

Abstract
Neuropeptides are abundant signaling molecules in the central nervous system. Yet remarkably little is known about their spatiotemporal spread and biological activity. Here, we developed an integrated optical approach using Plasmonic nAnovesicles and cell-based neurotransmitter fluorescent engineered reporter (CNiFER), or PACE, to probe neuropeptide signaling in the mouse neocortex. Small volumes (fL to pL) of exogenously supplied somatostatin-14 (SST) can be rapidly released under near-infrared light stimulation from nanovesicles implanted in the brain and detected by SST2 CNiFERs with nM sensitivity. Our measurements reveal reduced but synchronized SST transmission within 130 μm, and markedly smaller and delayed transmission at longer distances. These measurements enabled a quantitative estimation of the SST loss rate due to peptide degradation and binding. PACE offers a new tool for determining the spatiotemporal scales of neuropeptide volume transmission and signaling in the brain.
Keywords: Plasmonic nanovesicles, neuropeptide release, biosensors, neuropeptide transmission, brain
Graphical Abstract
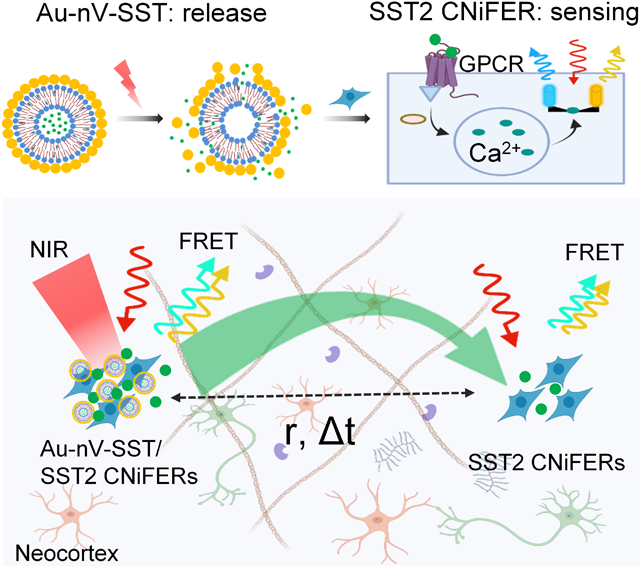
We developed an integrated optical approach by combining plasmonic nanovesicles (Au-nV-SST) and cell-based neurotransmitter fluorescent engineered reporter (CNiFER) for neuropeptide release and detection. This approach allows spatiotemporal mapping of neuropeptide volume transmission and signaling in the mouse cortex.
Introduction
Neuropeptides comprise a diverse class of signaling molecules that regulate brain states, modify neural activity, and control vascular tone.[1] Neuropeptides are synthesized through RNA translation, processed proteolytically, and then released from dense-core vesicles in peptide-expressing neurons.[2] In contrast to classical, fast synaptic transmission (nanometer distance), neuropeptides diffuse from their release sites and signal through G-protein coupled receptors (GPCRs) at relatively long distances (μm to mm). This diffusion-driven distribution is referred to as volume transmission, an extrasynaptic dispersion of the transmitter in the extracellular space.[3] One major obstacle to determining where and when a neuropeptide acts relative to its release site is the lack of tools to release peptides with high temporal resolution and detect nM levels of neuropeptide in vivo. Several factors affect the rate and extent of peptide signaling. First, neuropeptides activate GPCRs at low nanomolar (nM) concentrations, up to 1000-fold lower than the affinity for ionotropic receptors (e.g., μM for GABA or glutamate), making it difficult to detect neuropeptide release.[3] Second, peptide diffusion is impacted by the extracellular matrix, peptidases, and the geometry of the extracellular space in the brain. Several studies have accurately measured the diffusion properties in vivo but have relied on physiologically inert tracers.[4] These factors have made it challenging to measure neuropeptide volume transmission and signaling in vivo quantitatively.
To address this challenge, we developed a new integrated optical approach (PACE) by a synergistic combination of Plasmonic nAnovesicles with cell-based neurotransmitter fluorescent engineered reporter (CNiFER). PACE mimics and probes the neuropeptide transmission and signaling in vivo. The plasmonic nanovesicles consist of a phospholipid liposome core with a gold coating. They can be implanted in the mouse cortex and remotely stimulated by near-infrared laser pulses to release a small volume (fL to pL) of exogenously introduced and biologically active neuropeptides within several seconds. CNiFERs provide a functional readout on physiologically relevant concentrations of neurotransmitters.[5] Here, we engineered a new CNiFER to detect nM somatostatin (SST), a 14-amino acid cyclic peptide expressed in many GABA neurons throughout the brain. Under intravital two-photon imaging, we determined the time delay and maximal distance for photoreleased SST signal transmission in the mouse cortex. This measurement reveals reduced but synchronized transmission within 130 μm, with a smaller delayed transmission at larger distances. Our study provides the first quantitative estimation of the overall SST loss rate due to the degradation and binding in vivo. The results suggest that neuropeptide binding and degradation limit its extrasynaptic transmission at large distances (>100 μm). We further demonstrate that the neuropeptide transmission and GPCR signaling is significantly faster in mouse neocortex with a degraded extracellular matrix. Our method provides a robust and quantitative way to measure neuropeptide volume transmission and functional signaling in the brain that is beyond current state-of-the-art.
