

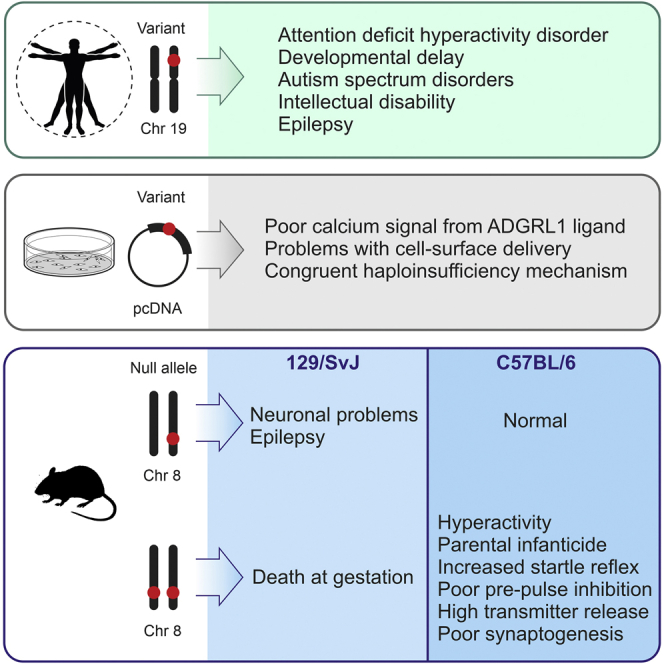
Summary
ADGRL1 (latrophilin 1), a well-characterized adhesion G protein-coupled receptor, has been implicated in synaptic development, maturation, and activity. However, the role of ADGRL1 in human disease has been elusive. Here, we describe ten individuals with variable neurodevelopmental features including developmental delay, intellectual disability, attention deficit hyperactivity and autism spectrum disorders, and epilepsy, all heterozygous for variants in ADGRL1. In vitro, human ADGRL1 variants expressed in neuroblastoma cells showed faulty ligand-induced regulation of intracellular Ca2+ influx, consistent with haploinsufficiency. In vivo, Adgrl1 was knocked out in mice and studied on two genetic backgrounds. On a non-permissive background, mice carrying a heterozygous Adgrl1 null allele exhibited neurological and developmental abnormalities, while homozygous mice were non-viable. On a permissive background, knockout animals were also born at sub-Mendelian ratios, but many Adgrl1 null mice survived gestation and reached adulthood. Adgrl1−/− mice demonstrated stereotypic behaviors, sexual dysfunction, bimodal extremes of locomotion, augmented startle reflex, and attenuated pre-pulse inhibition, which responded to risperidone. Ex vivo synaptic preparations displayed increased spontaneous exocytosis of dopamine, acetylcholine, and glutamate, but Adgrl1−/− neurons formed synapses in vitro poorly. Overall, our findings demonstrate that ADGRL1 haploinsufficiency leads to consistent developmental, neurological, and behavioral abnormalities in mice and humans.
Keywords: ADGRL1, Adgrl1 knockout mice, developmental delay, neuropsychiatric disorders, epilepsy, autism spectrum disorder, ASD, attention deficit hyperactivity disorder, ADHD, intellectual disability, variable expressivity, malfunctional behavior in mice
Graphical abstract
ADGRL1 encodes an adhesion G protein-coupled receptor implicated in synaptic development and activity. We conducted clinical and biochemical studies in humans with ADGRL1 variants and behavioral and cellular studies in Adgrl1 null mice. Our findings demonstrate that ADGRL1 haploinsufficiency leads to consistent developmental, neurological, and behavioral abnormalities in mice and humans.
Introduction
G protein-coupled receptors (GPCRs) are seven-transmembrane cell-surface receptors that mediate a plethora of cellular responses to a variety of stimuli, regulate many important physiological functions, and represent targets of 34% of all FDA-approved drugs.1, 2, 3, 4, 5 The members of the adhesion GPCR family (aGPCRs) interact with membrane-bound proteins, extracellular matrix components, or soluble molecules and play a critical role in central nervous system development,6, 7, 8, 9, 10, 11, 12, 13 synapse formation,14 myelination,15 and immunity.16 These receptors feature a distinctive structure composed of a large, adhesion-like N-terminal extracellular region containing a “GPCR autoproteolysis-inducing” (GAIN) domain and a signaling domain characterized by seven transmembrane helices with interconnecting loops and a cytosolic C-terminal tail. In most aGPCRs, the GAIN domain constitutively cleaves the receptor at the “GPCR proteolysis site” (GPS) into N- and C-terminal fragments (NTFs and CTFs) that are non-covalently linked to each other at the cell membrane.17, 18, 19
The aGPCR latrophilin 1 (LPHN1 or ADGRL1) (ADGRL1, MIM: 616416), also known as the calcium-independent receptor of α-latrotoxin 1 (CIRL1), is considered to be a prototypical aGPCR; studies of this receptor were seminal for the understanding of aGPCR structure and function.6,18 It is a member of the ADGRL subfamily comprising three paralog members, ADGRL1–3/LPHN1–3 (ADGRL2, MIM: 607018; ADGRL3, MIM: 616417), and a structurally divergent member, ADGRL4/ELTD1 (ADGRL4, MIM: 616419).20 ADGRL1 was initially isolated and described because of its affinity for an exogenous ligand, α-latrotoxin (αLTX), the main neurotoxin of black widow spider venom. αLTX causes massive spontaneous neurotransmitter release via a complex mechanism involving the activation of several distinct receptors, with ADGRL1 playing a major role, and the insertion of tetrameric toxin complexes into the plasma membrane with subsequent pore formation.21, 22, 23, 24, 25 The NTF of ADGRL1 (Figure 1A) has an adhesive function and consists of two adhesion modules, lectin and olfactomedin domains, followed by a hormone receptor motif adjacent to the GAIN domain, which self-cleaves the receptor. The CTF has a signaling function and a typical GPCR structure.26 Although ADGRL2 and 3 share extensive sequence similarity to ADGRL1, only ADGRL2 weakly binds αLTX, and all three are differentially expressed in tissues, which could reflect different functions.
Figure 1.

Individuals and variants identified in our cohort
(A) Schematic representation of ADGRL1 and distribution of the pathogenic variants reported in the study. Galactose-binding lectin domain (GL), olfactomedin-like domain (OLF), hormone receptor domain (HRM), GPCR-autoproteolysis-inducing domain (GAIN), GPCR proteolysis site domain (GPS), 7 transmembrane domain (7TM) and cytosolic latrophilin domain are depicted. Nonsense and frameshift variants are indicated in red. Missense variants are indicated in black.
(B) Variant segregation analysis in the families described in this cohort. Individuals with documented evaluation are indicated as i1 through i10. Arrows indicate the first family member coming to medical attention. E1 indicates exome results regarding ADGRL1. E2 refers to the pathogenic TAOK1 variant (encoding p.Trp188∗) identified in family F9 (Table 1). DD, developmental delay; ID, intellectual disability; ASD, autism spectrum disorder; ADHD, attention deficit hyperactivity disorder.
(C) Individuals with pathogenic ADGRL1 variants.
In the rat, the orthologous genes of human ADGRL1 and ADGRL3 are almost exclusively expressed in brain tissue, while Adgrl2 is expressed in most tissues, including liver, heart, and kidney, albeit with a significant level of cerebral expression.27,28 In humans, a similar expression pattern was observed, even though ADGRL1 was detected at low levels also in other non-neural tissues including heart, placenta, lung, liver, skeletal muscle, kidney, and pancreas.29 Alternative splicing of the NTF and CTF of ADGRLs and their protein ligands possibly contributes to the complexity of these interactions and the diversity of cellular responses.27,29, 30, 31, 32, 33 In rat brain, Adgrl1, the most abundantly expressed paralog, appears at early postnatal stages and peaks at the age of 2–3 weeks.26,27,29,31,34 Its expression is observed in all neurons, but not in glial cells, and is especially abundant in the cortex, hippocampus, dentate gyrus, and cerebellum, with an expression pattern similar to that of many synaptic proteins, including another αLTX receptor, NRXN1/neurexin 1α (NRXN1, MIM: 600,565).35
Several laboratories contributed to the untangling of the ADGRL1 interactome, showing its ability to bind a variety of ligands—including teneurin transmembrane protein 2 (TEN2, encoded by TENM2, MIM: 610119); neurexins 1α, 1β, and 2β (encoded by NRXN2, MIM: 600566); fibronectin leucine-rich transmembrane protein 1 and 3 (encoded by FLRT1, MIM: 604806; FLRT3, MIM: 604808); and contactins—and indicating its major role in regulating synaptic development and activity.30,31,36, 37, 38, 39, 40 Studies of Adgrl1 knockout (KO) mice showed that this gene is apparently dispensable for embryonic development, with homozygous mutant mice demonstrating normal appearance, life span, and fertility, but distinct synaptic physiology and inability to attend to their litters, a behavior that required further investigation.39,41
Few studies have so far directly implicated ADGRLs in human pathology, although this subfamily has been linked to several psychiatric, neurologic, or neurodevelopmental conditions, such as autism spectrum disorder (ASD), attention deficit and hyperactivity disorder (ADHD), bipolar disorder, schizophrenia, epilepsy, and substance use disorder (SUD).26 Individuals harboring overlapping 19q13.12 microdeletions with a common critical region including five genes, most prominently ADRGL1 and PKN1 (MIM: 601032), show intellectual disability, psychomotor and language delay, hearing impairment, and brachycephaly, as well as a behavioral phenotype characterized by hyperactivity and stereotyped movements.42 ADGRL2 has been associated with brain and craniofacial development disorders.43 A de novo heterozygous ADGRL2 missense variant was identified in a fetus with extreme microcephaly, rhombencephalosynapsis, and almost absent sulcation.44 Authors hypothesized that this variant was responsible for an excessive neuronal cell adhesion, leading to this severe phenotype. ADGRL3 polymorphisms have been associated with ADHD susceptibility and increased response to stimulant medication.45, 46, 47, 48, 49, 50, 51, 52, 53 Temporal and spatial expression of this gene appeared relevant at the earlier stages of brain development and in brain regions known to be associated with ADHD. Multiple ADGRL3 coding and non-coding variants within the gene or in nearby regulatory regions have been associated with variable-severity ADHD phenotypes.49,54 A large study of dyslexic families also showed an association between the chromosomal 4q13.1 region (encompassing ADGRL3) and dyslexia, by identifying a single-nucleotide polymorphism (SNP) located 707 kb upstream of ADGRL3.55 Recently, a significant association was observed between an ADGRL3 SNP and susceptibility to early-onset ADHD and ASD in a large cohort of male individuals.56
However, to date, ADGRL1 has not been clearly implicated in human disease, and behavioral phenotypes in Adgrl1 KO mice remain unclear. Here, we present a series of ten individuals with rare variants in ADGRL1 and provide molecular data demonstrating that their pathological functions are consistent with haploinsufficiency. Furthermore, we dissect the pathophysiological mechanisms leading to synaptic dysfunction in Adgrl1 KO mice and provide a detailed characterization of the associated behavioral phenotypes. Overall, our data demonstrate that ADGRL1 haploinsufficiency accounts for a spectrum of developmental, neurological, and behavioral features.
Material and methods
Individuals, exome sequencing analysis, and ethics statement
The procedures followed for genetic testing were in accordance with the ethical standards of the responsible committee on human experimentation, and proper informed consent was obtained from all individuals. Individuals underwent exome sequencing as part of their care, and several care providers were not required to obtain institutional review board (IRB) approval to participate to this study. The protocols 2016-A01347-44 (Discovery), EK302-16, and SJ-91 were deployed by the CHU Dijon Bourgogne, the Uniklinik RWTH Aachen, and the ethics committee in region Sjaelland/Denmark, respectively.
Publication of anonymized data from individual 2 was approved by the IRB of Baylor College of Medicine (protocol H-47546). Individual 5 was already reported in Guo et al., 2019.57 When needed, written consent for publication of photographs was obtained.
Exome sequencing was performed as previously described (supplemental information).58
All experimental procedures involving animals were approved by the Imperial College London and University of Kent Ethical Review Committees and performed in accordance with the Animals (Scientific Procedures) Act 1986 and the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes.
