowed to acclimate for a minimum of 30 min to achieve stable baseline blood pressure and heart rate levels. Rats were divided into four treatment groups: icv infusion of artificial cerebrospinal fluid (aCSF) + PVN microinjection of aCSF (n = 6); icv aCSF + PVN microinjection of BDNF (n = 7); icv losartan + PVN microinjection of BDNF (n = 7); and intravenous (iv) hexamethonium + PVN microinjection of BDNF (n = 4). Losartan (15 μg) or aCSF was infused icv in a volume of 5 μL over 20 s or hexamethonium (30 mg/kg bolus) was infused intravenously over 30 s. Fifteen minutes following icv aCSF or losartan infusion, or 5 min following intravenous hexamethonium administration, BDNF (12.5 ng) was infused into the PVN at 25 nL/s for 20 s. Blood pressure was recorded for an additional 15 min. At the end of the experiment, rats were sacrificed by decapitation under deep isoflurane anesthesia. Brains were collected, fixed in 4% paraformaldehyde, freeze-protected with 30% sucrose solution and sectioned for verification of the injection site (Fig. 1). All experiments were conducted between 1:00 pm and 5:00 pm in a quiet lit room.
Fig. 1.
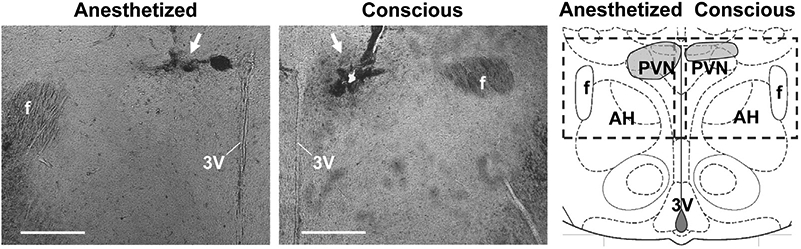
Representative PVN injection site verification images in coronal brain sections (~1.8 mm posterior to bregma) from rats involved in anesthetized (left) and conscious (center) microinjection experiments. Arrows indicate the position of the tip of the injector (conscious rat) or glass micropipette (anesthetized rat), bars = 500 μm. Right: Schematic representation of a coronal section containing the PVN at 1.8 mm posterior to bregma, modified from Paxinos and Watson (2014). All microinjection sites fell within the shaded regions of the pictogram. Dashed line-rectangles indicate the approximate position where images were taken. 3V, third ventricle; AH, anterior hypothalamic area; f, fornix.
2.1.2. Microinjections in anesthetized rats
To study specific intra-PVN BDNF signaling mechanisms, we performed PVN microinjection experiments in anesthetized rats. This approach allowed the use of double-barreled glass micropipettes for pretreatment injections of inhibitors followed by BDNF into the same region of the PVN. Femoral artery and vein catheters were implanted under isoflurane anesthesia. Rats were then placed in a stereotaxic apparatus and a sagittal incision was made to expose the dorsal surface of the skull. A cranial window (~3 × 4 mm) was created to expose an area of the brain surface dorsal to the PVN. As previously described (Cassaglia et al., 2014), animals were then transitioned from isoflurane to α-chloralose anesthesia (100 mg/kg/h), administered intravenously through the femoral vein catheter over a 30-min period during which isoflurane was gradually withdrawn. Blood pressure, heart rate, and toe-pinch and eye-blink reflexes were monitored closely to ensure the animal remained anesthetized. After complete withdrawal of isoflurane, anesthesia was maintained by intravenous α-chloralose infusion (25 mg/kg/h) for the remainder of testing. Oxygen flow remained constant for the duration of the experiment, and core body temperature and pO2 were continuously monitored using a PhysioSuite system (Kent Scientific, Torrington, CT).
A minimum of 60 min passed between cessation of isoflurane and the first microinjection to achieve stable baseline measurements of blood pressure and heart rate. A double-barreled glass micropipette (World Precision Instruments; 1 mm outer diameters, ~50 μm diameter at the tip) was lowered into the left or right PVN (1.8 mm posterior to bregma; 1.7 mm lateral to the midline; 7.64 mm below the brain surface at a 10° tilt toward the midline). Each rat was injected with one of the following pretreatments: aCSF (n = 6), the TrkB-selective inhibitor ANA-12 (2 μg; n = 6), the AT1R antagonist losartan (10 μg; n = 6), or the angiotensin converting enzyme (ACE) inhibitor lisinopril (200 ng; n = 5), followed by BDNF (12.5 ng) 15 min later. Each drug was infused at a rate of 50 nL/s over 5 s. Drug doses were based on previous studies (Busnardo et al., 2014; Cazorla et al., 2011) and our preliminary tests. An additional experimental group received intravenous hexamethonium (30 mg/kg; n = 4) through the femoral vein catheter split by a 3-way stopcock, followed by intra-PVN BDNF (12.5 ng) 5 min later. Recording continued for an additional 15 min after BDNF microinjection. At the end of experiments, rats were sacrificed by decapitation under deep isoflurane anesthesia. Brains were then collected, fixed in 4% paraformaldehyde, freeze-protected with 30% sucrose solution and sectioned for verification of injection sites (Fig. 1).
