

Abstract
Background -
The FLNC gene has recently garnered attention as a likely cause of arrhythmogenic cardiomyopathy (ACM), which is considered an actionable genetic condition. However, the association with disease in an unselected clinical population is unknown. We hypothesized that individuals with loss-of-function variants in FLNC (FLNCLOF) would have increased odds for ACM-associated phenotypes versus variant-negative controls in the Geisinger MyCode cohort.
Methods -
We identified rare, putative FLNCLOF among 171,948 individuals with exome sequencing linked to health records. Associations with ACM phenotypes from available diagnoses and cardiac evaluations were investigated.
Results -
Sixty individuals (0.03%; median age 58 years [47–70 IQR], 43% male) harbored 27 unique FLNCLOF. These individuals had significantly increased odds ratios (OR) for dilated cardiomyopathy (OR:4.9, [95% confidence interval: 2.6–7.6]; p<0.001), supraventricular tachycardia (OR:3.2, [1.1–5.6]; p=0.048), and left-dominant ACM (OR:4.2, [1.4–7.9]; p=0.03). Echocardiography revealed reduced left ventricular ejection fraction (52±13% vs. 57±9%; p=0.001) associated with FLNCLOF. Overall, at least 9% of FLNCLOF patients demonstrated evidence of penetrant disease.
Conclusions -
FLNCLOF variants are associated with increased odds of ventricular arrhythmia and dysfunction in an unselected clinical population. These findings support genomic screening of FLNC for actionable secondary findings.
Keywords: FLNC gene, arrhythmogenic cardiomyopathy, dilated cardiomyopathy, sudden cardiac death, genotype-phenotype correlation
Introduction
The FLNC gene, which encodes the filamin-C sarcomeric protein, has recently garnered attention as a potential cause of arrhythmogenic cardiomyopathy (ACM).1 Early reports have shown convincing associations of FLNC variants with an overlapping phenotype of dilated cardiomyopathy (DCM) and ACM, which is typically left-dominant (also called arrhythmogenic left ventricular cardiomyopathy (ALVC)).2–5 Commonly cited characteristics from clinical evaluation of individuals with FLNC variants have included frequent ventricular arrhythmias or sudden cardiac death, reduced left ventricular (LV) systolic function, and regional structural abnormalities such as non-ischemic late gadolinium enhancement (LGE) patterns on cardiac MRI (CMR).6–10
To date, these studies have primarily investigated gene-phenotype associations in individuals or families with known cardiomyopathy. However, little is known of the disease burden and phenotype characteristics associated with FLNC variants when identified through broad exome sequencing within an unselected clinical population. Such data are critical for understanding the disease penetrance of these variants when discovered as a secondary or incidental finding, and to thus guide clinical management of identified patients. This consideration is particularly timely given the recent inclusion of FLNC in the American College of Medical Genetics and Genomics (ACMG) recommendation for clinical return of secondary findings.11
The advent of large genomic screening initiatives provides opportunities to identify FLNC variants—and characterize the associated phenotype—in a relatively unselected population. With one of the largest sequenced biobanks linked to a long-standing electronic health records (EHR) system, Geisinger’s MyCode Community Health Initiative enables such an assessment. Using this resource, the phenotypic burden in individuals with rare, putative loss-of-function FLNC variants (FLNCLOF) was retrospectively investigated and compared to the remaining sequenced MyCode cohort using EHR-based phenotypes. Additionally, blinded manual chart review of available cardiac evaluations in these individuals was performed and compared with matched controls. We hypothesized that MyCode participants with FLNCLOF would have increased odds for ACM-associated phenotypes compared to controls.
Methods
A full description of the methods used in this study is available in the supplement. Institutional review board approval was obtained for this study; all individuals previously provided informed consent to participate in the MyCode Community Health Initiative (MyCode). The data that support the findings of this study will not be made available.
Results
A total of 171,948 MyCode participants had available sequencing data linked to their EHR. Of these, 60 individuals (0.03%) were identified harboring 27 unique FLNCLOF variants, comprising frameshift (n=14), splice site (10), and stop gained (3). Variant details are presented in Table 1. Basic demographics of FLNCLOF individuals were comparable to the remaining MyCode cohort lacking a LOF FLNC variant (Table 2). Of note, these 60 individuals comprised 50 distinct families, with 15 (25%) of these individuals having at least one related family member in the cohort. The remaining MyCode cohort comprised 117,232 distinct families.
Table 1.
