

Abstract
Recent studies in both humans and animal models call into question the completeness of recovery after chronic sleep disruption. Studies in humans have identified cognitive domains particularly vulnerable to delayed or incomplete recovery after chronic sleep disruption, including sustained vigilance and episodic memory. These findings, in turn, provide a focus for animal model studies to critically test the lasting impact of sleep loss on the brain. Here, we briefly summarize the human response to sleep disruption and then discuss recent findings in animal models examining recovery responses in circuits pertinent to vigilance and memory. We then propose pathways of injury common to various forms of sleep disruption and consider the implications of this injury in aging and in neurodegenerative disorders.
Keywords: Sleep deprivation, sleep restriction, sleep fragmentation, locus coeruleus, neurodegeneration, amyloid
Chronic sleep disruption in humans: evolving thoughts on recovery
Chronic sleep curtailment is common in modern society and is related in part to increased work demands, lifestyle choices, the development and use of medications and substances that suppress or disrupt sleep, in addition to the increased use of artificial light-emitting devices that delay sleep. A generally held presumption has been that while chronic sleep disruption results in neurobehavioral impairments, performance deficits are reversed with limited-period recovery sleep (e.g. over the weekend). A collection of studies examining the human response to sleep loss, however, challenges this belief and suggests that impairments persist, and that additionally, individuals may be poor judges of their incurred sleep loss impairments over time. One to two weeks of sleep restriction to <7 hours sleep/night in adults has been shown repeatedly to result in cumulative increases in sleep propensity, along with decrements in mood and vigilance[1–3]. Objective and subjective discrepancies, however, were evident, as study participants were unaware of the progressive deterioration in performance across sleep restriction[1,2]. While subjective impairments (sleepiness and mood) typically normalized with 1–2 nights of recovery sleep, objective measures of vigilance showed persistent deficits, relative to baseline performance, after 2–3 nights of recovery sleep[3–5]. A similarly delayed recovery from sleep loss was evident in adolescents with incomplete recovery in sustained vigilance after 2 nights of recovery sleep[6]. In young adults, sustained vigilance was impaired across 5 days of 4-hour/night sleep restriction, and the increased frequency of lapses in performance was not reversed despite 3 recovery nights[7]. More recently, a field study was conducted in which sleep time was reduced by one third for 10 consecutive days in young adults[8]. Neither accuracy in a cognitive interference assay (Stroop) nor eyes-open alpha power spectra had normalized after a 7-day recovery period[8]. When adults were deprived of all sleep for 40 consecutive hours, subjective sleepiness resolved after one night of recovery sleep, while performance of tasks requiring higher cognitive function (reading comprehension, serial addition, go-no-go tasks) did not normalize after two recovery nights[9]. As shown for chronic sleep restriction, adults across 3 days of recovery after total sleep deprivation overestimate their own vigilance performance[10]. In addition to impaired vigilance, one night of total sleep deprivation impairs both episodic memory and hippocampal connectivity to the prefrontal cortex and default mode network[11]. While two nights of recovery sleep restored hippocampal connectivity, episodic memory impairments persisted. Collectively, these lines of work strongly support the notion that humans are vulnerable to protracted recovery responses after chronic sleep disruption and manifest imperceptions of the incurred impairments. The latter finding may have contributed to the false sense of security upon weekend recovery sleep.
Neurobehavioral deficits in response to sleep disruption in both humans and animal models have been largely attributed to homeostatic wake-induced increases in brain extracellular adenosine, levels of which readily reverse upon recovery sleep after short-term sleep loss[12–16]. With longer durations of sleep disruption, however, adenosine levels do not necessarily parallel the protracted neurobehavioral impairments. In a study on individuals undergoing 40 hours of total sleep deprivation, extracellular adenosine did not increase in any of the brain regions in which measurements were made, including the hippocampus[17]. In this study, however, participants had histories of epilepsy, and values were not compared to samples in individuals allowed to sleep to determine expected drift in adenosine measures over time. In rats exposed to three days of sleep restriction, adenosine levels measured in hippocampal slices were reduced, rather than elevated, and after a two-week recovery opportunity, levels in slice remained below baseline levels two weeks into recovery[18]. In contrast, after 4 hours of sleep deprivation, adenosine levels increased as expected in hippocampal slices[18]. Thus, adenosine may very well contribute to sleepiness and performance decrements caused by acute sleep loss. However, the presence of incomplete recoveries from chronic partial sleep loss in the absence of evidence of protracted elevations of adenosine in chronic sleep disruption, raises the possibility of sleep loss-induced neural injury.
Neural injury in response to sleep loss has been difficult to assess without pre-defined indices indicative of lasting neuronal loss, glial modification, and/or dysfunction. This challenge is illustrated by a series of experiments performed over three decades ago, which aimed to assess potential brain cell damage in rats following sleep deprivation using methodologies available at the time. Adult rats were exposed to an extreme form of sleep deprivation: total sleep deprivation for 2–3 weeks. The sleep deprivation resulted in profound systemic changes (severe weight loss, malnutrition, bacterial sepsis, hormonal dysregulation, and ultimately death), yet examination of the brains immediately after sleep loss for general histology, apoptosis and necrosis, yielded no significant brain abnormalities[19]. Does this mean that sleep loss does not injure the brain? We would argue that when injury is defined as neuronal loss or lasting behavioral impairment or circuit dysfunction, additional analyses are needed to exclude sleep loss neural injury.
With improved definitions, assays and strategies to assess neural injury following sleep loss, a paradigm shift is emerging, with a transition from considering the effects of sleep loss as a readily reversible response, to greater appreciation that sleep loss can result in lasting neuron loss and dysfunction. In this review, we begin by highlighting recent findings in animal models, emphasizing the more protracted neural effects of sleep disruption. We will discuss how impairment and reversibility are influenced, not only by the chronicity of the sleep disruption but also by the age at the time of exposure, the interval after sleep loss used for assessment, neuronal subtypes or brain regions, and the propensity for protein aggregation. We then review what has been learned regarding mechanisms of neural injury following sleep loss and suggest future directions to ultimately identify therapies to lessen brain injury incurred during the commonly encountered scenarios of sleep disruption.
Neural effects of chronic sleep disruption in animal models
Consideration of sleep disruption paradigms
Various modalities of sleep disruption have been developed to determine the effects of total sleep deprivation, sleep restriction, rapid-eye-movement (REM) sleep deprivation and sleep fragmentation. There is some inherent overlap across modes of sleep disruption. For example, total sleep deprivation, by definition, includes REM sleep deprivation, and methods of REM sleep deprivation can impart partial disruption of non-rapid-eye-movement (NREM) sleep[20,21]. A simple technique of platform-over-water may be used to prevent all sleep or REM sleep, depending on the size of the platforms relative to the size of animals studied. The platform size should be adjusted so that when animals initiate the sleep state to be disrupted, state-dependent reductions in postural muscle tone result in the animal falling from the platform into water and abruptly waking up. Larger platforms have been used to control for the platform environment; however, animals on these control platforms can evidence partial sleep restriction[22]. Platform approaches in some but not all studies result in elevated plasma corticosterone levels [20,23,24]. Additionally, each of these methods disrupts the continuum of NREM and REM sleep patterns across a 24-hour cycle.
Most studies to date, including those discussed here, have implemented physical means to disrupt sleep, although it is now possible to elicit sleep disruption with chemogenetic activation of wake circuits[25] or, potentially, inactivation of sleep circuits. Commonly used paradigms for the various types of targeted sleep disruption are illustrated in Fig. 1, along with potential advantages, disadvantages and confounds. Additionally, there are differences in the level of arousal and the amount of learning experienced across methods of total sleep deprivation, where paradigms of exploratory wakefulness induce more robust immediate early gene response in wake-activated neurons, relative to use of gentle handling across the same duration[26]. Exploratory wakefulness results in increased locomotor activity, and by providing a continuously enriched and changing environment, exploratory wakefulness may involve more learning than other approaches with constant (or expected) environments. Techniques that require continuous experimenter vigilance to disrupt sleep, like gentle handling, may allow more breakthrough sleep than automated systems (e.g., rotating platform) that intervene as soon as sleep electroencephalographic patterns are detected by a computer algorithm[27,28]. Sleep fragmentation can be elicited with either a bar sweeping across the floor of the cage at set intervals or by way of a rotor table that briskly moves cages for a fraction of every minute or two. Methods to fragment sleep can also shorten 24-hour sleep amounts early on in the exposure[29]. Sleep fragmentation by either approach is generally effective for long continuous periods (weeks) of sleep disruption and seems not to influence either corticosterone levels or body weight[30,31]. The rotor table technique for sleep fragmentation allows group housing, ad lib eating and drinking, and access to undisturbed nests. By contrast, in the sweeper bar technique nests cannot be maintained, and the animals are not group-housed. As each sleep disruption approach has its own strengths and limitations, we will focus largely on sleep disruption findings for which use of multiple techniques have provided similar results.
