

Abstract
Extrachromosomal circular DNA (ecDNA), first described in the 1960’s, is emerging as a prevalent, yet less characterized oncogenic alteration in cancer. EcDNA is a reservoir for oncogene amplification and associated with aggressive tumor phenotype with poor patient outcome. Despite the long-held knowledge of its existence, little is known about how ecDNA affects tumor cell behavior. Recent data reveals that ecDNA hubs are mobile transcriptional enhancers which can trans-activate gene expression through chromatin interactions. Together with its prevalence, structural complexity and non-equally segregation inheritance, ecDNA can offer selective growth advantages, contribute to intratumor heterogeneity and accelerate tumor evolution. Future technology development is expected to transform the current paradigm for studying ecDNA and therapeutic strategies targeting ecDNA vulnerability.
Keywords: Chromatin interactions, Extrachromosomal circular DNA, EcDNA hubs, Super enhancers, Mobile activators, Cancer genome variation
Extrachromosomal circular DNA is an oncogenic alteration frequently found in cancer
In eukaryotic nuclei, DNA is organized in the form of chromosomes folded into highly organized chromatin architectures [1]. Extrachromosomal DNA circles, existing outside the twenty-three pairs of linear chromosomes, have been observed in the nuclei of many different cells and tissue types across multiple eukaryotic species [2–4]. The extrachromosomal circular DNAs detected across a wide range of somatic tissues and species are referred to as eccDNAs (see Glossary). They are generally less than 10 Kb in size [3, 5]. EccDNA sequences can be derived from regions across entire genomes [6] with variable abundance [2, 7] and are enriched in repetitive elements [3, 8]. They have many suggested functions including environmental adaptation [9], drug resistance [10], genome stability [6, 11] and stimulation of immune responses [12].Extrachromosomal DNA circles specifically found in the nuclei of cancer cells are generally referred to as ecDNAs [13]. Their sizes range from hundreds of kilobases to several megabases and comprise of complete oncogenes and regulatory elements [14]. Apart from their circular conformation and extrachromosomal nature, these two types of extrachromosomal DNA molecules do not appear to be related in respect to their origin, structure, or function.
First described in 1965 as ‘double-minute’ (DM) by microscopic imaging in the karyotypes of neuroblastoma [15], ecDNA has emerged as a prevalent, oncogenic alteration in cancer genomes [16]. From in-depth surveys in 17 different cancer types utilizing genomic sequencing, cytogenetic and computational approaches, ecDNA is found in as many as 50% of primary tumors and constitutes a bona fide mechanism for oncogene amplification [13]. The presence of ecDNA is associated with aggressive tumor behavior, characterized by poor patient prognosis [16, 17] and therapeutic resistance [18–20]. Analysis of whole genome sequence data from 3,212 cancer patients’ tumors across 29 cancer types from The Cancer Genome Atlas (TCGA) revealed that patients whose tumors harboring ecDNAs had significant worse five-year survival outcomes compared to patients whose tumors classified as non-circular amplifications. And these circular amplicons were more frequently found in aggressive cancer types like glioblastoma (GBM), sarcoma and esophageal carcinoma [17]. Moreover, ecDNA amplification is associated with more lymph node spread at initial diagnosis as well as gene expression signatures of cell proliferation and immune infiltration [16].
Despite the awareness of ecDNA for several decades and the recent recognition of its functional relevance to tumorigenesis, we are only beginning to understand the mechanism(s) by which ecDNA modulates tumor growth and contributes to tumor evolution. Structurally, ecDNA is a circular chromatin particle comprising of DNA, histones, and their modifications but lacking centromeres and telomeres [21]. It can rapidly accumulate in cancer cells through uneven inheritance [22] (Figure 1), offering a competitive advantage in response to the selective pressures from the tumor microenvironment and anti-cancer drugs [18, 23]. Extrachromosomal BRAF oncogene amplification is a mechanism by which tumors acquire drug resistance as seen in patient-derived tumor xenografts from lung cancer and melanoma treated with the extracellular signal regulated kinase inhibitor (ERKi) [23] and a melanoma model treated with a dual mitogen-activated protein kinase inhibitor (MAPKi) [24]. Tumor clones with high levels of extrachromosomal BRAF copies confer a fitness advantage and can selectively grow in the presence of drugs.
Figure 1. Segregation of ecDNA and chromosomes during cell division.
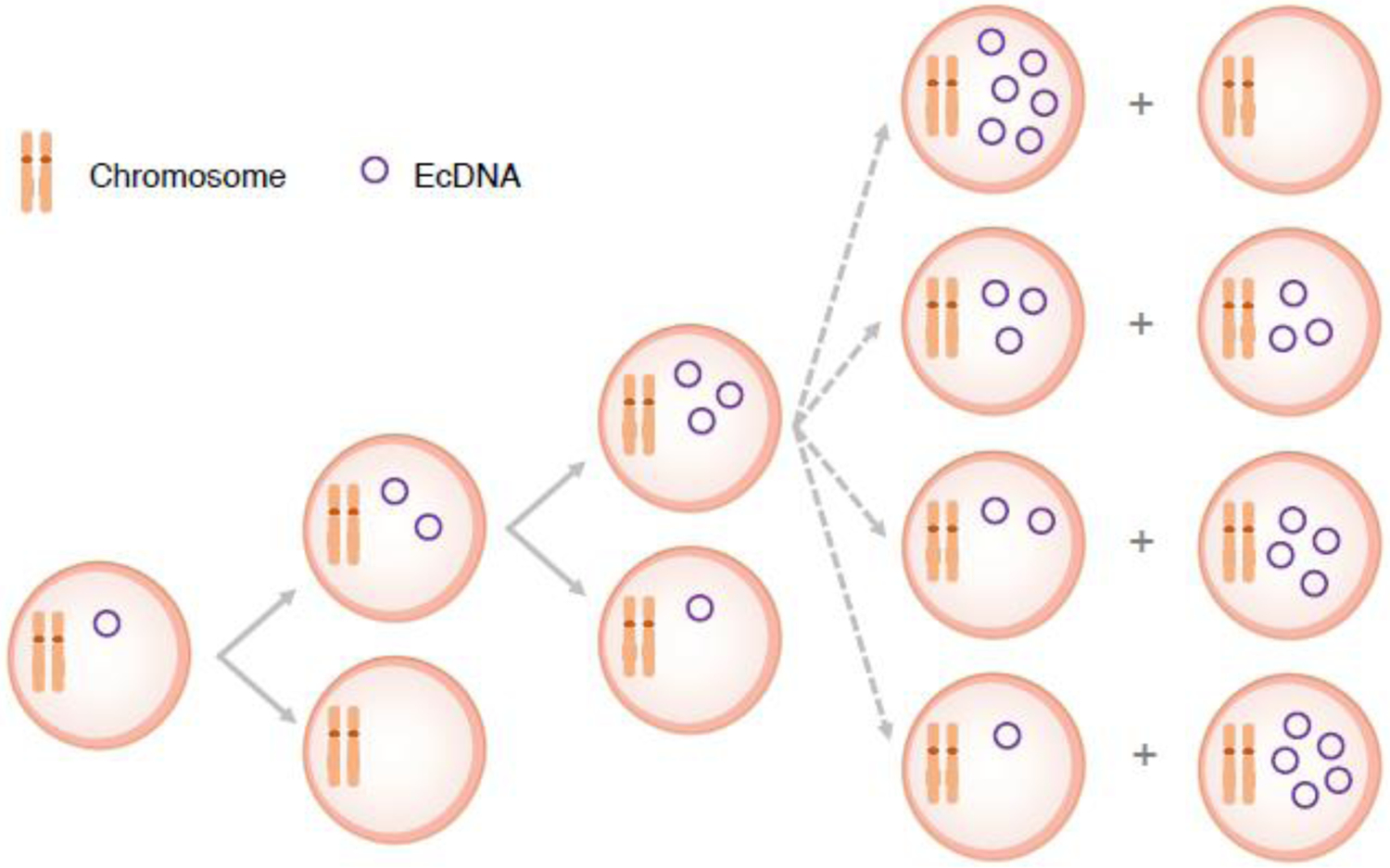
Chromosomes have centromeres and are evenly distributed into daughter cells. EcDNAs lack centromeres and are randomly inherited by daughter cells in mitosis.
