

Abstract
Cancer cells are plastic – they can assume a wide range of distinct phenotypes. Plasticity is integral to cancer initiation and progression, as well as to the emergence and maintenance of intra-tumoral heterogeneity. Further, plastic cells can rapidly adapt to and evade therapy, which poses a challenge for effective cancer treatment. As such, targeting plasticity in cancer holds tremendous promise. Yet, the principles governing plasticity in cancer cells remain poorly understood. Here, we provide an overview of the fundamental molecular and cellular mechanisms that underlie plasticity in cancer and in other biological contexts, including development and regeneration. We propose a key role for high-plasticity cell states (HPCSs) as crucial nodes for cell state transitions and enablers of intra-tumoral heterogeneity.
Keywords: Plasticity, differentiation, intra-tumoral heterogeneity, tumor evolution, cell state transition, cancer therapy
Plasticity is a fundamental feature of biological systems
Plasticity refers to the ability of cells to acquire new states (phenotypes) via differentiation programs. Cellular plasticity is an integral feature of biological systems that is dictated by changes in gene expression patterns. For instance, bacteria can acquire and discard distinct cellular states through horizontal gene transfer, which can occur even between distantly related unicellular organisms. Eukaryotes have similarly evolved mechanisms enabling plasticity. However, rather than permitting exchange of genetic material, eukaryotic cells maintain high genome integrity and instead rely on processes such as epigenetic regulation, mRNA splicing, alternative promoter usage, and selective utilization of 3’ untranslated regions in mRNA. These mechanisms endow metazoan cells with tremendous flexibility to diversify cellular functions and acquire distinct phenotypic states. Consistent with this notion, all cells within metazoan organisms harbor the genetic information required for acquiring plasticity. Perhaps the most striking example of such acquired plasticity is the ability of the “Yamanaka factors” OCT4, SOX2, KLF4, and MYC to induce pluripotency – a cell state with particularly high plasticity – in differentiated cells [1].
Cell state transitions enabled by plasticity are essential for normal development as well as adult tissue homeostasis and regeneration. Plasticity is also invariably co-opted in cancer pathogenesis, where it promotes tumor initiation and progression by enabling cell states with distinct functional properties such as a high capacity to proliferate or metastasize [2–4]. Furthermore, plasticity contributes significantly to failure of chemo-, targeted- and immunotherapies across cancers [3, 5–8], permitting cancer cells to acquire phenotypic states that are adapted to therapy via non-genetic mechanisms [9]. In addition to directly enabling adaptation to therapies, plasticity is a requisite for the emergence and maintenance of intra-tumoral heterogeneity, which increases the likelihood of cancer cell states harboring intrinsic resistance to therapy. Given the critical importance of plasticity in tumor progression and treatment resistance, targeting plasticity is garnering increasing interest [7–9]. However, it is not clear conceptually or practically how targeting cancer plasticity would be best achieved – i.e. whether to pursue selective elimination of highly plastic cancer cell subsets or rather target specific molecular programs that drive plasticity. Here, we discuss central concepts of both the cellular and molecular facets of plasticity in cancer.
Plasticity in development and normal tissue homeostasis
Cellular plasticity has been studied for decades in the context of embryonic development and tissue regeneration. Development starts from conception and continues in postnatal life, with the most drastic phenotypic conversions occurring during embryogenesis. Classic experiments employing transplantation of embryonic cells into ectopic sites within the embryo have demonstrated that they harbor the capacity to differentiate into other cell types within their lineage, directed by locally available morphogens [4]. However, this plasticity is highly dependent on embryonic stage. For example, presumptive neural ectoderm from an early gastrula amphibian embryo will assume the fate of its host environment, whereas presumptive neural tissue from a late gastrula will retain its neural fate [10]. Thus, the progression of development is accompanied by an overall decrease in plasticity.
Nevertheless, fully formed adult tissues retain plasticity (Fig. 1A). Under normal physiological conditions most tissues in adult metazoans undergo continuous loss and replacement of differentiated cells. The rate of cellular turnover during normal homeostasis varies dramatically across tissues: the frontal lobe of our brain is unlikely to turn over during our lifespan, whereas the skin is regenerated every two weeks and the intestinal epithelium every 3–4 days. Most organs rely on adult stem cells (ASCs) to replace dead cells during normal homeostasis and in response to injury. For instance, the rapid cell turnover of the intestinal epithelium is fueled by a relatively small pool of ASCs expressing leucine-rich G-protein coupled receptor-5 (LGR5) that are located at the base of glandular crypts. The intestinal stem cells display a high degree of plasticity, being capable of differentiating to all the cell types within the intestinal epithelium [11]. Similar constitutively active ASCs are found in the skin and the hematopoietic system.
Figure 1. Plasticity in normal physiology vs. cancer in the context of the Waddington landscape.