2.1.3. Isolation and incubation of hypothalamic tissue punches
The purpose of these experiments was to study the effect of BDNF on TrkB receptor activation and autophosphorylation in hypothalamic tissue samples including the PVN extracted from SD rats. Rats were deeply anesthetized under 5% isoflurane and decapitated. Brains were quickly extracted and placed in a brain block for dissection of 2-mm thick coronal sections corresponding to the location of the PVN. Slices were immediately placed in Dulbecco's Modified Eagle Medium (DMEM; Thermo-Fisher, Waltham, MA) containing 20 mM HEPES over ice and bilateral 1-mm-diameter punches containing the PVN were isolated under a dissection microscope. Tissue punches were then placed in 500-μL centrifuge tubes containing 40 μL of 37 °C DMEM-HEPES and one of the following pretreatments: aCSF (n = 6), losartan (1 μM final concentration; n = 5) or ANA-12 (100 μM final concentration; n = 7). The punches were incubated with gentle shaking for 30 min at 37 °C. Then, 10 μL of either aCSF or BDNF (to reach final concentration of 25 ng/mL) was added to each tube and tissue samples were incubated for an additional 30 min. Samples were then homogenized in modified RIPA buffer containing 150 mM NaCl, 50 mM Tris (pH 8.0), 5 mM EDTA, 0.5% deoxycholate, 1% Nonidet P-40 and 10% glycerol mixed with a protease inhibitor cocktail (Sigma-P2714) according to the manufacturer's instructions and phosphatase inhibitors (Na-pyrophosphate and Na-orthovanadate – 2 mM each). Samples were stored at −80 °C until further processing. Although this technique provides a sample that consists mostly of PVN tissue, hypothalamic regions adjacent to the PVN that also express TrkB, such as the medial preoptic nucleus and area (Merlio et al., 1992), may also be partially included in the brain punches. Each rat served as its own control: the same pretreatment was applied to both hypothalamic punches from each animal and then one sample was treated with aCSF and the other with BDNF.
2.2. Western blot
Protein content of hypothalamic punch homogenates was determined by Bradford assay, and equal amount of protein (24 μg) for each sample was separated by SDS-PAGE on 8% gel. Protein was then transferred to nitrocellulose membrane and probed overnight with specific antibodies for TrkB (1:400, Alomone Labs) and anti-phospho-(Y490/Y515)-Trk (1:1000, Cell Signaling Technology). An anti-β-tubulin (1:1000, Cell Signaling Technology) antibody was used to correct for loading errors. Blots were quantified using densitometry and data were expressed as the ratio of phospho-Trk to total TrkB expression.
2.3. Drugs
The recipe for aCSF was obtained from ALZET (Cupertino, CA) and contained 148 mM NaCl, 3 mM KCl, 1.4 mM CaCl2, 0.8 mM MgCl2, 1.5 mM Na2HPO4 and 0.18 mM NaH2PO4. Human mature BDNF was purchased from Phoenix Pharmaceuticals (Burlingame, CA), losartan and ANA-12 were purchased from Sigma-Aldridge (St. Louis, MO), and lisinopril was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.4. Data collection and statistical analysis
Blood pressure and heart rate were recorded using Powerlab (8/35) and LabChart software version 8.0.7 (ADInstruments, Dunedin, New Zealand) at a 1000 Hz sampling rate and condensed to 30-second moving averages for data analysis and presentation. Mean arterial pressure (MAP) and heart rate responses were assessed by averaging the values of the 30-s moving average segments over 15 min following BDNF microinjections, and peak responses were defined as the highest 30-s moving average segment value. Data are presented as mean ± SEM. Average and peak changes in MAP and heart rate from microinjection experiments were analyzed by one-way ANOVA with Tukey's post-hoc test for further differences among treatments. Western blot data were analyzed by two-way ANOVA and Tukey's post-hoc test. Statistical tests were performed using Prism 6.0 software (GraphPad, San Diego, CA). The criterion for statistical significance was P < 0.05.
3. Results
3.1. Cardiovascular responses to BDNF microinjections in conscious rats
Baseline MAP and heart rate were unaffected by icv infusions of either aCSF or losartan (Table 1). Following icv aCSF infusion, PVN microinjection of aCSF had no effect on MAP and heart rate, while BDNF microinjection into the PVN caused significant increases in MAP and heart rate producing peak changes of 27 ± 1 mm Hg (P < 0.001 vs aCSF) and 106 ± 17 bpm (P < 0.001 vs aCSF), respectively, that returned to baseline after approximately 15 min (Fig. 2). Rats pretreated with icv losartan displayed significantly reduced average and peak MAP responses to BDNF compared to rats pretreated with icv aCSF (P < 0.01), although these responses were significantly higher compared to responses to PVN microinjections of aCSF (Fig. 2, B and C). In contrast, losartan pretreatment had no effect on BDNF-induced heart rate elevations (Fig. 2, B and C). Peripheral autonomic blockade with hexamethonium reduced baseline MAP (Table 1) and lowered average and peak increases in MAP to BDNF to a level similar to MAP responses recorded in rats pretreated with losartan (Fig. 2, B and C). However, in contrast to losartan pretreatment, the heart rate response to BDNF was abolished by hexamethonium (Fig. 2, B and C).
Table 1.
MAP and heart rate values at baseline and 15 min after icv infusion of aCSF or losartan, or 5 min after intravenous hexamethonium in conscious rats.
| Group | N | MAP (mm Hg) | Heart rate (bpm) |
|---|---|---|---|
| aCSF | 7 | ||
| Baseline | 95 ± 4 | 379 ± 14 | |
| 15 min | 99 ± 6 | 383 ± 10 | |
| Losartan | 7 | ||
| Baseline | 97 ± 3 | 397 ± 7 | |
| 15 min | 97 ± 3 | 393 ± 17 | |
| Hexamethonium | 4 | ||
| Baseline | 96 ± 4 | 391 ± 11 | |
| 5 min | 62 ± 5** | 386 ± 21 |
P < 0.01 vs baseline.