Aided by the advances in 3-dimensional (3D) chromatin conformation analyses, long-read DNA sequencing, genome editing technologies and live-cell high-resolution imaging, recent studies have revealed the genomic composition, chromatin states, and 3D spatial configuration of ecDNA, which are critical for its function in cancer [21, 22, 25–28]. Particularly, ecDNA possesses enhanced chromatin accessibility, is active in aggregated conformation and modulates gene expression through enhancer-mediated chromatin interactions [21, 25–27]. These findings have provided valuable insights into the regulatory activities of ecDNA and their potential implications in designing cancer therapeutic strategies. As such, ecDNA has moved beyond a phenomenological description to become a unique genetic alteration that is functionally significant in cancer. Together with its prevalence, structural complexity and uneven segregation inheritance, there is a renewed interest in the biology of this enigmatic molecule and its roles in promoting cancer progression. In this review, we focus on the recent progress in understanding ecDNA and how such knowledge can be applied to benefit clinical and therapeutic approaches of cancer.
EcDNA carries oncogene amplification with highly complex and rearranged molecular structures
As an extrachromosomal circular chromatin particle, the most recognized feature of ecDNA has been its role as a genetic entity carrying oncogene amplification. The most frequently found oncogenes on ecDNAs are MDM2, MYC, EGFR, CDK4, ERBB2, SOX2, TERT, CCND1, E2F3, and CCNE1 [16]. While these genes are mostly found individually, multiple oncogenes can be co-amplified on the same ecDNA molecule [18, 20, 21]. In addition to oncogenes encoding full-length open reading frames, chimeric fusions or splicing variants are also found, presumably resulting from circle-derived rearrangements [5, 11, 29]. For example, an extrachromosomal allele encoding a truncated EGFR fused between exons 1 and 8, known as EGFRvIII, is the predominant form of EGFR found in primary glioblastoma [19]. EGFRvIII lacks the ligand-binding domain while still retaining intact catalytic structural domains, creating a constitutively active and highly oncogenic form of EGFR [30]. In addition to protein-coding genes, non-coding regulatory elements such as enhancers can be co-amplified on ecDNA [21, 25, 26], exemplifying the regulatory function of ecDNA in transcription. These co-amplified enhancers presumably are acquired by ecDNA through cycles of somatic genome rearrangement [21, 26]. In glioblastoma, almost all extrachromosomal EGFR amplicons harbor two enhancers located 130 Kb upstream of the EGFR promoter [26].
Current knowledge about the molecular structure of ecDNA is mostly derived from DNA sequencing, fluorescent in-situ hybridization (FISH), and super-resolution microscopic imaging analyses [20, 31, 32]. As an ecDNA molecule can span up to several megabases in size, specialized computational tools like AmpliconArchitect (AA) [32] and AmpliconReconstructor (AR) [28] are needed to reconstruct full ecDNA circles from short-read sequencing data and/or optical mapping data. EcDNA amplicon reconstruction by AA from sequencing data of 117 cancer samples covering 13 cancer types reveals the highly rearranged and heterogenous patterns of ecDNA [32]. While about half of these structures are defined as single-interval amplicons, which was previously observed to be 35% (8/23) in acute myeloid leukemia cases [33], fragments from up to three chromosomes were found to fuse into one ecDNA amplicon [32]. Overall, the average number of rearrangements detected per ecDNA is 4.9, with a maximum up to 49 [32]. In the lung cancer cell line NCI-H460, both oncogenes MYC and PVT1 are found in multiple copies in a complex arrangement on individual ecDNA molecules assembled by AR [28]. Furthermore, structural patterns of ecDNA can be heterogeneous, differing in their complexity and segments, within a population of cancer cells derived from the same tumor [34]. In a brain tumor, five ecDNA amplicons covering 44 segments and 27 structural variations (SVs) were identified with lengths ranging from 640 Kb to 966 Kb and SV numbers ranging from one to 16 [20]. Such a highly segmented arrangement comprised of oncogenes and regulatory enhancers found in a circular configuration represents a new dimension of SV within cancer genomes (Figure 2a). When examined between paired primary and relapsed tumors, copy numbers and segment boundaries of ecDNA were longitudinally shifted in relapse tumors [18, 20]. In some cases, new extrachromosomal amplified oncogenes emerged [18], indicating a highly dynamic and unstable nature of ecDNA structures that are subjected to longitudinal clonal evolution.
Figure 2. EcDNA structure complexity and dynamics.