FLNCLOF variant details in the MyCode cohort
| CHR:POS | HGVSc (NM_001458.5) |
HGVSp (NP_001449.3) |
Consequence | ClinVar Review Status | N Pts |
|---|---|---|---|---|---|
| 7:128835324 | c.353–2A>G | -- | Splice | . | 1 |
| 7:128837255 | c.697C>T | p.Gln233* | Nonsense | P, 1-star | 1 |
| 7:128841281 | c.1926_1947del | p.Ile643Glufs*21 | Frameshift | . | 2 |
| 7:128841364 | c.2007+1G>A | -- | Splice | . | 1 |
| 7:128841529 | c.2084del | p.Arg695Leufs*8 | Frameshift | P, 1-star | 1 |
| 7:128841565 | c.2119C>T | p.Gln707* | Nonsense | P, 1-star | 7 |
| 7:128842375 | c.2265+1G>A | -- | Splice | LP, 1-star | 4 |
| 7:128842375 | c.2265+1G>T | -- | Splice | LP, 1-star | 4 |
| 7:128843418 | c.2652_2653insG | p.Lys886Glufs*34 | Frameshift | . | 2 |
| 7:128843861 | c.2878del | p.Val960Trpfs*29 | Frameshift | . | 1 |
| 7:128844656 | c.3193–2A>G | -- | Splice | LP, 1-star | 2 |
| 7:128845989 | c.3791–1G>C | -- | Splice | P/LP, 2-star | 2 |
| 7:128846094 | c.3901_3904del | p.Asp1301Profs*3 | Frameshift | . | 1 |
| 7:128846744 | c.4128–1G>C | -- | Splice | . | 8 |
| 7:128848974 | c.4926_4927insACGTCACA | p.Val1643Thrfs*26 | Frameshift | P, 1-star | 1 |
| 7:128849329 | c.4952–2A>T | -- | Splice | LP, 1-star | 1 |
| 7:128851361 | c.5668+1G>C | -- | Splice | . | 4 |
| 7:128851539 | c.5754del | p.Leu1919Cysfs*34 | Frameshift | . | 1 |
| 7:128852730 | c.5983del | p.Arg1995Alafs*21 | Frameshift | P, 1-star | 1 |
| 7:128853517 | c.6258del | p.Asn2087Thrfs*40 | Frameshift | . | 1 |
| 7:128854163 | c.6676del | p.Leu2226Trpfs*25 | Frameshift | . | 1 |
| 7:128855206 | c.7143T>G | p.Tyr2381* | Nonsense | . | 2 |
| 7:128856630 | c.7365del | p.Tyr2455* | Nonsense | LP, 1-star | 4 |
| 7:128856636 | c.7371del | p.Glu2458Serfs*71 | Frameshift | P, 1-star | 4 |
| 7:128856856 | c.7496_7497insTGCT | p.Gln2499Hisfs*46 | Frameshift | P, 1-star | 1 |
| 7:128857312 | c.7757del | p.Ser2586Thrfs*29 | Frameshift | . | 1 |
| 7:128858006 | c.7781–2A>C | -- | Splice | . | 1 |
Coordinates are with respect to GRCh38. ClinVar review as of April 2021. P: pathogenic; LP: likely pathogenic.
Table 2.
Demographics of FLNCLOF group vs. variant-negative controls in MyCode
| FLNC LOF | Variant-negative controls | Unadjusted p-value | |
|---|---|---|---|
| Individuals | 60 | 171,888 | -- |
| Distinct families | 50 (83%) | 117,232 (68%) | -- |
| Male sex | 26 (43%) | 67,818 (39%) | 0.60 |
| European ancestry | 57 (95%) | 162,191 (94%) | >0.99 |
| Age at last encounter [years] | 58 (47–70) | 58 (42–70) | 0.52 |
| Follow up duration [years] | 16 (8–19) | 15 (8–19) | 0.68 |
| Vital status - living | 53 (88%) | 157,610 (92%) | -- |
| Body mass index | 30 (26–35) | 30 (26–35) | 0.61 |
Values are presented as n (%) or median (IQR). FLNCLOF: FLNC loss-of-function variant group
FLNCLOF are associated with increased odds of disease and ventricular remodeling/dysfunction diagnoses in the EHR
Comparing the EHR-derived phenotypes of individuals with FLNCLOF to the rest of the MyCode cohort, FLNCLOF was associated with significantly greater odds ratios (OR) of heart disease diagnoses, including DCM (OR:4.9 [95% CI 2.6–7.6]) and supraventricular tachycardia (SVT; OR:3.2 [95% CI 1.1–5.6]), as shown in Table 3. Need for an implantable cardioverter defibrillator (ICD; OR:4.6 [1.9–8.4]) and heart failure (HF; OR:2.2 [0.96–3.9]) had similar associations, though not statistically significant. Ventricular dysfunction (with or without arrhythmia) was observed in 15/60 (25%) FLNCLOF individuals vs. 19,461/171,888 (11%) variant-negative controls (OR:3.0 [1.6–5.0]; p=0.006). Arrhythmia was present in 9/60 (15%) FLNCLOF vs. 13,808/171,888 (8%) controls (OR:2.1 [0.9–3.6]; p=0.09). Together, 6/60 (10%) FLNCLOF patients had history of both ventricular dysfunction and arrhythmia—suggestive of ALVC—as compared to 4,903/171,888 (3%) of the remaining variant-negative cohort (OR:4.2 [1.4–7.9]; p=0.03; Table 3; Figure 1A).
Table 3.
Associations of cardiomyopathy phenotypes in FLNCLOF group vs. variant-negative controls in MyCode
| Phenotype | FLNCLOF (n=60) | Variant-negative controls (n=171,888) | OR | 95% CI | *p-value |
|---|---|---|---|---|---|
| DCM | 9 (15%) | 6,341 (4%) | 4.9 | 2.6 – 7.6 | <0.001 |
| HF | 9 (15%) | 14,333 (8%) | 2.2 | 0.96 – 3.90 | 0.09 |
| CM | 9 (15%) | 6,890 (4%) | 4.5 | 2.4 – 7.0 | 0.006 |
| AF | 7 (12%) | 16,408 (10%) | 1.3 | 0.6 – 2.3 | 0.41 |
| SVT | 6 (10%) | 6,171 (4%) | 3.2 | 1.1 – 5.6 | 0.048 |
| PVCs | 1 (2%) | 3,828 (2%) | 1.1 | 0.5 – 2.1 | 0.70 |
| ICD | 4 (7%) | 2,907 (2%) | 4.6 | 1.9 – 8.4 | 0.051 |
| ALVC dysfunction | 15 (25%) | 19,461 (11%) | 3.0 | 1.6 – 5.0 | 0.006 |
| ALVC arrhythmia | 9 (15%) | 13,808 (8%) | 2.1 | 0.9 – 3.6 | 0.09 |
| ALVC | 6 (10%) | 4,903 (3%) | 4.2 | 1.4 – 7.9 | 0.03 |
FLNCLOF: FLNC loss-of-function variant group; OR: odds ratio; CI: confidence interval; DCM: dilated cardiomyopathy; HF: heart failure; CM: Cardiomyopathy; AF: atrial fibrillation; VT: ventricular tachycardia; VF: ventricular fibrillation; SVT: supraventricular tachycardia; PVCs: premature ventricular contractions; ICD: implantable cardioverter defibrillator; ALVC: arrhythmogenic left ventricular cardiomyopathy.
p-values based on bootstrapped Firth’s logistic regression model adjusted for age, sex, and ancestry and adjusted for multiple comparisons (p<0.05 and OR 95% CI not crossing 1 considered significant).