Fig. 2.
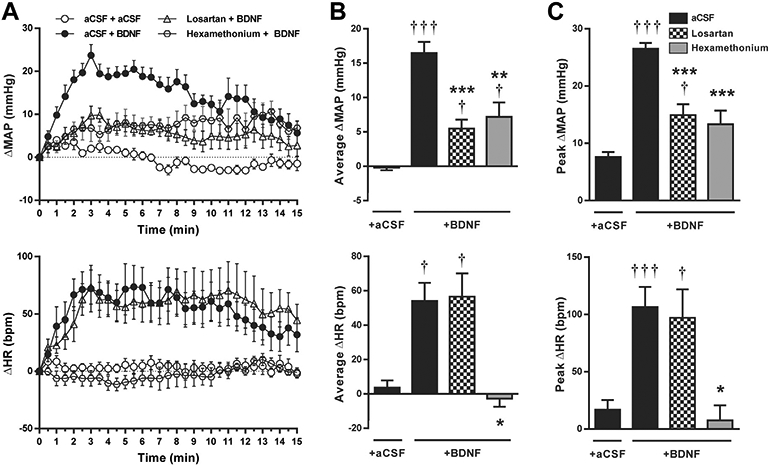
Changes in MAP (top panels) and heart rate (bottom panels) following PVN microinjections in conscious SD rats. A: Time course of changes in MAP and heart rate after PVN microinjections of aCSF or BDNF (at 0 min) following pretreatment with icv aCSF (n = 7), icv losartan (n = 7), or intravenous hexamethonium (n = 4). B: Changes in MAP and heart rate averaged over 15 min following BDNF or aCSF microinjections. C: Amplitude of peak MAP and heart rate responses to BDNF or aCSF microinjections. †P < 0.05, and †††P < 0.001 intra-PVN BDNF vs aCSF; *P < 0.05, **P < 0.01, and ***P < 0.001 pretreatment effectvs aCSF.
3.2. Cardiovascular responses to BDNF microinjections in anesthetized rats
MAP and heart rate were unchanged from baseline by pretreatment with PVN microinjection of aCSF, ANA-12, losartan or lisinopril (Table 2). Similar to conscious rats, PVN microinjection of BDNF in anesthetized rats pretreated with aCSF elicited significant increases in MAP (31 ± 4 mm Hg, P < 0.001 vs aCSF) that recovered toward baseline within 15 min (Fig. 3A). However, in contrast to conscious rats, BDNF lowered heart rate by a peak change of −33 ± 7 bpm (P < 0.001 vs aCSF; Fig. 3C). Blockade of PVN TrkB receptors by microinjection of ANA-12 significantly attenuated MAP and heart rate responses to BDNF and these responses were not significantly different from those elicited by aCSF microinjection alone (Fig. 3). Pretreatment with intra-PVN losartan or lisinopril also blocked MAP increases to BDNF (Fig. 3). However, while losartan significantly reduced heart rate responses, the effects of lisinopril on average and peak heart rate changes were not statistically significant (P = 0.19 and P = 0.24; Fig. 3B and 3C, respectively). Pretreatment with intravenous hexamethonium lowered baseline MAP without affecting heart rate (Table 2) and abolished average and peak MAP and heart rate responses to BDNF (Fig. 3). BDNF-induced MAP and heart rate responses seemed to be selective to the PVN, since BDNF injections into adjacent regions of the hypothalamus (juxtaparaventricular or peduncular parts of the lateral hypothalamus) or the thalamus (ventrolateral area of the reuniens thalamic nucleus) had no effect on MAP and heart rate (average changes over 15 min were 1 ± 1 mm Hg and −4 ± 3 bpm, n = 4).
Table 2.
MAP and heart rate values at baseline and 15 min after PVN infusion of aCSF, ANA-12, losartan, lisinopril, or 5 min after intravenous hexamethonium in anesthetized rats.
| Group | N | MAP (mm Hg) | Heart rate (bpm) |
|---|---|---|---|
| aCSF | 6 | ||
| Baseline | 88 ± 3 | 296 ± 15 | |
| 15 min | 89 ± 4 | 281 ± 16 | |
| ANA-12 | 6 | ||
| Baseline | 82 ± 3 | 271 ± 13 | |
| 15 min | 83 ± 3 | 272 ± 13 | |
| Losartan | 6 | ||
| Baseline | 86 ± 3 | 293 ± 9 | |
| 15 min | 92 ± 4 | 284 ± 10 | |
| Lisinopril | 5 | ||
| Baseline | 88 ± 4 | 284 ± 5 | |
| 15 min | 88 ± 4 | 291 ± 6 | |
| Hexamethonium | 4 | ||
| Baseline | 81 ± 2 | 276 ± 9 | |
| 15 min | 62 ± 1** | 286 ± 13 |
P < 0.01 vs baseline.
Fig. 3.