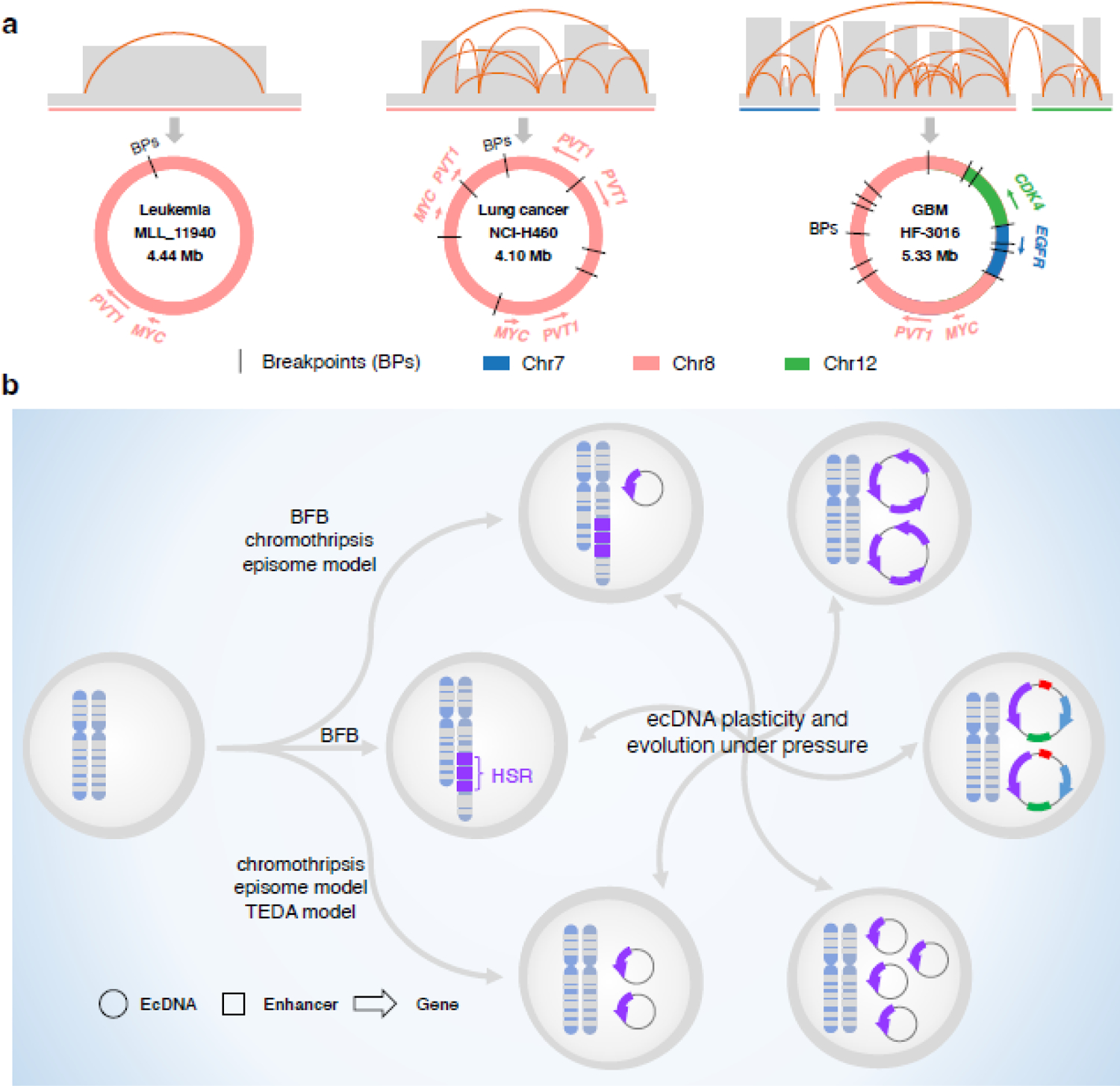
A. EcDNA complex structures across different cancer types. EcDNA molecules can differ in sizes, oncogenes and segment composition. Schematic structures of a single-interval amplicon in leukemia (Left, PMID: 29467491), a rearranged amplicon in lung cancer with single chromosome origination (Middle, PMID: 32873787) and an amplicon in glioblastoma with segments from multiple chromosomes (Right, PMID: 29686388) are shown. B. EcDNA biogenesis, plasticity and evolution. EcDNA can be formed in multiple nonexclusive processes including chromothripsis, episome model, breakage-fusion-bridge (BFB) cycles, and translocation-excision-deletion-amplification (TEDA). Selection pressure may result in copy number changes and structure alterations of ecDNA as well as switch between ecDNAs and HSRs.
The segmented configuration of ecDNA and its large size confound our ability to use short-read sequencing data to fully elucidate their complexity. Recent advances in single molecule long-read genomic technologies like ONT nanopore sequencing [35], PacBio Single Molecule Real-Time (SMRT) sequencing [36] and optical mapping [28] have offered significantly longer read lengths (from 10 Kb to Mb) directly from native DNA templates. Coupled with long-read specialized computational methods, they can resolve multiple complex segments and breakpoints from a single DNA molecule to assemble ecDNA structures [18, 21, 37]. Further data integration of both short-read and long-read sequences with optical mapping of ultra-long DNA fragments through advanced computational methods can enhance the accuracy and enable the reconstruction of high-fidelity ecDNA structures at single-nucleotide resolution [28]. Given the importance of non-coding regulatory elements in ecDNA function, a full reconfiguration of ecDNA structures will infer the selectivity of enhancer elements, which is expected to corroborate the enhancer activity of ecDNA function and offer valuable insights into the structural basis of ecDNA’s regulatory function.
EcDNA biogenesis
EcDNA originates from chromosomes. While the molecular mechanism(s) of how ecDNA is formed are unclear, several nonexclusive models have been proposed, such as chromothripsis [38], breakage-fusion-bridge (BFB) cycles [39], the translocation-excision-deletion-amplification model [40], and the episome model [41] (Figure 2b). Chromothripsis is a process that triggers the shattering of one or multiple chromosomes and thus produces many DNA segments. During DNA repair, these segments can be either randomly re-ligated with each other to generate complex rearrangement sequences or circularized into ecDNA [11, 29]. The BFB cycle is a mechanism of genomic instability [42, 43]. It initiates when anaphase bridge formation links broken ends of sister chromatids or different chromosomes resulting in the generation of a dicentric chromosome, which is pulled in opposite directions during mitosis, leading to chromosome breakage and triggering of the next BFB cycle. As the breakage is random, the repeat of BFB cycles can generate multiple genomic aberrations including chromosomal and extrachromosomal gene amplifications, depending on the location of breakage. The translocation-excision-deletion-amplification model is used to explain the co-amplification of genes from different chromosomes or amplification of fusion genes [40]. DNA fragments near the translocation breakpoint are unstable and can be deleted, circularized to form ecDNA, or locally amplified to generate a homogeneously staining region (HSR), a uniform staining of intense segment characterized in chromosomal karyotyping. The episome model suggests that a DNA fragment is excised from a chromosome and then circularized into circular DNA, named as ‘episome’, which is a precursor of ecDNA [33, 34, 41]. Episomes can further self-replicate into multiple copies and mutually reassemble to generate a large ecDNA carrying complex rearrangement [44]. Recent evidence suggests that ecDNA can be produced initially through BFB cycles followed by chromothripsis [11]. Under low level of methotrexate treatment, BFB cycles triggered by chromosome breaks leads to HSR formation. When drug dose is increased, the resultant dicentric chromosomes with HSR are shattered into pieces, some of which are circularized into highly rearranged ecDNA molecules [11] (Figure 2b).