Fig 1.
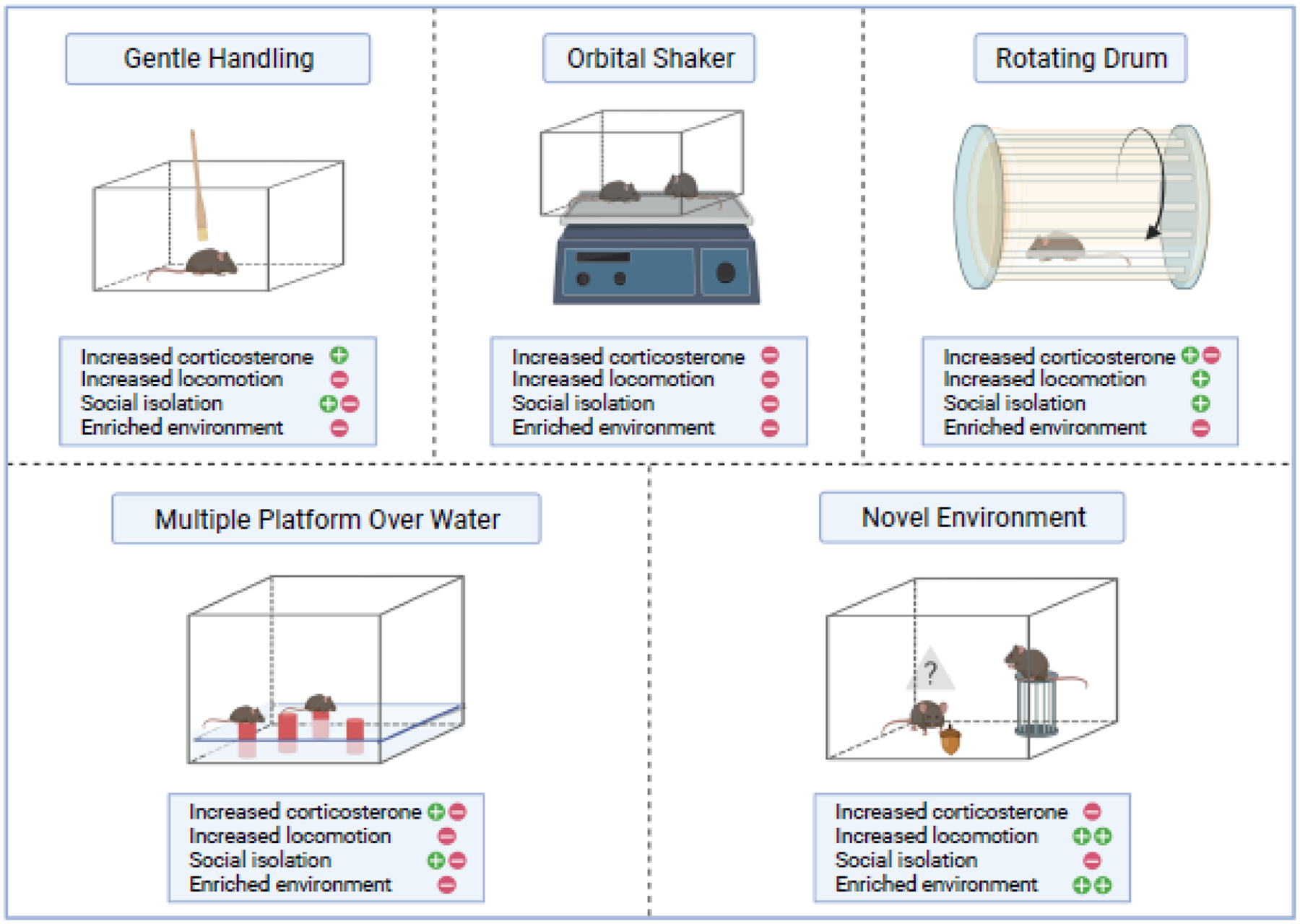
Chronic sleep disruption (CSD) paradigms. Approaches to chronic disruption in animal models vary not only by the way in which sleep is disrupted, but also by environment (including stress), intermittency or constancy of wakefulness, elicited motor activity and learning opportunities. Shown are specific attributes and potential confounders that may influence results. For each CSD paradigm, phenomena that were observed in multiple studies are indicated by a green plus sign, and phenomena that were absent in some or most of the relevant studies are indicated by a red negative sign.
Locus coeruleus: an early hit
Delayed recovery of sustained vigilance in the human studies supports the concept that chronic sleep disruption could affect neurons within the vigilance circuit, including locus coeruleus and anterior cingulate cortical neurons[32]. Locus coeruleus neurons (LCn) are wake-activated noradrenergic pontine neurons with firing rates highest during unexpected uncertainties[32,33]. While firing rates do not change across prolonged wakefulness, the duration of wakefulness influences metabolic responses in LCn. Upon brief wakefulness (3 hours) LCn in adult mice upregulate mitochondrial deacetylase sirtuin type 3 (SirT3), which then initiates a mitochondrial antioxidant response to maintain redox homeostasis in LCn [34], as illustrated in Fig 2. However, when sleep loss is prolonged to 8 hours of wakefulness/day for three consecutive days in the same enriched environment paradigm, SirT3 and its activating enzymes are not upregulated and LCn develop oxidative stress, increased mitochondrial protein acetylation, including acetylation and inactivation of electron transport chain proteins, and LCn counts are reduced[34]. Whether these changes observed immediately after the longer duration of sleep loss represent adaptation or neuronal dysfunction and/or injury, requires examination at a time point further from the termination of chronic sleep disruption. For example, the observed loss of LCn outlined above could represent an adaptive response in LCn to temporarily down-regulate mitochondrial activity and tyrosine hydroxylase, the marker used for LCn cell counts in the forementioned study. Of note, one of the noradrenaline metabolites, 3,4-dihydroxyphenylglycoaldehyde, modifies tau, increasing its propensity to aggregate and propagate[35]. Thus, reducing tyrosine hydroxylase when faced with metabolic stress may be an adaptive response in chronic sleep restriction. According to this scenario, once metabolic homeostasis is re-instated, neuronal counts and sirtuin activity are expected to resume. However, when this enriched wakefulness paradigm was continued across 12 weeks, and mice were then allowed one year to normalize metabolics and counts, LCn counts remained reduced [36], indicating irreversible injury to LCn. Similarly, whole-cell patch clamp LCn recordings showed reduced frequency responses to step currents and increased afterhyperpolarization amplitudes two weeks after sleep fragmentation exposure, supporting delayed functional recovery[37]. In addition, sleep fragmentation in mice for 4 weeks results in a loss of LCn and a loss of SirT3 in remaining LCn, which is evident 4 weeks after sleep disruption, further supporting the notion of lasting LCn injury and loss in response to chronic sleep fragmentation[38]. Not all sleep disruption studies, however, have shown loss of LCn. One study in rats, for instance, used a rotating drum paradigm that alternated 3 hours of enforced ambulation with one hour rest, for 4 days/week during 4 weeks. In this paradigm, LCn loss was not observed relative to cell counts in rats in non-rotating drums [39]. LCn cell count variability was high in controls, but it is also conceivable that the brevity of each sleep disruption period (3-hours) prevented LCn injury, and even engaged a SirT3 protective response across periods of brief wakefulness. In addition to loss of LCn, there is evidence of reduced LCn noradrenergic output in the adult rat in response to chronic sleep loss. Rats exposed to 3 weeks of sleep fragmentation using a sweeping bar have reduced levels of extracellular noradrenaline in the hippocampus in response to a spatial learning task, relative to control animals[40]. Because levels were measured immediately after sleep disruption, it is unclear whether this reduction in noradrenaline represents persistent dysfunction.
Fig 2.

Duration-dependent effects of sleep loss on locus coeruleus neurons (LCn). Short-term wakefulness (awake for three consecutive hours across the habitual sleep (lights-on) period) upregulates mitochondrial sirtuin type 3 (SirT3) activity, which then results in nuclear translocation of FoxO3a and transcriptional activation of anti-oxidants and PGC-1α to enhance mitochondrial biogenesis. In contrast, extended wakefulness (for 8 hours a day in the lights-on period for three consecutive days) reduces SirT3 protein and its NAD+-synthesizing enzymes, thereby reducing SirT3 activity and increasing mitochondrial acetylation of (and thereby inactivating) anti-oxidant enzymes, electron transport chain proteins, and FoxO3a. Mechanisms by which LCn switch from an adaptive to maladaptive response are not known.