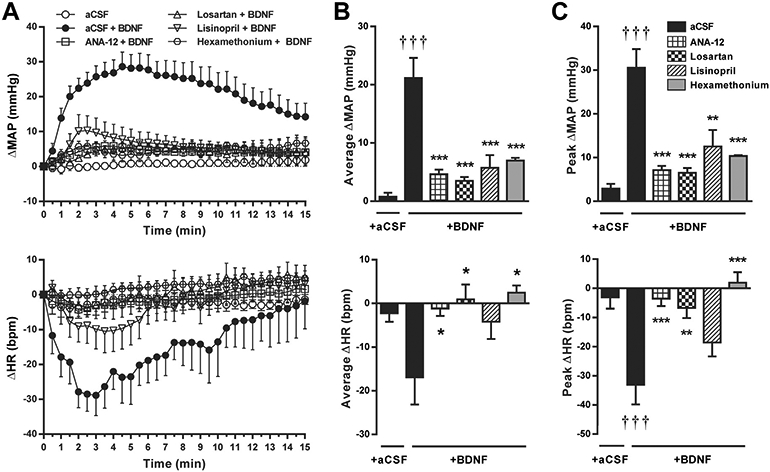
Changes in MAP (top panels) and heart rate (bottom panels) following PVN microinjections in anesthetized SD rats. A: Time course of changes in MAP and heart rate after PVN microinjections of aCSF or BDNF (at 0 min) following pretreatment with intra-PVN injections of aCSF (n = 6), ANA-12 (n = 6), losartan (n = 6), lisinopril (n = 5), or intravenous hexamethonium (n = 4). B: Changes in MAP and heart rate averaged over 15 min following aCSF or BDNF microinjections. C: Amplitude of peak MAP and heart rate responses to aCSF or BDNF microinjections. †††P < 0.001 intra-PVN BDNF vs aCSF; *P < 0.05, **P < 0.01, and ***P < 0.001 pretreatment effect vs aCSF.
3.3. BDNF-induced TrkB receptor phosphorylation in the PVN
Neither total TrkB expression nor the phospho-Trk to TrkB ratio was significantly altered by aCSF, ANA-12 or losartan pretreatments. However, BDNF significantly increased the ratio of phospho-Trk to TrkB in PVN samples pretreated with aCSF (P < 0.05; Fig. 4) indicating BDNF-induced activation of TrkB receptors. In contrast, the addition of BDNF to PVN samples pretreated with ANA-12 or losartan did not significantly change the phospho-Trk to TrkB ratio (Fig. 4).
Fig. 4.
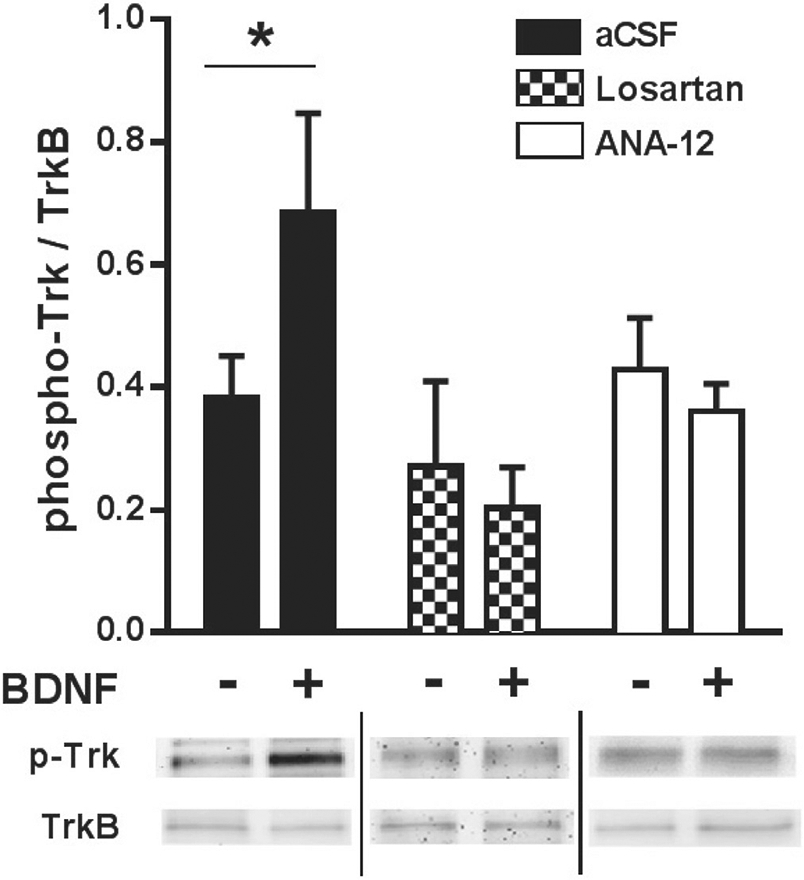
Ratio of phosphorylated Trk (p-Trk) to total TrkB protein expression in PVN tissue samples after in vitro pre-incubation in DMEM-HEPES + aCSF (n = 6), + losartan (n = 5) or + ANA-12 (n = 7) with or without addition of BDNF. *P < 0.05.
4. Discussion
In this study, we demonstrate for the first time that microinjection of exogenous BDNF into the PVN significantly elevates blood pressure both in conscious and anesthetized rats and that these responses are mediated by TrkB receptors, Ang II-AT1R signaling and the autonomic nervous system. in addition, we provide the first evidence that BDNF-induced TrkB receptor activation and autophosphorylation are prevented by AT1R blockade. Collectively, these results suggest an interaction between BDNF and Ang ii signaling in the PVN that contributes to short-term elevations in blood pressure and establishes BDNF within the PVN as a novel mediator of acute hypertensive responses.