Figure 1.
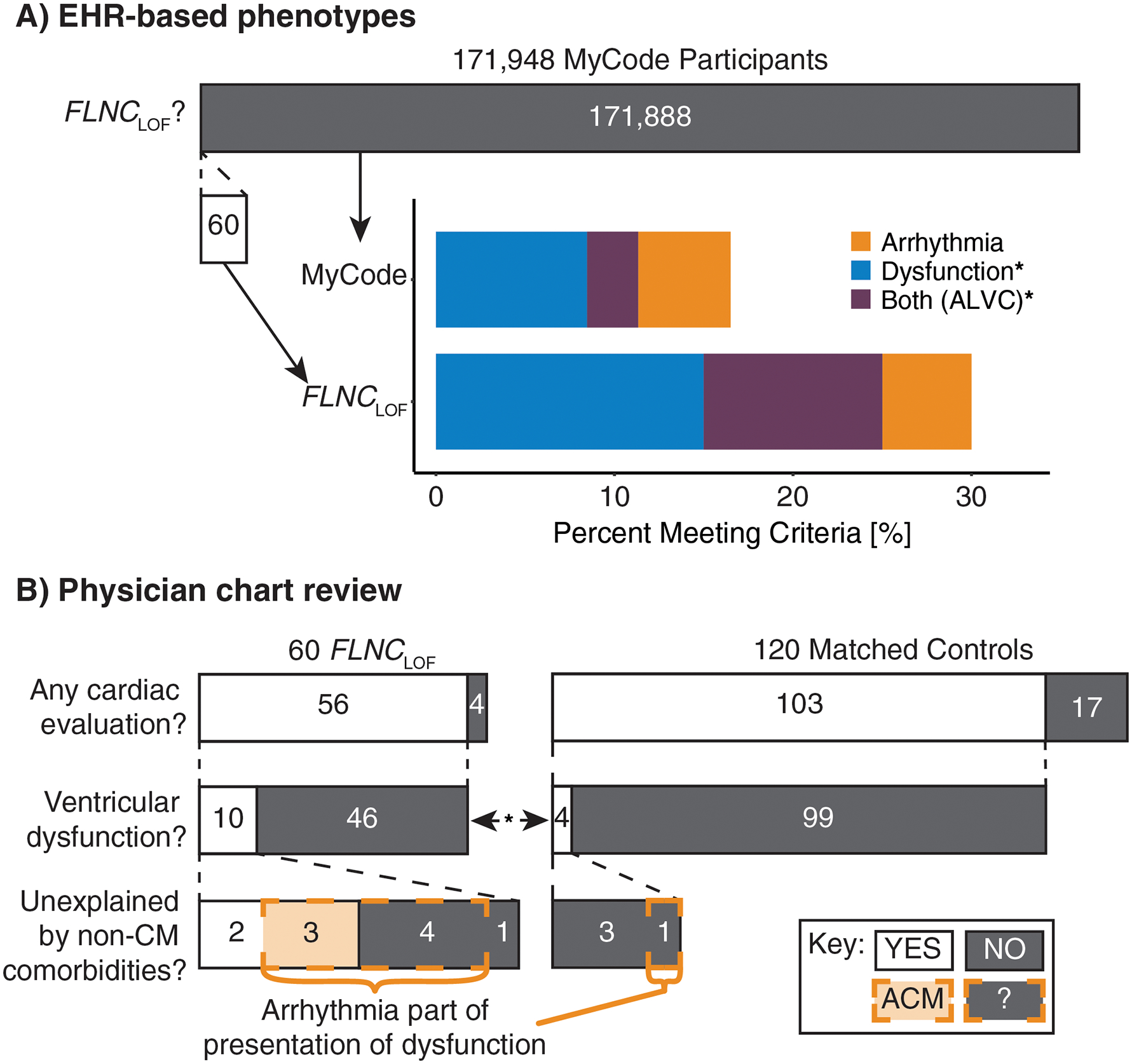
Summary of phenotypic findings in FLNCLOF individuals. (A) Compared to the rest of the MyCode population, individuals with FLNCLOF were significantly enriched for EHR-based phenotypes of ventricular dysfunction (p<0.001) and both dysfunction and arrhythmia (ALVC; p=0.003), but not for arrhythmia criteria (p=0.06). (B) ACM 2019 criteria from matched chart review revealed that individuals with FLNCLOF were enriched for ventricular dysfunction (p=0.04). Similar apparent enrichment was observed for unexplained dysfunction and having arrhythmia as part of the presentation of the dysfunction, however these group differences were not significant. EHR: electronic health records; ALVC: arrhythmogenic left ventricular cardiomyopathy; FLNCLOF: FLNC loss-of-function variant group; CM: cardiomyopathy; ACM: arrhythmogenic cardiomyopathy-associated phenotype; ?: represents those with dysfunction confounded by other co-morbidities and arrhythmia, not classified as ACM. *p<0.05 for groupwise comparisons.
In addition to the diagnostic data, echocardiograms were available for 32/60 (53%) FLNCLOF individuals and 75,837/171,888 (44%) variant-negative controls (Table 4). Those in the FLNCLOF group had significantly reduced LVEF (52 ± 13% FLNCLOF vs. 57 ± 9% controls; p=0.001; Figure 2) and increased left ventricular end-diastolic inner diameter (LVIDd; 4.9 ± 0.8 cm FLNCLOF vs. 4.6 ± 0.7 cm; p=0.02) compared to controls. However, the indexed LVIDd was not significantly different (2.5 ± 0.4 cm/m2 FLNCLOF vs. 2.4 ± 0.4 cm/m2; p=0.10). Interventricular septum thickness in diastole (IVSd) was also slightly increased (1.12 ± 0.21 cm FLNCLOF vs. 1.08 ± 0.24 cm controls; p=0.02).