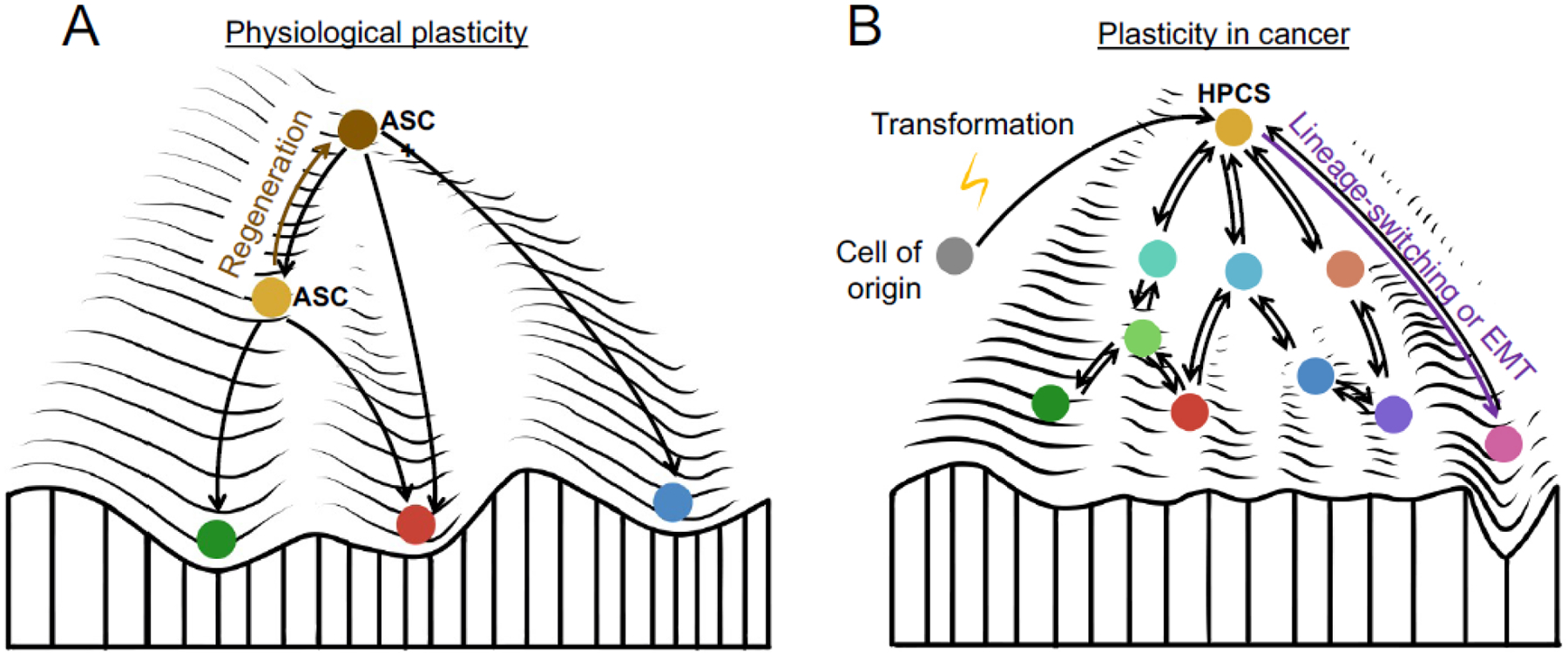
(A) During normal tissue homeostasis, differentiated cells arise from a multipotent adult stem cell (ASC, orange). Upon injury, stem cells acquire plasticity, which manifests in increased differentiation potential towards related cellular lineages (ASC+, brown state). (B) Cellular transformation of the cell of origin (gray) is associated with the acquisition of increased plasticity. Within established tumors, cancer cell differentiation states are less defined and capable of rapid transitions. We propose that plasticity concentrates within molecularly distinct cell states (orange) that have significantly increased capacity for cell state transitions. These high-plasticity cell states (HPCSs) enable drastic phenotypic transitions, such as lineage-switching or epithelial-mesenchymal transition (EMT). The environment (peaks and valleys) in cancer also changes to facilitate the phenotypic transitions, compared to normal cells.
On the other hand, multiple adult cell types do not arise from ASCs, but rather harbor high proliferative potential and plasticity in their differentiated state. For example, endothelial cells in all tissue types can become activated to form vascular networks by growth factor signals, which stimulate differentiation into migratory “tip cells” that lead the formation of vascular sprouts and “stalk cells” that follow the tip cells and proliferate [12]. Once the vessel network is complete, endothelial cells re-enter quiescence and assume a differentiated state as a functional component of the vessel wall [12], demonstrating that plasticity is not a feature confined to ASCs.
Plasticity in tissue regeneration and repair
Most mammalian tissues harbor substantial potential to repair and regenerate following injury. Regenerative programs are associated with an increase in ASC plasticity (Fig. 1A). As a result, the lineage choices available to ASCs become more flexible. Upon skin wounding, hair follicle stem cells exhibit lineage plasticity by adopting features characteristic of epidermal stem cells while retaining some distinct differentiation features [13, 14]. This is in part mediated by loss of canonical homeostatic transcription factors (TFs) and associated changes in chromatin accessibility, as well as co-expression of TFs specific for other stem cell lineages. These changes endow ASCs with greater capacity to repair tissue damage. The increased ASC plasticity associated with regeneration or cellular stress permits drastic lineage reversions in experimental systems. In skin sweat glands, ASCs can form either milk-producing glandular structures or epidermal tissue when transplanted into either mammary fat pads or skin, respectively [15].
Acquired plasticity is observed in other tissues, including during pancreas [16, 17], liver [18], airway [19], and nerve regeneration [20]. Injured pancreatic acinar cells become proliferative and plastic, exhibiting both acinar and ductal markers and regenerating acini by transitioning through a ductal phenotype. This acinar-to-ductal metaplasia (ADM) is crucial for pancreas regeneration [16, 17]. Interestingly, acinar cells retain an epigenetic memory of this metaplasia. Under normal conditions, this persistence of acquired plasticity protects the pancreas by facilitating reactivation of ADM upon subsequent injury [21]. Similarly upon liver injury, hepatocytes can undergo a Notch-dependent transition to biliary epithelial cells [18]. During Wallerian degeneration after nerve injury, c-Jun reprograms Schwann cells to a phenotype dedicated to supporting nerve repair [20]. In the lung, where cellular turnover is slow during normal homeostasis, alveolar type 2 (AT2) cells function as facultative stem cells of the alveoli by both self-renewing and replacing alveolar type 1 (AT1) cells when necessary [22–24]. Significant functional heterogeneity exists within AT2 cells: AT2 cells that reside in a niche formed by WNT-producing fibroblasts and that express the WNT target gene Axin2 are particularly efficient stem cells for maintaining and regenerating alveoli during normal homeostasis and in response injury [22, 23]. Upon damage to the alveoli, AT2 cells expressing interleukin-1 receptor 1 (IL-1R1) are primed by IL-1β produced by interstitial macrophages to acquire a damage-associated transient progenitor (DATP) state [25] (also described as alveolar differentiation intermediate (ADI) [26] and pre-alveolar type-1 transitional cell state (PATS) [27]). This cell state represents a plastic transition state with the capacity to produce fully differentiated AT2 or AT1 cells and does not exist in uninjured healthy lung [25–27]. Chronic inflammation mediated by IL-1β prevents exit from the DATP state, leading to impaired AT1 differentiation and impaired alveolar regeneration [25].
Plasticity in tumor initiation
Both intrinsic and acquired plasticity play an important role in tumor initiation. This is evident in the frequency of tumor development across various tissues, as cancer arises more commonly in tissues with greater plasticity and proliferative potential. For instance, highly regenerative tissues such as the liver and colon are common sites of cancer [28]. In contrast, tissues such as the heart, mostly comprised of terminally differentiated cells with little regenerative potential and plasticity, are largely resistant to tumorigenesis [29]. These observations suggest that the cellular plasticity involved in healthy tissue regeneration contributes to tumor initiation. Progenitors and stem cells transform readily, whereas differentiated cells residing in fixed phenotypic states are resistant to transformation. For example, Nestin+ adult neural stem cells, Ascl1+ bipotent neuronal progenitors, and Cspg4+ (NG2+) oligodendrocyte precursors give rise to glioblastomas with high efficiency upon targeted inactivation of the tumor suppressors Nf1, Trp53, and Pten. However, Dlx1+ progenitors that represent a later stage in neuronal differentiation harbor significantly lower potential for gliomagenesis, and fully differentiated Neurod1+ newly born neurons or Camk2a+ post-mitotic neurons never give rise to gliomas upon initiation of the same mutations [30]. The plasticity that enables malignant transformation of certain cell states, but not of others, is likely established by specific chromatin states. Identical driver mutations engineered into neural crest progenitor cells and melanoblasts produce melanomas, whereas terminally differentiated melanocytes do not transform [31]. This difference in oncogenic competence is due to greater transcriptional plasticity in progenitor cells, which is maintained by chromatin-modifying enzymes such as ATAD2 [31].