Why would LCn be vulnerable to increased mitochondrial stress in response to chronic sleep disruption? The metabolic status of LCn may dictate vulnerability to sleep loss. Mice deficient in SirT3 have lower LCn counts at baseline, yet counts do not decline further with prolonged sleep loss[34], suggesting that LCn with higher mitochondrial metabolic activity are more susceptible to loss upon extended wake. A second possibility is that exploratory wakefulness further raises LCn firing rates because of repeated exposures to unexpected uncertainties[41]. LCn are most active in response to novelty or immediately upon arousal from sleep when autoreceptor activity is lowest, particularly in REM sleep[42]. Opening of L-type calcium channels on LCn, upon arousal, or across extended periods of exploratory wakefulness is expected to increase calcium in mitochondria which would then activate calcium-dependent mitochondrial nitric oxide synthesis, impairing electron transport and increasing superoxide production in mitochondria[43]. While identification of the mechanisms underlying LCn vulnerability requires further work (see Outstanding Questions), we propose that the vulnerability of LCn will be determined in part by the underlying metabolic activity of LCn, the mode and duration of sleep disruption implemented, and the ability of recovery sleep to resolve the metabolic perturbance.
Outstanding questions.
What approaches could be used to refine characterization and extend analysis of neural injury and dysfunction incurred following sleep loss in both humans and animal models? How should recovery be characterized in both humans and animal models of chronic sleep disruption?
What events underly LCn’s conversion from adaptive to maladaptive responses in short and long duration sleep loss? Does a similar conversion occur in other neuronal populations? To what extent does sleep loss-induced LCn injury contribute to forebrain responses to sleep loss?
What are the determinants of differential susceptibility to sleep loss across neuronal populations? Is the heightened susceptibility in the developing brain a consequence of increase sleep need, increased plasticity or other factors?
Are responses of glial cells modified across the course of sleep loss and/or after, and if so, to what extent do modified microglial and astrocyte profiles contribute to neuronal loss? Are glial responses to sleep loss determined by local neuronal injury and/or by intrinsic properties of regional glia?
Is sleep loss a significant contributing factor to common neurodegenerative processes, including Alzheimer’s disease? How does sleep loss influence the processing of Aβ and tau in synapses and are these effects significant contributors to neurodegeneration?
Hippocampal responses to chronic sleep disruption
In humans, chronic sleep disruption impairs hippocampal-dependent episodic memory, and deficits persist after two nights of recovery sleep[11]. The question arises, is there evidence in animal models that chronic sleep disruption imparts residual injury to the hippocampus? Two studies highlight the importance of timing in the assessment after sleep loss in addressing this question. REM sleep deprivation in rats for 72 hours impairs LTP as assessed in hippocampal slices, yet when the rats are allowed a 24-hour recovery after sleep loss, LTP normalizes[44], suggesting rapid reversal of hippocampal dysfunction. A later study, however, examined in vivo hippocampal LTP in adult rats after 120 hours of REM sleep deprivation and also found no LTP impairments after the first recovery day, yet after the second recovery day LTP impairments were evident[45], suggesting the sleep loss hippocampal dysfunction may progress after sleep loss.
Several studies have explored longer time frames for recovery of hippocampal neurons and memory function to more fully address the question of lasting injury and/or dysfunction. Adult rats exposed to 3 weeks of chronic sleep restriction using a platform technique (18 hours/day) develop spatial memory impairments that persist at least 3 weeks after the sleep restriction[46]. Consistent with behavioral impairments, apical and basal dendritic arborization of CA1 hippocampal neurons was significantly reduced and remained reduced across the 3 weeks of recovery sleep[46]. In this study, corticosterone levels were unchanged across sleep disruption, supporting the notion that minimal stress was experienced across the exposure. In a separate study, adult rats exposed to 72 hours of REM sleep deprivation showed spatial memory impairments that persisted 3 weeks after sleep disruption[47]. TUNEL analyses in the hippocampal dentate gyrus showed no effect immediately yet revealed a marked increase at 2 and 3 weeks after sleep disruption[47], lending further support that some sleep loss injury is evident only after some duration beyond sleep loss. The optimal time after sleep disruption to examine neural injury has yet to be determined and may vary with the mode of sleep disruption and the behavioral or morphologic assessment. However, significant injury can be seen well after sleep loss exposures. When young adult mice were exposed to 12 weeks of chronic short sleep with enriched wakefulness, CA1 volume loss was evident one year after sleep loss[36]. Additional changes observed one year after sleep loss included: impaired spatial memory, reduced CA1 neuron counts, an increase in microglial and astrocyte markers across CA1, and accumulation of Aβ42 in CA1 [36]. LCn were also lost in this model and within animal LCn counts predicted CA1 cell counts[36], suggesting that the two processes may either be dependent or that factors unique to specific animals predict vulnerability in both neuronal groups. Chronic sleep disruption (using enforced treadmill ambulation or sleep fragmentation has also been shown to reduce neurogenesis and cell proliferation in the dentate gyrus, [48–52]. It remains to be determined whether hippocampal neurogenesis recovers after chronic sleep loss.
What mechanisms underlie hippocampal responses to chronic sleep disturbances? In the hippocampus, in response to active wakefulness, cytosolic calcium activity in excitatory CAMKII neurons increases significantly[53], which suggests that like LCn, CAMKII neurons in the hippocampus may experience mitochondrial stress upon extended periods of active wakefulness. Electron cryo-tomography has been used to identify mitochondrial morphologic alterations in the hippocampus in response to chronic sleep loss in adult rats[54]. REM sleep deprivation by platform for 72 hrs resulted in loss of mitochondrial cristae with less internal membrane connectivity in mitochondria isolated from homogenized hippocampal or cortical tissue[54]. The morphological changes were observed in parallel with reduced basal and maximal oxygen consumption rates in hippocampal, but not cortical, mitochondria after REM sleep deprivation, supporting regional or neuronal differential susceptibility to mitochondrial dysfunction in response to chronic sleep disruption. In support of mitochondrial dysfunction, a number of groups have shown increased oxidative stress (reduced glutathione, increased oxidized:reduced glutathione, increase malondialdehyde) in hippocampal tissue of rats and mice exposed to total sleep deprivation, REM sleep deprivation and sleep fragmentation[21,46,55–58]. Whole brain also shows similar measures of oxidative stress in response to chronic sleep loss[59]. Various antioxidant therapies administered in animal studies across the diverse chronic sleep disruption paradigms mitigate neurobehavioral impairments, mitochondrial injury and apoptosis[55,57,58,60], supporting an active role for oxidative stress in sleep disruption hippocampal responses. It remains unclear, however, how specifically mitochondria and metabolics are challenged by chronic sleep disruption.
One clue may reside in the synapses, which are particularly vulnerable to sleep loss. Synaptic transmission consumes a substantial portion of brain energy[61,62]. Waking and learning increase excitatory synapse number and activity, and in contrast, sleep enables a downscaling of these excitatory synapses, in part by reducing AMPA receptor availability on the post-synapse and shrinking the axon-spine interface (ASI)[63–66].
How specifically the ASI (and synaptic strength) are influenced by behavioral state is not known. As mentioned above, active wakefulness increases calcium transients in excitatory (CAMKIIα positive) hippocampal neurons [53]. It has been proposed that mitochondria serve as a homeostatic read out of neuronal activity[67], as intracellular calcium upon neuronal activation will be transferred primarily to mitochondria via the mitochondrial channel uniporter[68], which is under homeostatic regulation[69]. The mitochondrial dihydroorotate dehydrogenase enzyme, which is activated by wakefulness[70], can modify the metabolic set point of this homeostasis in hippocampal and cortical neurons. Thus, prolonged exploratory wakefulness could modify the homeostatic set point for neuronal activity and synaptic strength, an effect that could contribute to continued oxidative and inflammatory injury.