4.1. BDNF-mediated mechanisms in cardiovascular regulatory nuclei
Recent investigations indicate that BDNF within medullary nuclei such as the NTS and RVLM is involved in the neuronal control of blood pressure and heart rate. For example, BDNF is highly expressed at synapses along the arterial baroreceptor reflex pathway in adult rats, including in baroreceptor afferent neurons of the nodose ganglion, first-order baroreceptor neurons in the NTS and terminal projections in the lower NTS (Martin et al., 2009, 2012). Furthermore, inhibition of BDNF TrkB receptors in the NTS decreases, whereas microinjection of BDNF increases blood pressure, heart rate, and lumbar sympathetic nerve activity in anesthetized rats by modulating activity of excitatory glutamatergic neurons (Clark et al., 2011). In addition, BDNF microinjection into the rat RVLM elicits rapid increases in blood pressure without changing heart rate (Wang and Zhou, 2002). Thus, BDNF exerts hypertensive effects in brainstem nuclei mediating cardiovascular pressor responses and sympathetic activity, suggesting a role for BDNF in central blood pressure regulation. However, the acute cardiovascular effects of BDNF within the PVN have not been previously studied, despite previous reports that BDNF expression increases significantly in the PVN in response to hypertensive stimuli including stress and hyperosmolarity (Aliaga et al., 2002; Hammack et al., 2009; Smith et al., 1995a, 1995b).
Our present study demonstrates that BDNF microinjections in the PVN elicit hypertensive effects in both conscious and anesthetized SD rats. In conscious rats, PVN microinjection of BDNF transiently but markedly increased MAP and heart rate, whereas in anesthetized rats BDNF microinjections increased MAP but decreased heart rate. Thus, although the effect of BDNF microinjections on MAP is of comparable magnitude to studies in the NTS and RVLM in anesthetized rats (Clark et al., 2011; Wang and Zhou, 2002), heart rate responses to BDNF vary considerably among the NTS, RVLM and PVN, as well as between conscious and anesthetized animals in the PVN. Furthermore, there are conflicting reports on the effect of icv BDNF infusion, which may increase blood pressure without changing heart rate in rats (Wang et al., 2012) but decrease heart rate without changing blood pressure in mice (Wan et al., 2014). Together, these results suggest that the actions of BDNF with respect to blood pressure and heart rate regulation are likely region-specific. During our studies in conscious rats, a noticeable increase in locomotor activity and grooming behavior accompanied the hypertensive responses to BDNF infusion (unpublished observation) consistent with our previous finding that long-term overexpression of BDNF in the PVN is associated with significantly increased nighttime physical activity (Erdos et al., 2015). Hexamethonium pretreatment, which significantly reduced the pressor response and abolished the heart rate response to BDNF, also noticeably reduced locomotor activity in conscious rats (unpublished observation) possibly as a side effect of reduced resting blood pressure (Shyu and Thoren, 1986). Hypothalamic BDNF is a known regulator of locomotor activity (Kernie et al., 2000), so we cannot discount that tachycardic responses elicited by BDNF in the PVN may be driven indirectly by increased physical activity in conscious rats. In contrast, the bradycardic response elicited by BDNF in anesthetized rats may be interpreted as a baroreceptor reflex response to increased blood pressure in the absence of increased physical activity. In addition, we cannot exclude the possibility that differences in injection techniques, such as the larger diameter of the stainless steel injector compared to the glass micropipette tip and the slower infusion rate but larger infusion volume used in conscious rats vs anesthetized rats may have resulted in different distribution of BDNF within the PVN potentially leading to altered cardiovascular responses.
4.2. Cross-talk between Ang II- and BDNF-mediated cardiovascular regulatory mechanisms
Mounting evidence supports an interaction between BDNF and AT1R signaling mechanisms that may play a significant role in acute and long-term central blood pressure regulation, including in the PVN as our results demonstrate. For instance, Ang II acutely upregulates BDNF protein expression and induces TrkB signaling in the RVLM of Wistar-Kyoto rats and in human and rat adrenocortical cells through AT1R mechanisms (Chan et al., 2010; Szekeres et al., 2010). As we previously demonstrated, overexpression of BDNF in the PVN markedly increases PVN AT1R mRNA while decreases Mas receptor mRNA expression (Erdos et al., 2015), which may create an imbalance between the hypertensive Ang II-AT1R and counterbalancing Ang-(1–7)-Mas receptor axes as a long-term consequence (Dupont and Brouwers, 2010; Xu et al., 2011). Notably, a recent study in cultured CATH.a neurons found that inhibiting BDNF attenuates Ang II-induced suppression of the voltage-gated rapidly inactivating K+ current IA (Becker et al., 2015), a known Ang II-mediated mechanism for increased neuronal excitability that augments sympathetic outflow (Ma et al., 2006), including in sympathoregulatory neurons in the PVN (Li and Ferguson, 1996; Luther and Tasker, 2000).
Here, we provide further evidence for complex interaction between BDNF and Ang II mechanisms. Inhibition of central AT1Rs by pretreatment with icv losartan significantly attenuated but did not abolish the increase in blood pressure by PVN infusion of BDNF in conscious rats, mirroring the effect of chronic icv losartan infusion on blood pressure elevations following BDNF overexpression in the PVN (Erdos et al., 2015). However, icv losartan had no effect on the heart rate responses to BDNF microinjection, suggesting that central AT1Rs are involved in mediating the MAP but not the heart rate responses to acute BDNF infusions in the PVN. Interestingly, these findings are in line with a recent observation that losartan microinjected into the PVN reduces the increase in MAP during acute restraint stress without changing the heart rate response (Busnardo et al., 2014), suggesting that PVN AT1R signaling is not involved in mediating acute tachycardic responses. Acute heart rate responses to intra-PVN BDNF may instead be mediated indirectly by increases in physical activity, although the pathways producing these responses to stress or BDNF infusion have yet to be elucidated.