Results and Discussion
Somatostatin-14 (SST) is an endogenous cyclic 14-amino acid peptide widely expressed in GABAergic cortical neurons,[6] but little is known about the extent of extrasynaptic transmission in the brain.[1b] Prior studies focused on measuring the diffusion properties with physiologically inert tracers[4] and did not consider whether the peptide retained its biological activity. Here, we developed tools for releasing and measuring the SST signaling in vivo through exogenously introduced and biologically active SST. First, we encapsulated SST in nanovesicles containing 1,2-dipalmitoyl-sn-glycerol-3-phosphocholine (DPPC) and cholesterol (4:3 molar ratio). We then examined the photorelease with femtosecond laser excitation (Figure 1A). Most SST-loaded nanovesicles (nV-SST) have unilamellar morphology (Figure S1). Gold-coating endows nanovesicles with strong absorption in the near-infrared range (700–900 nm) due to the plasmonic coupling between the small gold nanoparticles[7] and also increases the hydrodynamic diameter to ~200 nm (Figure 1B, Figure S1). To confirm this, we used transmission electron microscopy (TEM, Figure 1C and Figure S1) and observed clusters of small gold nanoparticles on the nanovesicle surface and close to each other. The near-infrared absorption of gold-coated nanovesicles decreased upon 720 nm femtosecond laser pulse irradiation. This decrease could be due to the fragmentation or ablation of gold nanoparticles,[8] confirmed by the TEM images (Figure S1). The SST content in gold-coated nanovesicles remained stable (~75%) after 24 hours of incubation with a peptidase, α-chymotrypsin, compared to the half-life of degradation for free SST of ~16 min (Figure S2). Gold-coated nVs containing SST (Au-nV-SST) showed good colloidal stability in artificial cerebrospinal fluid (ACSF) and 10% fetal bovine serum at 37°C for 4 h and under storage conditions in 0.01 M phosphate-buffered saline (PBS) at 4°C over two weeks (Figure S3). To characterize the SST release efficiency, Au-nV-SST flowing in a capillary tube were stimulated by femtosecond laser pulses. To focus the stimulation laser on a specific subregion, we used a multi-photon imaging system with two lasers tuned to different wavelengths, referred to as imaging laser and a stimulation laser with tornado scans, respectively (Figure S4). Each tornado scan (200 μm, 35 ms) has a dwell time of 10 μs on a pixel, leading to 2.8 × 106 pulses per scan (80 MHz laser repetition rate). The released SST concentration was measured using an ELISA kit for SST. Figure 1D shows that the photorelease efficiency increases with laser power and scan duration. To analyze the photorelease kinetics in real-time, we loaded gold-coated nanovesicles containing calcein dye (Au-nV-Cal) into 1 nL microwells (Figure S5). We packaged calcein at a self-quenching concentration (75 mM), which increases in fluorescence upon release and dequenching. Calcein fluorescence increased significantly under the laser pulses at 700–820 nm (100 mW, 0.65 s) while showing no change with imaging laser pulses at 840–1000 nm, even at prolonged durations (15 mW, 8 min) (Figure 1E, Figure S5). Importantly, the imaging laser at lower power and longer wavelength did not photorelease encapsulated compounds, which is ideal for long-term imaging. Real-time fluorescent imaging showed that the average calcein fluorescence in a microwell (diameter: 60 μm) reached a maximal value at 0.15 s after a 65 ms tornado scan (Figure 1F, Figure S5). Thus, the photorelease from Au-nV-Cal has a fast temporal resolution, completing within 0.15 s in the 60-μm-diameter tornado-scanned area. Although SST is slightly larger than fluorescent calcein, we anticipate that SST release follows a similar profile and can be controlled by stimulation power and duration.
Figure 1.
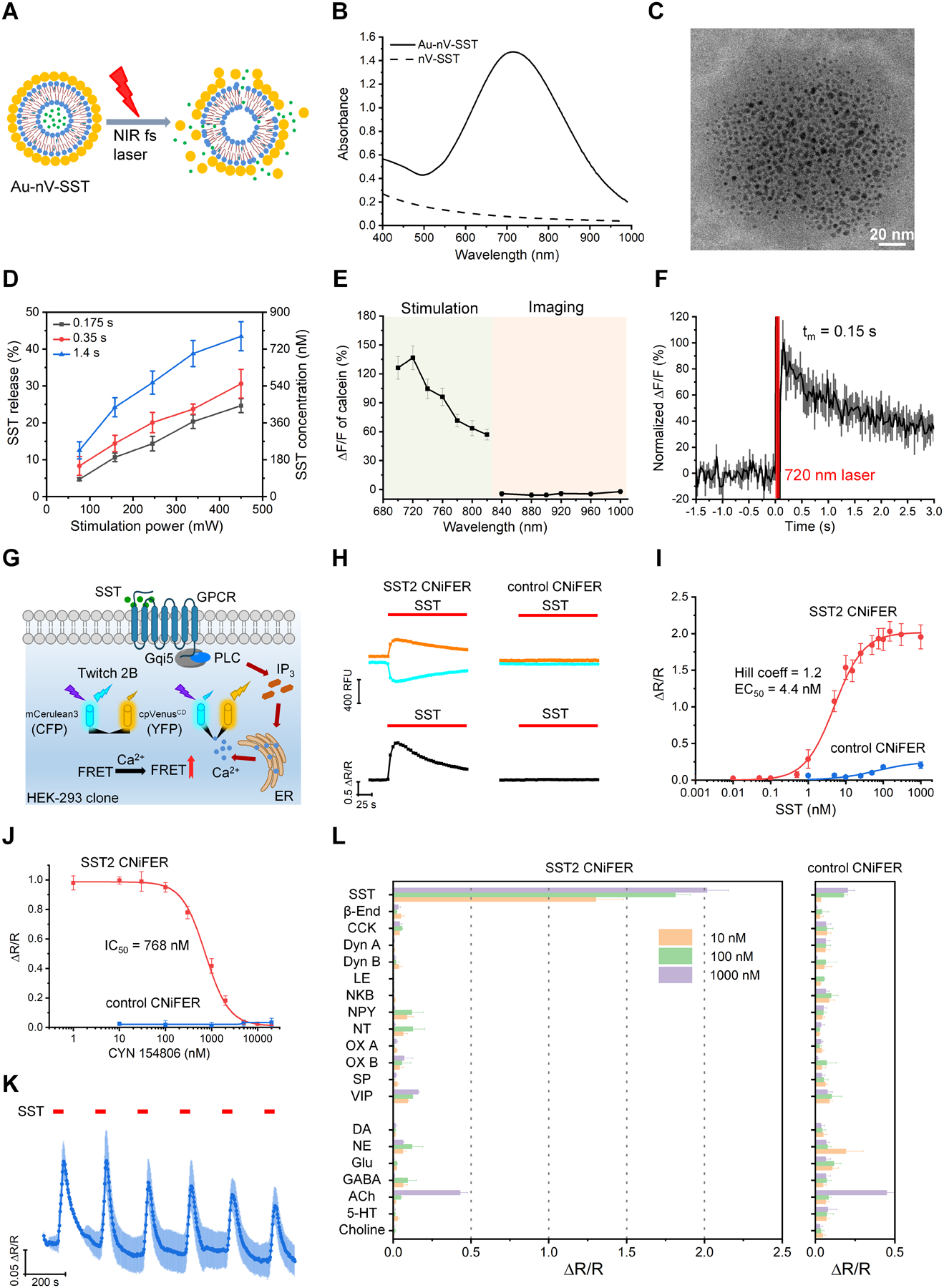
Development and characterization of neuropeptide photoreleasing and sensing techniques. A) Schematic of near-infrared (NIR) femtosecond (fs) laser pulses triggered release from gold-coated somatostatin-14 (SST) loaded nanovesicles (Au-nV-SST). B) UV-Vis spectra of Au-nV-SST and nV-SST. C) Transmission electron microscopy (TEM) image of Au-nV-SST. Scale bar: 20 nm. D) The efficiency and amount of SST released from nanovesicles under the irradiation of 720 nm fs laser pulses (n = 3). Duration (0.175 s, 0.35 s, 1.4 s) of photostimulation refers to the scan number (5, 10, 40). E) Release of calcein under stimulation conditions (100 mW, 10 tornado scans, 0.65 s, 700–820 nm) and imaging conditions (15 mW, resonant scans, 8 minutes, 840–1000 nm) (n = 5). F) Release kinetics of calcein from nanovesicles stimulated at 720 nm for 65 ms (n = 5). G) Schematic of SST2 CNiFER signaling pathway. SST activates SST2 GPCR to induce Ca2+ cytoplasmic influx detected by the FRET-based genetically encoded Ca2+ detector (Twitch 2B). Twitch 2B contains mCerulean3 (CFP) and cpVenusCD (YFP). H) FRET response from SST2 and control CNiFERs after application of 100 nM SST (red bar). Response in CFP (cyan) and YFP (yellow) fluorescence (top) leads to the change in FRET ratio (ΔR/R, bottom). I) Dose-response curve for SST2 (n = 4) and control (n = 3) CNiFERs. The smooth curve shows the best fit with the Hill equation, with the indicated EC50 and Hill coefficient. J) The FRET response is shown for SST2 CNiFERs and control CNiFERs activated by 5 nM SST in the presence of different concentrations CYN 154806, a SST2 receptor antagonist. K) FRET response for SST2 CNiFER with repeated 30 s applications of SST (5 nM, n = 4). L) FRET response with the indicated peptides and classical neurotransmitters (NTs) at three different concentrations for SST2 CNiFERs (n = 3–6) and control CNiFERs (n = 3–6). Data are expressed as Mean ± S.D.