Effects of sleep loss on the developing brain
Chronological age at the time of sleep loss exposure may also influence neural responses. Studies in animal models indicate that younger animals may be more vulnerable to the neural effects of sleep loss, and under certain circumstances can develop lifelong impairments. When younger, post-natal day 16 (PND16) and older (PND44) rats are exposed to REM sleep restriction for 3 days, the younger rats show more pronounced hippocampal LTP impairment and greater loss of synaptic proteins in hippocampal tissue[71]. These effects persist at least 3–7 days after sleep disruption[71]. Similar age responses are observed for total sleep deprivation (for 24 hours), where young adult rats evidence reduced CA1 spine densities, while spine densities are unchanged in older rats (22 months old)[72]. An age-dependence in response is also observed for short-term sleep loss, where spatial memory consolidation impairments in response to short-term sleep loss in young mice (3 months old), relative to older mice (14 months old)[73] are more pronounced. In adolescent rats, chronic sleep restriction (gentle handling for just 4 hours/day for 10 non-consecutive days) is sufficient to impair hippocampal-dependent memory impairment, although the effect was not compared to adult rats. [74]. The effect persisted 4 weeks after sleep restriction, without impairing performance in a hippocampal-independent memory task at any time point, supporting the concept that even mild sleep restriction in young animals can have enduring effects in the hippocampus.
Protracted effects of sleep loss beyond the hippocampus have also been observed in young animals. Prairie voles exposed to chronic sleep fragmentation from 2 to 3 weeks of age, evidence as adults impaired novel object recognition and poor social bonding as adults, particularly in males[75]. These lasting behavioral changes occur with increased numbers of parvalbumin-immunolabeled cells in the sensory cortex in adulthood[75], yet whether this phenomenon underlies the behavioral changes has not been determined. As in adult mice, REM sleep deprivation in 1-month old kittens for 1 week resulted in a loss of LCn, as measured with stereology[76]. In this experiment, sleep recordings confirmed successful REM suppression across the sleep disruption period. Chronic sleep restriction in the adolescent rat has also been shown to reduce the thickness of myelin in two white matter tracts, with incomplete reversal after 36 hours of recovery opportunity [77].
While most chronic sleep disturbance studies have been performed in mammals, studies in drosophila have revealed lifelong effects of sleep disruption as well. Newly eclosed flies deprived of sleep for just 24 hours show impairments in a contextual memory test and in courtship behaviors when older[78]. Another study in flies also provided insights into the mechanisms underlying the effects of sleep loss. The study compared the effects of 36 hours of sleep deprivation in young (0–2 days) and adult (7–9 days) flies, showing that early life sleep loss impairs courtship behaviors 3 days after sleep loss in the young flies, but not in older flies[79]. In young flies, dopaminergic signaling is suppressed by sleep-active neurons across sleep. Wake-induced increased activity of dopaminergic neurons across sleep deprivation in younger flies, prevents proper development of olfactory glomeruli, which are critical for courtship behavior[79]. Thus, inappropriately timed wake-activated dopamine neurotransmission in flies impairs brain plasticity/development and results in lifelong mating dysfunction.
Glial response to chronic sleep disruption
An increase in inflammatory markers is observed in response to chronic sleep disruption paradigms, suggesting a glial role in exacerbating this injury, and in the protracted effects. In PND19 rats exposed to 6–8 hours/day of chronic sleep fragmentation for 14 days, markers for both oxidative and inflammatory changes were increased in the prefrontal cortex immediately following the sleep disruption on PND33 [80]. The increase in oxidative markers, however, was no longer present by PND90, while the inflammation persisted [80]. This suggests that following an initial oxidative environment induced by neuronal activity during chronic sleep restriction, chronic inflammation persists, which may then contribute to lasting effects after sleep disruption.
A study of chronically sleep deprived wild-type mice (gentle handling for 6 hours/day for 4 weeks) showed that extracellular levels of ATP, measured by microdialysis, were increased throughout the sleep deprivation protocol[81]. This extracellular ATP can bind to purinergic P2X7 receptors found on both microglia and astrocytes [82]. In samples from the cortex and hippocampus, activation of the P2X7 receptor during chronic sleep deprivation was shown to contribute to an increase in astrocytic nucleotide-binding protein and leucine rich repeat protein-3 (NLRP3) inflammasomes which promoted the production of pro-inflammatory IL-1β and IL-18 [83]. The same study found increases in cortical neuron apoptosis in response to chronic sleep disruption [83]. The production of these NLRP3 inflammasomes has also been shown to contribute to reductions in astrocytic BDNF levels, which may then contribute to the neuron loss seen in sleep deprivation protocols [84].
In the aforementioned study looking at PND33 and 90 rats post chronic sleep restriction, AMPAR subunit GluA1, NMDAR subunit GluN2b, PSD95 and phosphorylated CAMKII protein levels were decreased in rats at both PND33 and PND90 suggesting sustained decreases in synaptic density in the PFC [80]. The inflammatory profile seen in chronic sleep disruption models has detrimental effects on neighboring neurons and may explain the decrease in synapses seen in chronic sleep disruption models. When sufficiently increased, the pro-inflammatory compounds IL-1β and TNF-α can induce synapse loss and neuronal death, respectively [85,86]. Additionally, IL-1β and TNF-α can trigger the activation and translocation of NF- kB to the nucleus in astrocytes[87]. This activation can initiate excessive production of nitric oxide and lead to neuronal death [88,89]. Similarly, inflammatory microglia can amplify glutamate release by astrocytes via TNF-α leading to neuronal excitotoxicity and apoptosis[90].
Another possible feed-forward mechanism involves the fact that noradrenaline, which may be reduced in sleep disruption injury to LCn, reduces microglial release of nitric oxide, IL-6 and TNF-α [91]. Additionally, microglia do not appear to phagocytose post-synaptic proteins, but can influence pre-synapses through a process termed trogocytosis (selective partial phagocytosis)[92]. Microglial surveillance, phagocytosis, and presumably trogocytosis, are suppressed by noradrenaline and waking[93–95]. We propose that the chronic sleep loss injury is a multiple hit injury where loss of LCn causes a subsequent decrease in noradrenaline to the hippocampus and cortices which prolongs or even exacerbates glial inflammation, furthering synaptic dysfunction, and promotes inappropriate synaptic pruning during wakefulness. Also, direct injury in the hippocampus and cortices results in metabolic dyshomeostasis, as illustrated in Fig. 3.
Fig 3.
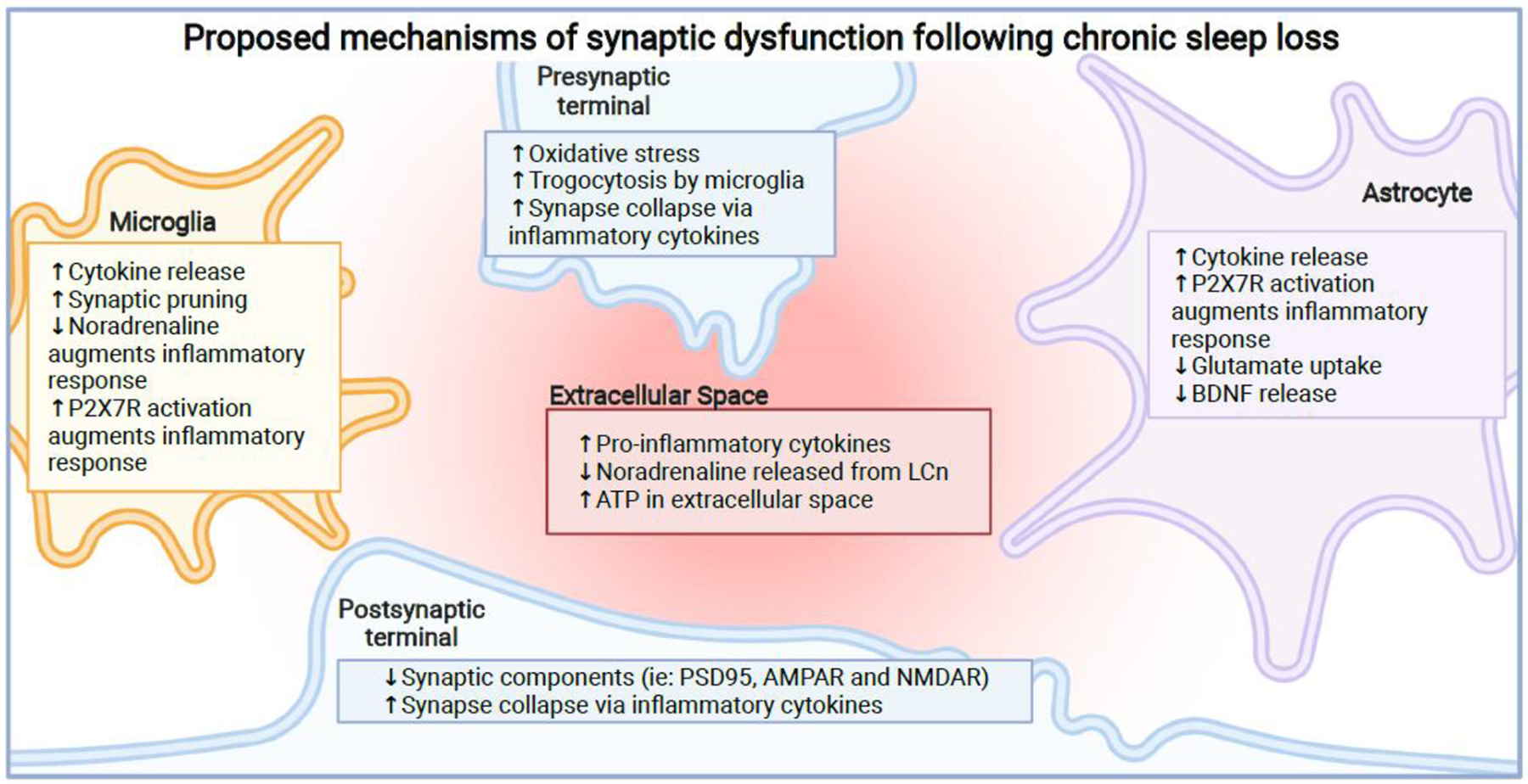
Proposed mechanisms of synaptic dysfunction following chronic sleep loss.