Table 4.
Associations of echocardiogram measures with FLNCLOF
| Echocardiogram measure | FLNC LOF | Variant-negative control | p-value* |
|---|---|---|---|
| Individuals with available studies (% of group) | 32 (53%) | 75,837 (44%) | |
| LVEF [%] | 52 ± 13 | 57 ± 9 | 0.001 |
| LVIDd [cm] | 4.9 ± 0.8 | 4.6 ± 0.7 | 0.02 |
| IVSd [cm] | 1.12 ± 0.21 | 1.08 ± 0.24 | 0.02 |
FLNCLOF: FLNC loss-of-function variant group; LVIDd: left ventricular internal diameter and end-diastole; IVSd: interventricular septum dimension at end-diastole; LVEF: left ventricular ejection fraction.
p-values based on bootstrapped linear model adjusted for age, sex, and ancestry and adjusted for multiple comparisons (p<0.05 considered significant).
Figure 2.
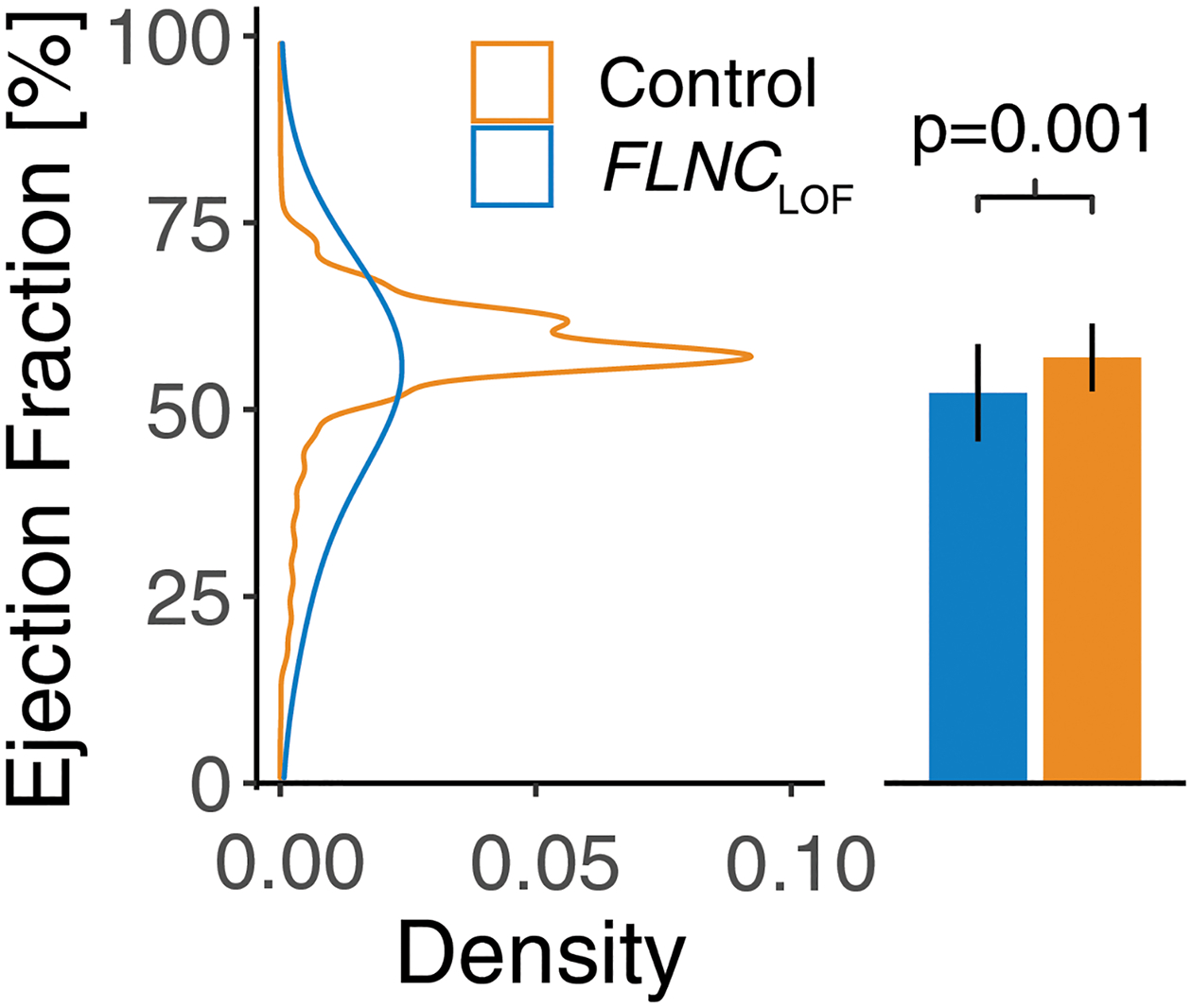
Most recent left ventricular ejection fraction in individuals with FLNCLOF variants and variant-negative controls. Ejection fraction was significantly reduced in the FLNCLOF group compared to controls, as observed using a bootstrapped linear regression model (p=0.001). FLNCLOF: FLNC loss-of-function variant group.
Chart review confirms association with arrhythmias and cardiomyopathies and identification of ACM
Results from matched chart reviews are summarized in Table 5. Overall, the burden of cardiomyopathy, ventricular tachycardia (VT), and sudden cardiac arrest (SCA) was observed to be higher (OR >3) in the FLNCLOF group than matched controls—consistent with the EHR-based analysis—but these observations were not independently statistically significant in this smaller sample.
Table 5.