Adgrl1 knockout in mice
To inactivate Adgrl1 in 129/SvJ mice using homologous recombination in embryonic stem cells (ESCs), a large fragment of mouse genome was first isolated from a mouse genomic library in a BAC vector (BACPAC Genomics, USA), using a 150-bp fragment of the Adgrl1 cDNA (which included exon 1) as a hybridization probe. Three BAC clones (120–150 kbp) containing overlapping fragments of Adgrl1 were isolated, mapped, and partially sequenced. To create a targeting vector for homologous recombination, a 12.8-kbp fragment of the gene, containing the promoter and exons 1–3, was subcloned into the pBlueScript plasmid. The 3.3-kbp intron between exons 1 and 2 (including small parts of these exons) was replaced with a neomycin (Neo) resistance gene under the 3-phosphoglycerate kinase promoter (1.8 kbp), which disrupted the open reading frame and served as a positive selection marker. The Neo cassette was flanked by two loxP sequences for potential Cre-mediated excision and Adgrl1 expression rescue. The vector also contained the gene encoding the A chain of diphtheria toxin (DTA) under the RNA polymerase II promoter for negative selection against random incorporation of the whole vector into the ESC genome. A linearized targeting vector was used to generate a stably transfected 129/SvJ ESC line. The successful homologous recombination was verified by Southern blot hybridization and PCR. Using ESC clones, which carried the mutant Adgrl1− allele, and standard transgenic techniques, chimeric 129/SvJ mice were generated. Mice transmitting the inactivated Adgrl1 allele through the germline (strain designation AG148-2) were selected, inter-crossed, and then backcrossed to C57BL/6J mice (Charles River, UK). Mice from the colony maintained on the C57BL/6 background were used in most experiments.
Western blot analysis
Immediately after extraction, prefrontal cortices from wild-type (WT), heterozygous (HET), and KO mouse brains were used to prepare synaptosomes, as described previously.59 The samples containing equal amounts of protein were dissolved in sample buffer, containing 2% SDS, 100 mM DTT, 60 mM Tris-HCl, pH 6.8, and 6% glycerol. To avoid irreversible precipitation of the CTF of ADGRL1 due to boiling in SDS, all samples were heated for 30 min at 50°C but never boiled. The samples were separated by electrophoresis in SDS-Tris-glycine gels containing 4% (for TEN2) or 8% (for all other proteins) polyacrylamide (National Diagnostics, Atlanta, GA, USA). Separated proteins were blotted onto polyvinylidene fluoride membranes (Immobilon-P, IPVH00010, Merck) in Tris-glycine transfer buffer containing 20% methanol at 100 V for 90 min (120 min for TEN2). The membranes were blocked in 5% fat-free milk and immunostained with the following primary antibodies: rabbit polyclonal antibodies against the NTF (RL1)60 and the CTF of rat ADGRL1 (R4);61 rabbit polyclonal IgGs against peptides from the NTFs of ADGRL1 (PAL1), ADGRL2 (PAL2), and ADGRL3 (PAL3);62 a mouse polyclonal antibody against human TEN2 (dmAb, made in-house against the C-terminal amino acids 2412–2637 of LASSO, JF784343);30 a rabbit polyclonal antibody against rat NRXN1α and β (#116; made in-house using the C-terminal peptide CSANKNKKNKDKEYYV);61 a rabbit anti-V5 antibody (to stain the NTF of ADGRL1 variant constructs; Sigma-Aldrich, cat# V8137, RRID: AB_261889); and a mouse monoclonal antibody against β-actin (Abcam, Cambridge, UK). Prior to their use for immunostaining, some of these antibodies were affinity-purified, using the following procedures: (1) to purify RL1, R4, PAL1, PAL2, PAL3, and dmAb, ∼100 μg of their respective recombinant proteins expressed in NB2a cells were separated by SDS-gel electrophoresis, transferred onto immobilon membranes, and incubated overnight with their respective IgG fractions or the immune sera; the membranes were then washed with 25 mL of 1 M NaCl; the bound antibodies were eluted from the antigen with 50 mM triethylamine (pH 12) and neutralized with 1 M Tris-HCl, pH 8.3; (2) NRXN1 antibody was purified from the immune serum by affinity chromatography on the cognate peptide conjugated to SH-sepharose. The blots were then incubated with their respective secondary antibodies conjugated with horseradish peroxidase, followed by chemiluminescent detection using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and the LAS3000 (FUJIFILM) gel documentation system. To determine the linear signal range for each protein, different signal development and detection times were used. For protein quantification, ImageJ (NIH, Bethesda, MD, USA; RRID: SCR_003070) was employed.
Genotyping
Tail biopsies obtained from 21-day-old mice (or tissue fragments of partially cannibalized newborns) were used to extract genomic DNA. Two PCR-amplification reactions were set up for each sample and included 1 μg of genomic DNA and one of the two primer pairs (Figure S2A): N252 (5′-AGG CCG TGG TAC CCT GGT GAT GCG GGG CGA GG) and N253 (5′-GCG TGT GCA GGA TCC CAG GCC AGA GCC GGG TAA TTA CTT GTT TT), or N252 and N255 (5′-CGA GAC TAG TGA GAC GTG CTA CTT CCA TTT GTC), which were specific for the WT or KO Adgrl1 allele and produced amplification fragments of 522 and 459 bp, respectively. Hot-start PCR reactions were performed on a Mastercycler (Eppendorf, UK), using the following program: initial denaturation (10 min at 94°C), 34 cycles of amplification (60 s denaturation at 94°C, 90 s annealing at 60°C, 45 s extension at 72°C), and final extension (10 min at 72°C). The PCR products were analyzed by agarose gel electrophoresis.
Neuronal cell cultures
Hippocampi from 1-day-old mice were dissected under a binocular microscope in aseptic conditions, placed into vials with HAB buffer (Hibernate A, 2% B27, 0.5 mM GlutaMAX), and kept at 4°C until plating (no later than 10 days after dissection). The hippocampi were placed in dissociation buffer (Hibernate A without Ca2+, containing 2 mg/mL papain) and incubated for 10 min at 37°C. The buffer was then replaced with HAB buffer, and the tissue was triturated using a silanized Pasteur pipette. The undispersed pieces were allowed to settle for 1 min; the supernatant containing the dispersed cells was transferred into a 15-mL tube and centrifuged at 700 rpm for 2 min. The pellet was resuspended in Neurobasal A medium supplemented with 2% B27 and 0.5 mM GlutaMAX; the cells were plated at a density of 10,000 cells/well on coverslips precoated with 50 μg/mL poly-D-lysine and 10 μg/mL laminin and placed into 24-well plates. Neurons were incubated at 37°C, 5% CO2, and half of the medium was changed every 3–4 days. The cultures were maintained until 60 days in vitro (DIV) and used for electrophysiological recordings at different times.
Synaptic activity in cultured hippocampal neurons
Recordings of spontaneous postsynaptic currents in hippocampal neurons were carried out after 14–60 DIV. Coverslips with neurons were transferred into a perfusion chamber (Harvard Biosciences, Inc.) mounted on the stage of an inverted microscope and perfused at room temperature with the continuously oxygenated External solution (2 mM CaCl2, 3 mM CsCl, 11 mM glucose, 4.8 mM HEPES, pH 7.4 adjusted with NaOH, 160 mM NaCl, 1 μM tetrodotoxin) at a flow rate of 1 mL/min. Patch pipettes were prepared from filamented borosilicate glass capillaries (Harvard Biosciences) to achieve a DC resistance of 3–7 MΩ and filled with the Internal solution (150 mM CsCl, 10 mM EGTA, 5 mM HEPES, pH 7.4 adjusted with KOH, 10 mM NaCl, 4.5 mM ATP-Mg, 0.1 mM GTP). Cells were observed using a color video camera, and pipettes were positioned using a PatchStar motorized micromanipulator (Scientifica, UK). The recordings were carried out in the whole-cell configuration, on cells voltage-clamped at −70 mV. To block current, signals were amplified (10 mV/pA), filtered at 2.9 kHz, and digitized at 20 kHz using a recording system including a Model 2400 patch-clamp amplifier (A-M Systems, Inc., USA), an LPF202A filter/amplifier (Warner Instruments, USA), a HumBug harmonic frequency quencher (Quest Scientific, USA), and a Digidata 1322A digitizer (Axon Instruments, USA). Data acquisition was controlled using pClamp (Axon Instruments), and the traces were analyzed with MiniAnalysis (Synaptosoft, USA)
Synaptic activity at the NMJ
Flexor digitorum brevis muscles were dissected from P21 mice (Adgrl1+/+ or Adgrl1−/−) and pinned to the bottom of Petri dishes coated with Sylgard (Dow Corning). In this set of experiments, we systematically tested male mice to avoid any effects of the estrous cycle; however, several experiments conducted on adult females in metestrus produced similar results. The recording buffer contained 137 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.4, 5.6 mM glucose, and 1 μM tetrodotoxin. Sharp electrodes were manufactured from borosilicate glass (Harvard Biosciences) with a tip diameter <0.5 mm and ∼70 MOhm impedance and filled with a 5 M ammonium acetate solution. Spontaneous presynaptic activity was recorded using an Axoclamp 2B pre-amplifier (Axon Instruments) in the current clamp mode, an LPF202A secondary filter/amplifier, a HumBug harmonic frequency quencher, a Digidata 1322A digitizer, and a microcomputer running AxoScope software (Axon Instruments). The recorded signals were analyzed using MiniAnalysis.
Loss of righting reflex
The righting reflex was tested between postnatal days P3 and P10. No pre-test learning was required. Pups were gently held on their backs on a flat surface for 5 s and then released. The time required for the pup to return to prone position was recorded in three trials, each of which lasted up to 60 s.
Locomotor activity and stereotypy
WT, HET, and KO mice aged 2–6 months were housed individually, with food and water provided ad libitum. Cages were fitted with running wheels, whose revolutions were recorded in 1-min bins and analyzed using the Chronobiology Kit (Stanford Software Systems, Santa Cruz, CA, USA). Before commencement of experiments, mice were kept under a 12:12 h light-dark cycle for a minimum of 14 days, and the same regime was maintained during the experiments. Environmental room lighting consisted of white fluorescent strip lights providing 350 lux at cage level.
Stereotypic behaviors (grooming, excessive jumping, tonic immobility, excessive digging, etc.) were observed and timed for 10 min after mice were individually transferred into new cages.
Pre-pulse inhibition
The acoustic startle response apparatus was designed in-house and consisted of an acoustically insulated 35 × 35 × 35 cm chamber equipped with a ventilation system and a test platform. The latter included a plastic tubular rodent holder (Kent Scientific Corporation, USA) magnetically positioned on a 14 × 18 cm plastic plate, which rested on a piezoelectric force transducer (MLT1010, ADInstruments, Australia) attached to a heavy base; the transducer was used to detect animal motion inside the holder. Broad-band acoustic stimuli and background noise (BN) were delivered via two speaker drivers placed 5 cm above the animal holder and connected to a computer-controlled audio amplifier. Acoustic signal patterns for various trials were designed and replayed using the Audacity software (Audacity Team, USA) and calibrated to the required sound intensity using a sound level meter (815, Testo, Germany) placed near the animal holder. A BN was maintained in the chamber at 60 dB during the experiment. Animals were habituated to the experimental conditions in preliminary sessions that included no acoustic stimuli. Test experiments were designed according to previously published studies and consisted of 5 min acclimatization, followed by ten no-prepulse/startle (NS) trials, then by a random series of three prepulse/startle (PS) trials, five NS trials, and five no-prepulse/no-startle (NN) trials, and finally by another ten NS trials.63, 64, 65, 66 All trials were interspaced by 15-s BN intervals and consisted of the following segments: 100 ms BN; 20 ms pre-pulse (75 dB, 15 dB above BN) or BN; 100 ms interstimulus interval; 40 ms startle stimulus (120 dB, 60 dB above BN) or BN; 300 ms response period. Animal motion signals from each trial were rectified, digitized at 1,000 Hz, and recorded using a PowerLab data acquisition system (ADInstruments, Australia). Startle responses were quantified by calculating the area under response curves (AOC). Risperidone (1 mg/kg body weight, in 150 μL) was administered intraperitoneally 30 min prior to the pre-pulse inhibition (PPI) test; control mice were injected with vehicle; each mouse was used only once.