To measure the release of SST in vivo, we engineered a new neuropeptide CNiFER that expresses the type 2 SST GPCR (SST2) along with a chimeric Gqi5 G protein and a high signal-to-noise FRET-based calcium sensor, Twitch 2B (Figure 1G).[5b, 9] We first conducted a series of in vitro experiments to determine the sensitivity and specificity of the SST2 CNiFERs. Application of SST to SST2 CNiFERs induces a FRET response, whereby the mCerulean3 (referred to as ‘CFP’) emission decreases and cpVenusCD (referred to as ‘YFP’) emission increases with receptor activation (Figure 1H). We converted the CFP and YFP emissions to a FRET response, calculated as ΔR/R.[9] The SST2 CNiFER (clone #5F3) exhibited high affinity binding for SST (EC50 = ~4 nM) and a large ΔR/R (~2), while the control CNiFER (clone #128C3) without the SST2 receptor showed little or no response with up to 1 μM SST (Figure 1I). To confirm the involvement of the SST2 receptor, we applied a selective SST2 receptor antagonist (CYN 154806) and observed complete inhibition of the SST-dependent CNiFER activation (Figure 1J). To assess whether SST2 CNiFERs can respond to the repetitive release of SST, we measured the FRET response of SST2 CNiFERs to six 30 s pulses of SST. The amplitude of the FRET response (ΔR/R) gradually decreased after the first two pulses, suggesting some moderate desensitization of SST2 GPCRs, but importantly reached a stable plateau (Figure 1K). We next screened for potential non-specific responses of SST2 CNiFERs using a large number of different neuropeptides and neurotransmitters, at three different concentrations (10, 100, and 1000 nM) (Figure 1L). For most agonists, the SST2 CNiFERs showed little or no FRET response, except for that with SST. There was a small response with acetylcholine (ACh) at the highest concentration (1000 nM) for both SST2 and control CNiFERs. The background levels of ACh in the brain, however, do not contribute to the SST activated SST2 CNiFER response as confirmed below (Figure 2G and Figure S11). Taken together, these results show that the SST2 CNiFER (#5F3) has nanomolar sensitivity, exhibits a large FRET ratio response, and is highly specific for SST detection.
Figure 2.
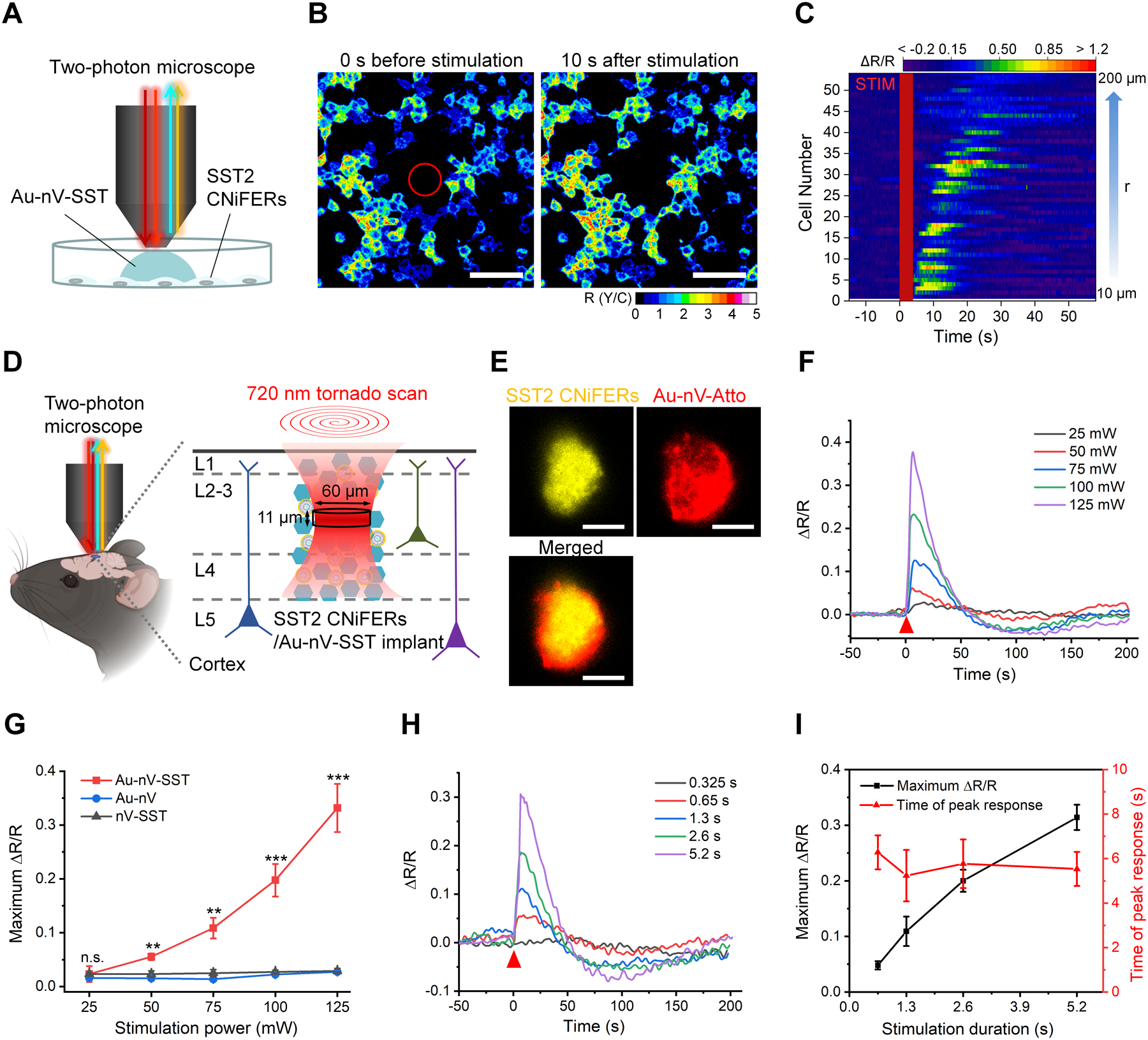
Real-time photorelease and monitoring of SST. A) Schematic of photoreleasing and monitoring SST on cultured SST2 CNiFERs with a two-photon microscope. B) Representative images of the ratio of YFP and CFP (R(Y/C)) of SST2 CNiFERs before and after laser stimulation on Au-nV-SST (720 nm, 300 mW, 4 s). Tornado scans with a diameter of 60 μm were performed in the center (marked as a red circle) at 0 s. Scale bar: 100 μm. C) Heat map of FRET response (ΔR/R) for activated CNiFERs. STIM represents the photo-stimulation at 0 s. Cell number was sorted by the distance (r) from the center. D) Schematic of implantation of co-mingled SST2 CNiFERs/Au-nV-SST mixture into mouse cortex. Au-nV-SST was stimulated by tornado scans (diameter: 60 μm) at 720 nm with an axial resolution of 11 μm. E) Representative two-photon fluorescent images of SST2 CNiFERs (Ex: 900 nm; Em: 520–560 nm) and Atto 647N-labeled Au-nV (Ex: 1100 nm; Em: 575–645 nm) at the depth of 200 μm in mouse cortex. Scale bar: 50 μm. F, G) FRET change (ΔR/R) trace (F) and maximum ΔR/R (G) of the SST2 CNiFERs implant under 720 nm laser stimulation with different laser power (2.6 s, n = 3 implants from 3 mice per group). The red triangle indicates the photo-stimulation at 0 s. Au-nV: gold-coated empty nanovesicles. nV-SST: SST-loaded nanovesicles without gold coating. Statistical analysis was conducted by a two-sample Student’s t-test between the Au-nV-SST and control groups (Au-nV, nV-SST). H) FRET change (ΔR/R) for SST2 CNiFERs implants plotted for different scan durations with 720 nm laser stimulation (100 mW). I) The maximum ΔR/R and time of peak response are plotted as a function of stimulation duration (n = 3 implants from 3 mice). No significant differences were found between the two times of peak response at different scan numbers by a two-sample Student’s t-test. Data are expressed as Mean ± S.D.; **p < 0.01; ***p < 0.001; n.s., not significantly different.