Once formed, ecDNA can be subjected to further clonal evolution [11, 29], reintegration into chromosomes as a means to stabilize oncogene amplification [5, 11], or elimination by being trapped inside of micronuclei [45]. In some cases, ecDNA reintegration into chromosomes can result in HSRs [29], as DM and HSR can co-exist in cells and have similar amplified segments [24, 46]. Reintegration of ecDNA can be triggered by DNA damage and occur at the free ends of DNA resultant from double-strand breaks (DSBs) [11]. EcDNA reintegration can disrupt functional chromatin domains or cis-regulatory elements, thereby affecting gene transcription, like repressing tumor suppressor genes or activating oncogenes, which promotes oncogenic events that lead to cancer [5]. Therefore, extrachromosomal amplification is a dynamic state (Figure 2b) with adaptation to selection pressure and considered as a source of somatic rearrangements with diverse functional impact on cancer initiation.
EcDNA functions as a mobile enhancer to activate transcription in cancer
The recent characterization of ecDNA molecular structures and chromatin states collectively point to a transcription regulatory function of ecDNA. EcDNA is packaged in the form of nucleosomes with core histone proteins [47]. The chromatin of ecDNA is largely open and accessible, and enriched for active histone modification marks like H3K4me1 and H3K27ac [21]. This open chromatin conformation permits higher accessibility to transcriptional activators and RNA polymerases, which facilitate the massive transcription of the oncogenes residing on ecDNA [21, 22, 25]. Consequently, the expression of oncogenes on ecDNA is often the highest among all the transcripts in these cells [21, 25].
Chromosomes are extensively folded into chromatin loops that occupy distinct chromatin territories [1]. This highly organized, 3D chromatin organization provides a topological basis for many genome functions, including transcription, by bringing distal regulatory elements like enhancers and their cognate genes into close spatial proximity [48–50]. In contrast to chromosomes, which are confined within their respective chromatin territories, ecDNA can move more freely and contact chromosomes due to its smaller and circular conformation. Hence, the mobility of ecDNA, coupled with co-amplified enhancers, exerts a unique regulatory influence on promoting both chromosomal and extrachromosomal gene expression. EcDNA-associated chromatin conformations have been interrogated through different chromatin interaction mapping approaches [51–53] (See Box1) in tumor cells containing well-characterized ecDNA-harbored oncogenes [21, 25–27]. EcDNA exhibits dense and widespread chromatin interactions with actively transcribed genes and regulatory elements residing both within ecDNAs (in cis) and on chromosomes (in trans) [25]. The predominant interaction sites exhibit the key characteristics of super-enhancers, a class of regulatory regions bound by high levels of transcription factors and coactivators [54].Super enhancers form extensive long-range chromatin interactions with gene promoters and drive their transcription activation [55, 56]. In GBM and prostate cancer models, which harbor well-characterized ecDNAs, chromosomal genes that are brought into contact spatially with ecDNA super enhancers exhibit higher expression, consistent with their transcriptional activation by mobile ecDNA elements [25].
BOX1: Epigenetic and chromatin interaction assays in ecDNA analyses.
Various chromatin interaction profiling assays including Hi-C, 4C, ChIA-PET and HiChIP coupled with chromatin status profiling methods such as ATAC-seq, MNase-seq and ChIP-seq, were applied in ecDNA analysis, which have greatly improved the understanding of ecDNA functions.
ATAC-seq (Assay for transposase-accessible chromatin with high throughput sequencing) can profile genome-wide chromatin accessibility by randomly digesting open genomic DNA with Tn5 transposase and simultaneously inserting adapters into DNA. The DNA fragments with adapters were amplified and sequenced. Identified open chromatin regions are usually enriched with active promoters and DNA regulatory elements that are bound by transcription factors and contribute to gene expression [72].
MNase-seq (Micrococcal nuclease digestion with deep sequencing) is to map genome-wide nucleosome positioning and occupancy by digesting the chromatin with micrococcal nuclease (MNase) [73]. MNase-seq sequences genomic regions bound by histones or other chromatin-bound proteins as those regions are protected from cutting.
ChIP-seq (Chromatin immunoprecipitation sequencing) is a widely used method to reveal genomic binding sites of a specific protein or histone modification landscape by immunoprecipitation of chromatins with antibody targeting these proteins or modifications [74].
Hi-C is a high-throughput chromosome conformation capture approach for unbiased identification of genome-wide long-range chromatin interactions. Hi-C is built on the proximity ligation of restriction enzyme digested chromatins and marking of digested DNA ends with biotin during overhangs filling. Ligation junctions are enriched for sequencing and interaction analysis [75].
ChIA-PET (Chromatin interaction analysis with paired-end tag sequencing) [76, 77] and HiChIP, a variant of ChIA-PET [51], incorporate ChIP based enrichment with chromatin proximity ligation to enrich specific protein-associated chromatin interactions. The proximity ligation of ChIA-PET is mediated by a biotinylated bridge linker while HiChIP uses the same way as Hi-C.
Using live cell imaging approaches, multiple ecDNA molecules are found to cluster together into hubs where RNA polymerase II is co-localized and genes within ecDNA hubs can be trans-activated through inter-molecular enhancer-promoter interactions [22, 27]. Key components crucial for the spatial aggregation of ecDNA into hubs are enhancers and BRD4, a bromodomain and extraterminal domain (BET) protein preferentially bound at enhancers. Inhibition of BRD4 leads to disperse hubs and the reduction of ecDNA-mediated oncogene overexpression, cell proliferation and viability [27]. Hence, aggregated ecDNA chromatin hubs can act as potent and versatile mobile transcriptional activators to trans-activate genes localized on both ecDNAs and chromosomes, and thereby offer a selective growth advantage in ecDNA-carrying cancer cells (Figure 3). It is worth noting that the trans-activating mechanism, although less studied, has been demonstrated in both normal physiological contexts and during viral infection to modulate gene expression programs [57], such as the expression of olfactory receptors (OR) in the sensory neurons by interchromosomal enhancer–promoter interactions [58, 59] and the modulation of the viral transcriptome by the circular hepatitis B virus (HBV) DNA when infecting human hepatocytes [60, 61]. This model raises an intriguing possibility that the rewiring of enhancer–promoter interactions by extrachromosomal chromatin particles could underlie a new type of aberrant transcriptional control that expands the current views of how eukaryotic genes are regulated and how genetic structure and epigenetics promote tumorigenesis.
Figure 3.