Neurotransmitter release from central synapses
Synaptosomes were prepared from prefrontal cortices of mouse brains as described previously and resuspended in physiological buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, 20 mM HEPES, pH 7.3) at a concentration of 1 mg protein/mL.67 To measure spontaneous and evoked release of dopamine and noradrenaline, the synaptosomes were first equilibrated in physiological buffer supplemented with 2 mM Ca2+, 1 mM ascorbic acid, and 0.1 mM pargyline (Sigma-Aldrich, UK) for 10 min, at 37°C. The synaptosomes were then incubated with 2.5 μCi [7,8-3H]-dopamine or [7,8-3H]-noradrenaline (Amersham, UK) as outlined.67 To load with [14C]-glutamate, synaptosomes in basal buffer containing 2 mM Ca2+ and 0.1 mM aminooxyacetic acid (Fisher, UK) were preincubated for 10 min, at 37°C, then supplemented with 2–5 μCi/mL [14C]-glutamic acid (Amersham, UK) for 5 min, washed, resuspended in buffer with 2 mM Ca2+, incubated for 1 h at 37°C, washed, and used to measure release. All buffers used to prepare synaptosomes and study release were oxygenated. Release of neurotransmitters was determined in multiple identical experiments, each involving triplicate samples of 25 μg loaded synaptosomes/experimental point.
Recombinant ADGRL1 constructs
The variant ADGRL1 expression constructs were created on the basis of rat Adgrl1 (GenBank: U78105) in the pcDNA3.1 vector, using a site-directed mutagenesis system (NEBaseChanger, New England Biolabs) and the following primer pairs: p.Trp9∗, 5'-TGCAGCACTCTAGAGTCTCTGTGTGACGAC-3', 5'-GCCAAGCGGGCCATGGCG-3' (annealing temperature [Ta], 72°C); p.Tyr346Cys, 5'-CCGCGTGGACTGTGCCTTTAACA-3', 5'-TTGCCTGCTGCCTCACTG-3' (Ta, 69°C); p.Trp1005Arg, 5'-CTATTTCATCAGGAGCTTCATTGGG-3', 5'-TTATCCACCCTCAGCCAG-3' (Ta, 62°C); p.Met1152Thr, 5'-AATCCGGAGGACGTGGAATGACAC-3', 5'-CGGCTCTGGGTCCCTGTG-3' (Ta, 67°C); and p.Ser1164Phe, 5'-GACAGAGTCGTTCTTTATGGCAG-3', 5'-TGCTTCCTCACGGTGTCA-3' (Ta, 64°C). The correct base substitutions were confirmed by DNA sequencing. To obtain stable cell lines expressing these constructs, NB2a neuroblastoma cells were transfected using Escort III (Sigma-Aldrich) with WT Adgrl1 (positive control), its variants (described above), or an empty vector (negative control) and selected using Geneticin (Thermo Scientific, UK).
Measurements of cytosolic Ca2+
When expressed in neuroblastoma cells, ADGRL1 is known to react to the binding of LTXN4C by triggering the intracellular Ca2+ signaling cascade.68,69 To monitor cytosolic Ca2+ changes, a fluorescent Ca2+ indicator dye was used as described previously.68,69 When stimulating ADGRL1 with toxin, it was important to use the mutant LTXN4C, which does not form membrane pores and thus allows study of the receptor-mediated signals only.25,70 As a positive control of LTXN4C-induced Ca2+ signaling, the WT Adgrl1 was used; two negative controls were employed: cells transfected with an empty vector and stimulated with LTXN4C, and ADGRL1-expressing cells stimulated with buffer. Cells were grown in Dulbecco’s modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS) and L-glutamine in 30-mm dishes to a required density. One day prior to the experiment, the medium was replaced with serum-free DMEM. The cells were then incubated in the dark with physiological buffer containing 0.2 mM EGTA, 0.5 mg/mL BSA, 2.5 mM Fluo-4 acetomethoxy ester (Fluo-4-AM, Thermo Fisher Scientific) and 10% pluronic F-127 for 30 min, then washed and further incubated for 30 min to allow for dye de-esterification. The cells loaded with the fluorescent dye were observed under a confocal microscope (LSM510, Zeiss, United Kingdom) using a 40× Achroplan water-dipping objective, a 488-nm laser, and a band-pass emission filter of 505–550 nm. Confocal images were acquired every 5 s for the duration of the experiment, which typically lasted 55–60 min, according to the following protocol: (1) Baseline fluorescence (F0) was initially recorded. (2) After 5 min recording, 1 nM LTXN4C (or control buffer) was added in the absence of Ca2+e to avoid asynchronous signaling while the receptors are independently activated in individual cells (this allowed the toxin to bind and activate the receptors without inducing Ca2+ signals).68 (3) At 30 min, Ca2+ was added to a final concentration of 2 mM to induce synchronous receptor signaling. (4) At 50 min, 1 nM WT αLTX was added to identify all cells able to bind the toxin and measure the maximal Ca2+ fluorescence (Fmax) of each individual cell by inducing Ca2+-permeable pores in them. (6) The recording continued for another 5 min. The fluorescence profile of each cell was adjusted to the baseline drift using controls and normalized between the specific F0 and Fmax.
Immunostaining of cultured hippocampal neurons
Neuronal cultures on glass coverslips were fixed with 4% paraformaldehyde/1% glutaraldehyde, quenched with 0.7 M Tris, blocked with 10% goat serum, and then incubated overnight with rabbit anti-VGAT or mouse monoclonal anti-VGLUT antibodies (ThermoFisher Scientific, UK), at 4°C, before being stained for 1 h with Alexa Fluor 488-conjugated anti-rabbit or Alexa Fluor 568-conjugated anti-mouse secondary antibodies (ThermoFisher Scientific). The coverslips were mounted on glass slides and imaged under the LSM-510 confocal microscope using a Plan-Neofluar 40×/1.3 oil-immersion objective.
Statistical analysis
The data were initially assessed using a Lillefors normality test, and normally distributed sets of data were compared using a one-way ANOVA, with the Bonferroni correction for multiple pairwise comparisons where necessary. Non-normally distributed data were compared using a Wilcoxon (Mann-Whitney) non-parametric test for unpaired data. For binomial distributions, Jeffreys 95% and 99% confidence intervals were calculated. The level of statistical significance was set at p = 0.05, and the following indicators of probability levels were used throughout the paper: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. All data in the figures are the means ± SEM. In behavioral experiments, littermates or age-matched mice were used, and the experimenter was always blind to mouse genotype.
Results
In an 8-year-old boy with a neurodevelopmental disorder, including mild to moderate intellectual disability, global developmental delay, and behavioral disorders (individual 1, F1-II-1, Figure 1 and Table 1 and supplemental information), we performed array-comparative genomic hybridization (CGH) and trio exome sequencing (ES) and identified a de novo HET missense variant (g.14267505A>G [GenBank: NC_000019.9]; c.3013T>C [GenBank: NM_001008701.2]; p.Trp1005Arg) in ADGRL1. No additional candidate single-nucleotide, indel, or copy-number variants accounting for his phenotype were identified. ADGRL1 is mainly expressed in the brain (The Human Protein Atlas, www.proteinatlas.org) and is highly intolerant to loss-of-function (LoF) and missense variants according to gnomAD (v2.1.1), with a probability of LoF intolerance (pLI) of 1 and a missense Z (misZ) score of 3.43.71 There were only 12 high-confident LoF alleles in gnomAD, all at the heterozygous state with a frequency ranging from 9.95 × 10−5 to 8.81 × 10−6 in ethnically matched allele counts. The p.Trp1005Arg variant is absent from the gnomAD database and located within the CTF of ADGRL1, in a position highly intolerant to substitutions (Figure S1, metadome score 0.5). Furthermore, two individuals with de novo missense variants (c.1037A>G [p.Tyr346Cys] and c.1637G>C [p.Ser546Thr]) and ASD were reported in a meta-analysis cohort of 3,871 autism cases and 9,937 ancestry-matched or parental controls (Figure S1A).72
Table 1.
Clinical features and variants found in the nine individuals of the cohort
| Sex | Individual 1 |
Individual 2 |
Individual 3a |
Individual 4a |
Individual 5 (Guo et al., 2019)57 |
Individual 6 |
Individual 7 |
Individual 8 |
Individual 9 |
Individual 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Male | Male | Female | Female | Male | Male | Female | Male | Male | Male | |
| Age at last examination | 11 years | 3 years 3 months | 12 years 1 month | 11 years 1 month | 19 years | 10 years | 13 years | 10 years | 8 years | 43 years |
| Birth measurements | ||||||||||
| Weight in g (centile) | 3,780 (83rd) | N/Ab | 2,950 (14th) | 3,720 (87th) | N/A | N/A | 3,000 (2–9th) | 4,470 (>97th) | 4,100 (90th) | N/A |
| Length in cm (centile) | 53 (95th) | N/A | 50 (55th) | 52 (94th) | N/A | N/A | 54 (50–75th) | 57 (>97th) | 51 (75th) | N/A |
| OFCc in cm (centile) | 39 (99th) | N/A | 34.5 (52nd) | 37 (98th) | N/A | N/A | N/A | N/A | 36.5 (75th) | N/A |
| Developmental features | ||||||||||
| Age at sitting (months) | 18 | delayed | 12 | 7 | N/A | N/A | N/A | 7 | 9 | 12 |
| Age at walking (months) | 23 | delayed | 13 | 14 | N/A | 17 | 11 | 30 | 16 | 17 |
| Age at first words (months) | 15 | delayed | 24 | 12 | N/A | 24 | 14 | 36 | 36 | 15 |
| Developmental delay | + | + | + | + | – | + | + | + | + | + |
| Intellectual disability degree (IQ)d | mild to moderate | N/A | mild | mild (IQ 57) | IQ 96 | no IDe | borderline (IQ 74) | IQ 109 | mild to moderate | mild |
| ASDf | + | N/A | + | – | + | – | – | + | – | – |
| ADHDg | + | N/A | + | – | suspected | – | + | – | – | – |
| Neurological features | ||||||||||
| Hypotonia | + | N/A | + | + | – | – | – | + | – | – |
| Stereotypies | + | N/A | – | – | – | – | N/A | + | – | – |
| Epilepsy | – | N/A | – | – | – | – | + | + | – | – |
| Sleep disturbance | + | N/A | + | + | – | + | + | – | – | – |
| Brain imaging | Normal | N/A | N/A | N/A | N/A | nodular heterotopia | normal | normal | Normal | N/A |
| Facial dysmorphism | – | +h | – | + | – | + | + | + | + | + |
| Additional features | ||||||||||
| Macrocephaly | Congenital | N/A | Postnatal (93rd centile) | + | N/A | – | N/A | N/A | + (97th centile) | – |
| Overweight | – | N/A | + (99th centile) | + | + | – | – | – | – | + |
| Joint hypermobility | + | N/A | + | – | – | + | N/A | + | – | – |
| Others | Hyperopia, temporary regression, megadolichocolon | Learning disabilities | Hyperkinetic disorder, small hands and feet |
Small hands and feet | Poor adaptive skills Learning disabilities |
Migraine,dermatological issues, genital abnormalities, borderline short stature, advance bone age, clinodactyly | Delayed puberty |
Dermatological issues, scoliosis | Learning disabilities | 2 Neuroendocrine pancreatic tumors at 43 years |
|
ADGRL1 variant DNA change GRCh37/hg19 GenBank: NC_000019.9 (chr19) |
g.14267505A>G | g.14263628G>A | g.14273794C>T | g.14273 794C>T |
g.14294389C>T | g.14263409A>G | g.142633 73G>A |
g.14270002dup | g.14263332G>A | g.14273591T>C |
| cDNA change GenBank: NM_001008701.2 |
c.3013T>C | c.3406C>T | c.834G>A | c.834G>A | c.26G>A | c.3455T>C | c.3491C>T | c.2064dup | c.3532C>T | c.1037A>G |
| Amino acid chance | p.Trp1005Arg | p.Arg1136∗ | p.Trp278∗ | p.Trp278∗ | p.Trp9∗ | p.Met1152Thr | p.Ser1164Phe | p.Glu689Argfs∗113 | p.Arg1178∗ | p.Tyr346Cys |
| Inheritance | de novo | de novo | paternally inherited | paternally inherited | de novo | de novo | de novo | de novo | de novo | Paternally inherited |
| Additional findings | – | 9p13.3 deletion | – | – | – | 5q14.3 deletion |
– | – | 6p12.3 duplication | TAOK1 |
| Variant GRCh37/hg19 |
– | (34655686–34662203)x1 | – | – | – | (83112653–85261932)x1 | – | – | (47075799–48930453)x3 | c.564G>A (GenBank: NM_020791.1) |
| Amino acid change | – | – | – | – | – | – | – | – | – | p.Trp188∗ |
| OMIM gene phenotypes | – |
IL11RA Craniosynostosis and dental anomalies AR,i MIM: 614188 |
– | – | – | – | – | – |
CD2AP Glomerulosclerosis, focal segmental 3, MIM: 607832 |
Developmental delay with or without intellectual impairment or behavioral abnormalities AD,jMIM: 619575 |
| ACMGk classification | – | VUSl | – | – | – | VUS | – | – | VUS | pathogenic |
| Inheritance | – | unknown | – | – | – | maternally inherited | – | – | maternally inherited |
paternally inherited |
Siblings.