Chronic sleep loss impairs the functionality of the synapse, with consequences seen in both the pre- and post-synapse in addition to the surrounding glial cells. With most of these effects seen in the hippocampus or cortex, these effects likely impair learning and memory. Microglia can release cytokines contributing to the inflammatory environment[105]. The loss of LCn leads to loss of anti-inflammatory effects of noradrenaline and increased microglial-mediated synaptic pruning[91,93–95]. Elevated levels of ATP in the extracellular space activates microglial P2X7 receptors, amplifying inflammation[81,82]. In astrocytes, there is an increase in cytokine production contributing to the inflammatory environment[83]. Since astrocytes also express P2X7 receptors, elevated ATP in the extracellular space activates these receptors and amplifies the inflammatory response[80,82]. In addition, the ability of astrocytes to regulate glutamate levels in the synapse is impaired, which can lead to neuronal excitotoxicity [90]. Astrocytes also release less BDNF, which may contribute to synapse loss[84]. In the extracellular space, there is an increase in cytokines[83], a decrease in noradrenaline likely due to LCn loss, and an increase in ATP[80]. In the pre-synapse, there is increased oxidative stress[80], increase in inappropriately-timed trogocytosis [93–95] and inflammation-mediated synaptic loss[85,86]. Finally, in the post-synapse, there is loss of synaptic components due to synapse loss[80], which may be mediated by the inflammatory environment [85,86]. In summary, chronic sleep loss creates a pro-inflammatory environment at the synapse level which is characterized by impaired glial functionality and synapse loss.
Effects of sleep loss on neurodegenerative processes
The specific groups of neurons shown to be particularly vulnerable to sleep loss, LCn and hippocampal neurons, are cell groups with heightened susceptibility to neurodegenerative disorders, including Alzheimer’s disease, other tauopathies, and Parkinson’s disease. An important question to address, therefore, is whether chronic sleep loss can prompt or worsen neurodegenerative disease. There are several lines of evidence to support both acute and chronic sleep loss effects on proteostasis in wild type mice that might influence neurodegenerative processes. Using in vivo microdialysis in wild type mice, it was found that total Aβ peptides increase in the hippocampal interstitial fluid in response to spontaneous waking or short-term (6 hours) sleep deprivation relative to levels in sleep[96]. A similar sleep-wake effect for Aβ1–40 is observed in healthy humans, measured in cerebrospinal fluid [96], but whether sleep loss increases the more aggregable peptide, Aβ1–42, remains to be determined. Wild type mice exposed to chronic sleep restriction by enriched wakefulness accumulate non-fibril Aβ42 in the hippocampus, in parallel with spatial memory impairment, pronounced glial responses and CA1 neuron loss[97]. Fig. 4 highlights (i) commonalities between effects of sleep disruption as observed in wild type mice and neural or cognitive changes observed in Alzheimer’s disease, and (ii) Alzheimer’s pathological hallmarks which are not observed in wild type mice (whether exposed to sleep loss or not).
Fig 4.
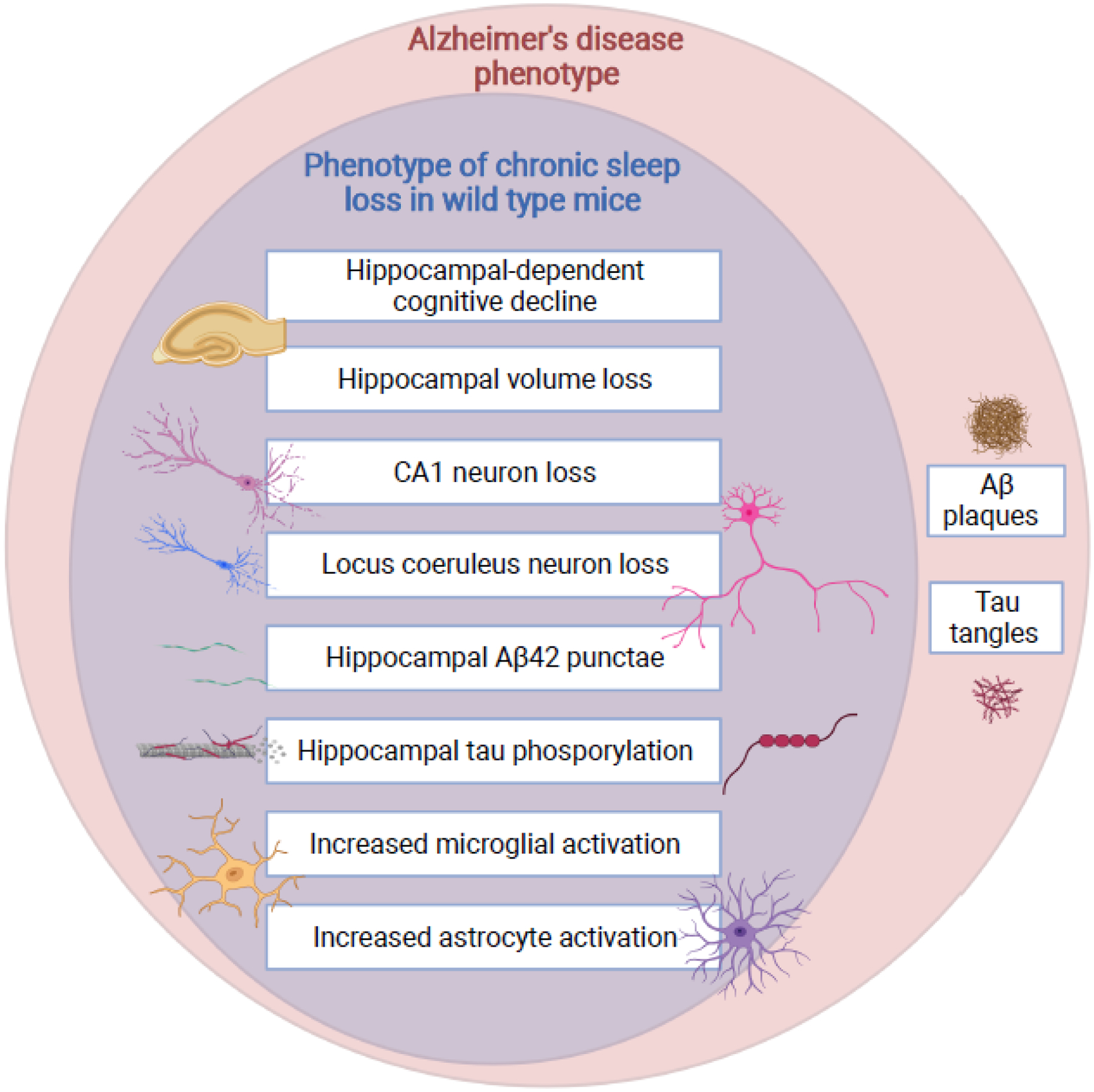
Phenotypic overlap between chronic sleep disruption and Alzheimer’s disease. Commonalities in the neural response to chronic sleep disruption in wild type mice are shown as a subset of the neural and cognitive changes observed in Alzheimer’s disease. Of note, wild type mice do not naturally develop amyloid plaques or tau tangles. In animal models with expression of human amyloid precursor protein and/or tau mutations observed in familial degenerative disorders, sleep disruption increases plaque and tangle formation, as well as tau propagation.