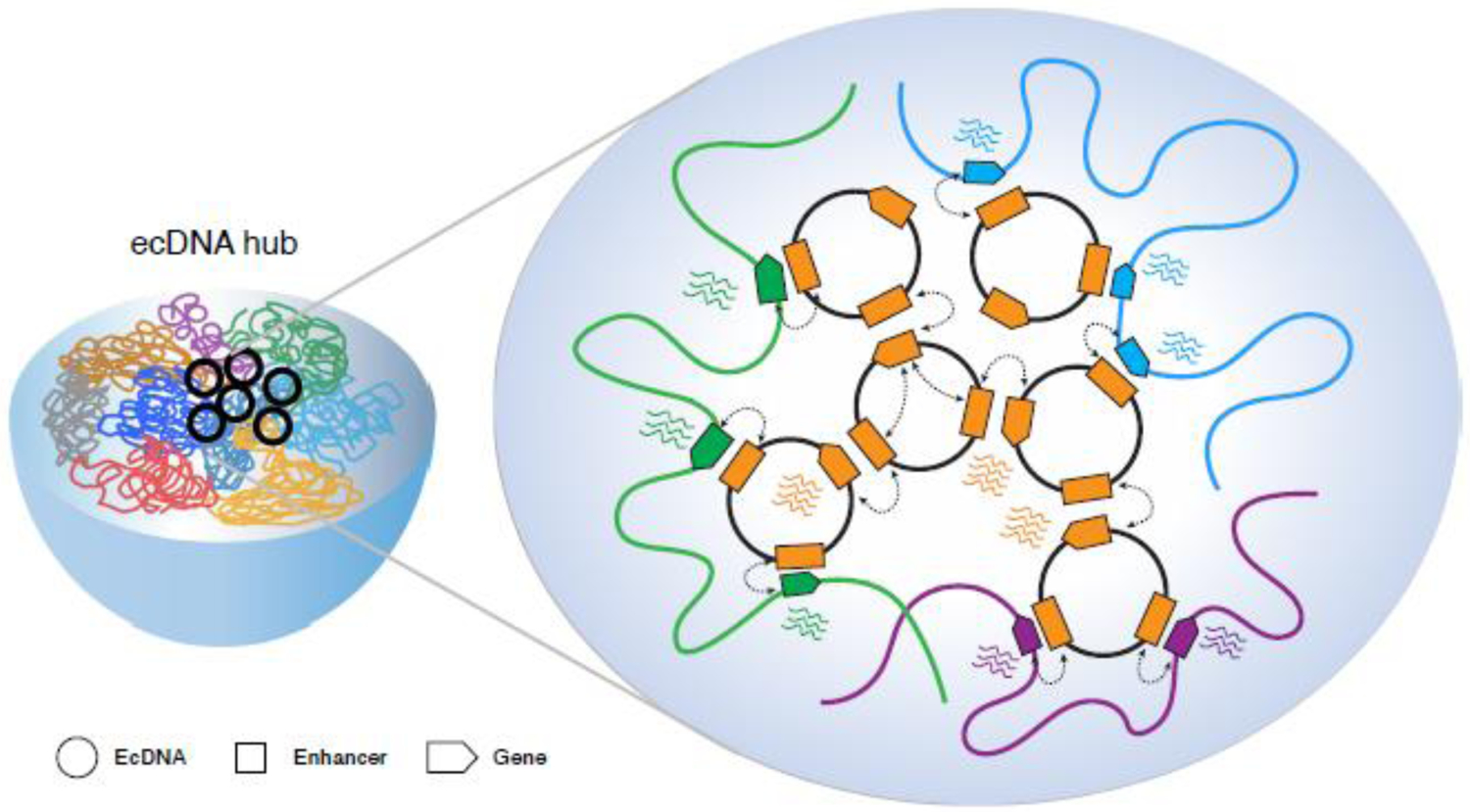
EcDNA-associated transcriptional regulation hub. EcDNA molecules cluster together to form a highly active transcriptional hub, in which genes localized on ecDNAs and chromosomes are trans-activated by mobile ecDNA enhancers through ecDNA-ecDNA or ecDNA-chromosome interactions.
EcDNA contributes to intratumor heterogeneity and can be explored for clinical utility
The impacts from uneven segregation of inheritance, complex molecular structures, and transcription regulatory activities, suggest ecDNA confers a selective advantage to promote tumor cell heterogeneity and accelerate tumor evolution [16, 21, 25, 37] (Figure 4). EcDNA persisted during both in vitro and in vivo tumor growth of GBM [18], indicating its importance during tumor propagation. In both GBM tumors and their derivatives, the evolution of tumor subclones marked by chromosomal single nucleotide variants (SNVs) diverged from subclones marked by ecDNA’s SNVs. This finding suggests that the selection dynamics of ecDNA-harboring cells differs from that of cells harboring chromosomal variants. As ecDNA lacks centromeres, it is subjected to unequal segregation during mitosis and exhibits highly variable copy number distribution within individual tumors [16, 18] (Figure 1). The non-Mendelian inheritance pattern allows for rapid accumulation of ecDNA molecules in some cells, resulting in high-level expression of oncogenes or drug-resistance genes and subsequent growth advantages. Moreover, ecDNA molecules are structurally diverse within a tumor population [13, 20]. Hence, ecDNA copy number heterogeneity and structural diversity can drive intratumor heterogeneity (ITH), which can be subjected to further clonal evolution through microenvironmental changes or continuous drug selection [11, 18] (Figure 2b). Continuous treatment with methotrexate, a DHFR enzyme inhibitor, can promote the clonal selection of cells with extrachromosomal DHFR amplification and confer tumor cell tolerance to methotrexate [11]. Such a selective nature could have important implications in tumor behavior and vulnerability. Elimination of ecDNA could offer an avenue of clinical intervention for cancer treatment. To this end, nucleotide analogs or inhibitors of DNA replication have been exploited. For example, hydroxyurea, an inhibitor of DNA replication, has been shown to eliminate ecDNA and result in reduced tumorigenicity and increased drug sensitivity [62, 63]. Radiation therapy can also induce the loss of extrachromosomal DNA harboring drug-resistant genes or oncogenes [64]. Nevertheless, the loss of ecDNA can also contribute to drug resistant mechanisms. In primary glioblastoma, EGFRvIII is the most prevalent variant of EGFR. Tumor cells with EGFRvIII-bearing ecDNAs are more sensitive to standard EGFR tyrosine kinase inhibitor (TKI) treatment and the elimination of these ecDNAs enables the tumor cells to develop resistance to EGFR TKI [65, 66]. Further study of the mechanism(s) by which ecDNA contributes to tumor evolution could provide insight into tumor-cell fitness and targeted therapies for ecDNA-driven cancers.
Figure 4. EcDNA driven intratumor heterogeneity and tumor evolution.