Not available.
Occipital frontal circumference.
Intellectual quotient.
Intellectual disability.
Autism spectrum disorder.
Attention-deficit hyperactivity disorder.
Probably not linked to ADGRL1 variant.
Autosomal recessive.
Autosomal dominant.
American College of Medical Genetics and Genomics classification.
Variant of unknown significance.
Through international data sharing (GeneMatcher, ERN-ITHACA, and personal collaborators), we ascertained nine additional individuals from eight unrelated families with congruent phenotypes and de novo or inherited HET candidate ADGRL1 variants (Table 1 and Figure 1), namely four nonsense (among which one was previously included in Guo et al., 201957), one frameshift, and four missense variants (Table 1 and Figures 1A and S1B). Seven of them were de novo, while two, c.834G>A (p.Trp278∗) and c.1037A>G (p.Try346Cys), were inherited from an affected parent with intellectual disability and learning difficulties (Families F3 and F9, respectively; Figure 1). In the p.Trp278∗ family, the variant segregated in six affected cases (F3, Figure 1B), but only two individuals (individuals 3 and 4, F3-II-5 and F3-II-6, respectively) were available for clinical evaluation. In the p.Tyr346Cys family (F9), the variant co-segregated with a pathogenic TAOK1 variant (developmental delay with or without intellectual impairment or behavioral abnormalities—autosomal dominant, MIM: 619575) in three affected cases (Figure 1B), but only one individual (individual 10, F9-II-2) was available for clinical evaluation.
Retrospective phenotyping of the ten individuals of our cohort delineated a consistent neurodevelopmental disorder characterized by global developmental delay, intellectual disability, ASD, and/or ADHD. Age in the cohort ranges from 3 to 43 years (median age: 11 years). Nine individuals presented with a developmental delay, including poor motor skills and speech delay. Among them, two benefited from a significant psychomotor catch-up during their development (individuals 6 and 8). Walking acquisition age ranged from 11 to 30 months (median age: 16.5 months), and first pronounced words age ranged from 12 to 36 months (median age: 14.5 months).
Five individuals presented with mild to moderate intellectual disability, one presented with a borderline intelligence quotient (IQ; individual 7; Figure 1C), and three had average IQs. Individual 2 (F2-II-1, Figure 1) was 3 years and 3 months old at last consultation, and therefore too young for a neuropsychological assessment. Five underwent brain imaging, which was unremarkable, except for individual 6, who showed bilateral focal nodular heterotopias of the frontal horns of the lateral ventricles.
In our cohort, 5/9 individuals presented with neuropsychiatric disorders (detailed clinical data were unavailable for individual 2). Among them, three matched diagnosis criteria for ADHD and four for ASD ranging from atypical autism to Asperger syndrome. Two individuals met the criteria for both ASD and ADHD (individuals 1 and 3, F1-II-1 and F3-II-5, respectively; Figure 1), and individual 5 (F4-II-1, Figure 1), presenting with Asperger syndrome, also presented with hyperactivity, impulsivity, inattention, and executive functioning deficits, which was strongly suggestive of ADHD. Stereotypies were present in two cases.
Epileptic features with EEG abnormalities were present in 2/9 individuals: individual 7 (F6-II-1, Figure 1) presented focal hypermotor seizures with secondary generalization to tonic-clonic seizures; individual 8 (F7-II-1, Figure 1) presented developmental delay and behavioral abnormalities at 5 years with the diagnosis of epileptic aphasia. His EEG revealed bilateral, asynchronous slow and spike waves on the central and posterior regions. He was prescribed carbamazepine (15 mg/day), with an effect on both EEG abnormalities and speech problems. Additional neurological features included inconstant hypotonia (4/9). No spasticity or ataxia was noted in the cohort.
Facial dysmorphism was observed in 7/10 cases, but individual 2 (F2-II-1, Figure 1) presented with frontonasal dysplasia probably not linked to the ADGRL1 variant. Dysmorphic features included broad nasal tip, deep philtrum, thin upper lip, and donwslanted palpebral fissures. Macrocephaly (i.e., head circumference above 2 standard deviations of the 97th percentile) was present in 4/6 individuals with available data.
Other findings include sleep disturbance (5/9) such as non-organic insomnia (3/5) and nocturnal awakenings (2/5), mild extremity anomalies (3/9), overweight (4/9), joint hypermobility (4/8), dermatological issues (2/9), hyperphagia (2 individuals from the same family), anxiety (1/9), megadolichocolon (1/9), hyperopia (1/9), genital anomalies (1/9), delayed puberty (1/9), scoliosis (1/9), migraine (1/9), and neuroendocrine pancreatic tumors (1/9). Overall, our data indicated that ADGRL1 should be considered as a candidate gene for a neurodevelopmental disorder including ASD and ADHD with a variable spectrum.
To understand the pathophysiological mechanisms underpinning the ADGRL1 variants identified in our cohort, we produced Adgrl1 constructs encoding the p.Trp9∗, p.Tyr346Cys, p.Trp1005Arg, p.Met1152Thr, and p.Ser1164Phe variants, and expressed them in mouse neuroblastoma cells. As shown in Figure 2A, four variant constructs were relatively well expressed, while the p.Trp9∗ construct produced no protein, indicating that this variant introduces a functional premature stop codon. Delivery of the expressed proteins to the cell surface was assessed using αLTX and anti-LTX antibodies. As expected, the cells expressing p.Trp9∗ showed no detectable αLTX binding (Figures 2A and 2B), whereas the p.Tyr346Cys- and p.Trp1005Arg-expressing cells bound the toxin proportionately to the amount of protein produced (Figures 2A and 2B). Interestingly, the two constructs with variants in the intracellular tail of the CTF (p.Met1152Thr and p.Ser1164Phe) bound substantially less toxin (Figures 2A and 2B). As αLTX only binds to the extracellular NTF, this result was likely due to inefficient delivery of the p.Met1152Thr and p.Ser1164Phe constructs to the cell surface, and this was confirmed by labeling of the surface-exposed ADGRL1 proteins with a membrane-impermeable biotin reagent and subsequent staining with streptavidin (Figure 2A, right).
Figure 2.

In vitro analysis of mutations in ADGRL1
(A) Expression and surface delivery of the mutated constructs in NB2a cells. Cells transfected with the vector expressing WT ADGRL1, its variants (as indicated), or no ADGRL1 (Vector) were incubated with αLTX, lysed, and immunoblotted with antibodies against the NTF of ADGRL1, αLTX, and β-actin. In a separate experiment, surface exposure of p.Met1152Thr and p.Ser1164Phe was detected by biotinylation of live cells and staining of the lysate with streptavidin. The blots represent n = 3 experiments, with similar results. M, molecular mass markers.
(B) Quantification of surface expression of the ADGRL1 mutants relative to native ADGRL1 (n = 3).
(C) Cytosolic calcium signaling induced in individual NB2a cells transfected with ADGRL1, its variants, or an empty vector and detected using confocal microscopy and an intracellular fluorescent Ca2+ sensor, Fluo-4. As indicated by arrowheads, the cells were first stimulated by buffer or 2 nM LTXN4C in the absence of extracellular calcium, then 2 mM Ca2+e was added, and at the end of the procedure, cells were treated with 1 nM αLTX to form membrane pores and detect maximal fluorescence. Two exemplary traces are shown for each variant; the number of cells analyzed was 72–93 in n = 3–7 independent experiments.
(D) Quantification of integrated calcium signals in the cells expressing ADGRL1 or its mutants.
All data are the means ± SEM; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; NS, non-significant.
Physiological activity of the ADGRL1 variants was probed by recording their intracellular Ca2+ signaling in response to stimulation with LTXN4C, a mutant αLTX unable to form membrane pores (as described in material and methods).25,68, 69, 70 The WT ADGRL1 showed a large response to extracellular Ca2+ (Ca2+e) influx, followed by strong and persistent oscillatory calcium signaling in most cells. Similar to vector-transfected cells, cells expressing p.Trp9∗ did not react to LTXN4C or native αLTX. Cells expressing p.Tyr346Cys demonstrated high influx of Ca2+e but very few calcium oscillations. The p.Trp1005Arg construct only reacted to LTXN4C by Ca2+e influx, without subsequent calcium waves. The two constructs with intracellular variants (p.Met1152Thr and p.Ser1164Phe) produced a low Ca2+e signal, followed by infrequent calcium oscillations in some cells. Thus, all ADGRL1 variants studied displayed impairments in LTXN4C-induced intracellular calcium signaling (Figure 2D), indicating that they should be considered as pathogenic. Overall, our data indicate that the molecular consequences of ADGRL1 mutations are consistent with haploinsufficiency.
To model the pathophysiological effects of human ADGRL1 haploinsufficiency, we inactivated Adgrl1 in mice (described in material and methods) (Figure S2A). When the colony was established, we noted a pronounced sub-Mendelian distribution of the Adgrl1 null allele in the offspring and a lack of KO progeny on the original 129/SvJ genetic background (Figure S2B), which suggested that the deletion of Adgrl1 was embryonically lethal (for additional data, see supplemental results, Figure S2). On this non-compensatory background, many HET pups showed neurodevelopmental abnormalities. More than 15% of HET offspring were unable to move in a coordinated manner, failing to suckle or right themselves (Figures 3A and 3B). Some less-affected HET animals that survived until weaning also demonstrated neurological deficits, e.g., seizures upon transfer to a new environment (Figure 3A). On this genetic background, WT animals displayed no abnormalities (Figure 3B). For a more detailed description of behavioral data, see supplemental information.
Figure 3.

Cognitive and behavioral abnormalities in Adgrl1−/− mice
(A and B) Neurological deficits in the offspring on the mixed 129/SvJ-C57BL/6 background.
(A) Left: An example of a loss of righting reflex in a P4 HET pup, compared to its WT littermate. Center: P3 HET pup underdeveloped due to suckling problems, with its WT littermate. Right: An example of a P21 HET pup experiencing arrest and seizures after transfer into a new environment (open space).
(B) The frequency of neurodevelopmental deficits in WT, HET, and KO pups (circles, mean values; bars, ± 95% confidence intervals, CI; whiskers, ± 99% CIs. WT, n = 23; HET, n = 28 normal, 6 compromised; KO, n = 1 normal, estimated 23 dead in utero).
(C–J) Behavioral abnormalities in the Adgrl1−/− colony on the compensatory C57BL/6 background.
(C) Percent of breeding pairs of specified genotype committing parental infanticide (X = WT or HET; circles, mean values; bars, ± 95% CIs; whiskers, ± 99% CIs).
(D) Consecutive litters killed by parents of indicated genotype.
(E) Parity in breeding pairs of specified genotypes (X = WT or HET; circles, mean values; bars, ± 95% CI; all KO pairs n = 156, nulliparous n = 72; all WT pairs n = 20, nulliparous n = 4).
(F) Examples of running wheel activity of WT and KO littermates (ticks correspond to revolutions per min).
(G) Average wheel-running activity in mice of indicated genotypes (KO animals are plotted as two groups of high- or low-locomotor activity; WT or HET, n = 8; KO, n = 4 and 4).
(H) Typical auditory startle reflex responses in WT and KO mice under indicated protocols (respective individual traces overlaid). Trial types: PN, pre-pulse, no startle stimulus; NS, no pre-pulse, startle stimulus; PS, pre-pulse, startle stimulus; RIS, 1 mg/kg risperidone, 30 min prior to test. (WT animals: vehicle group, n = 6; risperidone group, n = 4; KO animals: vehicle group, n = 4; risperidone group, n = 4).
(I) Quantification of startle responses as in (H).
(J) PPI in PS trials in control and risperidone-treated animals.
(K) Overall time spent self-grooming by WT and KO mice over a 10-min period in a new environment. (WT animals, n = 9; KO animals, n = 10).
All data are the means ± SEM; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; NS, non-significant.