In transgenic mouse models of Alzheimer’s disease (AD), amyloid plaques and/or tau tangles increase in density in response to chronic sleep restriction[96,98,99]. Chronic sleep disruption in the APPSWE/PS1ΔE9 transgenic mouse model also worsens spatial memory performance, increases pathogenic phosphorylated tau (pT231-tau) in the cortex, and may increase neuronal apoptosis as suggested by increased cleaved poly(ADP-ribose) polymerase-1 and cleaved caspase-3 [98]. When this same transgenic strain was exposed to platform sleep disruption for 20 hours for 3 weeks, the sleep disruption aggravated spatial memory impairments, increased microglial marker Ionized calcium-binding adaptor molecule 1 (Iba1) and prevented in vivo LTP induction in the hippocampus[100].Thus, many of the pathological and behavioral sequelae of Alzheimer’s have been demonstrated to worsen in response to chronic sleep loss in various AD mouse models. Of note, neuron loss in response to chronic sleep disruption has not been assessed, to the authors’ knowledge, in any of the amyloid transgenic mouse studies.
Like amyloid, tau levels increase in the interstitial space in the hippocampus of wild type mice in response to wakefulness and short-term sleep loss[25]. In the P301S mouse model of tauopathy, chronic sleep disruption hastens the progression of biochemical effects (increased tau oligomers), behavioral impairments, and pathological hallmarks, including neuron loss in at least two neuronal populations, the LC and basolateral amygdala, and an increase in tau tangles and gliosis[101]. These findings demonstrate that sleep loss exerts comprehensive effects in this tauopathy model. Chronic sleep disruption can also hasten the spread of tau fibrils injected into the hippocampus to the LCn and the entorhinal cortex in the P301S mouse model [25], suggesting that chronic short sleep could influence LCn tau accumulation through sleep loss tau-mediated effects in the hippocampus. It can even be speculated that the presence of early tau in LCn in Alzheimer’s disease may in fact originate in the hippocampus.
Sleep disruption in a time-dependent manner may also negatively influence dopaminergic neurons and thereby Parkinson’s disease. In mice, sleep disruption prior to administration of the dopaminergic neurotoxin, 1-methyl-2-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was not found to influence motor outcomes or dopaminergic neuron loss [102]. In contrast, sleep loss after administration of a dopaminergic neurotoxin worsens loss of dopaminergic neurons [103]. Of note, enhancing sleep may be protective in neurodegenerative processes. In murine models of Parkinson’s disease, enhancing slow-wave sleep with sodium oxybate reduces synuclein aggregates; however, motor deficits were not affected, and other aspects of Parkinson’s pathology were not assessed[104]. In these models, however, sleep enhancement was initiated relatively late in the course of the disease. Whether early life sleep enhancement can prevent or delay motor deficit progression remains to be examined.
Concluding remarks
New findings in humans and animal models showing protracted and in some cases incomplete neurobehavioral recovery after sleep loss have prompted a reconsideration of neural consequences of chronic sleep disturbances. The phenotyping of susceptible cell populations and brain regions at ages across the lifespan should be extended, and our understanding of mechanisms underlying sleep loss neural injury must be furthered, particularly mechanisms underlying the early oxidative stress and persistent inflammatory responses. Finally, there is sufficient evidence that chronic sleep loss can exacerbate amyloid, tau and a-synuclein neurodegenerative processes in animal models. It remains to be examined, however, whether these links translate to humans, and their mechanistic understanding must be furthered to identify potential therapies to prevent neural injury due to sleep loss, as sleep disruption is an inevitable occurrence in modern societies.
Highlights.
Recent evidence in humans reveals that chronic sleep disruption can lead to protracted recovery of neurobehavioral performance, particularly sustained vigilance and episodic memory.
Studies in animal models of chronic sleep disruption demonstrate protracted and even incomplete recovery, including neuron loss in brain areas critical for vigilance and episodic memory, specifically, the locus coeruleus and hippocampus.
The severity of neural injury incurred by chronic sleep disruption varies with duration and type of sleep disruption, age at which sleep loss exposure occurs, neuronal populations being assessed, and genetic predisposition to neurodegenerative processes.
Early oxidative stress and sustained inflammation contribute to a metabolic resetting, behavioral impairment, and pathologic findings associated with chronic sleep disruption.
Acknowledgements
This work was supported by NIH grants AG054104 and AG064231 to S.V. The artwork was created in Biorender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Dinges DF et al. (1997) Cumulative sleepiness, mood disturbance, and psychomotor vigilance performance decrements during a week of sleep restricted to 4–5 hours per night. Sleep 20, 267–277 [PubMed] [Google Scholar]
- 2.Van Dongen HP et al. (2003) The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 26, 117–126 [DOI] [PubMed] [Google Scholar]
- 3.Belenky G et al. (2003) Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J Sleep Res 12, 1–12. 337 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Banks S et al. (2010) Neurobehavioral dynamics following chronic sleep restriction: dose-response effects of one night for recovery. Sleep 33, 1013–1026. 10.1093/sleep/33.8.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pejovic S et al. (2013) Effects of recovery sleep after one work week of mild sleep restriction on interleukin-6 and cortisol secretion and daytime sleepiness and performance. Am J Physiol Endocrinol Metab 305, E890–896. 10.1152/ajpendo.00301.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lo JC et al. (2017) Neurobehavioral Impact of Successive Cycles of Sleep Restriction With and Without Naps in Adolescents. Sleep 40. 10.1093/sleep/zsw042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Axelsson J et al. (2008) Sleepiness and performance in response to repeated sleep restriction and subsequent recovery during semi-laboratory conditions. Chronobiol Int 25, 297–308. 10.1080/07420520802107031 [DOI] [PubMed] [Google Scholar]
- 8.Ochab JK et al. (2021) Observing changes in human functioning during induced sleep deficiency and recovery periods. PLoS One 16, e0255771. 10.1371/journal.pone.0255771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikegami K et al. (2009) Recovery of cognitive performance and fatigue after one night of sleep deprivation. J Occup Health 51, 412–422. 10.1539/joh.l8127 [DOI] [PubMed] [Google Scholar]
- 10.Boardman JM et al. (2018) The ability to self-monitor cognitive performance during 60 h total sleep deprivation and following 2 nights recovery sleep. J Sleep Res 27, e12633. 10.1111/jsr.12633 [DOI] [PubMed] [Google Scholar]
- 11.Chai Y et al. (2020) Two nights of recovery sleep restores hippocampal connectivity but not episodic memory after total sleep deprivation. Scientific reports 10, 8774. 10.1038/s41598-020-65086-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porkka-Heiskanen T et al. (1997) Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276, 1265–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalinchuk AV et al. (2011) The time course of adenosine, nitric oxide (NO) and inducible NO synthase changes in the brain with sleep loss and their role in the non-rapid eye movement sleep homeostatic cascade. J Neurochem 116, 260–272. 10.1111/j.1471-4159.2010.07100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna JT et al. (2007) Sleep fragmentation elevates behavioral, electrographic and neurochemical measures of sleepiness. Neuroscience 146, 1462–1473. S0306–4522(07)00329–6 [pii] 10.1016/j.neuroscience.2007.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porkka-Heiskanen T et al. (2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99, 507–517. 10.1016/s0306-4522(00)00220-7 [DOI] [PubMed] [Google Scholar]
- 16.Portas CM et al. (1997) Role of adenosine in behavioral state modulation: a microdialysis study in the freely moving cat. Neuroscience 79, 225–235. 10.1016/s0306-4522(96)00640-9 [DOI] [PubMed] [Google Scholar]
- 17.Zeitzer JM et al. (2006) Extracellular adenosine in the human brain during sleep and sleep deprivation: an in vivo microdialysis study. Sleep 29, 455–461. 10.1093/sleep/29.4.455 [DOI] [PubMed] [Google Scholar]
- 18.Clasadonte J et al. (2014) Chronic sleep restriction disrupts sleep homeostasis and behavioral sensitivity to alcohol by reducing the extracellular accumulation of adenosine. J Neurosci 34, 1879–1891. 34/5/1879 [pii] 10.1523/JNEUROSCI.2870-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cirelli C et al. (1999) No evidence of brain cell degeneration after long-term sleep deprivation in rats. Brain Res 840, 184–193. S0006–8993(99)01768–0 [pii] [DOI] [PubMed] [Google Scholar]