We next investigated the feasibility of simultaneously photoreleasing and measuring SST with these tools. To test in vitro, SST2 CNiFERs were grown in a monolayer and imaged with a two-photon microscope (Figure 2A). Au-nV-SST (loaded with 300 nM SST) were diluted directly in the extracellular solution of the CNiFERs and stimulated by 60-μm-diameter tornado scans at 720 nm (300 mW, 64 scans) in the center of the CNiFER culture. Real-time two-photon fluorescent images showed that the FRET response (ΔR/R) in the SST2 CNiFERs near the stimulation area dramatically increased following photo stimulation (Figure 2B). We observed ~38% of CNiFERs activated in a 200 μm × 200 μm area (Figure S6). We further found that SST2 CNiFERs at longer distances from the tornado scan area had a delayed response (Figure 2C, Figure S6). In addition, the FRET response from SST2 CNiFERs increased with the stimulation power and scan number (Figure S7 and S8). For controls, there was no CNiFER response following photo-stimulation of SST-loaded nanovesicles lacking gold (nV-SST) or gold-coated empty nanovesicles (Au-nV). Together, these results suggest that SST2 CNiFERs can detect the SST release and transmission in vitro in real-time.
We further tested SST photorelease and signaling in vivo by implanting a mixture of SST2 CNiFERs and fluorescent dye-labeled Au-nV (Au-nV-Atto, Atto refers to Atto 647N) into the mouse somatosensory cortex and imaging 2–4 hours later (Figure 2D). Here, we used the YFP emission (520–560 nm) to locate SST2 CNiFERs and the red fluorescence (575–645 nm) to monitor potential movement of Au-nV-Atto. The local distribution of Au-nV-Atto overlapped well with that of the CNiFERs (Figure 2E), suggesting limited diffusion of nanovesicles in the brain. The limited diffusion of nanovesicles is likely due to the physical barrier of CNiFERs and narrow extracellular space in the brain (images at different depths in Figure S9).[4c, 10] To further examine the localization of nanovesicles, we incubated nanovesicles with SST2 CNiFERs for 4 hours in vitro and observed no significant endocytosis of nanovesicles by the CNiFERs (Figure S10). This result suggests that Au-nV-SST remained predominantly in the extracellular space in the brain. Upon 720 nm light stimulation of Au-nV-SST, we observed a substantial increase in the FRET response (ΔR/R) that depended on the laser power and scan duration (Figure 2F, 2G and 2H), indicative of SST release. Interestingly, with irradiations of high power or long duration, the ΔR/R for the FRET response dropped below baseline at ~100 s but recovered. This drop might be due to SST2 receptor desensitization at high concentrations of SST, but further investigation is required. Note a monotonic increase in the amplitude of ΔR/R with increasing stimulation durations, while the time of peak response shows no significant differences (Figure 2I). The peak response of SST2 CNiFERs takes several seconds, mostly due to the time needed for activation of the GPCR and downstream signaling processes. Taken together, these results indicate a fast photorelease of SST and robust CNiFER detection (within several seconds), when Au-nV-SST are co-mingled with SST2 CNiFERs. We also conducted a series of controls. Photo-stimulation on Au-nV (gold-coated empty nanovesicles) or nV-SST (SST-loaded nanovesicles without gold coating) did not produce a FRET response in the SST2 CNiFER implants, even at the highest laser power and scan numbers (125 mW and 5.2 s). This result suggests that the photo-stimulation alone or encapsulated SST cannot trigger a SST2 CNiFER response (Figure 2G and Figure S11). Thus we conclude the SST2 CNiFER response is specific to the photoreleased SST.