Results of manual chart review in FLNCLOF group and matched controls
| FLNCLOF (n=60) | Matched controls (n=120) | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Any cardiac evaluation | 56 (93%) | 103 (86%) | -- | -- | -- |
| CM Dx | 8 (14%) | 5 (5%) | 3.2 | 0.9 – 13.3 | 0.10 |
| VT | 7 (13%) | 3 (3%) | 4.7 | 1.0 – 29.5 | 0.08 |
| SCA | 3 (5%) | 1 (1%) | 5.7 | 0.4 – 305.5 | 0.15 |
| ECG features | |||||
| ECG available | 54 (90%) | 97 (81%) | -- | -- | -- |
| CRBBB | 5 (9%) | 4 (4%) | 2.4 | 0.5 – 12.5 | 0.42 |
| Any TWI V3-V6 | 10 (19%) | 6 (6%) | 3.4 | 1.0 – 12.2 | 0.11 |
| PVCs† | 9 (17%) | 5 (5%) | 3.6 | 1.0 – 14.7 | 0.11 |
| ARVC Repolarization Major Criteria | 5 (9%) | 4 (4%) | 2.4 | 0.5 – 12.5 | 0.42 |
| ARVC Repolarization Minor Criteria | 7 (13%) | 12 (12%) | 1.1 | 0.3 – 3.1 | >0.99 |
| ARVC Depolarization Major Criteria | 0 | 0 | -- | -- | -- |
| ARVC Depolarization Minor Criteria | 4 (7%) | 4 (4%) | 1.9 | 0.3 – 10.4 | 0.55 |
FLNCLOF: FLNC loss-of-function variant group; OR: odds ratio; CI: confidence interval; CM Dx: Cardiomyopathy diagnosis; VT: ventricular tachycardia; SCA: sudden cardiac arrest; CRBBB: complete right bundle branch block, TWI: T-wave inversion; PVCs: premature ventricular contractions; ARVC: arrhythmogenic right ventricular cardiomyopathy;
As observed on resting 12-lead ECG or >0.5% PVC burden on Holter. Adjusted p<0.05 considered significant.
The most recent non-paced, interpretable ECG was evaluated for each patient (54/60 available for FLNCLOF group and 97/120 for matched controls) to identify the presence of any depolarization, repolarization, or arrhythmia abnormalities, such as those commonly used to diagnose arrhythmogenic right ventricular cardiomyopathy (ARVC). ARVC-specific task force criteria (TFC) were uncommon (4–13%) and comparable in frequency between groups (Table 5). Other characteristics, such as T-wave inversions in any anterior or lateral precordial leads (V3-V6) and premature ventricular contractions (PVC) on ECG or Holter (>0.5% burden) were observed more frequently (OR >3) in the FLNCLOF group, but those differences were not statistically significant (Table 5; p=0.11 for both).
Finally, a detailed evaluation of the presence, form, and etiology of any cardiomyopathy present in the subset of these patients with any cardiac evaluation (56/60 FLNCLOF and 103/120 matched controls) was performed. Ventricular dysfunction was present in 10/56 (18%) FLNCLOF individuals vs. 4/103 (4%) matched controls (OR:5.3 [1.4–24.5]; p=0.04; Figure 1B), and arrhythmia was part of the presentation of the dysfunction for seven of these patients in the FLNCLOF group (7/10; 70%) vs. 1/4 (25%) controls (OR:6.0 [0.3–417.2]; p=0.24). Moreover, for 50% of the 10 FLNCLOF patients with dysfunction (5/56 (9%) overall), the ventricular dysfunction was unexplained by ischemia, hypertension, or valvular disease compared with 0/4 controls with dysfunction (p=0.24). Thus, by strict application of the 2019 consensus statement, 3/56 FLNCLOF individuals (5%) satisfied the criteria for classification as ACM vs. 0/103 controls (p=0.10).
The three individuals identified as having an ACM-associated phenotype had the following characteristics. All three were male. One was diagnosed with DCM in his 40s, with concurrent ventricular tachycardia and resultant syncope. An ICD was placed in this patient for secondary prevention (with a subsequent appropriate shock), and the pathogenic FLNC variant in this individual was known via clinical genetic testing. Another individual had New York Heart Association class II heart failure in his 70s with atrial fibrillation and frequent PVCs noted on ECG, with an ICD placed for primary prevention. The third (in his 70s) had a globally hypokinetic LV, with atypical late gadolinium enhancement in the midwall on CMR, with non-sustained VT and polymorphic PVCs.
Of the remaining two FLNCLOF individuals with unexplained ventricular dysfunction (and no concomitant arrhythmia), one was a female diagnosed with DCM in her 40s, with later development of non-sustained VT, ICD and left ventricular assist device placement. The likely pathogenic FLNC variant was known via clinical genetic testing in this individual. The other was also female, with normal LVEF but diffuse right ventricular hypokinesis in her 50s, confounded by lung cancer with chronic malignant pericardial effusion. This patient also later developed supraventricular tachycardia and AF, with frequent PVCs noted on ECG.
Discussion
Genetic variants associated with ‘high-risk’ arrhythmogenic conditions—such as pathogenic variants in LMNA, PKP2, and SCN5A—are increasingly being considered medically actionable and recommended for clinical genomic screening.11 Loss-of-function variants in FLNC have been recognized as a potential cause of DCM/ACM and heart failure from disease-based cohorts,2,7,9,12 and indeed, this recognition has led to the recent addition of FLNC to such professional society recommendations for secondary genomic findings.11 However, the disease burden associated with FLNC variants in broader clinical populations (i.e., not ascertained through clinical symptoms or family history) have not been described to inform clinical management in relation to such secondary findings. This work begins to address this gap by evaluating the prevalence and associated phenotype of individuals with FLNCLOF variants in the MyCode cohort—a large, healthcare-seeking population. In this cohort, individuals carrying FLNCLOF variants have increased odds for heart disease-associated phenotypes (cardiomyopathy and arrhythmia) as well as quantitative changes in cardiac structure and function compared with controls. Moreover, detailed cardiologist chart review confirmed a significantly increased burden of ventricular dysfunction associated with FLNCLOF, half of which was not explained by ischemia, hypertension, or valve disease; and with a large proportion (70%) presenting with concurrent arrhythmia. In contrast, few individuals had ventricular dysfunction in age- and sex-matched controls, all of which was explained by ischemia, hypertension, or valve disease, and only one of which (25%) had concurrent arrhythmia. Furthermore, the remaining two with unexplained dysfunction in the FLNCLOF group eventually developed arrhythmia. These data provide strong evidence that incidental/secondary identification of FLNCLOF is both medically relevant and actionable via medication, arrhythmia monitoring, or if warranted, ICD placement.