After backcrossing to C57BL/6, some KO offspring were finally obtained, as demonstrated by polymerase chain reaction (PCR) and western blotting for ADGRL1 protein (Figures S2C and S2D). Although the expected Mendelian Adgrl1−/− frequency was still not achieved (Figure S2E), the C57BL/6 genetic background provided sufficient compensation of Adgrl1 deletion for some KO animals to survive gestation (see also supplemental results, Figures S2F and S2G). On this more permissive genetic background, HET animals developed normally and revealed no behavioral or neurological deviations.
By contrast, most KO animals successfully completing their development demonstrated a host of aberrant behaviors. The main abnormality was persistent maternal infanticide: 71% of KO mothers killed their newborn pups (Figure 3C). The dead neonates never showed milk spots, indicating a lack of nursing, but the promptness of the attack (usually within minutes postpartum) and its exhaustive character (all pups killed) suggested a deliberate act of aggression due to an affective dysfunction rather than litter abandonment, passive cannibalism, or inability to bond. The KO dams showed no adaptation to litters’ demands with consecutive parturitions (Figure 3D), which also indicates some cognitive dysfunction.
KO male mice also frequently killed their offspring; however, they did it ∼2.5 times less frequently than KO dams (Figure 3C). Similar to KO females, KO males showed no adaptation to periodic appearances of litters, killing the same percent of litters after each parturition taking place in their home cage (Figure 3D). The fact of paternal infanticide argued against maternal postpartum psychosis being the main reason of infanticide but was consistent with faulty sensory gating in KO animals. When both parents lacked Adgrl1, they always killed their offspring (Figures 3C and 3D). Importantly, WT-HET and WT-WT pairs on the compensatory genetic background never killed their offspring, while only one HET-HET pair ever committed infanticide (1 out of 31).
KO animals also demonstrated aberrant social and sexual interactions: 50% of matings involving at least one KO animal remained nulliparous, while only 20% of WT/HET breeding pairs produced no progeny (Figure 3E). Although many reasons could underlie sexual dysfunction, we noted that the KO animals demonstrated a bimodal distribution. While some KO mice conceived/sired repeatedly, albeit regularly killing their pups, other animals were consistently averse to reproductive behavior. These two modalities suggested the presence of either a bipolar affective disorder or a bimodal cognitive impairment, such as schizophrenia.
To begin distinguishing between these possibilities, we studied locomotor activity in the Adgrl1−/− mice. Given their exaggerated response to novel stressful stimuli, we initially assessed their routine behavior using home-cage running wheels. As shown in Figure 3F, KO mice often demonstrated hyperactive behavior, greatly exceeding their WT or HET littermates in locomotion. Intriguingly, the KO animals fell into two opposite categories: either strongly hyperactive or strongly hypoactive (Figure 3G), which resembled manic and depressive states. WT and HET mice showed much less variance in these tests (Figure 3G). However, we did not observe any spontaneous switching between the two polar behavioral types, which may suggest that the KO animals had either one disorder with two invariable modalities or a spectrum of separate disorders.
Stress response and extremes of locomotion in the KO mice were consistent with impaired sensorimotor gating; therefore, we tested them in acoustic startle and PPI experiments, designed to assess animals’ attention and sensory gating deficits in relation to salient environmental stimuli.63,73 The KO mice demonstrated a clearly increased startle response (Figure 3H, NS protocol; Figure 3I). When the startle stimulus was shortly preceded by a sub-threshold acoustic pre-pulse (PS protocol) (Figure 3H), the startle response was inhibited by 40% in WT mice but only by 10% in KO mice (Figures 3I and 3J). To assess the involvement of monoaminergic neurotransmission in the startle responses observed, we injected WT and KO mice with 1 mg/kg risperidone, an atypical antipsychotic and an antagonist at dopamine and 5-HT receptors. This increased the PPI to ∼60% for WT and to ∼45% for KO mice (Figures 3I and 3J), while also attenuating the difference between these cohorts. These results confirm the abnormal sensorimotor gating in Adgrl1−/− mice and suggest an involvement of dopaminergic and/or serotonergic receptors.
Finally, the HET animals on the non-compensatory background and the KO mice on the compensatory genotype displayed persistent stereotypic behaviors: compulsive grooming, repetitive serial movements, excessive digging, tonic immobility, etc., especially when transferred into a new cage. As an example, self-grooming in a new environment was two times longer, or more frequent, in KO mice than WT animals (Figure 3K). For additional results of behavioral tests, see supplemental information. These behaviors are similar to those of mutant mice with neuropsychiatric dysfunctions or increased release of dopamine in the brain.74,75
We then investigated the molecular and synaptic characteristics of the Adgrl1 KO mice. The amount of ADGRL1 detected in the brain of these mice corresponded well to the dose of the Adgrl1 allele (Figures 4A and 4B). Interestingly, these changes in ADGRL1 affected the abundance of other related proteins, in particular the ADGRL1 paralogs, ADGRL2 and ADGRL3, and the ADGRL1 ligand, TEN2 (or LASSO).27,30 Specifically, the amounts of ADGRL2 and TEN2 in KO brains were, respectively, 12% and 19% lower than those in WT brains (Figures 4A and 4B). In contrast to these moderate changes, the expression of the more distant paralog ADGRL3 was dramatically affected (Figures 4A and 4B): HET and KO brains contained, respectively, 20% and 60% less ADGRL3 than WT brains. These data indicate that not only TEN2, which directly interacts with ADGRL1, but also ADGRL3 display coordinated expression with ADGRL1 and thus may interact with it physically, functionally, or both.30,31
Figure 4.

Protein expression, synaptic activity, and synapse formation in Adgrl1−/− mice
(A) Expression of the ADGRL family proteins and TEN2 in WT, HET, and KO mouse brains. A representative western blot of brain membranes, stained for the NTFs of ADGRL proteins and the C-terminal fragment of TEN2. β-actin was used to control gel loading.
(B) Quantification of receptor expression, as in (E) (ADGRL1, n = 5; ADGRL2, n = 5–9; ADGRL3, n = 5–6; TEN2, n = 3–5; NRXN1, n = 3–6).
(C) Increased spontaneous release of acetylcholine (electrophysiologically recorded as mEPPs) at KO mouse NMJs. Left, representative raw traces; right, quantification of mEPP frequency at WT and KO mouse NMJs (p < 0.012; WT, n = 8; KO, n = 11).
(D) Increased release of glutamate (Glu) and dopamine (DA) from synaptosomes isolated from KO mouse brain. Synaptosomes were preloaded with [14C]Glu and [3H]DA and incubated for 15 min without stimulation (Glu release, p < 0.0008, n = 6; DA release, p < 0.0041, n = 6). NT, neurotransmitter.
(E) Specific Ca2+-independent binding of 125I-LTX to cerebrocortical synaptosomes from the WT and KO mice (n = 6).
(F) LTXN4C (1 nM) increases glutamate release from the WT but not KO synaptosomes in the presence of 2 mM Ca2+ (n = 6).
(G) LTXN4C increases the mEPP frequency at the NMJs of WT and HET mice, but not KO mice. Left, representative original recordings; right, frequencies of LTXN4C-evoked mEPPs (WT, n = 6; HET, n = 3; KO, n = 5 independent animals).
(H) Spontaneous synaptic activity is greatly decreased in KO hippocampal cultures. Patch-clamp recordings demonstrate a regular occurrence of both mIPSCs (upward spikes) and mEPSCs (downward spikes) in WT hippocampal cultures, and a much rarer detection of mIPSCs and especially mEPSCs in KO cultures.
(I) The amplitudes and shapes of average miniature postsynaptic currents are similar in hippocampal cultures from WT and KO mice.
(J) The frequencies of mIPSCs and mEPSCs in WT and KO neuronal cultures (n = 28 cells from 6 experiments for each condition).
(K) KO neurons in culture form a lower number of inhibitory and especially excitatory synapses compared to WT neurons. Hippocampal cultures from P1 WT and KO brains were grown for 21 days and stained with antibodies against VGAT (inhibitory synapses) or VGLUT (excitatory synapses). The images are representative of n = 5 experiments.
(L) Quantification of inhibitory and excitatory synapse formation in WT and KO hippocampal cultures (n = 5 for both conditions).
All data are the means ± SEM; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; NS, non-significant.
ADGRL1 has been hypothesized to regulate the activity of both central and peripheral synapses.30,67,76 Therefore, we studied spontaneous release of acetylcholine, glutamate, and dopamine in KO mice. Surprisingly, KO mice displayed an increased basal release of these neurotransmitters (Figures 4C and 4D). Thus, spontaneous miniature end-plate potentials (mEPPs) were 2.5 times more frequent at KO neuromuscular junctions (NMJs) than at WT NMJs. This effect was purely presynaptic, because the mEPP amplitude was unchanged (Figure S2H). Likewise, the release of glutamate and dopamine from isolated synaptic terminals (synaptosomes) prepared from KO mouse brains was significantly ameliorated compared to WT mouse synaptosomes (Figure 4D). Although unexpected, the increased neurotransmitter release in KO mice was consistent with their hyperactivity and dysfunctional sensorimotor gating.
Given that ADGRL1 was identified as the major Ca2+-independent receptor for αLTX,23,25,60 we tested whether ADGRL1 ablation led to any changes in αLTX binding and activity. Indeed, the disruption of the Adgrl1 allele clearly decreased toxin binding (Figure 4E). However, about 30% of Ca2+-independent αLTX binding still remained in the KO brains. To determine whether this residual binding could be responsible for any effects of αLTX, we treated cerebrocortical synaptosomes and NMJs from the Adgrl1−/− mice with LTXN4C.25,67,77,78 This toxin strongly stimulated spontaneous exocytosis of glutamate from WT synaptosomes (Figure 4F) and acetylcholine from WT and HET NMJs (Figure 4G), while affecting only mEPP frequency, but not the amplitudes (Figure S2I), and thus acting purely presynaptically. By contrast, despite substantial toxin binding (Figure 4E and data not shown), KO synaptosomes and NMJs did not react to LTXN4C (Figures 4F and 4G). These results demonstrate that ADGRL1 is indeed the main mediator of the receptor-dependent effects of LTXN4C and that any changes in KO mouse brain functions most likely reflect ADGRL1 removal.
Combined, the above data suggested dysfunctional neuronal transmission at least in some KO synapses. To study synaptic transmission in central synapses in more detail, we prepared dissociated neuronal cultures from the hippocampi of WT and KO neonates. Using whole-cell patch-clamp recordings, we simultaneously recorded spontaneous activity of excitatory and inhibitory synapses formed by hippocampal neurons in culture (Figure 4H). The features of both excitatory and inhibitory miniature postsynaptic currents (mEPSCs and mIPSCs, respectively) were very similar in WT and KO mice, including their shapes and amplitudes (Figures 4I and S2J). However, synapses formed by KO neurons in vitro showed a much lower frequency of spontaneous firing than WT neurons: the mIPSC frequency was ∼4 times lower and the mEPSC frequency 83-fold lower in KO cultures than in WT cultures (Figures 4H and 4J).
This dramatic loss of spontaneous synaptic activity in cultured KO neurons was in stark contrast to an increased spontaneous synaptic activity in ex vivo synapses. We hypothesized that KO neurons form synapses in vitro much less efficiently than WT neurons, and this results in a lower recorded activity. This hypothesis was tested by immunostaining KO and WT neuronal cultures with antibodies against vesicular transporters of glutamate (VGLUT) and γ-aminobutyric acid, GABA (VGAT). As demonstrated in Figures 4K and 4L, inhibitory synapses (identified by the anti-VGAT antibody) were much less numerous in KO neuronal cultures than in WT cultures. Likewise, and in agreement with the recorded synaptic activity, excitatory synapses (identified by the anti-VGLUT antibody) were exceptionally rare in KO neuron cultures compared to WT cultures. Thus, the decreased synaptic activity in KO hippocampal cultures was most probably caused by poor synapse formation (or survival) in cultured KO neurons.
In summary, the lack of ADGRL1 significantly enhances basal spontaneous synaptic activity in cholinergic, glutamatergic, and dopaminergic synapses but blocks LTXN4C-induced neurotransmitter release in vivo while decreasing the rate of synapse formation in vitro.