- 20.Arthaud S et al. (2015) Paradoxical (REM) sleep deprivation in mice using the small-platforms-over-water method: polysomnographic analyses and melanin-concentrating hormone and hypocretin/orexin neuronal activation before, during and after deprivation. J Sleep Res 24, 309–319. 10.1111/jsr.12269 [DOI] [PubMed] [Google Scholar]
- 21.Silva RH et al. (2004) Role of hippocampal oxidative stress in memory deficits induced by sleep deprivation in mice. Neuropharmacology 46, 895–903. 10.1016/j.neuropharm.2003.11.032 S0028390803004684 [pii] [DOI] [PubMed] [Google Scholar]
- 22.Machado RB et al. (2004) Sleep deprivation induced by the modified multiple platform technique: quantification of sleep loss and recovery. Brain Res 1004, 45–51. 10.1016/j.brainres.2004.01.019 [DOI] [PubMed] [Google Scholar]
- 23.Tobler I et al. (1983) The effect of sleep deprivation and recovery sleep on plasma corticosterone in the rat. Neurosci Lett 35, 297–300 [DOI] [PubMed] [Google Scholar]
- 24.Suchecki D et al. (1998) Increased ACTH and corticosterone secretion induced by different methods of paradoxical sleep deprivation. J Sleep Res 7, 276–281. 10.1046/j.1365-2869.1998.00122.x [DOI] [PubMed] [Google Scholar]
- 25.Holth JK et al. (2019) The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884. 10.1126/science.aav2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deurveilher S et al. (2013) Social and environmental contexts modulate sleep deprivation-induced c-Fos activation in rats. Behav Brain Res 256, 238–249. 10.1016/j.bbr.2013.08.029 [DOI] [PubMed] [Google Scholar]
- 27.Leenaars CH et al. (2011) A new automated method for rat sleep deprivation with minimal confounding effects on corticosterone and locomotor activity. J Neurosci Methods 196, 107–117. 10.1016/j.jneumeth.2011.01.014 [DOI] [PubMed] [Google Scholar]
- 28.Deboer T et al. (2007) Long term effects of sleep deprivation on the mammalian circadian pacemaker. Sleep 30, 257–262. 10.1093/sleep/30.3.257 [DOI] [PubMed] [Google Scholar]
- 29.Sinton CM et al. (2009) Validation of a novel method to interrupt sleep in the mouse. J Neurosci Methods 184, 71–78. S0165–0270(09)00405–1 [pii] 10.1016/j.jneumeth.2009.07.026 [DOI] [PubMed] [Google Scholar]
- 30.Li Y et al. (2014) Effects of chronic sleep fragmentation on wake-active neurons and the hypercapnic arousal response. Sleep 37, 51–64. 10.5665/sleep.3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallace E et al. (2015) Differential effects of duration of sleep fragmentation on spatial learning and synaptic plasticity in pubertal mice. Brain Res 1615, 116–128. 10.1016/j.brainres.2015.04.037 [DOI] [PubMed] [Google Scholar]
- 32.Gompf HS et al. (2010) Locus ceruleus and anterior cingulate cortex sustain wakefulness in a novel environment. J Neurosci 30, 14543–14551. 30/43/14543 [pii] 10.1523/JNEUROSCI.3037-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bliss-Moreau E et al. (2021) Anterior Cingulate Cortex Ablation Disrupts Affective Vigor and Vigilance. J Neurosci 41, 8075–8087. 10.1523/JNEUROSCI.0673-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J et al. (2014) Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. J Neurosci 34, 4418–4431. 10.1523/JNEUROSCI.5025-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang SS et al. (2020) Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest 130, 422–437. 10.1172/JCI130513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen JE et al. (2021) Late-in-Life Neurodegeneration after Chronic Sleep Loss in Young Adult Mice. Sleep. 10.1093/sleep/zsab057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, P. L, Zhang J, Zhu Y,Zhan G, Chou Y, Fenik P, Bhatnagar S, Piel D, Beck SG, Veasey S (2013) Effects of Chronic Sleep Fragmentation on Wake-Active Neurons and the Hypercapneic Arousal Response. Sleep in press, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Y et al. (2015) Degeneration in Arousal Neurons in Chronic Sleep Disruption Modeling Sleep Apnea. Front Neurol 6, 109. 10.3389/fneur.2015.00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deurveilher S et al. (2021) No loss of orexin/hypocretin, melanin-concentrating hormone or locus coeruleus noradrenergic neurons in a rat model of chronic sleep restriction. Eur J Neurosci 54, 6027–6043. 10.1111/ejn.15412 [DOI] [PubMed] [Google Scholar]
- 40.Lu HJ and Lv J (2021) beta-adrenergic Receptor Activity in the Hippocampal Dentate Gyrus Participates in Spatial Learning and Memory Impairment in Sleep-deprived Rats. Exp Neurobiol 30, 144–154. 10.5607/en20058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vankov A et al. (1995) Response to novelty and its rapid habituation in locus coeruleus neurons of the freely exploring rat. Eur J Neurosci 7, 1180–1187. 10.1111/j.1460-9568.1995.tb01108.x [DOI] [PubMed] [Google Scholar]
- 42.Aston-Jones G and Bloom FE (1981) Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1, 876–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanchez-Padilla J et al. (2014) Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci 17, 832–840. nn.3717 [pii] 10.1038/nn.3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDermott CM et al. (2003) Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci 23, 9687–9695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim EY et al. (2005) REM sleep deprivation inhibits LTP in vivo in area CA1 of rat hippocampus. Neurosci Lett 388, 163–167. 10.1016/j.neulet.2005.06.057 [DOI] [PubMed] [Google Scholar]
- 46.Konakanchi S et al. (2022) Effect of chronic sleep deprivation and sleep recovery on hippocampal CA3 neurons, spatial memory and anxiety-like behavior in rats. Neurobiol Learn Mem 187, 107559. 10.1016/j.nlm.2021.107559 [DOI] [PubMed] [Google Scholar]
- 47.Soto-Rodriguez S et al. (2016) Rapid Eye Movement Sleep Deprivation Produces Long-Term Detrimental Effects in Spatial Memory and Modifies the Cellular Composition of the Subgranular Zone. Frontiers in cellular neuroscience 10, 132. 10.3389/fncel.2016.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guzman-Marin R et al. (2007) Hippocampal neurogenesis is reduced by sleep fragmentation in the adult rat. Neuroscience 148, 325–333. S0306–4522(07)00643–4 [pii] 10.1016/j.neuroscience.2007.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guzman-Marin R et al. (2003) Sleep deprivation reduces proliferation of cells in the dentate gyrus of the hippocampus in rats. J Physiol 549, 563–571. 10.1113/jphysiol.2003.041665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzman-Marin R et al. (2005) Sleep deprivation suppresses neurogenesis in the adult hippocampus of rats. Eur J Neurosci 22, 2111–2116. 10.1111/j.1460-9568.2005.04376.x [DOI] [PubMed] [Google Scholar]
- 51.Mirescu C et al. (2006) Sleep deprivation inhibits adult neurogenesis in the hippocampus by elevating glucocorticoids. Proc Natl Acad Sci U S A 103, 19170–19175. 10.1073/pnas.0608644103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jean-Louis G et al. (2014) Associations between inadequate sleep and obesity in the US adult population: analysis of the national health interview survey (1977–2009). BMC Public Health 14, 290. 10.1186/1471-2458-14-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H et al. (2019) Cholinergic modulation of hippocampal calcium activity across the sleep-wake cycle. Elife 8. 10.7554/eLife.39777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu Z et al. (2021) Topological reorganizations of mitochondria isolated from rat brain after 72 hours of paradoxical sleep deprivation, revealed by electron cryo-tomography. Am J Physiol Cell Physiol 321, C17–C25. 10.1152/ajpcell.00077.2021 [DOI] [PubMed] [Google Scholar]
- 55.Zuo JX et al. (2020) Hydrogen Sulfide Prevents Sleep Deprivation-Induced Hippocampal Damage by Upregulation of Sirt1 in the Hippocampus. Front Neurosci 14, 169. 10.3389/fnins.2020.00169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khadrawy YA et al. (2011) Effect of oxidative stress induced by paradoxical sleep deprivation on the activities of Na+, K+-ATPase and acetylcholinesterase in the cortex and hippocampus of rat. Transl Res 157, 100–107. 10.1016/j.trsl.2010.11.005 [DOI] [PubMed] [Google Scholar]
- 57.Alzoubi KH et al. (2012) The neuroprotective effect of vitamin E on chronic sleep deprivation-induced memory impairment: the role of oxidative stress. Behav Brain Res 226, 205–210. S0166–4328(11)00686–3 [pii] 10.1016/j.bbr.2011.09.017 [DOI] [PubMed] [Google Scholar]
- 58.Alzoubi KH et al. (2013) Evaluation of the effect of pentoxifylline on sleep-deprivation induced memory impairment. Hippocampus 23, 812–819. 10.1002/hipo.22135 [DOI] [PubMed] [Google Scholar]
- 59.Arora S et al. (2021) Neurobehavioral alterations in a mouse model of chronic partial sleep deprivation. Metabolic brain disease 36, 1315–1330. 10.1007/s11011-021-00693-9 [DOI] [PubMed] [Google Scholar]