Phenotype Characteristics of FLNCLOF through Genome-First Approach
There has been considerable attention over the last 5 years to defining the disease characteristics associated with FLNC variants identified in large disease-based cohorts. These studies have broadly found that cardiomyopathy in individuals with FLNCLOF is characterized by LV dysfunction/dilation, a high burden of malignant arrhythmia/sudden cardiac arrest, frequent findings of myocardial fibrosis, and reported high rates of penetrance.2,3,6,10,13,14 Despite the stark differences in clinical context, the characteristics of this genome-first cohort generally recapitulate these findings. For example, FLNCLOF variants were associated with LV dilatation (increased LVIDd) and decreased LVEF, with 25% of FLNCLOF patients being classified as having ventricular dysfunction from EHR-based phenotypes (18% via chart review). Similarly, while affecting relatively small proportions of FLNCLOF patients, this analysis revealed evidence of potentially severe arrhythmias based on significantly increased use of implanted defibrillators, significantly increased odds of SVT, and 13% and 5% of patients with history of VT and sudden cardiac arrest, respectively.
Characterizing the risk of severe arrhythmia for individuals with FLNCLOF is particularly important given that FLNC is included in the Heart Rhythm Society’s list of genes that warrant consideration for ICD placement for primary prevention of ACM-associated sudden cardiac death.15 Furthermore, emerging data suggest that, as for LMNA and DSP,16 significant arrhythmia risk may be present with FLNCLOF even in the setting of only mild reductions in LVEF,10,14 potentially requiring alternate criteria for use. The application of these criteria to the specific case of secondary findings warrants particular attention and additional study.
In that regard, consideration of the apparent penetrance observed in this cohort is relevant. While prior studies have reported disease penetrance as high as 70% in FLNCLOF families with known cardiomyopathy,2 the proportion of individuals with FLNCLOF from MyCode exhibiting penetrant disease is considerably lower, which is consistent with similar genome-first observations in other cardiac-associated genes.17–19 Specifically, 9% of patients had ventricular dysfunction not explained by ischemia, hypertension, or valve disease—arguably representing a conservative estimate of penetrant disease. For instance, it is worth noting that, in an additional 7% of cases, arrhythmia was part of the disease presentation of dysfunction potentially attributable to other co-morbidities. In such cases, the potential modulating effect of the variant on the disease course in those individuals is difficult to quantify. Furthermore, this estimate of penetrance is based on disease history to date, whereas some individuals may yet develop disease during their lifetime. Indeed, of those in the FLNCLOF group, affected individuals were apparently older (current median age 71 years [IQR:55–73]) than those unaffected (median 58 [45–71]; p=0.09). Despite this challenge, the observed penetrance estimate is comparable, if not higher, than prior analyses of other cardiomyopathy-associated genes in the MyCode cohort, such as the genes for ARVC (6%)17 and TTN (5–12%).20,21
An additional consideration is that FLNCLOF are highly constrained (i.e., LOF variants are not well tolerated in the population), with a loss-of-function observed/expected upper bound fraction (LOEUF) of 0.17 [95% CI 0.12–0.25].22 It follows logically that disease penetrance may be higher than the estimate observed herein. However, as pointed out above, we have shown in individuals from the same cohort, similar penetrance estimates for other cardiomyopathy-associated genes with LOEUF in the same range, specifically for ARVC-associated desmosome genes (LOEUF range 0.18 (DSP) – 0.79 (PKP2)) and TTN (0.33). These observations suggest that additional study regarding the relationship between variant tolerance and disease penetrance is warranted.
FLNC as an ACM-Associated Gene
Whereas some studies have reported FLNC associations with ARVC3,4,6,7,9; that is, the specific right ventricle-affecting subtype of ACM, the 2010 ARVC TFC exhibited apparently low sensitivity in our population-based cohort, though incomplete testing may have contributed to this low yield. In particular, repolarization and depolarization ECG criteria from the 2010 ARVC TFC were indistinguishable between FLNCLOF individuals and controls, a finding consistent with other reports, even in patients meeting other (non-ECG) TFC.6 Prospective studies with comprehensive evaluations for TFC will help clarify the prevalence of these and other ARVC TFC in FLNCLOF.