Discussion
Previous molecular and physiological studies using mammalian models have demonstrated preferential brain expression of ADGRL1 and suggested that it plays an important role in neuronal functions and maturation during early brain development and adult life. However, very few studies have addressed ADGRL1 in humans; it has never been directly implicated in human pathology, and its role remains poorly defined. Here, we first describe a cohort of individuals presenting with a neurodevelopmental disorder and carrying pathogenic HET variants in ADGRL1. Furthermore, our results also show an important correlation between the disruption of Adgrl1 in KO mice and a neurodevelopmental phenotype.
We show here that on two distinct genetic backgrounds, the deletion of this gene is either lethal or partially lethal, leading to a sub-Mendelian ratio of the null allele in offspring genotypes (Figures S2B and S2E). This was unexpected, because a previous more limited study did not report another Adgrl1−/− mouse line to deviate from Mendelian genotype distribution.41 However, our extensive breeding data indicate that, on even a compensatory background, the disruption of the Adgrl1 allele still causes ∼50% lethality of KO embryos (Figure S2E). Those KO animals that survive gestation generally develop normally but demonstrate profound behavioral abnormalities.
The most obvious phenotype in Adgrl1 KO animals was frequent, deliberate parental neonaticide (Figure 3C). One factor known to cause aggression in mice is the maintenance conditions.79, 80, 81 However, the neonaticide was committed by KO parents only; the colony was maintained in a standard rodent facility; and the conditions were explicitly altered several times during the study, which had no effect on the frequency of infanticide. The specific circumstances of this behavior suggest that KO mice were in a state of hypervigilance/over-arousal, and when the major disturbance of parturition occurred, the mother’s sensory endurance was overloaded by physical demands from the pups, leading her to experience intense distress in the presence of the feared stimulus (her own litter). This indicates a failure to respond properly to environmental intrusions, i.e., a dysfunction of sensory processing.
Similarly, KO males also frequently killed their offspring, but they demonstrated extreme aggression more rarely than KO females for several possible reasons: (1) only some of the pups’ demands affected the fathers, thus rarely exceeding their sensory threshold; (2) the WT/HET females that were usually paired with KO males always nurtured their pups and possibly protected them; (3) the normal maternal behavior of the WT/HET females could have a calming effect on the distressed KO males. The stochastic nature of the infanticide suggested a consistent, but not completely penetrant, behavioral pattern in both female and male KO mice that could be attenuated by unknown factors, occasionally allowing the parents to spare and rear some of their litters. On the other hand, a lack of habituation to the repeated appearance of an offspring (Figure 3D) indicated the presence of general cognitive/learning deficits.
Faulty sensorimotor gating in the Adgrl1 KO mice was directly confirmed in the acoustic startle response and PPI experiments (Figures 3H–3J). PPI is predominantly used to probe gating deficits in schizophrenia,63 and the decreased PPI in our Adgrl1−/− mice was augmented by the antipsychotic drug risperidone (Figures 3I and 3J), implicating dopaminergic and/or serotonergic brain circuits in this phenotype and suggesting a link to schizophrenia. Indeed, direct measurements demonstrated a significantly upregulated release of dopamine in prefrontal cortices of Adgrl1−/− mice (Figure 4D). Furthermore, ADGRL1 has been implicated in fine regulation of neurotransmitter release, while Adgrl1 has been indirectly linked to schizophrenia.45,82 On the other hand, individuals with bipolar disorder also demonstrate a reduced PPI,83 and this is consistent with the bimodal pattern of locomotor activity in our Adgrl1−/− mice (Figures 3F and 3G) and the two modalities of behavior observed in their infanticide chronology (Figure 3D). Thus, in our study, the PPI test most likely detected a variable spectrum of neurological conditions, similar to those found in humans with variants in ADGRL1.
The sensory overload and locomotor hyperactivity in Adgrl1 KO mice appeared to correlate with strongly increased spontaneous release of glutamate and dopamine in their prefrontal brain areas and of acetylcholine at their NMJs (Figures 4C and 4D). As the activation of ADGRL1 by LTXN4C, TEN2, or A1 ScFv antibody is known to cause a massive increase in spontaneous synaptic activity, this aGPCR has been hypothesized to regulate neurotransmitter release at presynaptic nerve terminals.30,39,84 Therefore, it was initially unclear why Adgrl1 deletion caused an increase (rather than a decrease) in unstimulated release of glutamate, dopamine, and acetylcholine in KO mice. However, both central and peripheral KO synapses were insensitive to LTXN4C (Figures 4E–4G), which demonstrates the role for ADGRL1 in controlling synaptic activity and suggests that KO synapses, not activated via ADGRL1, have developed increased spontaneous transmitter secretion to compensate for ADGRL1 absence.
Adgrl1 deletion also led to impaired synapse formation in neuronal cultures, where it affected excitatory synapses more profoundly (Figures 4H–4L). This is in line with the previous observation that ADGRL2 and 3 may be involved in specifying the pattern of excitatory synapse formation in vitro, which depends on simultaneous binding of two ADGRL1 ligands, TEN2 and FLRT3.85 On the other hand, Adgrl1 deletion also caused dramatic Adgrl3 hypomorphism (Figures 4A and 4B). Thus, some of the phenotypical manifestations of Adgrl1 deletion could be due to the lower expression of Adgrl3 rather than the lack of Adgrl1. Indeed, mutations in ADGRL3 have been linked to ADHD in humans.45,50 However, the previous studies did not look at the possibility that mutations in ADGRL3 could cause ADGRL1 hypomorphism, an effect reciprocal to that described here for the Adgrl1−/− mice. Our observations suggest that the molecular basis of any neurodevelopmental disorders caused by mutations in ADGRL1 or ADGRL3 in humans may be complex, involving not only both of these proteins but also their interacting partners (TEN2, FLRT3).
One interesting question from our study is: which genetic change(s) could mitigate the embryonic lethality caused by Adgrl1 inactivation in mice? This question requires further in-depth studies, but one likely candidate is the already-mentioned hypomorphic Adgrl3 allele, which was a serendipitous consequence of extensive breeding and selection for an Adgrl1−/−-permissive background. The resultant decrease in Adgrl3 expression could indeed represent such a compensatory response, offsetting the absence of ADGRL1. If ADGRL1 and ADGRL3 are interdependent and physiologically antagonistic, their functions would normally remain in balance, but a loss of one protein would lead to an unabated activity of the other and thus jeopardize animals’ development/viability. Interestingly, the lack of ADGRL1 in mouse brain did not lead to any appreciable changes in the expression of NRXN1, suggesting that these two αLTX receptors are probably not functionally connected.
As shown above, the behavioral phenotypes of mice lacking ADGRL1, in many aspects, resembled those that are displayed by humans with pathogenic variants in ADGRL1. Neuropsychiatric and cognitive phenotypes in mice do not always perfectly match human behaviors and are not easy to interpret; however, it is clear from these data that the loss of Adgrl1 in mice is responsible for a broad neurodevelopmental phenotype. This provides a strong argument for extrapolating these data to humans.
The occurrence of variants in ADGRL1 in ten individuals from nine unrelated families with overlapping neurodevelopmental and neurological phenotypes, corroborated by the functional studies, strongly implicates ADGRL1 variants in intellectual disability and developmental delay. We could not identify any genotype-phenotype correlations. Emblematically, individuals 3 and 4 (F3-II-5 and F3-II-6, respectively) from the same family showed a variable expressivity of the symptoms associated with the same variant (Figure 1B). Among the most interesting variants identified are the p.Trp9∗ and p.Trp278∗ nonsense variants, which would be expected to result in a lack of the full-size ADGRL1. Indeed, our in vitro data indicate that at least the p.Trp9∗ mutant does not express any ADGRL1 (Figure 2A). Intriguingly, 5 out of 8 individuals, including individuals 3 and 5 (with the p.Trp278∗ and p.Trp9∗ variant, F3-II-5 and F4-II-1, respectively), demonstrate ASD/ADHD symptoms (Figure 1B), indicating that the lack of ADGRL1 expression could be directly associated with ADHD. This is further corroborated by the fact that several unrelated individuals, lacking different segments of chromosome 19p that include ADGRL1, also present with hyperactivity and intellectual disability.42 In addition, mutations that affect the ADGRL1 function in humans (p.Trp1005Arg and p.Ser1164Phe) (Figures 2C and 2D) also lead to ASD/ADHD symptoms (Figure 1B). Finally, the Adgrl1−/− mice described above demonstrate hyperactivity and cognitive deficits, consistent with human phenotypes.
Thus, although the effect of individual ADGRL1 variants will have to be studied further in detail, we can already conclude that perturbations in the function of ADGRL1 and its deletion seem to be associated mainly with cardinal symptoms, including developmental delay with delayed speech development, intellectual disability, ASD, ADHD, and—less frequently—epilepsy. However, these symptoms appear to be of variable expressivity, in line with our model studies in Adgrl1 KO mice, where the Adgrl1−/− phenotype strongly depended on the genetic background. In addition, several other inconstant features were identified in our work, such as nonspecific facial dysmorphism, macrocephaly, generalized hypotonia, joint hypermobility, dermatological issues, genital anomalies, and delayed puberty. It would be premature to conclude on any association between these secondary features and the ADGRL1 variants based on a single human cohort, although due to the significant expression of ADGRL1 in multiple tissues, these symptoms could be part of the ADGRL1-associated phenotype.
In conclusion, we provide a first overview of the phenotypes associated with ADGRL1 variants in humans. These phenotypes are strongly supported by the data from Adgrl1−/− mice. Additional functional studies and identification of a larger cohort of individuals carrying pathogenic ADGRL1 variants through international data sharing will be necessary to clarify the phenotypic spectrum associated with this gene and its counterpart, ADGRL3. We hope this information will help to achieve a better interpretation of any new ADGRL1 variants that may be identified after exome/genome sequencing, leading to a better understanding and diagnosis of individuals with neurodevelopmental disorders.
Acknowledgments
We thank the individuals and their families for their participation. We thank the Integragen society and the French National Center of Human Genomics Research (CNRGH) for exome analysis in some cases. We thank Evan E. Eichler and his laboratory for sharing clinical data for individual 5. This work was supported by grants from the Regional Council of Burgundy (PARI) and the European Regional Development Fund (FEDER) (to C.T.-R.). This work was also in part supported by Wellcome Trust Project Grants GR074359MA and WT083199MF, Biotechnology and Biological Sciences Research Council Core Support Grants BBD523078 and BBF0083091, and core funding from the University of Kent School of Pharmacy (to Y.U.).
Declaration of interests
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. A.C. is an employee of GeneDx, Inc.
Published: July 25, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.06.011.
Contributor Information
Antonio Vitobello, Email: antonio.vitobello@u-bourgogne.fr.
Yuri Ushkaryov, Email: y.ushkaryov@kent.ac.uk.
Web resources
gnomAD, https://gnomad.broadinstitute.org
MetaDome, https://stuart.radboudumc.nl/metadome
NHLBI Grand Opportunity Exome Sequencing Project (ESP), https://esp.gs.washington.edu/drupal
The Human Protein Atlas, https://www.proteinatlas.org
Supplemental information
Data and code availability
The published article includes all data generated or analyzed during the study.
Publicly available data utilized in this work were obtained from NHLBI GO Exome Sequencing Project (see URLs) and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), the Heart GO Sequencing Project (HL-103010), and the Rare Disease Initiative Zürich (RADIZ), Clinical Research Priority Program for Rare Diseases of the University of Zurich.