To estimate the amount of SST released, we analyzed the scattering of the stimulation beam in the brain by numerical simulation (see Methods) and obtained a stimulation volume of ~10 fL (0.93 μm × 0.93 μm × 11.3 μm) with a single pulse. A 60-μm-diameter tornado scan gives a stimulation volume of 32 pL (Figure S12 and Table S1). Stimulation of the 32 pL volume at 100 mW and 40 scans leads to an estimated photorelease of 1.2 × 108 molecules and 100 nM concentration (Table S2). These results demonstrate the capability of PACE to release and detect physiological concentrations (nM) of SST in the brain.[11]
Next, we designed an experiment to measure SST diffusion and signaling directly in mouse brain cortex in real-time. We hypothesized that SST diffusion could be detected by implanting two distinct SST2 CNiFER clusters and measuring the FRET response as SST diffuses from one cluster to the other. We injected one cluster of SST2 CNiFERs co-mixed with Au-nV-SST (defined as the core implant) in the mouse cortex (Figure 3A). We then implanted additional SST2 CNiFER clusters at adjacent sites (defined as satellite implants). Figure 3B shows a core implant and two satellite implants (Sat-1, Sat-2) at a ~200 μm depth in the mouse somatosensory cortex. Photo-stimulation of Au-nV-SST in the core implant triggered SST release that was detected first by the core implant and then by satellite implants (Figure 3C). The peak CNiFER responses at satellite implants (Sat-1 and Sat-2) occurred after a delay and were smaller than in the core implant, consistent with neuropeptide dilution and diffusion, respectively. As shown previously, photo-stimulating satellite implants lacking Au-nV-SST did not evoke a FRET response in SST2 CNiFERs (Figure 3C). To determine the maximal diffusion distance to activate GPCRs, we extended the distances between the implants to 200–300 μm (Figure 3D). Figure 3E shows a significant FRET signal from the satellite implant 220 μm away from the core implant, while no CNiFER response was detectable at longer distances (Figure S13). Thus, SST2 CNiFERs could detect SST (i.e., < 0.5 nM) at distances ≤ 220 μm from the area of photo-stimulated Au-nV-SST. We then systemically varied the distances of the satellite implants from 50–220 μm. The amplitude of the CNiFERs response decreased with distance, indicating a spatial concentration gradient with reduced SST concentrations at longer distances from the core (Figure 3F). We measured the time of the peak SST2 CNiFER response following photostimulation. The time of the peak response for SST2 CNiFERs co-mingled with Au-nV-SST (core implant) revealed the time for GPCR activation and subsequent Ca2+ signaling (~ 5 s) (Figure 3G). The time to diffuse and activate SST2 CNiFERs positioned at short distances (less than 130 μm) was similar (~ 15 s), suggesting transmission is mainly synchronous. By contrast, the transmission time dramatically increased at larger distances (150–220 μm). For example, SST took approximately 46 s to reach the maximum diffusion distance and induce a CNiFER response, while it took ~23 s to reach CNiFERs at 160 μm (i.e., less synchronous). We examined whether variation in the shape or size of the implant would affect our measurements. By comparing two scenarios with the same core/satellite implant distance but two different implant sizes, we found that while the response amplitude varied, the time of peak response was not significantly different (Figure S14). We conducted a simulation using the exact distribution (uniform, inner or outer) of Au-nV-SST within the core cluster and found this would not significantly affect the response time of the satellite cluster (Figure S15). Taken together, these results suggests that the time delay of the peak response provides a robust indicator of SST transmission and signaling, and reveals the spatiotemporal scale of SST transmission and signaling in the neocortex.
Figure 3.
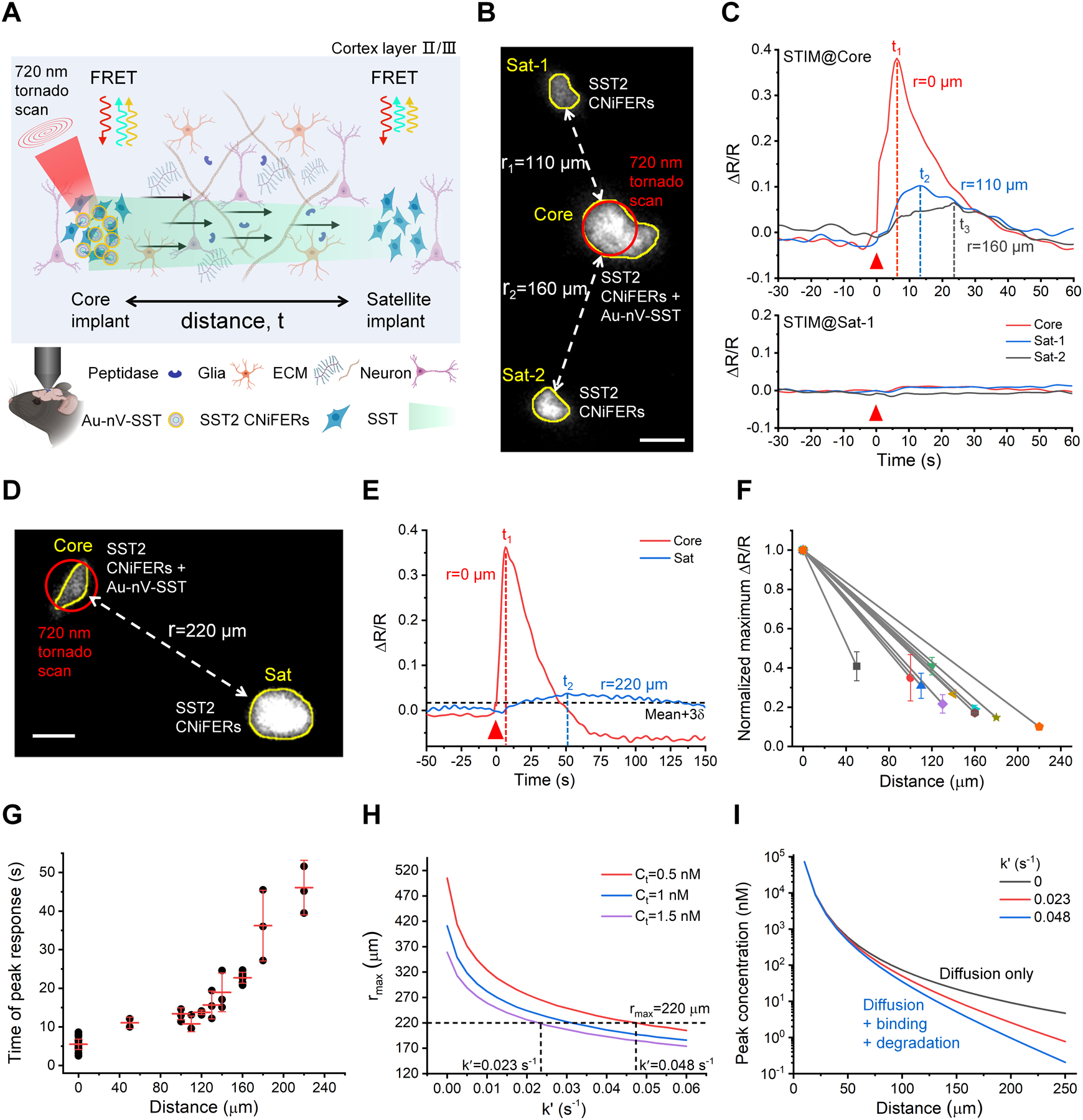
Measurement of SST volume transmission in vivo. A) Schematic of SST transmission measurement by implanting two clusters of SST2 CNiFERs, in which the core implant (left) is mixed with Au-nV-SST. B) Two-photon fluorescent image of SST2 CNiFERs at a depth of 200 μm in mouse cortex (Ex: 900 nm; Em: 520–560 nm). The implant in the center (Core) was mixed with Au-nV-SST, while the other two satellite implants (Sat-1, Sat-2) were SST2 CNiFERs alone. Scale bar: 50 μm. C) The response curves of SST2 CNiFERs when stimulating (STIM) at different regions (720 nm, 100 mW, 2.6 s). The red triangle indicates the photo-stimulation at 0 s. D) Two-photon fluorescent image of SST2 CNiFER clusters at a distance of 220 μm in mouse cortex. Scale bar: 50 μm. E) The response curves of SST2 CNiFERs when stimulating at the core implant (Core). The threshold of the valid signal is set as Mean + 3δ (δ: the standard deviation of baseline). F) Normalized maximum ΔR/R of paired SST2 CNiFERs implants at different distances in untreated mice brains (n = 10 pairs of implants in 9 mice, 2–4 repeated measurements for a pair of implants). G) Time of peak response from SST2 CNiFER implants at defined distances in untreated mice brains. Photo-stimulation is at 0 s. The centerline represents the mean value. H) Predicted maximal diffusion distance (rmax) as a function of loss rate (k′) from point-source diffusion model (Equation 2) with total SST released number Q = 1.2 × 108 and effective diffusion coefficient D* = 8.9 × 10−7 cm2•s−1 (from optical integrative imaging measurement). The horizontal dashed line represents the experimental rmax = 220 μm. I) Predicted concentration as a function of distance from Equation 2. Data are expressed as Mean ± S.D.