Instead, this work provides strong evidence that FLNCLOF variants primarily lead to left-sided disease. For example, 10% of FLNCLOF individuals demonstrated evidence (both ventricular dysfunction and arrhythmia) of ALVC in their EHR. This classification is challenged by the fact that ALVC, as yet, has no formal widely recognized diagnostic criteria.23 Some studies have proposed such criteria, which include LV systolic global or regional dysfunction, LGE, evidence of depolarization or repolarization abnormalities on ECG, and evidence of ventricular arrhythmias.13,24,25 However, in the current absence of consensus criteria, our definition represents a pragmatic approach that is both conducive to our EHR-based phenotyping and consistent with the broad definition of ACM by current HRS guidelines. These findings also highlight the potential utility of genotype-first approaches to the diagnosis, as well as the management, of ACM.15
Of note, several proposals for ALVC criteria have emphasized findings of LGE by CMR in the diagnostic scoring. For example, ring-like LGE patterns in the LV have been preferentially associated with DCM in individuals with desmoplakin (DSP) and FLNC variants.13,26 Unfortunately, the availability of contrast-enhanced CMR data was limited in these patients, precluding evaluation of LGE patterns or detailed regional ventricle structure/function, though this finding was observed in at least one FLNCLOF individual with ventricular dysfunction. Based on the presumed notion that myocardial fibrosis is the substrate for ventricular arrhythmias in ACM, such an assessment may uncover additional structural evidence of ALVC in FLNCLOF. Consistent LGE patterns have been reported in multiple disease-based FLNC cohorts already.6,10,13
Limitations
This study was from a single healthcare system, representing primarily European ancestry. There is an inherent possibility of survival bias in genomic screening ascertainment, which may have favored unaffected individuals. Variants detected via exome sequencing were not confirmed by orthogonal methods. Only rare, putative loss-of-function variants were investigated herein, so the potential disease association with rare missense variants has not been established and must be investigated. Despite supplementation with cardiologist chart review, the retrospective nature of this study, and the inherent potential for errors or missing data in the EHR, resulted in incomplete testing for ACM phenotypes, particularly Holter monitor data, so there may be additional evidence of disease in either group that was not observed herein. CMR findings were not investigated in this work due to the limited number of studies available, so the prevalence of LGE findings or other structural or functional abnormalities in these individuals is unknown. Finally, the effects of environmental factors, such as exercise history, may contribute to variable disease penetrance, and should be investigated in future studies.
Conclusion
In this retrospective study, individuals with FLNC loss of function variants identified via genome screening had increased odds of DCM and ACM phenotypes, including reduced systolic LV function and common ventricular arrhythmias, compared to variant-negative controls from the MyCode cohort. Physician chart review confirmed these EHR-based associations with cardiomyopathy and arrhythmia, with an estimated disease penetrance of at least 9%. The growing evidence of these associated disease phenotypes in individuals with FLNC variants supports the inclusion of this gene for surveillance as part of clinical genomic screening for earlier disease detection and potential prevention of serious adverse outcomes, which may include sudden cardiac death.
Supplementary Material
Acknowledgments:
The authors gratefully acknowledge the participation of MyCode participants and the sequencing efforts of Regeneron Genetics Center. This work was done in conjunction with Regeneron Genetics Center, as part of the DiscovEHR collaboration.
Sources of Funding:
Research reported in this publication was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R01HL141901. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures:
Dr. Calkins receives research support from Boston Scientific Corp. Ms. Tichnell and Dr. James receive salary support from this grant. Dr. Calkins is a consultant for Medtronic, Inc., Biosense Webster, Pfizer, StrideBio, and Abbott. Dr. James is a consultant for Pfizer and StrideBio, Inc. Ms. Murray is a consultant for MyGeneCounsel. Dr. Fornwalt is an employee of Tempus, Inc.
Nonstandard Abbreviations and Acronyms
- ACM
arrhythmogenic cardiomyopathy
- DCM
dilated cardiomyopathy
- ALVC
arrhythmogenic left ventricular cardiomyopathy
- LV
left ventricular
- LGE
late gadolinium enhancement
- CMR
cardiac magnetic resonance imaging
- ACMG
American College of Medical Genetics and Genomics
- EHR
electronic health records
- FLNC LOF
loss-of-function FLNC variant(s)
- RGC
Regeneron Genetics Center
- VCR
VCRome
- IDT
Integrated DNA Technologies
- ICD9/10
International Classification of Disease, 9th or 10th edition
- VT
ventricular tachycardia
- VF
ventricular fibrillation
- SVT
supraventricular tachycardia
- ICD
implantable cardioverter-defibrillator
- HF
heart failure
- AF
atrial fibrillation
- LVEF
left ventricular ejection fraction
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- TFC
task force criteria
- PVC
premature ventricular contraction
- IQR
inter-quartile range
- OR
odds ratio
- LVIDd
left ventricular internal diameter at end-diastole
- IVSd
interventricular septum thickness in diastole
- SCA
sudden cardiac arrest
- LOEUF
loss-of-function observed/expected upper bound fraction
Footnotes
References:
- 1.Corrado D, Zorzi A. Filamin C: A New Arrhythmogenic Cardiomyopathy–Causing Gene? JACC Clin Electrophysiol 2018;4:515–517. doi: 10.1016/j.jacep.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padrón-Barthe L, Duro-Aguado I, Jiménez-Jáimez J, Hidalgo-Olivares VM, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol 2016;68:2440–2451. doi: 10.1016/j.jacc.2016.09.927. [DOI] [PubMed] [Google Scholar]