These results highlight the importance of the cell of origin, and the extent of its plasticity, in tumor initiation. However, the cell of origin has not been clearly defined in many cancer types, including breast, prostate, and pancreatic cancer. One explanation for the lack of a defined cell of origin is that the plasticity involved in tumor initiation is induced through specific oncogenic mutations. For example, overexpression of the reprogramming factor SOX2 can drive the formation of lung squamous cell carcinomas (that often harbor SOX2 amplifications in patients) from multiple lung epithelial cell types, including tracheobronchial basal cells, club cells or AT2 cells [32]. Another possibility involves stressors such as tissue injury or inflammation, which can increase plasticity and facilitate tumor initiation even in cells that are resistant to transformation under homeostatic conditions. In the intestine, LGR5+ ASCs efficiently give rise to intestinal adenomas and adenocarcinomas, whereas differentiated intestinal epithelium do not transform readily [33, 34]. However, with inflammation induced by epithelial injury or genetic NF-κB gain-of-function, adenomas can arise from differentiated intestinal epithelial cells [33]. This phenomenon provides a mechanistic explanation for the high incidence of colon cancer in individuals with chronic inflammatory bowel disease [33]. In the pancreas, the confluence of pancreatitis induced by tissue injury and oncogene activation produce a chromatin state uniquely poised for cellular transformation [35]. Inflammation associated with the injury triggers acinar-to-ductal metaplasia. During tissue repair associated with pancreatitis, oncogenic KRAS can co-opt this transient increase in plasticity to drive tumor initiation by stabilizing the acinar-to-ductal transition [21, 35], underscoring the importance of plasticity in malignant transformation.
Plasticity in established tumors
A hierarchy of plasticity
Plasticity is a central feature of fully formed tumors. Cancer cells are endowed with higher plasticity than terminally differentiated normal cells (Fig. 1B). This plasticity is driven by selection of epigenetic states permitting transitions within a wide range of cellular phenotypes. Furthermore, mutations in tumor suppressors such as p53 or RB enable a greater range of chromatin states [7, 8, 36]. However, abundant evidence indicates that, as in healthy tissues, certain cancer cell subsets are more plastic than others even in genetically homogenous tumors. According to the cancer stem cell (CSC) model, the CSCs reside at the apex of a hierarchy and harbor the capacity for self-renewal, persistent proliferation, and differentiation into other cancer cell states that do not retain these stemness properties [37, 38]. In this model, CSCs possess the most plasticity, having the capacity to give rise to all cancer cell states within the tumor along a unidirectional differentiation trajectory, similar to the function of ASCs in healthy tissues. In multiple adenocarcinomas, including colon [39, 40] and lung [41], LGR5 marks a population with CSC properties, which include high differentiation and proliferative potential as well as a greater capacity to initiate transplant tumors when compared to non-CSCs. However, recent evidence indicates that the LGR5+ CSC state can be acquired by LGR5− cells. When LGR5+ cells are ablated in models of colon adenocarcinoma, KRT20+/LGR5− differentiated cancer cells can convert to a LGR5+ state that is functionally indistinguishable from the cells that were ablated [42]. This suggests that KRT20+/LGR5− cells may enter the niche vacated by the ablated LGR5+ cells, causing them to de-differentiate and acquire stemness. Similarly, in lung adenocarcinoma (LUAD) the LGR5+ state emerges in allografts established from cells that did not express LGR5 at the time of transplantation [41]. These studies suggest that the LGR5+ state is dependent on a defined niche within the 3-dimensional context of tumors. In LUAD, the LGR5+ cells neighbor another subset of cancer cells expressing porcupine, an enzyme responsible for an essential post-translational modification of WNT ligands. Genetic or pharmacological targeting of porcupine suppressed WNT signaling and abrogated the LGR5+ cells, highlighting the importance of niche-derived signals as drivers of stemness and plasticity in cancer [41].
Acquired plasticity in cancer
Recent evidence indicates that a highly plastic cell state distinct from CSCs and ASCs can emerge during tumor evolution. Single-cell transcriptomic analysis of cancer cell states during the evolution of mouse LUAD identified a cancer cell subpopulation present in all mouse adenomas and adenocarcinomas [43] (Fig. 2A), suggesting that this subset may have unique significance. A mathematical model, Waddington’s Optimal Transport (WOT) [44], predicted that this subset represents a high-plasticity cell state (HPCS), having the capacity to acquire the largest number of distinct downstream cancer cell states out of the 12 states detected [43] (Fig. 2B). Isolated HPCS cells gave rise to more phenotypic heterogeneity in tumor sphere cultures and orthotopic transplants than the remainder of the tumor cell pool. The HPCS shares functional features of ASCs and CSCs, including robust growth and differentiation potential [38, 45], but it was distinct when compared to 1,197 published ASC and CSC transcriptional signatures. Instead, it harbored features of unrelated lineages, such as trophoblast stem cells, kidney epithelium and chondroblasts, as well as stress response signatures [43]. Rather than ASCs, the HPCS transcriptionally resembles the DATP/PATS/ADI state that emerges after lung injury [25–27] and is conceptually similar to the state described in skin ASCs during wound repair [46]. Unlike CSCs, the HPCS is predicted to form an intersection, or transition hub, that connects other cancer cell states [43] rather than be at the apex of a hierarchy (Fig. 2C). The HPCS model may apply to other cancers, as a pan-cancer analysis of the HPCS gene signature revealed patients with this signature had a worse prognosis [43], though a formal exploration of the HPCS in other cancer types has yet to be performed. However, single-cell transcriptomic analysis in pancreatic ductal adenocarcinoma (PDAC) identified a HPCS-like cell state expressing stress response programs, which was transcriptionally distinct from well-defined classical and basal phenotypes but co-expressed subsets of classical and basal genes [47]. Tumors harboring a greater mix of classical and basal cells also had a higher proportion of this intermediate population, suggesting it may important in cell state transitions between the basal and classical phenotypic extremes.
Figure 2. Role of a high-plasticity cell state (HPCS) in lung adenocarcinoma evolution.