Neuropeptides can bind to receptors on neurons as well as undergo peptidase degradation when diffusing in the extracellular space. We compared our distance-dependent signaling measurements with a theoretical point-source diffusion model to evaluate the loss of SST caused by these two factors during diffusion. We first measured the effective diffusion coefficient (D*) of fluorescently labeled SST, fluorescein-5(6)-carbonyl-somatostatin-14 (5(6)-FAM-SST), by the integrative optical imaging (IOI) method in acute brain slices (Figure S16, D* = (8.9 ± 1.3) × 10−7 cm2•s−1).[12] Since the cyclic structure of SST remains the same in 5(6)-FAM-SST, this method gives a reasonable estimate of the effective diffusion coefficient (D*) for SST in brain tissue. Note that the loss of SST was not considered here in IOI measurements since the SST concentration was 10–100 times higher (1.5 mM of 5(6)-FAM-SST) than physiological concentrations. We defined the maximal signaling distance (rmax) as the farthest location where the peak SST concentration reaches the threshold for CNiFER detection (Ct). With the effective diffusion coefficient (D*), nM sensitivity of CNiFERs (Ct = 0.5–1.5 nM, see Methods), and an estimated number of photoreleased SST molecules (1.2 × 108), the point-source diffusion model predicts the theoretical rmax as a function of SST loss rate constant (k′, Figure 3H). Here the loss rate constant k′ is determined by a combination of neuropeptide binding degradation during diffusion in extracellular space (Figure 3A). The comparison of experimental and theoretical rmax yields the k′ in the range of 0.023 s−1 – 0.048 s−1 (Figure 3H). Using these transport parameters, we calculated the SST concentration at different distances based on the point-source diffusion model (Equation 2). The SST loss from the binding and degradation becomes more significant at large distances (> 100 μm, Figure 3I). These results suggest that receptor binding and degradation in concert with dilution in the extracellular space limit the maximal distance that SST can signal to receptor-expressing neurons. We also compared the experimental diffusion time with the time for the released neuropeptide to diffuse and reach the threshold concentration (Equation 2) and peak concentration (Equation 4) at different distances. The overlap in the experimental data (Δtexp) and the predicted times, which are inclusive of the range of times for CNiFERs to sense and respond the released neuropeptide (Figure S17), further validates the estimate of SST loss rate.
To examine whether the geometry of brain extracellular space and the composition of the extracellular matrix play essential roles in controlling the local diffusion of endogenous signaling molecules and nutrients,[4a, 13] we applied the PACE approach to probe SST volume transmission in mice with an altered extracellular environment. We predicted that degradation of the extracellular matrix would decrease the resistance and enhance peptide diffusion. Hyaluronic acid (HA) is one of the major components of the brain extracellular matrix and forms high molecular weight long-chain molecules, a physical barrier for molecular diffusion.[14] To alter the extracellular environment, we injected hyaluronidase (hyase) into the lateral ventricles of adult mouse brain and measured SST diffusion two days after the hyase injection. The immunostaining for hyaluronan binding protein (HABP) confirms HA reduction (Figure 4A and 4B). Quantitative analysis shows that the intensity of HABP decreased to 23% of that in untreated brains (Figure 4C). Notably, there were no differences in cell nucleus number or cell size in the cortex compared to untreated brains (Figure 4D, Figure S18), indicating no significant cell loss.
Figure 4.
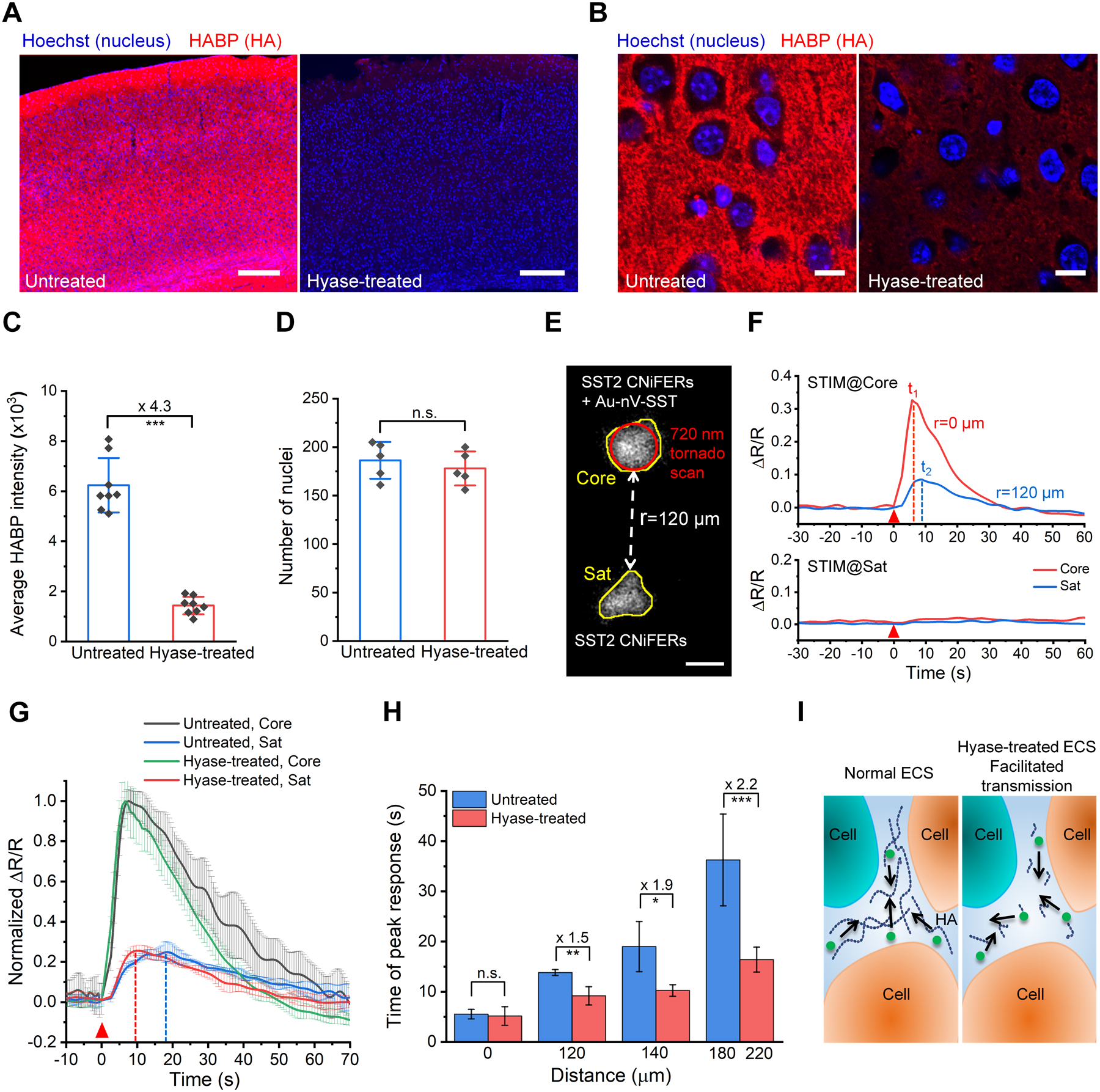
SST volume transmission in hyaluronan-deficient brains. A) Representative images of HABP-labeled (red) brain cortex sections from untreated mouse and hyaluronidase (hyase)-treated mouse. Nuclei were stained by Hoechst 33342 (blue). Scale bar: 200 μm. B) Confocal images of the brain cortex in the left hemisphere with high magnification. Scale bar: 10 μm. C) Average fluorescence intensity for HABP in mouse cortex (0.8 mm × 0.8 mm) of untreated and hyase-treated brains (n = 8 slices). D) Number of nuclei in the cortex of untreated and hyase-treated brains (n = 5 slices). E) Two-photon fluorescent image of SST2 CNiFERs implanted at a depth of 200 μm in mouse cortex. The top implant (Core) has SST2 CNiFERs mixed with Au-nV-SST, while the bottom implant (Sat) contains only SST2 CNiFERs. Scale bar: 50 μm. F) The response curves of SST2 CNiFERs when stimulating (STIM) at different regions (720 nm, 100 mW, 2.6 s). G) Comparison of normalized ΔR/R traces of the core implants (Core) and satellite implants (Sat) at the distance of 140 μm in untreated and hyase-treated brains (n = 3). H) Comparison of time of peak response over different distances in hyase-treated brains (n ≥ 4 measurements for each distance, 6 mice in total) and in untreated brains (acquired from Figure 3G). I) Schematic of the change in brain extracellular space (ECS) and SST volume transmission under different conditions. Data are expressed as Mean ± S.D.; *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significantly different.