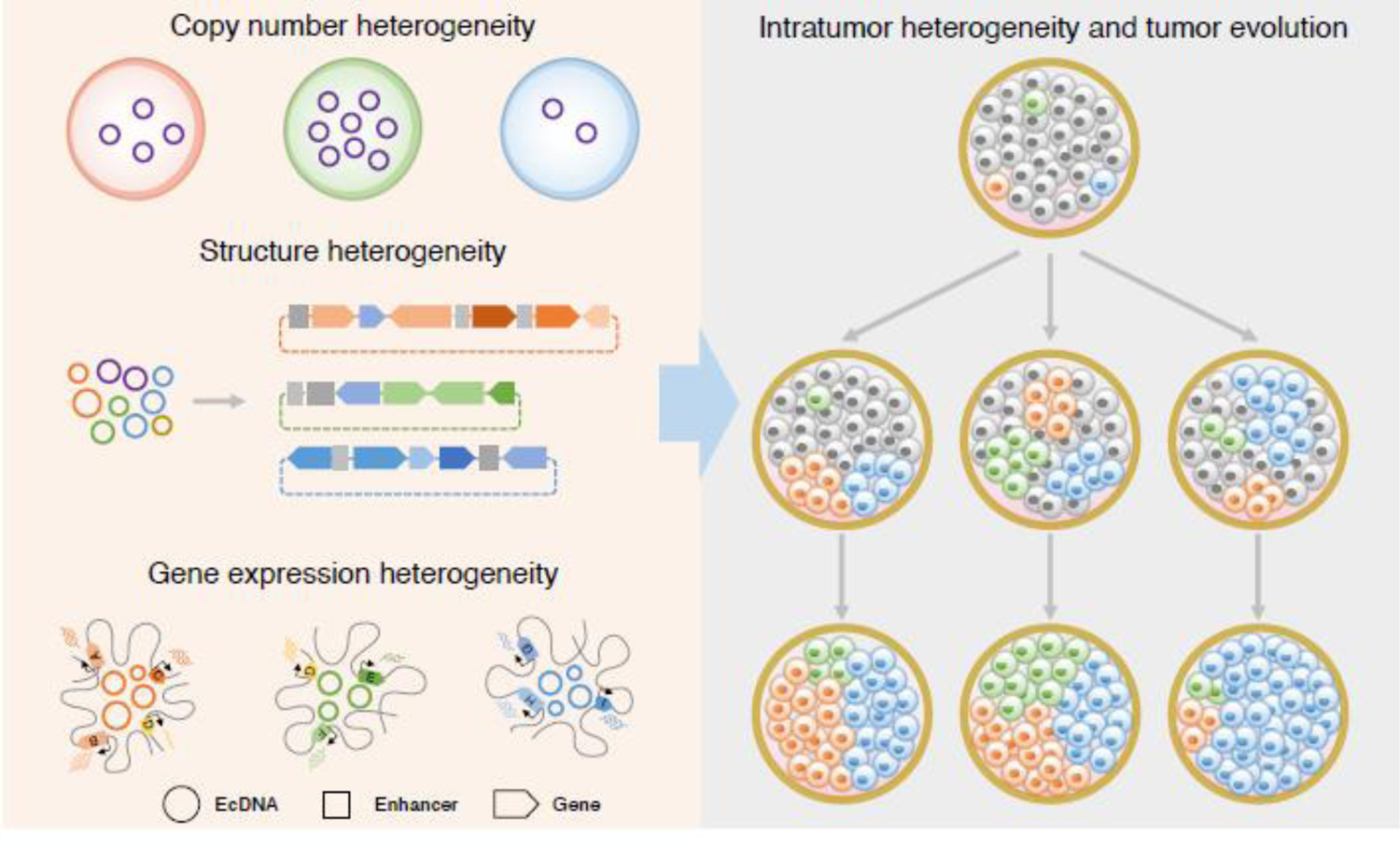
Heterogenous ecDNA copy numbers and structures together with distinct ecDNA-associated transcriptional regulation pattern among cells resulted in intratumor heterogeneity and boost tumor evolution.
Based on its strong association with aggressive cancer phenotypes and drug responses, ecDNA could be adopted as a biomarker for cancer diagnosis, classification, and monitoring of therapeutic responses. In GBM, the states of malignant cells are classified into four major types (neural-progenitor-like, oligodendrocyte-progenitor-like, astrocyte-like, and mesenchymal-like) in TCGA glioblastoma cohort analyses [67]. Three of them are represented by high-level amplifications of CDK4 (neural-progenitor-like), EGFR (astrocyte-like), and PDGFRA (oligodendrocyte-progenitor-like), respectively, which are also among the top amplified genes on ecDNAs in GBM [16, 67]. Circular DNA can be released from circulating tumor cells into human plasma [68–70] and circulating circular DNA exhibits larger sizes than linear cell-free DNA (cfDNA) [68]. In GBM, amplification of genes like ERBB2, MET, and EGFR that are commonly found on ecDNA has been detected in cell-free circulating tumor DNA [71]. These amplicons carry cancer-specific genetic and epigenetic information, presenting as potential avenues for developing non-invasive biomarkers for cancer diagnosis and monitoring.
Concluding remarks and future perspectives
With the advances in genomic technologies, ecDNA has been systematically studied in recent years through a variety of in vitro and in vivo cancer models. Its prevalence and functional importance are now well-recognized, and we have just started to appreciate the structural complexity and unique chromatin features of ecDNA. Beyond serving as reservoirs for oncogene amplification, ecDNA can function as a mobile enhancer to modulate chromatin interactions and regulate gene expression. Combined with its copy number heterogeneity and rearranged structures, these features manifest as a basis for intratumor heterogeneity and a driving force for tumor clonal evolution. Despite these significant advancements, knowledge of the molecular functions and regulatory activities of ecDNA that lead to aggressive tumor phenotypes remain incompletely understood (see Outstanding questions). We also do not fully understand the processes and components that control ecDNA biogenesis, replication, maintenance, and homeostasis as well as the dynamics of histone modifications, DNA methylation and 3D epigenome structures of ecDNA, which could inform on approaches to eliminate ecDNA for therapeutic purpose. Future efforts to reveal the dynamics of ecDNA chromatin conformation and complexity of molecular structures will glean insight into its contributions to cellular fitness and treatment response, specifically, the longitudinal evolution of ecDNA molecular structures subjected to the courses of tumor clonal evolution. New approaches that can exploit the unique 3D chromatin features of ecDNA and the chromosomal targeting specificity as cancer-specific traits should yield potential translational value.
Outstanding Questions Box.
What’s the molecular mechanism(s) and components driving ecDNA biogenesis and replication?
How is ecDNA selectively maintained or eliminated in response to DNA damage and repair?
What’s the dynamics of histone modifications, DNA methylation and 3D epigenome structures of ecDNA?
What’s the molecular structure and diversity of ecDNA, their common features and source of variation?
How does ecDNA evolve across longitudinal tumor growth and throughout metastasis?
What is the nature of ecDNA mediated chromatin hubs and how it elicits ecDNA’s regulatory activities leading to aggressive tumor phenotypes?
What is the vulnerability of ecDNA transcriptional regulatory activity that can be clinically exploited for therapeutic and diagnostic value?