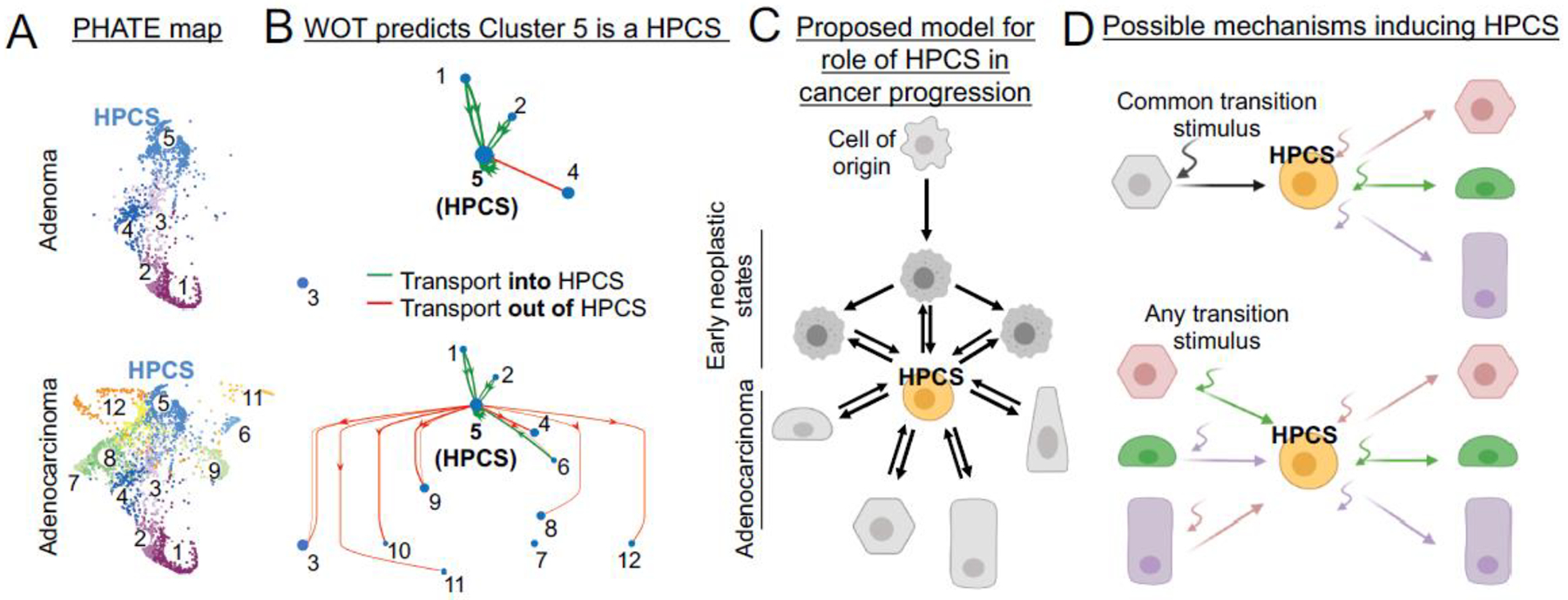
(A) Single-cell RNA-sequencing analysis of cell states in lung atypical adenomatous hyperplasias and adenomas (top), as well as adenocarcinomas (bottom), visualized by a potential of heat-diffusion for affinity-based trajectory embedding (PHATE) map [43]. (B) Waddington’s Optimal Transport analysis of cell state transitions. Note that only connections involving Cluster 5 (HPCS) are shown (full map is published [43]). Green HPCS-upstream (green) and HPCS-downstream (red) states are indicated. (C) Proposed model for the role of HPCS in tumor evolution: The cell of origin gives rise to early neoplastic cell states that harbor limited plasticity. The emergence of the HPCS (orange) enables a plethora of downstream cell states. In established adenocarcinomas, the HPCS forms the center of a network of discrete cancer cell states, enabling cell state transitions and promoting heterogeneity. Transitions from early neoplastic stages into the HPCS, or from the HPCS to late neoplastic states have increased probabilities (bold arrows), whereas reverse transitions can happen with lower probability or when triggered by a transition stimulus (light arrow). (D) Two conceptual models for the induction of the high-plasticity cell state in established tumors. Top: A common transition stimulus (black curved arrow) triggers activation of the HPCS program in a differentiated cancer cell. Bottom: Specific transition stimulus engenders the HPCS program, which is a necessary dedifferentiation intermediate in a phenotypic transition. In both models, the cell acquires the ability to differentiate via various trajectories once residing in the HPCS.
We propose that cancer cells must acquire the HPCS to transition between cell states (Fig. 2D). In early stages of LUAD evolution, tumors are homogenous, but emergence of the HPCS enables an explosion of cell state heterogeneity observed in fully formed adenocarcinomas. These findings are surprising, as they do not support an intuitive model of tumor evolution whereby acquisition of new cancer cell states occurs gradually from a “leading edge” of progressively more de-differentiated cells. In late-stage adenocarcinomas, the HPCS appears to be a precursor to epithelial-mesenchymal transition (EMT), a dramatic example of phenotypic transformation [48]. During tumorigenesis, EMT endows carcinoma cells with high invasive and metastatic potential [49] by allowing loss of intercellular adhesion and polarity, cytoskeletal reorganization, and acquisition of an invasive phenotype [50]. Cells with the most metastatic capacity in PDAC reside in a hybrid (or partial) EMT state characterized by co-expression of both mesenchymal and epithelial genes [51, 52]. Similarly, skin squamous cell carcinoma (SCC) cells occupy a spectrum of states along an epithelial-mesenchymal axis, where both ends of the spectrum are bridged by a hybrid EMT state. Whereas all mesenchymal SCC subpopulations had equivalent ability for tumor propagation, hybrid EMT states were more competent at forming metastases and had increased plasticity [53]. The HPCS appears to be similar, sharing features with both pure epithelial and EMT states, including the metastasis-associated TF RUNX2 [43, 54].
Molecular mechanisms of plasticity
Cell-intrinsic mechanisms
The molecular mechanisms that drive cell states, including highly plastic states, are perhaps best decoded through the lens of systems biology: The cell state is a sum of cell-extrinsic signals (inputs) into the cell and the cell-intrinsic epigenetic and biochemical state of the cell (system) (Fig. 3A). Plasticity is ultimately a function of a cell’s ability to access genetic information, which is determined by epigenetic mechanisms such as histone post-translational modifications, DNA methylation, and 3-dimensional chromatin organization. Distinct chromatin states define plastic cells among different tissue types and cancers, including breast, colon, and lung adenocarcinomas [43, 54–56]. In breast cancer, a subset of cells harbor “bivalent” (poised) chromatin, characterized by the simultaneous presence of repressive and activating histone post-translational modifications, at promoters of EMT driver genes [57]. This subset is highly plastic, being able to either maintain epithelial features or rapidly undergo EMT upon stimulation with transforming growth factor-β [57]. Lower levels of DNA methylation at enhancers and genomic insulators, and tight coupling of promoter methylation with gene expression correlated with high global plasticity in glioblastoma [58]. Conversely, gliomas driven by mutations in isocitrate dehydrogenase-1 (IDH1) [58], which promotes DNA hypermethylation [59], harbor significantly less global plasticity [59]. Plasticity in IDH1 mutant gliomas is confined to a small subset of CSCs with a specific DNA methylation pattern [59]. These results suggest that DNA methylation restricts cancer cell plasticity and safeguards differentiation states.