With the HA-deficient mouse brain model, we measured SST volume transmission using the Au-nV-SST and SST2 CNiFERs (Figure 4E), as described above. Photo-stimulation of the core implant containing both CNiFERs and Au-nV-SST resulted in a large FRET response in the core CNiFERs and after a delay in satellite CNiFERs. Similarly, no FRET response was observed upon the direct photo-stimulation of the satellite implant (Sat), which only contained SST2 CNiFERs (Figure 4F). However, the main difference observed in the HA-deficient mice was the peak of the FRET response had a faster rise and decay time for satellite implant (Sat) as compared with the untreated brain (Figure 4G). The difference between the peak response times in the untreated and hyase-treated brains was more apparent at longer distances (Figure 4H). For example, the transmission and signaling over 200 μm in hyase-treated brains was 2.2-fold faster than that in untreated brains. Thus, the results suggest that the speed of neuropeptide volume transmission is, in part, determined by the extracellular space and the extracellular matrix (Figure 4I).
Tissue heating is a common concern with multiphoton techniques for in vivo imaging.[15] An important consideration here is whether PACE has potential impact of local brain heating. To measure tissue heating, we inserted a thermocouple probe in the brain during laser stimulation and imaging (Figure S19). Elevations in temperature originated predominantly from tissue absorption of the near-infrared light (3°C/100 mW), consistent with previous reports.[15a] Importantly, the presence of gold-coated nanovesicles did not lead to an additional temperature increase. Furthermore, our experimental results showed no observable response to the stimulation of Au-nV or SST-nV (Figure 2G and Figure S11). Therefore, tissue heating is minimal and likely does not significantly impact the signaling measurement reported here.
PACE is a versatile approach to probe extrasynaptic signaling in different contexts. These combined technologies provide significant advantages over current neuropeptide release and sensing methodologies. First, optogenetic stimulation of neuropeptide expressing neurons can induce release of a peptide but classical neurotransmitters can be co-released (e.g., glutamate, GABA).[1a, 16] Photo-stimulation of nanovesicles releases a bolus of a specific neuropeptide. Second, compared with caged compounds,[17] nanovesicles allow in vivo measurement by protecting from rapid peptidase degradation. Third, CNiFERs provide in vivo neuropeptide real-time detection and signaling at physiologically relevant nanomolar neuropeptide concentrations. In contrast, microdialysis has a slow sampling rate (several minutes or longer),[18] while fast scanning cyclic voltammetry requires specific electroactive amino acid in the sequence of neuropeptides such as tyrosine.[19] GPCR-based genetically encoded fluorescent sensors for neuropeptides are emerging;[20] however, challenges remain in the brightness and sensitivity in vivo.
In this work, we have demonstrated that the PACE strategy provides new insights into neuropeptide volume transmission by precisely releasing and sensing neuropeptide transmission at defined distances. PACE measures the integrated process of neuropeptide release, diffusion, GPCR binding, and intracellular signaling, which includes the predominant steps in the neuropeptide extrasynaptic volume transmission. Using PACE, our study provides the first quantitative estimation of the overall SST loss rate (k′, 0.023–0.048 s−1) due to the degradation and binding in vivo. The results suggest that neuropeptide binding and degradation limit extrasynaptic transmission at long distances (>100 μm).
Conclusion
In summary, we report an integrated approach (PACE) to mimic and probe neuropeptide volume transmission in vivo by optical release and sensing. PACE provides the ability to package and release an exogenously supplied neuropeptide using small gold-coated nanovesicles, with high temporal and spatial resolution in the brain. A newly created SST2 CNiFER detects nM concentrations of photo-released SST in real-time in the brain. In the future, it may be possible to detect release of endogenous SST with SST2 CNiFERs. With the PACE technique, we measured the time for SST transmission and signaling across a range of distances and revealed distance-dependent signaling for neuropeptides (reduced but synchronized within 130 μm, while smaller and delayed at longer distances). Importantly, we provide the range of SST loss rate due to the binding and degradation during diffusion and determined that SST loss limits its transmission at longer distances (>100 μm). PACE is a useful tool to reveal important biological signaling processes in the brain and opens up new avenues for using nanotechnology to address fundamental questions in neuroscience. The findings in this work will advance the understanding of neuropeptide volume transmission and its functional role in brain circuits.