- 60.Wang X et al. (2021) Melatonin ameliorates anxiety-like behaviors induced by sleep deprivation in mice: Role of oxidative stress, neuroinflammation, autophagy and apoptosis. Brain Res Bull 174, 161–172. 10.1016/j.brainresbull.2021.06.010 [DOI] [PubMed] [Google Scholar]
- 61.Attwell D and Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21, 1133–1145. 10.1097/00004647-200110000-00001 [DOI] [PubMed] [Google Scholar]
- 62.Harris JJ et al. (2012) Synaptic energy use and supply. Neuron 75, 762–777. 10.1016/j.neuron.2012.08.019 [DOI] [PubMed] [Google Scholar]
- 63.Tononi G and Cirelli C (2014) Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34. 10.1016/j.neuron.2013.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Vivo L et al. (2017) Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510. 10.1126/science.aah5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diering GH et al. (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355, 511–515. 10.1126/science.aai8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyamoto D et al. (2021) Net decrease in spine-surface GluA1-containing AMPA receptors after post-learning sleep in the adult mouse cortex. Nat Commun 12, 2881. 10.1038/s41467-021-23156-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruggiero A et al. (2021) Mitochondria: new players in homeostatic regulation of firing rate set points. Trends Neurosci 44, 605–618. 10.1016/j.tins.2021.03.002 [DOI] [PubMed] [Google Scholar]
- 68.Marchi S and Pinton P (2014) The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol 592, 829–839. 10.1113/jphysiol.2013.268235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu J et al. (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4, 2034. 10.1038/ncomms3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bourdon AK et al. (2018) Metabolomic analysis of mouse prefrontal cortex reveals upregulated analytes during wakefulness compared to sleep. Sci Rep 8, 11225. 10.1038/s41598-018-29511-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopez J et al. (2008) Rapid eye movement sleep deprivation decreases long-term potentiation stability and affects some glutamatergic signaling proteins during hippocampal development. Neuroscience 153, 44–53. 10.1016/j.neuroscience.2008.01.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acosta-Pena E et al. (2015) Sleep deprivation induces differential morphological changes in the hippocampus and prefrontal cortex in young and old rats. Synapse 69, 15–25. 10.1002/syn.21779 [DOI] [PubMed] [Google Scholar]
- 73.Yuan RK et al. (2021) Differential effect of sleep deprivation on place cell representations, sleep architecture, and memory in young and old mice. Cell reports 35, 109234. 10.1016/j.celrep.2021.109234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howard KA and Hunter AS (2019) Immediate and long-lasting cognitive consequences of adolescent chronic sleep restriction. Behav Neurosci 133, 461–466. 10.1037/bne0000312 [DOI] [PubMed] [Google Scholar]
- 75.Jones CE et al. (2019) Early-life sleep disruption increases parvalbumin in primary somatosensory cortex and impairs social bonding in prairie voles. Sci Adv 5, eaav5188. 10.1126/sciadv.aav5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shaffery JP et al. (2012) Selective rapid eye movement sleep deprivation affects cell size and number in kitten locus coeruleus. Front Neurol 3, 69. 10.3389/fneur.2012.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bellesi M et al. (2018) Myelin modifications after chronic sleep loss in adolescent mice. Sleep 41. 10.1093/sleep/zsy034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seugnet L et al. (2011) Sleep deprivation during early-adult development results in long-lasting learning deficits in adult Drosophila. Sleep 34, 137–146. 10.1093/sleep/34.2.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kayser MS et al. (2014) A critical period of sleep for development of courtship circuitry and behavior in Drosophila. Science 344, 269–274. 10.1126/science.1250553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Atrooz F et al. (2019) Early Life Sleep Deprivation: Role of Oxido-Inflammatory Processes. Neuroscience 406, 22–37. 10.1016/j.neuroscience.2019.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xia M et al. (2020) Sleep Deprivation Selectively Down-Regulates Astrocytic 5-HT2B Receptors and Triggers Depressive-Like Behaviors via Stimulating P2X7 Receptors in Mice. Neurosci Bull 36, 1259–1270. 10.1007/s12264-020-00524-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Di Virgilio F et al. (2009) Purinergic signalling in inflammation of the central nervous system. Trends Neurosci 32, 79–87. 10.1016/j.tins.2008.11.003 [DOI] [PubMed] [Google Scholar]
- 83.Xia M et al. (2017) The ameliorative effect of fluoxetine on neuroinflammation induced by sleep deprivation. J Neurochem. 10.1111/jnc.14272 [DOI] [PubMed] [Google Scholar]
- 84.Li X et al. (2018) Leptin Increases Expression of 5-HT2B Receptors in Astrocytes Thus Enhancing Action of Fluoxetine on the Depressive Behavior Induced by Sleep Deprivation. Front Psychiatry 9, 734. 10.3389/fpsyt.2018.00734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Micheau O and Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. 10.1016/s0092-8674(03)00521-x [DOI] [PubMed] [Google Scholar]
- 86.Mishra A et al. (2012) Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol 7, 571–578. 10.1007/s11481-012-9342-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qian Y et al. (2007) The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8, 247–256. 10.1038/ni1439 [DOI] [PubMed] [Google Scholar]
- 88.Calabrese V et al. (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8, 766–775. 10.1038/nrn2214 [DOI] [PubMed] [Google Scholar]
- 89.Colombo E et al. (2012) Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J Exp Med 209, 521–535. 10.1084/jem.20110698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bezzi P et al. (2001) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4, 702–710. 10.1038/89490 [DOI] [PubMed] [Google Scholar]
- 91.Farber K et al. (2005) Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci 29, 128–138. 10.1016/j.mcn.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 92.Weinhard L et al. (2018) Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature communications 9, 1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stowell RD et al. (2019) Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci 22, 1782–1792. 10.1038/s41593-019-0514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu YU et al. (2019) Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci 22, 1771–1781. 10.1038/s41593-019-0511-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choudhury ME et al. (2020) Phagocytic elimination of synapses by microglia during sleep. Glia 68, 44–59. 10.1002/glia.23698 [DOI] [PubMed] [Google Scholar]
- 96.Kang JE et al. (2009) Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. 1180962 [pii] 10.1126/science.1180962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Owen JE et al. (2021) Late-in-life neurodegeneration after chronic sleep loss in young adult mice. Sleep 44. 10.1093/sleep/zsab057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qiu H et al. (2016) Chronic Sleep Deprivation Exacerbates Learning-Memory Disability and Alzheimer’s Disease-Like Pathologies in AbetaPP(swe)/PS1(DeltaE9) Mice. J Alzheimers Dis 50, 669–685. 10.3233/JAD-150774 [DOI] [PubMed] [Google Scholar]
- 99.Rothman SM et al. (2013) Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Abeta and pTau in a mouse model of Alzheimer’s disease. Brain Res 1529, 200–208. 10.1016/j.brainres.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang C et al. (2021) Chronic sleep deprivation exacerbates cognitive and synaptic plasticity impairments in APP/PS1 transgenic mice. Behav Brain Res 412, 113400. 10.1016/j.bbr.2021.113400 [DOI] [PubMed] [Google Scholar]
- 101.Zhu Y et al. (2018) Chronic Sleep Disruption Advances the Temporal Progression of Tauopathy in P301S Mutant Mice. J Neurosci 38, 10255–10270. 10.1523/JNEUROSCI.0275-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu M et al. (2019) Effects of sleep disruption on stress, nigrostriatal markers, and behavior in a chronic/progressive MPTP male mouse model of parkinsonism. J Neurosci Res 97, 1706–1719. 10.1002/jnr.24520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lima MM et al. (2012) Paradoxical sleep deprivation modulates tyrosine hydroxylase expression in the nigrostriatal pathway and attenuates motor deficits induced by dopaminergic depletion. CNS Neurol Disord Drug Targets 11, 359–368. 10.2174/187152712800792839 [DOI] [PubMed] [Google Scholar]
- 104.Morawska MM et al. (2021) Slow-wave sleep affects synucleinopathy and regulates proteostatic processes in mouse models of Parkinson’s disease. Sci Transl Med 13, eabe7099. 10.1126/scitranslmed.abe7099 [DOI] [PubMed] [Google Scholar]
- 105.Welser-Alves JV and Milner R (2013) Microglia are the major source of TNF-alpha and TGF-beta1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide, and vitronectin. Neurochem Int 63, 47–53. 10.1016/j.neuint.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]