Figure 3. Molecular mechanisms of plasticity.
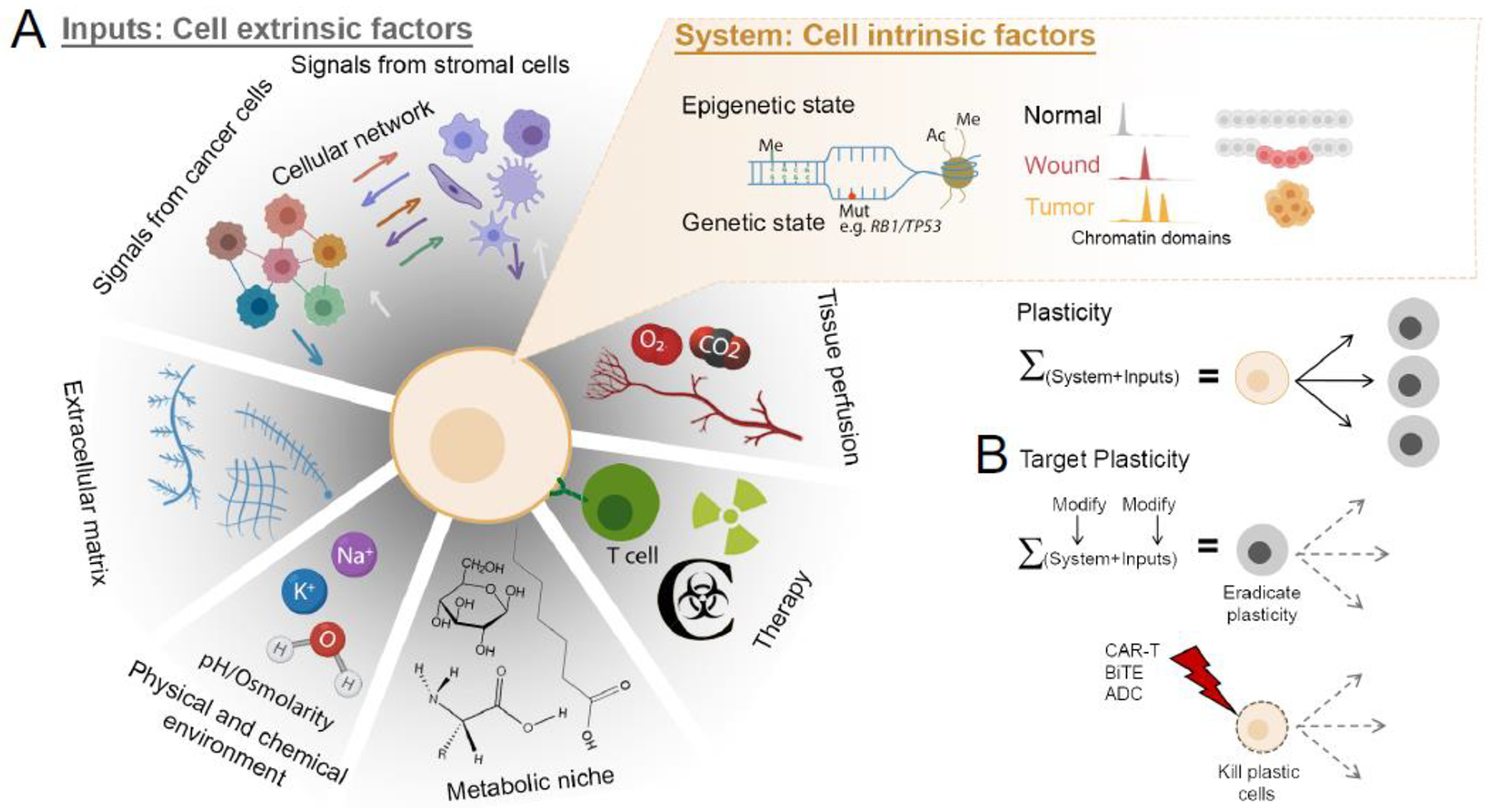
(A) Cellular plasticity is the sum of cell-intrinsic molecular features (System) and cell-extrinsic factors (Inputs). Cell-intrinsic features include epigenetic mechanisms such as DNA methylation (Me), mutational state (Mut), histone modifications such as acetylation (Ac) and methylation (Me), and chromatin accessibility differences. Chromatin accessibility changes associated with wound healing increase plasticity and cooperate with tumor initiation. (B) Targeting plasticity may involve modifying the System or the Inputs that establish plastic cell states, or by directing cytotoxicity to highly plastic cancer cells through cell surface markers.
Chromatin remodeling and gene expression are driven by coordinated activity of lineage-specific and globally active TFs. MYC and YAP/TAZ are prototypical examples of broadly active TFs. MYC becomes active in cells upon signals induced by growth factors in healthy tissues or upon oncogene activation [60]. High MYC activity promotes plasticity and lineage transformation in LUAD [61] and PDAC [62]. YAP/TAZ are effectors of the Hippo pathway, which controls organ size and epithelial integrity in normal tissues [63]. In cancer, YAP/TAZ promotes stemness in glioblastoma [64] and drives reversion of WNT-dependent intestinal cancer cells towards a fetal-like, WNT-independent state [65]. SOX family TFs are examples of lineage-restricted plasticity drivers: SOX2 activation promotes plasticity in squamous cell skin [66] and lung [32] cancers and SOX10 is activated in fetal-like breast cancer CSCs [55], whereas SOX2 and SOX9 maintain airway or alveolar-type progenitor-like states in LUAD, respectively [67]. Mutations in SWI/SNF complex members, such as ARID1A, ARID2, and SMARCA4, cause chromatin accessibility changes in a wide range of cancer types [68]. In LUAD, chromatin accessibility changes driven by Smarca4 mutations cause tumor cells to lose lung lineage markers and develop highly aggressive features [69].
p53 limits the ability of somatic cells to undergo epigenetic reprogramming into induced pluripotent stem cells [70, 71]. In adult tissues, p53 restricts cellular self-renewal in various stem and progenitor cells, especially those subject to oncogenic stress [72–74]. p53 safeguards lineage commitment and represses plasticity of cancer cells, which are important components of p53’s repertoire of tumor suppressive functions [36]. In mature hepatocytes, p53 restricts expression of the stem and progenitor-cell-associated protein nestin via Sp1/3 TFs [74]. Loss of p53 expands the lineage choices available to these cells, poising them for transformation into either hepatocellular carcinomas or cholangiocarcinomas in response to lineage-specific mutations that target either WNT or Notch signaling, respectively [74]. In LUAD, p53 loss is not required for acquisition of the HPCS, but appears to increase the range of available downstream phenotypic states [43]. In addition, a multitude of genetic mechanisms can promote plasticity in cancer, including point mutations, extrachromosomal DNA, and aneuploidy; these have been reviewed elsewhere [75, 76].