References
- 1.Hauser A.S., Attwood M.M., Rask-Andersen M., Schiöth H.B., Gloriam D.E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grammatopoulos D.K. Regulation of G-protein coupled receptor signalling underpinning neurobiology of mood disorders and depression. Mol. Cell. Endocrinol. 2017;449:82–89. doi: 10.1016/j.mce.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Leung C., Wong Y. Role of G Protein-Coupled receptors in the regulation of structural Plasticity and cognitive function. Molecules. 2017;22:1239. doi: 10.3390/molecules22071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kooistra A.J., Mordalski S., Pándy-Szekeres G., Esguerra M., Mamyrbekov A., Munk C., Keserű G.M., Gloriam D.E. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021;49:D335–D343. doi: 10.1093/nar/gkaa1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrne K.F., Pal A., Curtin J.F., Stephens J.C., Kinsella G.K. G-protein-coupled receptors as therapeutic targets for glioblastoma. Drug Discovery Today. 2021:2858–2870. doi: 10.1016/j.drudis.2021.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Bae B.-I., Tietjen I., Atabay K.D., Evrony G.D., Johnson M.B., Asare E., Wang P.P., Murayama A.Y., Im K., Lisgo S.N., et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science. 2014;343:764–768. doi: 10.1126/science.1244392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piao X., Hill R.S., Bodell A., Chang B.S., Basel-Vanagaite L., Straussberg R., Dobyns W.B., Qasrawi B., Winter R.M., Innes A.M., et al. G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004;303:2033–2036. doi: 10.1126/science.1092780. [DOI] [PubMed] [Google Scholar]
- 8.Nazarko O., Kibrom A., Winkler J., Leon K., Stoveken H., Salzman G., Merdas K., Lu Y., Narkhede P., Tall G., et al. A Comprehensive mutagenesis screen of the adhesion GPCR latrophilin-1/ADGRL1. iScience. 2018;3:264–278. doi: 10.1016/j.isci.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhnert F., Mancuso M.R., Shamloo A., Wang H.-T., Choksi V., Florek M., Su H., Fruttiger M., Young W.L., Heilshorn S.C., Kuo C.J. Essential regulation of CNS angiogenesis by the orphan G protein–coupled receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chae J., Kim M.J., Goo J.H., Collier S., Gubb D., Charlton J., Adler P.N., Park W.J. The Drosophila tissue polarity gene starry night encodes a member of the protocadherin family. Development. 1999;126:5421–5429. doi: 10.1242/dev.126.23.5421. [DOI] [PubMed] [Google Scholar]
- 11.Langenhan T., Prömel S., Mestek L., Esmaeili B., Waller-Evans H., Hennig C., Kohara Y., Avery L., Vakonakis I., Schnabel R., Russ A.P. Latrophilin signaling links anterior-posterior tissue polarity and oriented cell divisions in the C. elegans embryo. Dev. Cell. 2009;17:494–504. doi: 10.1016/j.devcel.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shima Y., Kengaku M., Hirano T., Takeichi M., Uemura T. Regulation of dendritic maintenance and growth by a mammalian 7-pass transmembrane cadherin. Dev. Cell. 2004;7:205–216. doi: 10.1016/j.devcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Usui T., Shima Y., Shimada Y., Hirano S., Burgess R.W., Schwarz T.L., Takeichi M., Uemura T. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell. 1999;98:585–595. doi: 10.1016/s0092-8674(00)80046-x. [DOI] [PubMed] [Google Scholar]
- 14.Bolliger M.F., Martinelli D.C., Südhof T.C. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. USA. 2011;108:2534–2539. doi: 10.1073/pnas.1019577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monk K.R., Naylor S.G., Glenn T.D., Mercurio S., Perlin J.R., Dominguez C., Moens C.B., Talbot W.S. A G protein–coupled receptor is essential for schwann cells to initiate myelination. Science. 2009;325:1402–1405. doi: 10.1126/science.1173474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu G.-M., Mai T.-L., Chen C.-M. Visualizing the GPCR network: classification and evolution. Sci. Rep. 2017;7:15495. doi: 10.1038/s41598-017-15707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Araç D., Boucard A.A., Bolliger M.F., Nguyen J., Soltis S.M., Südhof T.C., Brunger A.T. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis: cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012;31:1364–1378. doi: 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosa M., Noel T., Harris M., Ladds G. Emerging roles of adhesion G protein-coupled receptors. Biochem. Soc. Trans. 2021;49:1695–1709. doi: 10.1042/bst20201144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langenhan T., Aust G., Hamann J. Sticky signaling—adhesion class G protein–coupled receptors take the stage. Sci. Signal. 2013;6:re3. doi: 10.1126/scisignal.2003825. [DOI] [PubMed] [Google Scholar]
- 20.Krishnan A., Nijmeijer S., de Graaf C., Schiöth H.B. In: Adhesion G Protein-Coupled Receptors. Langenhan T., Schöneberg T., editors. Springer International Publishing; Cham: 2016. Classification, nomenclature, and structural aspects of adhesion GPCRs; pp. 15–41. [DOI] [PubMed] [Google Scholar]
- 21.Dunn H.A., Orlandi C., Martemyanov K.A. Beyond the ligand: extracellular and transcellular G protein–coupled receptor complexes in physiology and pharmacology. Pharmacol. Rev. 2019;71:503–519. doi: 10.1124/pr.119.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krasnoperov V.G., Beavis R., Chepurny O.G., Little A.R., Plotnikov A.N., Petrenko A.G. The calcium-independent receptor of α-latrotoxin is not a neurexin. Biochem. Biophys. Res. Commun. 1996;227:868–875. doi: 10.1006/bbrc.1996.1598. [DOI] [PubMed] [Google Scholar]
- 23.Davletov B.A., Shamotienko O.G., Lelianova V.G., Grishin E.V., Ushkaryov Y.A. Isolation and biochemical characterization of a Ca2+-independent α-Latrotoxin-binding protein. J. Biol. Chem. 1996;271:23239–23245. doi: 10.1074/jbc.271.38.23239. [DOI] [PubMed] [Google Scholar]
- 24.Krasnoperov V.G., Bittner M.A., Beavis R., Kuang Y., Salnikow K.V., Chepurny O.G., Little A.R., Plotnikov A.N., Wu D., Holz R.W., Petrenko A.G. α-Latrotoxin stimulates exocytosis by the interaction with a neuronal G-protein-coupled receptor. Neuron. 1997;18:925–937. doi: 10.1016/s0896-6273(00)80332-3. [DOI] [PubMed] [Google Scholar]
- 25.Volynski K.E., Capogna M., Ashton A.C., Thomson D., Orlova E.V., Manser C.F., Ribchester R.R., Ushkaryov Y.A. Mutant α-latrotoxin (LTXN4C) does not form pores and causes secretion by receptor stimulation. J. Biol. Chem. 2003;278:31058–31066. doi: 10.1074/jbc.m210395200. [DOI] [PubMed] [Google Scholar]
- 26.Moreno-Salinas A.L., Avila-Zozaya M., Ugalde-Silva P., Hernández-Guzmán D.A., Missirlis F., Boucard A.A. Latrophilins: a neuro-centric view of an evolutionary conserved adhesion G protein-coupled receptor subfamily. Front. Neurosci. 2019;13:700. doi: 10.3389/fnins.2019.00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushita H., Lelianova V.G., Ushkaryov Y.A. The latrophilin family: multiply spliced G protein-coupled receptors with differential tissue distribution. FEBS Lett. 1999;443:348–352. doi: 10.1016/s0014-5793(99)00005-8. [DOI] [PubMed] [Google Scholar]
- 28.Ichtchenko K., Bittner M.A., Krasnoperov V., Little A.R., Chepurny O., Holz R.W., Petrenko A.G. A novel ubiquitously expressed α-latrotoxin receptor is a member of the CIRL family of G-protein-coupled receptors. J. Biol. Chem. 1999;274:5491–5498. doi: 10.1074/jbc.274.9.5491. [DOI] [PubMed] [Google Scholar]
- 29.Sugita S., Ichtchenko K., Khvotchev M., Südhof T.C. α-Latrotoxin receptor CIRL/Latrophilin 1 (CL1) defines an unusual family of ubiquitous G-protein-linked receptors. J. Biol. Chem. 1998;273:32715–32724. doi: 10.1074/jbc.273.49.32715. [DOI] [PubMed] [Google Scholar]
- 30.Silva J.-P., Lelianova V.G., Ermolyuk Y.S., Vysokov N., Hitchen P.G., Berninghausen O., Rahman M.A., Zangrandi A., Fidalgo S., Tonevitsky A.G., et al. Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. USA. 2011;108:12113–12118. doi: 10.1073/pnas.1019434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boucard A.A., Maxeiner S., Südhof T.C. Latrophilins function as heterophilic cell-adhesion molecules by binding to teneurins. J. Biol. Chem. 2014;289:387–402. doi: 10.1074/jbc.m113.504779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Röthe J., Thor D., Winkler J., Knierim A.B., Binder C., Huth S., Kraft R., Rothemund S., Schöneberg T., Prömel S. Involvement of the adhesion GPCRs latrophilins in the regulation of insulin release. Cell Rep. 2019;26:1573–1584.e5. doi: 10.1016/j.celrep.2019.01.040. [DOI] [PubMed] [Google Scholar]
- 33.Li J., Xie Y., Cornelius S., Jiang X., Sando R., Kordon S.P., Pan M., Leon K., Südhof T.C., Zhao M., Araç D. Alternative splicing controls teneurin-latrophilin interaction and synapse specificity by a shape-shifting mechanism. Nat. Commun. 2020;11:2140. doi: 10.1038/s41467-020-16029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreienkamp H.-J., Zitzer H., Gundelfinger E.D., Richter D., Böckers T.M. The calcium-independent receptor for α-latrotoxin from human and rodent brains interacts with members of the ProSAP/SSTRIP/Shank family of multidomain proteins. J. Biol. Chem. 2000;275:32387–32390. doi: 10.1074/jbc.c000490200. [DOI] [PubMed] [Google Scholar]
- 35.Uchigashima M., Cheung A., Suh J., Watanabe M., Futai K. Differential expression of neurexin genes in the mouse brain. J. Comp. Neurol. 2019;527:1940–1965. doi: 10.1002/cne.24664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boucard A.A., Ko J., Südhof T.C. High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 Produces an Intercellular adhesion complex. J. Biol. Chem. 2012;287:9399–9413. doi: 10.1074/jbc.m111.318659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Sullivan M., de Wit J., Savas J.N., Comoletti D., Otto-Hitt S., Yates J.R., Ghosh A. FLRT proteins are endogenous latrophilin ligands and regulate excitatory synapse development. Neuron. 2012;73:903–910. doi: 10.1016/j.neuron.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burbach J.P.H., Meijer D.H. Latrophilin’s social protein network. Front. Neurosci. 2019;13:643. doi: 10.3389/fnins.2019.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vysokov N.V., Silva J.-P., Lelianova V.G., Suckling J., Cassidy J., Blackburn J.K., Yankova N., Djamgoz M.B., Kozlov S.V., Tonevitsky A.G., Ushkaryov Y.A. Proteolytically released Lasso/teneurin-2 induces axonal attraction by interacting with latrophilin-1 on axonal growth cones. Elife. 2018;7:e37935. doi: 10.7554/elife.37935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson V.A., del Toro D., Carrasquero M., Roversi P., Harlos K., Klein R., Seiradake E. Structural basis of latrophilin-FLRT interaction. Structure. 2015;23:774–781. doi: 10.1016/j.str.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tobaben S., Südhof T.C., Stahl B. Genetic analysis of α-latrotoxin receptors reveals functional interdependence of CIRL/latrophilin 1 and neurexin 1α. J. Biol. Chem. 2002;277:6359–6365. doi: 10.1074/jbc.m111231200. [DOI] [PubMed] [Google Scholar]
- 42.Bonaglia M.C., Marelli S., Novara F., Commodaro S., Borgatti R., Minardo G., Memo L., Mangold E., Beri S., Zucca C., et al. Genotype–phenotype relationship in three cases with overlapping 19p13.12 microdeletions. Eur. J. Hum. Genet. 2010;18:1302–1309. doi: 10.1038/ejhg.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y.R., Li J., Zhao S.D., Bradfield J.P., Mentch F.D., Maggadottir S.M., Hou C., Abrams D.J., Chang D., Gao F., et al. Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat Med. 2015;21:1018–1027. doi: 10.1038/nm.3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vezain M., Lecuyer M., Rubio M., Dupé V., Ratié L., David V., Pasquier L., Odent S., Coutant S., Tournier I., et al. A de novo variant in ADGRL2 suggests a novel mechanism underlying the previously undescribed association of extreme microcephaly with severely reduced sulcation and rhombencephalosynapsis. Acta Neuropathol. Commun. 2018;6:109. doi: 10.1186/s40478-018-0610-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arcos-Burgos M., Jain M., Acosta M.T., Shively S., Stanescu H., Wallis D., Domené S., Vélez J.I., Karkera J.D., Balog J., et al. A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol. Psychiatry. 2010;15:1053–1066. doi: 10.1038/mp.2010.6. [DOI] [PubMed] [Google Scholar]