Supplementary Material
Acknowledgments
We thank Dr. Oliver Griesbeck for providing the Twitch2B cDNA clone and discussions. Schematic figures were made with BioRender.com. This work was partially supported by National Science Foundation under award number 1631910 (Z.Q.), National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award number RF1NS110499 (Z.Q., P.A.S.), National Institute of Mental Health of NIH under award number R01MH111499 (P.A.S.), Human Frontier Science Program (HFSP) under award RGP0036/2020 (S.H.), and a postdoc research grant from the Phospholipid Research Center (Heidelberg, Germany) to H.X.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Hejian Xiong, Department of Mechanical Engineering, The University of Texas at Dallas, Richardson, Texas 75080 (USA).
Emre Lacin, Nash Family Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029-5674 (USA).
Hui Ouyang, Department of Mechanical Engineering, The University of Texas at Dallas, Richardson, Texas 75080 (USA).
Aditi Naik, Department of Cell Biology, SUNY Downstate Health Sciences University, Brooklyn, NY 11203 (USA).
Krutin Kumar, School of Behavioral and Brain Sciences, The University of Texas at Dallas, Richardson, TX 75080 (USA).
Xiuying Li, Department of Mechanical Engineering, The University of Texas at Dallas, Richardson, Texas 75080 (USA).
Joseph A. Zasadzinski, Department of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, MN 55455 (USA)
Sabina Hrabetova, Department of Cell Biology, SUNY Downstate Health Sciences University, Brooklyn, NY 11203 (USA).
Paul A. Slesinger, Nash Family Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029-5674 (USA).
Zhenpeng Qin, Department of Mechanical Engineering, The University of Texas at Dallas, Richardson, Texas 75080 (USA); Department of Bioengineering, The University of Texas at Dallas, Richardson, TX 75080 (USA); Department of Surgery, The University of Texas at Southwestern Medical Center, Dallas, TX 75390 (USA); Center for Advanced Pain Studies, The University of Texas at Dallas, Richardson, TX 75080 (USA).
References
- [1].a) Nusbaum MP, Blitz DM, Marder E, Nat. Rev. Neurosci 2017, 18, 389–403; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guillaumin MCC, Burdakov D, Front. Neurosci 2021, 15, 644313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Smith SJ, Sümbül U, Graybuck LT, Collman F, Seshamani S, Gala R, Gliko O, Elabbady L, Miller JA, Bakken TE, Elife 2019, 8, e47889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].van den Pol AN, Neuron 2012, 76, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Syková E, Nicholson C, Physiol. Rev 2008, 88, 1277–1340; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tønnesen J, Inavalli VVGK, Nägerl UV, Cell 2018, 172, 1108–1121; [DOI] [PubMed] [Google Scholar]; c) Godin AG, Varela JA, Gao Z, Danné N, Dupuis JP, Lounis B, Groc L, Cognet L, Nat. Nanotechnol 2017, 12, 238–243. [DOI] [PubMed] [Google Scholar]
- [5].a) Lacin E, Muller A, Fernando M, Kleinfeld D, Slesinger PA, J. Vis. Exp 2016, e53290; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Muller A, Joseph V, Slesinger PA, Kleinfeld D, Nat. Methods 2014, 11, 1245–1252; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Foo C, Lozada A, Aljadeff J, Li Y, Wang JW, Slesinger PA, Kleinfeld D, Curr. Biol 2021, 31, 4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C, Nat. Rev. Drug Discov 2003, 2, 999–1017; [DOI] [PubMed] [Google Scholar]; b) Jang HJ, Chung H, Rowland JM, Richards BA, Kohl MM, Kwag J, Sci. Adv 2020, 6, eaay5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Randrianalisoa J, Li X, Serre M, Qin Z, Adv. Optical Mater 2017, 5, 1700403. [Google Scholar]; b) Xiong H, Li X, Kang P, Perish J, Neuhaus F, Ploski JE, Kroener S, Ogunyankin MO, Shin JE, Zasadzinski JA, Wang H, Slesinger PA, Zumbuehl A, Qin Z, Angew. Chem., Int. Ed. Engl 2020, 59, 8608–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) González-Rubio G, Díaz-Núñez P, Rivera A, Prada A, Tardajos G, González-Izquierdo J, Bañares L, Llombart P, Macdowell LG, Palafox MA, Liz-Marzán LM, Peña-Rodríguez O, Guerrero-Martínez A, Science 2017, 358, 640–644; [DOI] [PubMed] [Google Scholar]; b) Plech A, Kotaidis V, Lorenc M, Boneberg J, Nat. Phys 2006, 2, 44–47. [Google Scholar]
- [9].a) Muller A, Joseph V, Slesinger PA, Kleinfeld D, Nat. Methods 2014, 11, 1245–1252; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thestrup T, Litzlbauer J, Bartholomäus I, Mues M, Russo L, Dana H, Kovalchuk Y, Liang Y, Kalamakis G, Laukat Y, Becker S, Witte G, Geiger A, Allen T, Rome LC, Chen T-W, Kim DS, Garaschuk O, Griesinger C, Griesbeck O, Nat. Methods 2014, 11, 175–182. [DOI] [PubMed] [Google Scholar]
- [10].Soria FN, Paviolo C, Doudnikoff E, Arotcarena M-L, Lee A, Danné N, Mandal AK, Gosset P, Dehay B, Groc L, Cognet L, Bezard E, Nat. Commun 2020, 11, 3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Russo AF, Headache 2017, 57, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Nicholson C, Tao L, Biophys. J 1993, 65, 2277–2290; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hrabe J, Hrabetova S, Biophys. J 2019, 117, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hrabetova S, Cognet L, Rusakov DA, Nägerl UV, J. Neurosci 2018, 38, 9355–9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z, Nat. Commun 2020, 11, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Picot A, Dominguez S, Liu C, Chen IW, Tanese D, Ronzitti E, Berto P, Papagiakoumou E, Oron D, Tessier G, Forget BC, Emiliani V, Cell Rep. 2018, 24, 1243–1253; [DOI] [PubMed] [Google Scholar]; b) Podgorski K, Ranganathan G, J. Neurophysiol 2016, 116, 1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Apergis-Schoute J, Burnstock G, Nusbaum MP, Parker D, Morales MA, Trudeau L-E, Svensson E, Front. Neural Circuits 2019, 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Banghart MR, Sabatini BL, Neuron 2012, 73, 249–259; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banghart MR, He XJ, Sabatini BL, ACS Chem. Neurosci 2018, 9, 684–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zestos AG, Kennedy RT, AAPS J. 2017, 19, 1284–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Calhoun S, Meunier C, Lee C, McCarty G, Sombers L, ACS Chem. Neurosci 2018, 10, 2022–2032; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Asai K, Ivandini TA, Einaga Y, Sci. Rep 2016, 6, 32429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abraham AD, Casello SM, Schattauer SS, Wong BA, Mizuno GO, Mahe K, Tian L, Land BB, Chavkin C, Neuropsychopharmacology 2021, 46, 2330–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.