Cell-extrinsic mechanisms
In addition to oncogenes and established lineage-specific programs, the activity of TFs that drive plasticity is controlled by cell-extrinsic inputs (Fig. 3A). Cellular stress is a key mechanism for activating plasticity in cancer cells. Computational modeling suggests that diverse tumor types, including triple-negative breast cancer, oligodendroglioma, and PDAC, harbor a subset of cells expressing stress response genes, including genes involved in DNA damage, temperature adaptation, oxidative damage, hypoxia, and starvation [77, 78]. This cell state exhibited a high capacity for metastatic seeding and drug resistance in functional experiments [78]. Inflammation is another common mediator of plasticity. In skin wound healing, inflammation activates stem cell transcriptional programs that differ significantly from homeostatic stem cells. These transcriptional programs are mediated by ETS2, STAT3, and AP-1, which are induced by cell-extrinsic cytokine inputs in target cells [14]. In breast cancer, inflammatory mediators such as TNFα and IL-6 drive cancer cell quiescence, therapeutic resistance, and EMT [79].
Hypoxia, a near-ubiquitous feature of solid tumors, can endow cancer cells with increased plasticity and drug resistance [80]. Through hypoxia-driven metabolic reprogramming of glycolysis, glutamine metabolism, and lipid synthesis by hypoxia-inducible factors, cancer cells can suppress apoptosis and proliferate under nutrient-deprived conditions [81]. Besides direct transcriptional changes, hypoxia induces translational reprogramming via phosphorylation of the translation initiation factor eIF2α, which leads to accumulation of NODAL, NANOG, and SNAIL that drive stemness and plasticity [82]. This translational stress response is also induced by multiple anti-cancer drugs [82], suggesting that cancer therapy may directly promote plasticity of cancer cells. Such examples of “what does not kill you, makes you stronger” are consistent with the concept of hormesis, the beneficial effects associated with an adaptive response to moderate (usually intermittent) stress [83]. Examples include ischemic preconditioning, exercise, dietary energy restriction, and exposures to low doses of ionizing radiation [83]. The mechanistic underpinnings of the hormesis response may represent entry points for manipulating cell fate determination and plasticity but remain poorly understood at present.
Various paracrine signals and other microenvironmental inputs mediate plasticity. Signals such as WNT and Notch ligands, produced in a spatially-constrained manner, support the development of CSCs with plastic properties [41, 84, 85]. WNT ligands are central to the maintenance and renewal of normal intestinal ASCs as well as colorectal CSCs [34, 39, 42, 86]. WNT-dependent cells and their niche also develop in LUAD [41] and basal cell carcinoma [87]. A cellular subset in small cell lung cancer undergoes a chemoresistant non-neuroendocrine fate switch in response to Notch signaling [88]. Exposing PDAC organoids to interferon-γ shifts cells into a highly plastic cell state co-expressing basal and classical transcriptional signatures [47].
Plasticity in treatment resistance and as a therapeutic target
Treatment resistance
In addition to a critical role in tumor progression, cancer cell plasticity contributes to treatment resistance by promoting adaptation to therapeutic pressure via differentiation to treatment-resistant states. LUAD treated with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) can lose dependence on EGFR signaling by undergoing a histological transformation from adenocarcinoma to either small cell lung cancer or squamous cell carcinoma [89]. Similarly, prostate adenocarcinomas develop resistance to next-generation antiandrogens by shifting phenotype from adenocarcinoma to neuroendocrine prostate cancer [90]. In both lung and prostate adenocarcinoma, loss of TP53 and RB1 greatly enhances lineage plasticity and the likelihood of histological transformation [8]. The robust and durable clinical response produced by EGFR TKIs and antiandrogens indicates that most cancer cells in lung and prostate adenocarcinomas, respectively, are sensitive to the drug. This suggests that only a subset of the cancer cells harbors the capacity for neuroendocrine transformation. In line with this, both prostate cancer [91] and LUAD [92], contain cell states that develop lineage infidelity under therapeutic pressure. In response to immunotherapy, PDAC cells undergo a phenotypic shift from a classical transcriptional state to a highly plastic intermediate state [47]. Thus, plasticity is also a major barrier to successful immunotherapy [6]. We speculate that cell states in the minimal residual disease that nucleate adaptation to therapy may constitute a HPCS, such as the one discovered in LUAD [43]. Supporting this, the HPCS is highly enriched following chemotherapy [43], indicating that it is intrinsically chemoresistant – or that chemotherapy engages the HPCS program in other cancer cell subsets. Whether the prostate cancer, PDAC, and LUAD cell states undergoing phenotypic shifts share similarity to the HPCS needs to be substantiated by further experimental work.
Therapeutic targeting
Drug resistant cancer cell states with high plasticity contribute to tumor repopulation following therapy. Skin basal cell carcinomas treated with the Smoothened inhibitor vismodegib contain a highly plastic LGR5+ cell state that repopulates tumors after therapy discontinuation [87]. This persister state relies on WNT-signaling to prevent differentiation into a drug-sensitive state. Similarly, glioblastomas treated with the chemotherapy agent temozolomide recur from a pool of Nestin+ CSCs. Depletion of this population in combination with temozolomide leads to longer survival, suggesting that plasticity plays a key role in tumor repopulation and treatment resistance [93].