- 46.Ribasés M., Ramos-Quiroga J.A., Sánchez-Mora C., Bosch R., Richarte V., Palomar G., Gastaminza X., Bielsa A., Arcos-Burgos M., Muenke M., et al. Contribution of LPHN3 to the genetic susceptibility to ADHD in adulthood: a replication study. Gene Brain Behav. 2011;10:149–157. doi: 10.1111/j.1601-183x.2010.00649.x. [DOI] [PubMed] [Google Scholar]
- 47.Acosta M.T., Swanson J., Stehli A., Molina B.S.G., MTA Team, Arcos-Burgos M., Martinez A.F., Arcos-Burgos M., Muenke M. ADGRL3 (LPHN3) variants are associated with a refined phenotype of ADHD in the MTA study. Mol. Genet. Genomic Med. 2016;4:540–547. doi: 10.1002/mgg3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruxel E.M., Moreira-Maia C.R., Akutagava-Martins G.C., Quinn T.P., Klein M., Franke B., Ribasés M., Rovira P., Sánchez-Mora C., Kappel D.B., et al. Meta-analysis and systematic review of ADGRL3 (LPHN3) polymorphisms in ADHD susceptibility. Mol. Psychiatry. 2021;26:2277–2285. doi: 10.1038/s41380-020-0673-0. [DOI] [PubMed] [Google Scholar]
- 49.Domené S., Stanescu H., Wallis D., Tinloy B., Pineda D.E., Kleta R., Arcos-Burgos M., Roessler E., Muenke M. Screening of human LPHN3 for variants with a potential impact on ADHD susceptibility. Am. J. Med. Genet. 2011;156:11–18. doi: 10.1002/ajmg.b.31141. [DOI] [PubMed] [Google Scholar]
- 50.Acosta M.T., Vélez J.I., Bustamante M.L., Balog J.Z., Arcos-Burgos M., Muenke M. A two-locus genetic interaction between LPHN3 and 11q predicts ADHD severity and long-term outcome. Transl. Psychiatry. 2011;1:e17. doi: 10.1038/tp.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jain M., Vélez J.I., Acosta M.T., Palacio L.G., Balog J., Roessler E., Pineda D., Londoño A.C., Palacio J.D., ArbelAez A., et al. A cooperative interaction between LPHN3 and 11q doubles the risk for ADHD. Mol. Psychiatry. 2012;17:741–747. doi: 10.1038/mp.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Labbe A., Liu A., Atherton J., Gizenko N., Fortier M.È., Sengupta S.M., Ridha J. Refining psychiatric phenotypes for response to treatment: contribution of LPHN3 in ADHD. Am. J. Med. Genet. 2012;159B:776–785. doi: 10.1002/ajmg.b.32083. [DOI] [PubMed] [Google Scholar]
- 53.Fallgatter A.J., Ehlis A.-C., Dresler T., Reif A., Jacob C.P., Arcos-Burgos M., Muenke M., Lesch K.-P. Influence of a Latrophilin 3 (LPHN3) risk haplotype on event-related potential measures of cognitive response control in attention-deficit hyperactivity disorder (ADHD) Eur. Neuropsychopharmacol. 2013;23:458–468. doi: 10.1016/j.euroneuro.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez A.F., Abe Y., Hong S., Molyneux K., Yarnell D., Löhr H., Driever W., Acosta M.T., Arcos-Burgos M., Muenke M. An Ultraconserved brain-specific enhancer within ADGRL3 (LPHN3) underpins attention-deficit/hyperactivity disorder susceptibility. Biol. Psychiatr. 2016;80:943–954. doi: 10.1016/j.biopsych.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Field L.L., Shumansky K., Ryan J., Truong D., Swiergala E., Kaplan B.J. Dense-map genome scan for dyslexia supports loci at 4q13, 16p12, 17q22; suggests novel locus at 7q36: high-density genome scan for dyslexia genes. Gene Brain Behav. 2013;12:56–69. doi: 10.1111/gbb.12003. [DOI] [PubMed] [Google Scholar]
- 56.Kappel D.B., Schuch J.B., Rovaris D.L., da Silva B.S., Müller D., Breda V., Teche S.P., Riesgo R., Schüler-Faccini L., et al. ADGRL3 rs6551665 as a common vulnerability factor underlying attention-deficit/hyperactivity disorder and autism spectrum disorder. Neuromol. Med. 2019;21:60–67. doi: 10.1007/s12017-019-08525-x. [DOI] [PubMed] [Google Scholar]
- 57.Guo H., Duyzend M.H., Coe B.P., Baker C., Hoekzema K., Gerdts J., Turner T.N., Zody M.C., Beighley J.S., Murali S.C., et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet. Med. 2019;21:1611–1620. doi: 10.1038/s41436-018-0380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nambot S., Thevenon J., Kuentz P., Duffourd Y., Tisserant E., Bruel A.-L., Mosca-Boidron A.-L., Masurel-Paulet A., Lehalle D., Jean-Marçais N., et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet. Med. 2018;20:645–654. doi: 10.1038/gim.2017.162. [DOI] [PubMed] [Google Scholar]
- 59.Berninghausen O., Rahman M.A., Silva J.-P., Davletov B., Hopkins C., Ushkaryov Y.A. Neurexin Iβ and neuroligin are localized on opposite membranes in mature central synapses. J. Neurochem. 2007;103:1855–1863. doi: 10.1111/j.1471-4159.2007.04918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davletov B.A., Meunier F.A., Ashton A.C., Matsushita H., Hirst W.D., Lelianova V.G., Wilkin G.P., Dolly J.O., Ushkaryov Y.A. Vesicle exocytosis stimulated by α-latrotoxin is mediated by latrophilin and requires both external and stored Ca2+ EMBO J. 1998;17:3909–3920. doi: 10.1093/emboj/17.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volynski K.E., Meunier F.A., Lelianova V.G., Dudina E.E., Volkova T.M., Rahman M.A., Manser C., Grishin E.V., Dolly J.O., Ashley R.H., Ushkaryov Y.A. Latrophilin, neurexin, and their signaling-deficient mutants facilitate α-latrotoxin insertion into membranes but are not involved in pore formation. J. Biol. Chem. 2000;275:41175–41183. doi: 10.1074/jbc.m005857200. [DOI] [PubMed] [Google Scholar]
- 62.Davydov I.I., Fidalgo S., Khaustova S.A., Lelyanova V.G., Grebenyuk E.S., Ushkaryov Y.A., Tonevitsky A.G. Prediction of epitopes in closely related proteins using a new algorithm. Bull. Exp. Biol. Med. 2009;148:869–873. doi: 10.1007/s10517-010-0838-y. [DOI] [PubMed] [Google Scholar]
- 63.Swerdlow N.R., Weber M., Qu Y., Light G.A., Braff D.L. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199:331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swerdlow N., Geyer M., Braff D. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- 65.Geyer M.A., Swerdlow N.R. Measurement of startle response, prepulse inhibition, and habituation. Curr. Protoc. Neurosci. 2001;Chapter 8:Unit 8.7. doi: 10.1002/0471142301.ns0807s03. [DOI] [PubMed] [Google Scholar]
- 66.Geyer M.A., McIlwain K.L., Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol. Psychiatry. 2002;7:1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- 67.Ashton A.C., Volynski K.E., Lelianova V.G., Orlova E.V., Van Renterghem C., Canepari M., Seagar M., Ushkaryov Y.A. α-Latrotoxin, acting via two Ca2+-dependent pathways, triggers exocytosis of two pools of synaptic vesicles. J. Biol. Chem. 2001;276:44695–44703. doi: 10.1074/jbc.m108088200. [DOI] [PubMed] [Google Scholar]
- 68.Volynski K.E., Silva J.-P., Lelianova V.G., Atiqur Rahman M., Hopkins C., Ushkaryov Y.A. Latrophilin fragments behave as independent proteins that associate and signal on binding of LTXN4C. EMBO J. 2004;23:4423–4433. doi: 10.1038/sj.emboj.7600443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Silva J.-P., Lelianova V., Hopkins C., Volynski K.E., Ushkaryov Y. Functional cross-interaction of the fragments produced by the cleavage of distinct adhesion G-protein-coupled receptors. J. Biol. Chem. 2009;284:6495–6506. doi: 10.1074/jbc.m806979200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capogna M., Volynski K.E., Emptage N.J., Ushkaryov Y.A. The α-latrotoxin mutant LTX N4C enhances spontaneous and evoked transmitter release in CA3 pyramidal neurons. J. Neurosci. 2003;23:4044–4053. doi: 10.1523/jneurosci.23-10-04044.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.De Rubeis S., He X., Goldberg A.P., Poultney C.S., Samocha K., Ercument Cicek A., Kou Y., Liu L., Fromer M., Walker S., et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Posner M.I., Dehaene S. Attentional networks. Trends Neurosci. 1994;17:75–79. doi: 10.1016/0166-2236(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 74.McFarlane H.G., Kusek G.K., Yang M., Phoenix J.L., Bolivar V.J., Crawley J.N. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 2008;7:152–163. doi: 10.1111/j.1601-183x.2007.00330.x. [DOI] [PubMed] [Google Scholar]
- 75.Wu W.-L., Cheng S.-J., Lin S.-H., Chuang Y.-C., Huang E.Y.-K., Chen C.-C. The effect of ASIC3 knockout on corticostriatal circuit and mouse self-grooming behavior. Front. Cell. Neurosci. 2019;13:86. doi: 10.3389/fncel.2019.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lelyanova V.G., Thomson D., Ribchester R.R., Tonevitsky E.A., Ushkaryov Y.A. Activation of α-latrotoxin receptors in neuromuscular synapses leads to a prolonged splash acetylcholine release. Bull. Exp. Biol. Med. 2009;147:701–703. doi: 10.1007/s10517-009-0600-5. [DOI] [PubMed] [Google Scholar]
- 77.Orlova E.V., Rahman M.A., Gowen B., Volynski K.E., Ashton A.C., Manser C., van Heel M., Ushkaryov Y.A. Structure of alpha-latrotoxin oligomers reveals that divalent cation-dependent tetramers form membrane pores. Nat. Struct. Biol. 2000;7:48–53. doi: 10.1038/71247. [DOI] [PubMed] [Google Scholar]
- 78.Ashton A.C., Rahman M.A., Volynski K.E., Manser C., Orlova E.V., Matsushita H., Davletov B.A., van Heel M., Grishin E.V., Ushkaryov Y.A. Tetramerisation of α-latrotoxin by divalent cations is responsible for toxin-induced non-vesicular release and contributes to the Ca2+-dependent vesicular exocytosis from synaptosomes. Biochimie. 2000;82:453–468. doi: 10.1016/s0300-9084(00)00199-1. [DOI] [PubMed] [Google Scholar]
- 79.Heiming R.S., Bodden C., Jansen F., Lewejohann L., Kaiser S., Lesch K.-P., Palme R., Sachser N. Living in a dangerous world decreases maternal care: a study in serotonin transporter knockout mice. Horm. Behav. 2011;60:397–407. doi: 10.1016/j.yhbeh.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 80.vom Saal F.S., Howard L.S. The regulation of infanticide and parental behavior: implications for reproductive success in male mice. Science. 1982;215:1270–1272. doi: 10.1126/science.7058349. [DOI] [PubMed] [Google Scholar]
- 81.Zafar T., Naik A.Q., Shrivastava V.K. Effect of cold stress on infanticide by female Swiss albino mice Mus musculus: a pilot study. J. Anim. Sci. Technol. 2018;60:7. doi: 10.1186/s40781-018-0168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen M.-L., Chen C.-H. Microarray analysis of differentially expressed genes in rat frontal cortex under chronic risperidone treatment. Neuropsychopharmacol. 2005;30:268–277. doi: 10.1038/sj.npp.1300612. [DOI] [PubMed] [Google Scholar]
- 83.Mao Z., Bo Q., Li W., Wang Z., Ma X., Wang C. Prepulse inhibition in patients with bipolar disorder: a systematic review and meta-analysis. BMC Psychiatr. 2019;19:282. doi: 10.1186/s12888-019-2271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vysokov N.V., Silva J.-P., Lelianova V.G., Ho C., Djamgoz M.B., Tonevitsky A.G., Ushkaryov Y.A. The mechanism of regulated release of Lasso/teneurin-2. Front. Mol. Neurosci. 2016;9:59. doi: 10.3389/fnmol.2016.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sando R., Jiang X., Südhof T.C. Latrophilin GPCRs direct synapse specificity by coincident binding of FLRTs and teneurins. Science. 2019;363:eaav7969. doi: 10.1126/science.aav7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during the study.
Publicly available data utilized in this work were obtained from NHLBI GO Exome Sequencing Project (see URLs) and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), the Heart GO Sequencing Project (HL-103010), and the Rare Disease Initiative Zürich (RADIZ), Clinical Research Priority Program for Rare Diseases of the University of Zurich.