Icv infusion of losartan may affect Ang II signaling in multiple sites besides the PVN. Therefore, we also conducted microinjection experiments in anesthetized rats, which allowed pretreatment injections of inhibitors and subsequent injections of BDNF within the same region of the PVN using double-barreled glass micropipettes. These studies demonstrate that BDNF-induced acute pressor responses depend on AT1R mechanisms within the PVN. Furthermore, the decrease in average and peak MAP responses to BDNF we observed following intra-PVN pretreatment with lisinopril indicates that the Ang II involved in the BDNF-induced pressor response is produced locally, consistent with previous findings that lisinopril in the PVN significantly reduced MAP increases during restraint stress to a level comparable to blockade of AT1Rs in the PVN (Busnardo et al., 2014).
One potential mechanism by which Ang II may interact with BDNF signaling is through transactivation of TrkB receptors by AT1Rs. Numerous G protein-coupled receptors, including AT1R, are known to transactivate receptor tyrosine kinases (RTKs) such as TrkB (see review by Lee et al., 2002). Indeed, Ang II is known to induce AT1R-mediated transactivation of several RTKs including insulin-like growth factor receptor and epidermal growth factor in vascular smooth muscle and cardiac myocytes (Takayanagi et al., 2015; Yin et al., 2003). These interactions result in RTK phosphorylation and recruitment of adaptor proteins and downstream tyrosine kinases necessary for activation of phospholipase C-γ and mitogen-activated protein kinase signaling pathways (Yin et al., 2003), which are also common downstream signaling targets shared by AT1R and TrkB receptors in the central nervous system (Numakawa et al., 2010; Wei et al., 2009). Although specific TrkB receptor transactivation by Ang II has yet to be confirmed, our data obtained from freshly isolated rat hypothalamic punches indicate that AT1R signaling is required for BDNF-induced activation and autophosphorylation of TrkB, suggesting that Ang II exerts a permissive role on BDNF signaling via TrkB possibly through an AT1R-TrkB interaction. However, further investigation is required to determine the functional and biochemical nature of a BDNF-Ang II interaction, in particular with respect to its role in central control of blood pressure during normo- and hypertensive conditions.
4.3. Downstream mechanisms potentially involved in BDNF-mediated cardiovascular regulation
The synergistic interaction between BDNF and Ang II signaling pathways could stimulate PVN sympathoregulatory neurons through a variety of ion channels, neuromodulators and receptors. For example, acute application of BDNF is known to increase depolarizing cation currents by activating transient receptor potential canonical-3 (TRPC3) channels (Li et al., 1999, 2010) and Nav1.9 sodium channels (Blum et al., 2002), and BDNF may also increase neuronal excitability by decreasing IA K+ currents (Becker et al., 2015). BDNF has also been shown to rapidly enhance NMDA receptor-mediated synaptic transmission (Lin et al., 1998; Song et al., 1998), which may activate sympathoregulatory PVN neurons that receive glutamatergic input (Dampney et al., 2005; Nunn et al., 2011). Finally, BDNF may increase vasopressin content (Aliaga et al., 2002) and could activate hypothalamic vasopressin-secreting neurons by diminishing GABAergic inhibition over these cells (Choe et al., 2015). The resulting vasopressin release into the circulation could increase blood pressure by peripheral vasoconstriction (Henderson and Byron, 2007), although a recent study suggests that vasopressin release within the PVN could also elevate sympathetic activity, heart rate and blood pressure via vasopressin V1b receptors (El-Werfali et al., 2015). In our studies, autonomic blockade significantly diminished but did not completely abolish blood pressure elevations by BDNF, indicating that both central V1b-mediated sympathoexcitation and circulating vasopressin-induced vasoconstriction could potentially be involved in mediating pressor responses.
5. Conclusions and perspectives
We have investigated changes in MAP and heart rate in response to acute PVN infusion of BDNF in conscious and anesthetized rats and conclude that exogenous BDNF in the PVN acutely increases blood pressure under the permissive control of local Ang II-AT1R mechanisms. These findings support a novel mechanism for central control of blood pressure at the level of the PVN by bidirectional interaction between BDNF and Ang II that may have important implications for understanding hypertensive responses to various conditions that influence hypothalamic levels of BDNF (Fig. 5) including stress (Givalois et al., 2004; Hammack et al., 2009; Smith et al., 1995a), aging (Smith and Cizza, 1996), increased dietary salt intake (Aliaga et al., 2002; Choe et al., 2015; Hasegawa et al., 2016), exercise (Nadel et al., 2013) and obesity (Wang et al., 2010).
Fig. 5.

Schematic working hypothesis for the bidirectional interaction between Ang II- and BDNF-signaling mechanisms and for potential downstream targets of BDNF and TrkB in PVN neurons. Hypertensive stimuli result in the release of excitatory mediators such as glutamate and Ang II leading to increased neuronal activity and activity-dependent synthesis and release of BDNF within the PVN. BDNF activates TrkB receptors on PVN neurons in a paracrine or autocrine fashion, but this activation is under permissive control of Ang II and AT1R. TrkB receptor activation in turn may alter ion channel function (TRPC3, Nav1.9 or potassium channels) and vasopressin expression acutely resulting in further increases in PVN neuronal activity, sympathetic tone and blood pressure, whereas long-term activation of TrkB may lead to elevations in AT1R expression. Thus, the role of BDNF-mediated mechanisms in the PVN may involve both acute amplification of hypertensive stimuli relayed to the PVN under permissive control of Ang II and chronic adaptive mechanisms that alter AT1R expression.
Acknowledgements
This work was supported by the American Heart Association (11SDG7560022 to B.E.), by the American Federation of Aging Research (AFAR Research Grant to B.E.), and by start-up funds from the University of Vermont.