Despite the importance of plasticity for tumor progression and treatment resistance, medicines that directly target cancer cell plasticity have not been clinically approved. The rapidly accumulating preclinical literature on cancer plasticity is becoming a rich source of candidate strategies for translational development (Fig. 3B) [3, 5–9]. As discussed above, TFs and epigenetic factors are central in enabling plasticity. For example, suppression of polycomb repressive complex-2 with EZH2 inhibitors restored androgen receptor expression and sensitivity to antiandrogen therapy in prostate cancer cells that had undergone a neuroendocrine transformation [94]. Bromodomain and extra-terminal motif (BET) protein inhibitors were found to reverse drug resistance and block the pro-tumorigenic activity exerted by YAP/TAZ binding to the epigenetic coactivator bromodomain-containing protein 4 (BRD4) [95]. Similarly, genetic or pharmacological suppression of YAP/TAZ partially suppressed acquisition of the fetal-like colorectal cancer cell state, sustaining sensitivity to WNT inhibitors [65]. Given that WNT ligands have been shown to promote plasticity and stemness in multiple cancers, the WNT pathway is an attractive target even in cancers where mutations in the pathway are not observed [41, 87]. Other therapeutically actionable signaling pathways that drive plasticity are emerging. Targeting JAK/STAT and FGFR signaling is promising for suppressing neuroendocrine transformation in prostate cancer [96]. Furthermore, a highly plastic cell state in small cell lung cancer depends on the activity of phospholipase-Cγ2 [97]. Distinct metabolic programs, such as the dependence of glioblastoma CSCs on mitochondrial complex V activity [98], may represent therapeutic vulnerabilities in plastic cells. Finally, targeting stroma may be a novel strategy for eliminating cancer cell plasticity, as inhibition of stromal remodeling with Smoothened inhibitors sensitized triple-negative breast cancer to chemotherapy [99]. Besides targeting molecular mechanisms that promote plasticity, directing cytotoxicity to cell states harboring high plasticity can be achieved via a cell state-specific surface receptors (Fig. 3B).
Concluding remarks
The critical importance of cellular plasticity in tumor initiation, progression, and treatment resistance is becoming increasingly apparent [2, 3, 100]. However, many fundamental questions regarding plasticity in cancer remain unanswered (see Outstanding Questions). Emerging evidence suggests that high plasticity within tumors is concentrated in a defined subset of cancer cells. Such high-plasticity cell states promote tumor progression by driving emergence and maintenance of cellular heterogeneity, including cell states with high metastatic potential or treatment-resistant states. The molecular mechanisms controlling plasticity in cancer appear to involve co-option of molecular programs that promote plasticity in development or tissue regeneration. Many of these programs and cell states are not active in normal tissues under homeostatic conditions, suggesting that plastic cancer cell states could be safely targeted in patients. This emerging fundamental understanding of cancer cell plasticity may translate into transformative therapeutic concepts.
OUTSTANDING QUESTIONS.
Do core programs exist that define plasticity across cancer types?
Are high-plasticity cancer cell states stable or merely short-lived transitions between cell states?
What is the relationship between plasticity and proliferative potential? In other words, can high-plasticity cell states be proliferative, or are they inherently quiescent?
Do all cancer cell states have the capacity to acquire high plasticity in response to the right cell-extrinsic inputs?
What are the consequences of ablating or targeting high-plasticity cell states for tumor progression or response to therapy?
HIGHLIGHTS.
Cellular plasticity is critical for cell state transitions in normal physiology and in cancer.
Tissue regeneration and cancer development are accompanied by increased plasticity.
High-plasticity cancer cell states promote tumor progression, intra-tumoral heterogeneity, and resistance to cancer therapy.
Targeting plasticity in cancer is a conceptually novel and potentially highly effective therapeutic approach.
ACKNOWLEDGEMENTS
The authors wish to thank Drs. Yoon-Chi Han and Lee Jones for critical reading of the manuscript. This work was supported by National Institutes of Health (NIH) / National Cancer Institute (NCI) Grant R37-CA244911 and Cancer Center Support Grant P30-CA08748 to MSKCC. S.T. was supported by a fellowship from NIH / NCI (F30-CA254120). S.T was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number: T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. J.C. was supported by the Linn Fund from Cycle for Survival. T.T. was supported by Josie Robertson, Rita Allen, American Cancer Society, and V Foundation Scholarships. The authors apologize to any colleagues whose work could not be cited due to space restrictions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None.
References
- 1.Takahashi K and Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4), 663–76. [DOI] [PubMed] [Google Scholar]
- 2.Marjanovic ND et al. (2013) Cell plasticity and heterogeneity in cancer. Clin Chem 59 (1), 168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta PB et al. (2019) Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24 (1), 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solini GE et al. (2017) Embryonic transplantation experiments: Past, present, and future. Trends Dev Biol 10, 13–30. [PMC free article] [PubMed] [Google Scholar]
- 5.Arozarena I and Wellbrock C (2019) Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer 19 (7), 377–391. [DOI] [PubMed] [Google Scholar]
- 6.Horn LA et al. (2020) Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 6 (5), 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beltran H et al. (2019) The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res 25 (23), 6916–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintanal-Villalonga A et al. (2020) Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marine JC et al. (2020) Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 20 (12), 743–756. [DOI] [PubMed] [Google Scholar]
- 10.Saxén L and Toivonen S (1962) Primary embryonic induction, Logos Press. [DOI] [PubMed] [Google Scholar]
- 11.Barker N et al. (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449 (7165), 1003–7. [DOI] [PubMed] [Google Scholar]
- 12.Potente M et al. (2011) Basic and therapeutic aspects of angiogenesis. Cell 146 (6), 873–87. [DOI] [PubMed] [Google Scholar]
- 13.Ito M et al. (2005) Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med 11 (12), 1351–4. [DOI] [PubMed] [Google Scholar]
- 14.Ge Y et al. (2017) Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 169 (4), 636–650.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu CP et al. (2012) Identification of stem cell populations in sweat glands and ducts reveals roles in homeostasis and wound repair. Cell 150 (1), 136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huch M et al. (2013) Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J 32 (20), 2708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storz P and Crawford HC (2020) Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 158 (8), 2072–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yanger K et al. (2013) Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 27 (7), 719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tata PR et al. (2013) Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature 503 (7475), 218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arthur-Farraj PJ et al. (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75 (4), 633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Poggetto E et al. (2021) Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 373 (6561), eabj0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nabhan AN et al. (2018) Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359 (6380), 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zacharias WJ et al. (2018) Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555 (7695), 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zepp JA and Morrisey EE (2019) Cellular crosstalk in the development and regeneration of the respiratory system. Nat Rev Mol Cell Biol 20 (9), 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi J et al. (2020) Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 27 (3), 366–382 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strunz M et al. (2020) Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun 11 (1), 3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi Y et al. (2020) Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol 22 (8), 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomasetti C et al. (2017) Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355 (6331), 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leja MJ et al. (2011) Primary cardiac tumors. Tex Heart Inst J 38 (3), 261–2. [PMC free article] [PubMed] [Google Scholar]
- 30.Alcantara Llaguno S et al. (2019) Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci 22 (4), 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baggiolini A et al. (2021) Developmental chromatin programs determine oncogenic competence in melanoma. Science 373 (6559), eabc1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferone G et al. (2016) SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 30 (4), 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwitalla S et al. (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152 (1–2), 25–38. [DOI] [PubMed] [Google Scholar]