Further breakthroughs in ecDNA biology requires continuous efforts in genomic technology development and innovation. Despite its prevalence in cancer, ecDNA detection is limited by the sensitivity, specificity, and resolution of the current methods. Molecular methods that can specifically isolate DNA molecules of circular conformation across wide size ranges without prior sequence information will be useful. Knowledge about ecDNA molecular structures is still hindered by their large sizes, high sequence similarity to chromosomal origins, and rearranged compositions. Given that individual ecDNA exists in a pool of molecules with highly similar sequences in each tumor, appropriate approaches that can decipher additional layers of sequence complexity are needed to discern individual ecDNA structures and fully appreciate their complexity, dynamics, and evolution. Improvements in third generation long-read sequencing, like PacBio SMRT sequencing and ONT nanopore sequencing, as well as single-molecule optical mapping, have offered potential solutions. Although initially error-prone and cost prohibitive, these technologies have been greatly enhanced in recent years and can be leveraged to characterize ecDNA structures. To fully maximize the value of long-read data for ecDNA reconstruction, continuous development of specialized computational tools is needed. In addition, genomic editing approaches that can specifically modulate the levels or perturb the composition of ecDNA are crucial to realize the exciting potential of manipulating ecDNA regulatory activities in cancer therapy. Innovative CRISPR/Cas9-mediated epigenome editing approaches will be useful in examining the functional significance of the regulatory modules within ecDNA and understanding the mechanism(s) by which ecDNA enhances transcription. Lastly, to fully evaluate ecDNA transcriptional impacts and how they promote ITH and tumor cell fitness, we can benefit from the progress made in single cell multi-omics analyses. Elucidation of the structures, functions and regulation of ecDNA is expected to transform the current paradigm for studying cancer genomes and profoundly expand the current views of how oncogenes are regulated, from which new clinical strategies can be realized.
Highlights.
EcDNA is an adaptive reservoir for oncogene amplification, composed of highly rearranged chromosomal sequences which can rapidly achieve high copy numbers in cancer cells through uneven segregation.
EcDNA exhibits enhanced chromatin accessibility with co-amplified enhancers to promote oncogene expression through inter- and intra-molecular chromatin interactions.
EcDNA aggregates in hubs which function as potent, versatile, and mobile transcriptional enhancers to trans-activate gene expression.
The trans-acting mechanism resulted aberrant transcriptional patterns, structural complexity and heterogenous copy numbers of ecDNA are driving intratumor heterogeneity and tumor evolution.
Advances in genomic technologies are expected to shed much-needed light on the roles of ecDNA in cancer progression and exploit its unique chromatin features as cancer-specific traits with potential translational value.
Acknowledgements
The authors thank Christine Goldfarb for her feedback and comments on the manuscript. C-L.W. is supported by 4DN (U54 DK107967), R01 GM127531 and ENCODE (UM1 HG009409) consortia, NCI under Award Number P30CA034196 and R33 CA236681 from the US National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- Breakpoint
a region or genomic location between two continuous DNA blocks on a genome, normally inferring where a rearrangement event occurs
- Double-minute (DM)
refers to double minute chromosomes, observed in the metaphase preparation as small extrachromosomal chromatin bodies by Giemsa staining in chromosomal karyotyping, normally appearing in pairs in cancer cells
- EcDNA
a class of large extrachromosomal circular DNA (usually with sizes of hundreds of kilobases to megabases) found specifically in cancer cells. They contain full genes and regulatory elements, and drive high expression of oncogenes
- Extrachromosomal circular DNA (eccDNA)
a class of small extrachromosomal circular DNA (usually with sizes of less than 10 Kb) found both in normal and tumor tissues exhibiting tissue specificity
- Homogeneously staining region (HSR)
a chromosomal region with a uniform staining of banding pattern in variable lengths when stained with Giemsa. Such pattern normally indicates amplification of a segment on a chromosome
- Intratumor heterogeneity (ITH)
refers to variation (molecular, genetic, epigenetic or phenotypical) observed in a distinct population of tumor cells within the same tumor specimen
- Micronuclei
small nuclei carrying DNA aggregates, which is expulsed from nucleus during cell division and appears in the cytoplasm
- Structural variation
Genomic variation affecting the absence, presence and abundance of nucleotide sequences, that is, deletions, insertions and copy number variations, as well as the direction and/or location alterations in chromosomal rearrangements, such as inversions, translocations and chromothripsis
- Tumor microenvironment
a highly complex and dynamic environment composed of cells and molecules in which tumor grows
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cremer T and Cremer M, Chromosome territories. Cold Spring Harb Perspect Biol, 2010. 2(3): p. a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paulsen T, et al. , Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet, 2018. 34(4): p. 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moller HD, et al. , Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun, 2018. 9(1): p. 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moller HD, et al. , Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A, 2015. 112(24): p. E3114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koche RP, et al. , Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet, 2020. 52(1): p. 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henriksen RA, et al. , Circular DNA in the human germline and its association with recombination. Mol Cell, 2022. 82(1): p. 209–217 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaubatz JW, Extrachromosomal circular DNAs and genomic sequence plasticity in eukaryotic cells. Mutat Res, 1990. 237(5–6): p. 271–92. [DOI] [PubMed] [Google Scholar]
- 8.Moller HD, et al. , Near-Random Distribution of Chromosome-Derived Circular DNA in the Condensed Genome of Pigeons and the Larger, More Repeat-Rich Human Genome. Genome Biol Evol, 2020. 12(1): p. 3762–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hull RM, et al. , Transcription-induced formation of extrachromosomal DNA during yeast ageing. PLoS Biol, 2019. 17(12): p. e3000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin C, et al. , Encoding gene RAB3B exists in linear chromosomal and circular extrachromosomal DNA and contributes to cisplatin resistance of hypopharyngeal squamous cell carcinoma via inducing autophagy. Cell Death Dis, 2022. 13(2): p. 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoshani O, et al. , Chromothripsis drives the evolution of gene amplification in cancer. Nature, 2021. 591(7848): p. 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, et al. , eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature, 2021. 599(7884): p. 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner KM, et al. , Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature, 2017. 543(7643): p. 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhaak RGW, Bafna V, and Mischel PS, Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer, 2019. 19(5): p. 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox D, Yuncken C, and Spriggs AI, Minute Chromatin Bodies in Malignant Tumours of Childhood. Lancet, 1965. 1(7402): p. 55–8. [DOI] [PubMed] [Google Scholar]
- 16.Kim H, et al. , Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet, 2020. 52(9): p. 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao XK, et al. , Focal amplifications are associated with chromothripsis events and diverse prognoses in gastric cardia adenocarcinoma. Nat Commun, 2021. 12(1): p. 6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.deCarvalho AC, et al. , Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet, 2018. 50(5): p. 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nathanson DA, et al. , Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science, 2014. 343(6166): p. 72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu K, et al. , Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol, 2019. 137(1): p. 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu S, et al. , Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature, 2019. 575(7784): p. 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi E, et al. , Live-cell imaging shows uneven segregation of extrachromosomal DNA elements and transcriptionally active extrachromosomal DNA hubs in cancer. Cancer Discov, 2021. 12(2): p. 468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Y, et al. , An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med, 2017. 23(8): p. 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song K, et al. , Plasticity of Extrachromosomal and Intrachromosomal BRAF Amplifications in Overcoming Targeted Therapy Dosage Challenges. Cancer Discov, 2022. 12(4): p. 1046–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, et al. , Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell, 2021. 39(5): p. 694–707 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morton AR, et al. , Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell, 2019. 179(6): p. 1330–1341 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hung KL, et al. , ecDNA hubs drive cooperative intermolecular oncogene expression. Nature, 2021. 600(7890): p. 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luebeck J, et al. , AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat Commun, 2020. 11(1): p. 4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosswog C, et al. , Chromothripsis followed by circular recombination drives oncogene amplification in human cancer. Nat Genet, 2021. 53(12): p. 1673–1685. [DOI] [PubMed] [Google Scholar]