Footnotes
Conflict of interest
None.
References
- Aliaga E, Arancibia S, Givalois L, Tapia-Arancibia L, 2002. Osmotic stress increases brain-derived neurotrophic factor messenger RNA expression in the hypothalamic supraoptic nucleus with differential regulation of its transcripts. Relation to arginine-vasopressin content. Neuroscience 112, 841–850. [DOI] [PubMed] [Google Scholar]
- Andero R, Choi DC, Ressler KJ, 2014. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog. Mol. Biol. Transl. Sci 122, 169–192. [DOI] [PubMed] [Google Scholar]
- Becker BK, Wang HJ, Tian C, Zucker IH, 2015. BDNF contributes to angiotensin II-mediated reductions in peak voltage-gated K+ current in cultured CATH.a cells. Physiol. Rep 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Medina JH, 2014. BDNF and memory processing. Neuropharmacology 76 (Pt C), 677–683. [DOI] [PubMed] [Google Scholar]
- Blum R, Kafitz KW, Konnerth A, 2002. Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature 419, 687–693. [DOI] [PubMed] [Google Scholar]
- Brahim JS, Thut PD, 1984. Hemodynamic changes during isoflurane anesthesia. Anesth. Prog. 31, 207–212. [PMC free article] [PubMed] [Google Scholar]
- Busnardo C, Tavares RF, Correa FM, 2014. Angiotensinergic neurotransmission in the paraventricular nucleus of the hypothalamus modulates the pressor response to acute restraint stress in rats. Neuroscience 270, 12–19. [DOI] [PubMed] [Google Scholar]
- Cassaglia PA, Shi Z, Li B, Reis WL, Clute-Reinig NM, Stern JE, Brooks VL, 2014. Neuropeptide Y acts in the paraventricular nucleus to suppress sympathetic nerve activity and its baroreflex regulation. J. Physiol 592, 1655–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D, 2011. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Invest 121, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Wu CW, Chang AY, Hsu KS, Chan JY, 2010. Transcriptional upregulation of brain-derived neurotrophic factor in rostral ventrolateral medulla by angiotensin II: significance in superoxide homeostasis and neural regulation of arterial pressure. Circ. Res 107, 1127–1139. [DOI] [PubMed] [Google Scholar]
- Chen QH, Toney GM, 2001. AT(1)-receptor blockade in the hypothalamic PVN reduces central hyperosmolality-induced renal sympathoexcitation. Am. J. Physiol. Regul. Integr. Comp. Physiol 281, R1844–R1853. [DOI] [PubMed] [Google Scholar]
- Choe KY, Han SY, Gaub P, Shell B, Voisin DL, Knapp BA, Barker PA, Brown CH, Cunningham JT, Bourque CW, 2015. High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron 85, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CG, Hasser EM, Kunze DL, Katz DM, Kline DD, 2011. Endogenous brain-derived neurotrophic factor in the nucleus tractus solitarius tonically regulates synaptic and autonomic function. J. Neurosci 31, 12318–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J, Killinger S, Sheriff MJ, Tan PS, McDowall LM, 2005. Long-term regulation of arterial blood pressure by hypothalamic nuclei: some critical questions. Clin. Exp. Pharmacol. Physiol 32, 419–425. [DOI] [PubMed] [Google Scholar]
- Davern PJ, Chen D, Head GA, Chavez CA, Walther T, Mayorov DN, 2009. Role of angiotensin II Type 1A receptors in cardiovascular reactivity and neuronal activation after aversive stress in mice. Hypertension 54, 1262–1268. [DOI] [PubMed] [Google Scholar]
- Dupont AG, Brouwers S, 2010. Brain angiotensin peptides regulate sympathetic tone and blood pressure. J. Hypertens 28, 1599–1610. [DOI] [PubMed] [Google Scholar]
- El-Werfali W, Toomasian C, Maliszewska-Scislo M, Li C, Rossi NF, 2015. Haemodynamic and renal sympathetic responses to V1b vasopressin receptor activation within the paraventricular nucleus. Exp. Physiol 100, 553–565. [DOI] [PubMed] [Google Scholar]
- Erdos B, Backes I, McCowan ML, Hayward LF, Scheuer DA, 2015. Brain-derived neurotrophic factor modulates angiotensin signaling in the hypothalamus to increase blood pressure in rats. Am. J. Physiol. Heart Circ. Physiol 308, H612–H622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givalois L, Naert G, Rage F, Ixart G, Arancibia S, Tapia-Arancibia L, 2004. A single brain-derived neurotrophic factor injection modifies hypothalamo-pituitary-adrenocortical axis activity in adult male rats. Mol. Cell. Neurosci 27, 280–295. [DOI] [PubMed] [Google Scholar]
- Hammack SE, Cheung J, Rhodes KM, Schutz KC, Falls WA, Braas KM, May V, 2009. Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology 34, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Nakagawa T, Uekawa K, Ma M, Lin B, Kusaka H, Katayama T, Sueta D, Toyama K, Koibuchi N, Kim-Mitsuyama S, 2016. Therapy with the combination of amlodipine and irbesartan has persistent preventative effects on stroke onset associated with BDNF preservation on cerebral vessels in hypertensive rats. Transl. Stroke Res 7, 79–87. [DOI] [PubMed] [Google Scholar]
- Henderson KK, Byron KL, 2007. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J. Appl. Physiol (1985) 102, 1402–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF, 2000. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal G, Afonso PM, Salazar IL, Duarte CB, 2015. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 1621, 82–101. [DOI] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Chao MV, 2002. Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 13, 11–17. [DOI] [PubMed] [Google Scholar]
- Li Z, Ferguson AV, 1996. Electrophysiological properties of paraventricular magnocellular neurons in rat brain slices: modulation of IA by angiotensin II. Neuroscience 71, 133–145. [DOI] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C, 1999. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 24, 261–273. [DOI] [PubMed] [Google Scholar]