- 34.Barker N et al. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457 (7229), 608–11. [DOI] [PubMed] [Google Scholar]
- 35.Alonso-Curbelo D et al. (2021) A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 590 (7847), 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kastenhuber ER and Lowe SW (2017) Putting p53 in Context. Cell 170 (6), 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meacham CE and Morrison SJ (2013) Tumour heterogeneity and cancer cell plasticity. Nature 501 (7467), 328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreso A and Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14 (3), 275–91. [DOI] [PubMed] [Google Scholar]
- 39.Schepers AG et al. (2012) Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337 (6095), 730–5. [DOI] [PubMed] [Google Scholar]
- 40.de Sousa e Melo F et al. (2017) A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 543 (7647), 676–680. [DOI] [PubMed] [Google Scholar]
- 41.Tammela T et al. (2017) A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 545 (7654), 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimokawa M et al. (2017) Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 545 (7653), 187–192. [DOI] [PubMed] [Google Scholar]
- 43.Marjanovic ND et al. (2020) Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38 (2), 229–246 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiebinger G et al. (2019) Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176 (4), 928–943 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Batlle E and Clevers H (2017) Cancer stem cells revisited. Nat Med 23 (10), 1124–1134. [DOI] [PubMed] [Google Scholar]
- 46.Ge Y et al. (2017) Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 169 (4), 636–650 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raghavan S et al. (2021) Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184 (25), 6119–6137 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J et al. (2020) Guidelines and definitions for research on epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology 21 (6), 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thiery JP et al. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139 (5), 871–90. [DOI] [PubMed] [Google Scholar]
- 50.Lamouille S et al. (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15 (3), 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simeonov KP et al. (2021) Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y et al. (2018) Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol Med 10 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pastushenko I et al. (2018) Identification of the tumour transition states occurring during EMT. Nature 556 (7702), 463–468. [DOI] [PubMed] [Google Scholar]
- 54.LaFave LM et al. (2020) Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 38 (2), 212–228 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dravis C et al. (2018) Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell 34 (3), 466–482 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim TH et al. (2014) Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506 (7489), 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaffer CL et al. (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154 (1), 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaligne R et al. (2021) Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet 53 (10), 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turcan S et al. (2018) Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet 50 (1), 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhanasekaran R et al. (2021) The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quintanal-Villalonga A et al. (2021) Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J Hematol Oncol 14 (1), 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farrell AS et al. (2017) MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun 8 (1), 1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zanconato F et al. (2016) YAP/TAZ at the Roots of Cancer. Cancer Cell 29 (6), 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castellan M et al. (2021) Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat Cancer 2 (2), 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han T et al. (2020) Lineage Reversion Drives WNT Independence in Intestinal Cancer. Cancer Discov 10 (10), 1590–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pastushenko I et al. (2021) Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 589 (7842), 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laughney AM et al. (2020) Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 26 (2), 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Centore RC et al. (2020) Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet 36 (12), 936–950. [DOI] [PubMed] [Google Scholar]
- 69.Concepcion CP et al. (2022) Smarca4 Inactivation Promotes Lineage-Specific Transformation and Early Metastatic Features in the Lung. Cancer Discov 12 (2), 562–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olivos DJ and Mayo LD (2016) Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci 17 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Menendez S et al. (2010) p53: guardian of reprogramming. Cell Cycle 9 (19), 3887–91. [DOI] [PubMed] [Google Scholar]
- 72.Friedmann-Morvinski D et al. (2012) Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338 (6110), 1080–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tosoni D et al. (2015) The Numb/p53 circuitry couples replicative self-renewal and tumor suppression in mammary epithelial cells. J Cell Biol 211 (4), 845–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tschaharganeh DF et al. (2014) p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158 (3), 579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verhaak RGW et al. (2019) Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nature Reviews Cancer 19 (5), 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pihan G and Doxsey SJ (2003) Mutations and aneuploidy. Cancer Cell 4 (2), 89–94. [DOI] [PubMed] [Google Scholar]
- 77.Flick K and Kaiser P (2012) Protein degradation and the stress response. Semin Cell Dev Biol 23 (5), 515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baron M et al. (2020) The Stress-Like Cancer Cell State Is a Consistent Component of Tumorigenesis. Cell Syst 11 (5), 536–546 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baram T et al. (2020) Inflammation-Driven Breast Tumor Cell Plasticity: Stemness/EMT, Therapy Resistance and Dormancy. Front Oncol 10, 614468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Connell MP et al. (2013) Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 3 (12), 1378–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie H and Simon MC (2017) Oxygen availability and metabolic reprogramming in cancer. J Biol Chem 292 (41), 16825–16832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jewer M et al. (2020) Translational control of breast cancer plasticity. Nat Commun 11 (1), 2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mattson MP (2008) Hormesis defined. Ageing Res Rev 7 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lim JS et al. (2017) Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 545 (7654), 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liau BB et al. (2017) Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 20 (2), 233–246 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Merlos-Suarez A et al. (2011) The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8 (5), 511–24. [DOI] [PubMed] [Google Scholar]
- 87.Sanchez-Danes A et al. (2018) A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 562 (7727), 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lim JS et al. (2017) Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 545 (7654), 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sutherland KD and Berns A (2010) Cell of origin of lung cancer. Mol Oncol 4 (5), 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mu P et al. (2017) SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355 (6320), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karthaus WR et al. , Reversal of lineage plasticity in RB1/TP53-deleted prostate cancer through FGFR and Janus kinase inhibition, Cold Spring Harbor Laboratory, 2021. [Google Scholar]
- 92.Quintanal-Villalonga A et al. (2021) Multiomic Analysis of Lung Tumors Defines Pathways Activated in Neuroendocrine Transformation. Cancer Discov 11 (12), 3028–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen J et al. (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488 (7412), 522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ku SY et al. (2017) Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355 (6320), 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zanconato F et al. (2018) Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24 (10), 1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karthaus WR et al. (2021) Reversal of lineage plasticity in RB1/TP53-deleted prostate cancer through FGFR and Janus kinase inhibition. bioRxiv, 2021.11.01.466615. [Google Scholar]
- 97.Chan JM et al. (2021) Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 39 (11), 1479–1496 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi Y et al. (2019) Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567 (7748), 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cazet AS et al. (2018) Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun 9 (1), 2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Massague J and Ganesh K (2021) Metastasis-Initiating Cells and Ecosystems. Cancer Discov 11 (4), 971–994. [DOI] [PMC free article] [PubMed] [Google Scholar]