- 30.Moscatello DK, et al. , Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res, 1995. 55(23): p. 5536–9. [PubMed] [Google Scholar]
- 31.Sanborn JZ, et al. , Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res, 2013. 73(19): p. 6036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshpande V, et al. , Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun, 2019. 10(1): p. 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.A LA, et al. , MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences. Leukemia, 2018. 32(10): p. 2152–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.L’Abbate A, et al. , Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res, 2014. 42(14): p. 9131–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong L, et al. , Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat Methods, 2018. 15(6): p. 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenger AM, et al. , Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol, 2019. 37(10): p. 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmsauer K, et al. , Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat Commun, 2020. 11(1): p. 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephens PJ, et al. , Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell, 2011. 144(1): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coquelle A, et al. , Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene, 2002. 21(50): p. 7671–9. [DOI] [PubMed] [Google Scholar]
- 40.Van Roy N, et al. , Translocation-excision-deletion-amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes Chromosomes Cancer, 2006. 45(2): p. 107–17. [DOI] [PubMed] [Google Scholar]
- 41.Vogt N, et al. , Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A, 2004. 101(31): p. 11368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hackett JA, Feldser DM, and Greider CW, Telomere dysfunction increases mutation rate and genomic instability. Cell, 2001. 106(3): p. 275–86. [DOI] [PubMed] [Google Scholar]
- 43.Cleal K and Baird DM, Catastrophic Endgames: Emerging Mechanisms of Telomere-Driven Genomic Instability. Trends Genet, 2020. 36(5): p. 347–359. [DOI] [PubMed] [Google Scholar]
- 44.Storlazzi CT, et al. , MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet, 2006. 15(6): p. 933–42. [DOI] [PubMed] [Google Scholar]
- 45.Ji W, et al. , Expulsion of micronuclei containing amplified genes contributes to a decrease in double minute chromosomes from malignant tumor cells. Int J Cancer, 2014. 134(6): p. 1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storlazzi CT, et al. , Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res, 2010. 20(9): p. 1198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith G, et al. , c-Myc-induced extrachromosomal elements carry active chromatin. Neoplasia, 2003. 5(2): p. 110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sexton T and Cavalli G, The role of chromosome domains in shaping the functional genome. Cell, 2015. 160(6): p. 1049–59. [DOI] [PubMed] [Google Scholar]
- 49.de Laat W and Duboule D, Topology of mammalian developmental enhancers and their regulatory landscapes. Nature, 2013. 502(7472): p. 499–506. [DOI] [PubMed] [Google Scholar]
- 50.Bertolini JA, et al. , Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell Stem Cell, 2019. 24(3): p. 462–476 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mumbach MR, et al. , HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods, 2016. 13(11): p. 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, et al. , Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat Protoc, 2017. 12(5): p. 899–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krijger PHL, et al. , 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods, 2020. 170: p. 17–32. [DOI] [PubMed] [Google Scholar]
- 54.Grosveld F, van Staalduinen J, and Stadhouders R, Transcriptional Regulation by (Super)Enhancers: From Discovery to Mechanisms. Annu Rev Genomics Hum Genet, 2021. 22: p. 127–146. [DOI] [PubMed] [Google Scholar]
- 55.Whyte WA, et al. , Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 2013. 153(2): p. 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loven J, et al. , Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell, 2013. 153(2): p. 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maass PG, Barutcu AR, and Rinn JL, Interchromosomal interactions: A genomic love story of kissing chromosomes. J Cell Biol, 2019. 218(1): p. 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bashkirova E and Lomvardas S, Olfactory receptor genes make the case for interchromosomal interactions. Curr Opin Genet Dev, 2019. 55: p. 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Monahan K, Horta A, and Lomvardas S, LHX2- and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature, 2019. 565(7740): p. 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreau P, et al. , Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat Commun, 2018. 9(1): p. 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hensel KO, et al. , Episomal HBV persistence within transcribed host nuclear chromatin compartments involves HBx. Epigenetics Chromatin, 2018. 11(1): p. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eckhardt SG, et al. , Induction of differentiation in HL60 cells by the reduction of extrachromosomally amplified c-myc. Proc Natl Acad Sci U S A, 1994. 91(14): p. 6674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Von Hoff DD, et al. , Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci U S A, 1992. 89(17): p. 8165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schoenlein PV, et al. , Radiation therapy depletes extrachromosomally amplified drug resistance genes and oncogenes from tumor cells via micronuclear capture of episomes and double minute chromosomes. Int J Radiat Oncol Biol Phys, 2003. 55(4): p. 1051–65. [DOI] [PubMed] [Google Scholar]
- 65.Vivanco I, et al. , Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov, 2012. 2(5): p. 458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mellinghoff IK, et al. , Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med, 2005. 353(19): p. 2012–24. [DOI] [PubMed] [Google Scholar]
- 67.Neftel C, et al. , An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell, 2019. 178(4): p. 835–849 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sin STK, et al. , Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc Natl Acad Sci U S A, 2020. 117(3): p. 1658–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu J, et al. , Molecular characterization of cell-free eccDNAs in human plasma. Sci Rep, 2017. 7(1): p. 10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar P, et al. , Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol Cancer Res, 2017. 15(9): p. 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Piccioni DE, et al. , Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol, 2019. 8(2): p. CNS34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corces MR, et al. , An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods, 2017. 14(10): p. 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pajoro A, et al. , Profiling Nucleosome Occupancy by MNase-seq: Experimental Protocol and Computational Analysis. Methods Mol Biol, 2018. 1675: p. 167–181. [DOI] [PubMed] [Google Scholar]
- 74.Park PJ, ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet, 2009. 10(10): p. 669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rao SS, et al. , A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 2014. 159(7): p. 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fullwood MJ, et al. , An oestrogen-receptor-alpha-bound human chromatin interactome. Nature, 2009. 462(7269): p. 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang P, et al. , In situ Chromatin Interaction Analysis Using Paired-End Tag Sequencing. Curr Protoc, 2021. 1(8): p. e174. [DOI] [PMC free article] [PubMed] [Google Scholar]