- Li Y, Calfa G, Inoue T, Amaral MD, Pozzo-Miller L, 2010. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2 + elevations in CA3 pyramidal neurons. J. Neurophysiol 103, 2846–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB, 1998. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res. Mol. Brain Res 55, 20–27. [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Lu Y, 2014. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb. Exp. Pharmacol 220, 223–250. [DOI] [PubMed] [Google Scholar]
- Luther JA, Tasker JG, 2000. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J. Physiol 523 (Pt 1), 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Bielefeldt K, Tan ZY, Whiteis CA, Snitsarev V, Abboud FM, Chapleau MW, 2006. Dual mechanisms of angiotensin-induced activation of mouse sympathetic neurones. J. Physiol 573, 45–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Jenkins VK, Hsieh HY, Balkowiec A, 2009. Brain-derived neurotrophic factor in arterial baroreceptor pathways: implications for activity-dependent plasticity at baroafferent synapses. J. Neurochem 108, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Brown AL, Balkowiec A, 2012. Glia determine the course of brain-derived neurotrophic factor-mediated dendritogenesis and provide a soluble inhibitory cue to dendritic growth in the brainstem. Neuroscience 207, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlio JP, Ernfors P, Jaber M, Persson H, 1992. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience 51, 513–532. [DOI] [PubMed] [Google Scholar]
- Nadel J, Huang T, Xia Z, Burlin T, Zametkin A, Smith CB, 2013. Voluntary exercise regionally augments rates of cerebral protein synthesis. Brain Res. 1537, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H, 2010. BDNF function and intracellular signaling in neurons. Histol. Histopathol 25, 237–258. [DOI] [PubMed] [Google Scholar]
- Nunn N, Womack M, Dart C, Barrett-Jolley R, 2011. Function and pharmacology of spinally-projecting sympathetic pre-autonomic neurones in the paraventricular nucleus of the hypothalamus. Curr. Neuropharmacol 9, 262–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C, 2014. The Rat Brain in Stereotaxic Coordinates. seventh ed. Academic Press, San Diego, CA, USA. [Google Scholar]
- Saavedra JM, Ando H, Armando I, Baiardi G, Bregonzio C, Jezova M, Zhou J, 2004. Brain angiotensin II, an important stress hormone: regulatory sites and therapeutic opportunities. Ann. N. Y. Acad. Sci 1018, 76–84. [DOI] [PubMed] [Google Scholar]
- Shyu BC, Thoren P, 1986. Circulatory events following spontaneous muscle exercise in normotensive and hypertensive rats. Acta Physiol. Scand 128, 515–524. [DOI] [PubMed] [Google Scholar]
- Smith MA, Cizza G, 1996. Stress-induced changes in brain-derived neurotrophic factor expression are attenuated in aged Fischer 344/N rats. Neurobiol. Aging 17, 859–864. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kim SY, Kvetnansky R, 1995a. Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology 136, 3743–3750. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM, 1995b. Effects of stress on neurotrophic factor expression in the rat brain. Ann. N. Y. Acad. Sci 771, 234–239. [DOI] [PubMed] [Google Scholar]
- Song DK, Choe B, Bae JH, Park WK, Han IS, Ho WK, Earm YE, 1998. Brain-derived neurotrophic factor rapidly potentiates synaptic transmission through NMDA, but suppresses it through non-NMDA receptors in rat hippocampal neuron. Brain Res. 799, 176–179. [DOI] [PubMed] [Google Scholar]
- Szekeres M, Nadasy GL, Turu G, Supeki K, Szidonya L, Buday L, Chaplin T, Clark AJ, Hunyady L, 2010. Angiotensin II-induced expression of brain-derived neurotrophic factor in human and rat adrenocortical cells. Endocrinology 151, 1695–1703. [DOI] [PubMed] [Google Scholar]
- Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, Davisson RL, Park JY, Eguchi S, 2015. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 65, 1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Weigand LA, Bateman R, Griffioen K, Mendelowitz D, Mattson MP, 2014. Evidence that BDNF regulates heart rate by a mechanism involving increased brainstem parasympathetic neuron excitability. J. Neurochem 129, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhou XF, 2002. Injection of brain-derived neurotrophic factor in the rostral ventrolateral medulla increases arterial blood pressure in anaesthetized rats. Neuroscience 112, 967–975. [DOI] [PubMed] [Google Scholar]
- Wang C, Godar RJ, Billington CJ, Kotz CM, 2010. Chronic administration of brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus reverses obesity induced by high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol 298, R1320–R1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MF, Chan YC, Lee HT, Hong LZ, 2012. Regulation of the intracerebroventricular administration of brain-derived neurotrophic factor on baroreflex function and insulin sensitivity in rats. Chin. J. Physiol 55, 184–191. [DOI] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB, 2009. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am. J. Physiol. Heart Circ. Physiol 296, H1425–H1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Sriramula S, Lazartigues E, 2011. ACE2/ANG-(1-7)/Mas pathway in the brain: the axis of good. Am. J. Physiol. Regul. Integr. Comp. Physiol 300, R804–R817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G, Yan C, Berk BC, 2003. Angiotensin II signaling pathways mediated by tyrosine kinases. Int. J. Biochem. Cell Biol 35, 780–783. [DOI] [PubMed] [Google Scholar]