- 3.Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, Gowan K, Jones KL, Brun F, Merlo M, et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC Clin Electrophysiol 2018;4:504–514. doi: 10.1016/j.jacep.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brun F, Gigli M, Graw SL, Judge DP, Merlo M, Murray B, Calkins H, Sinagra G, Taylor MR, Mestroni L, et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J Med Genet 2020:jmedgenet-2019–106394. doi: 10.1136/jmedgenet-2019-106394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ader F, De Groote P, Réant P, Rooryck-Thambo C, Dupin-Deguine D, Rambaud C, Khraiche D, Perret C, Pruny JF, Mathieu-Dramard M, et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin Genet 2019;96:317–329. doi: 10.1111/cge.13594. [DOI] [PubMed] [Google Scholar]
- 6.Hall CL, Akhtar MM, Sabater-Molina M, Futema M, Asimaki A, Protonotarios A, Dalageorgou C, Pittman AM, Suarez MP, Aguilera B, et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol 2020;307:101–108. doi: 10.1016/j.ijcard.2019.09.048. [DOI] [PubMed] [Google Scholar]
- 7.Oz S, Yonath H, Visochyk L, Ofek E, Landa N, Reznik-Wolf H, Ortiz-Genga M, Monserrat L, Ben-Gal T, Goitein O, et al. Reduction in Filamin C transcript is associated with arrhythmogenic cardiomyopathy in Ashkenazi Jews. Int J Cardiol 2020;317:133–138. doi: 10.1016/j.ijcard.2020.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Monda E, Frisso G, Rubino M, Caiazza M, Esposito A, Cirillo A, Fusco A, Palmiero G, Mazzaccara C, Pacileo R, et al. Potential role of imaging markers in predicting future disease expression of arrhythmogenic cardiomyopathy. Future Cardiol 2020:fca-2020–0107. doi: 10.2217/fca-2020-0107. [DOI] [PubMed] [Google Scholar]
- 9.Sveinbjornsson G, Olafsdottir EF, Thorolfsdottir RB, Davidsson OB, Helgadottir A, Jonasdottir A, Jonasdottir A, Bjornsson E, Jensson BO, Arnadottir GA, et al. Variants in NKX2–5 and FLNC Cause Dilated Cardiomyopathy and Sudden Cardiac Death. Circ Genomic Precis Med 2018;11:e002151. doi: 10.1161/CIRCGEN.117.002151. [DOI] [PubMed] [Google Scholar]
- 10.Akhtar MM, Lorenzini M, Pavlou M, Ochoa JP, O’Mahony C, Restrepo-Cordoba MA, Segura-Rodriguez D, Bermúdez-Jiménez F, Molina P, Cuenca S, et al. Association of Left Ventricular Systolic Dysfunction among Carriers of Truncating Variants in Filamin C with Frequent Ventricular Arrhythmia and End-stage Heart Failure. JAMA Cardiol 2021:1–11. doi: 10.1001/jamacardio.2021.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, Stewart DR, Amendola LM, Adelman K, Bale SJ, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021. doi: 10.1038/s41436-021-01172-3. [DOI] [PubMed] [Google Scholar]
- 12.Povysil G, Chazara O, Carss KJ, Deevi SVV, Wang Q, Armisen J, Paul DS, Granger CB, Kjekshus J, Aggarwal V, et al. Assessing the Role of Rare Genetic Variation in Patients With Heart Failure. JAMA Cardiol 2020. doi: 10.1001/jamacardio.2020.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, Akhtar MM, Protonotarios A, Gossios TD, Savvatis K, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Hear J - Cardiovasc Imaging 2019;44:1–11. doi: 10.1093/ehjci/jez188. [DOI] [PubMed] [Google Scholar]
- 14.Gigli M, Stolfo D, Graw S, Merlo M, Gregorio C, Chen SN, Dal Ferro M, Paldino A, De Angelis G, Brun F, et al. Phenotypic Expression, Natural History and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021. doi: 10.1161/CIRCULATIONAHA.121.053521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Hear Rhythm 2019;16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol 2019;74:1480–1490. doi: 10.1016/j.jacc.2019.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and Electronic Health Record-Based Phenotype of Loss-of-Function Genetic Variants in Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Genes. Circ Genomic Precis Med 2019;12:487–494. doi: 10.1161/CIRCGEN.119.002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation 2019;140:42–54. doi: 10.1161/CIRCULATIONAHA.119.039573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Begg CB. On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst 2002;94:1221–1226. doi: 10.1093/jnci/94.16.1221. [DOI] [PubMed] [Google Scholar]
- 20.Choi SH, Jurgens SJ, Weng LC, Pirruccello JP, Roselli C, Chaffin M, Lee CJY, Hall AW, Khera AV, Lunetta KL, et al. Monogenic and polygenic contributions to atrial fibrillation risk results from a national biobank. Circ Res 2020:200–209. doi: 10.1161/CIRCRESAHA.119.315686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ware JS, Cook SA. Role of titin in cardiomyopathy : from DNA variants to patient stratification. Nat Rev Cardiol 2017. doi: 10.1038/nrcardio.2017.190. [DOI] [PubMed] [Google Scholar]
- 22.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nat 2020 5817809 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-Dominant Arrhythmogenic Cardiomyopathy. An Under-Recognized Clinical Entity. J Am Coll Cardiol 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 24.Corrado D, Marra MP, Zorzi A, Beffagna G, Cipriani A, De Lazzari M, Migliore, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol 2020:108653. doi: 10.1016/j.ijcard.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Casella M, Gasperetti A, Sicuso R, Conte E, Catto V, Sommariva E, Bergonti M, Vettor G, Rizzo S, Pompilio G, et al. Characteristics of Patients with Arrhythmogenic Left Ventricular Cardiomyopathy: Combining Genetic and Histopathologic Findings. Circ Arrhythmia Electrophysiol 2020. doi: 10.1161/CIRCEP.120.009005. [DOI] [PubMed] [Google Scholar]
- 26.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020;141:1872–1884. doi: 10.1161/CIRCULATIONAHA.119.044934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, Murray MF, Smelser DT, Gerhard GS, Ledbetter DH. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18:906–13. doi: 10.1038/gim.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, Van Hout CV, Habegger L, Buckler D, Lai K-MV, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staples J, Maxwell EK, Gosalia N, Gonzaga-Jauregui C, Snyder C, Hawes A, Penn J, Ulloa R, Bai X, Lopez AE, et al. Profiling and Leveraging Relatedness in a Precision Medicine Cohort of 92,455 Exomes. Am J Hum Genet. 2018;102:874–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin MF, Rodeh O, Penn J, Bai X, Reid JG, Krsheninina O, Salerno WJ. GLnexus: joint variant calling for large cohort sequencing. bioRxiv 343970 (2018) doi: 10.1101/343970. [DOI] [Google Scholar]
- 31.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jing L, Ulloa Cerna AE, Good CW, Sauers NM, Schneider G, Hartzel DN, Leader JB, Kirchner HL, Hu Y, Riviello DM, et al. A Machine Learning Approach to Management of Heart Failure Populations. JACC Hear Fail. 2020;8:578–587. doi: 10.1016/j.jchf.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Raghunath S, Pfeifer JM, Ulloa-Cerna AE, Nemani A, Carbonati T, Jing L, van Maanen DP, Hartzel DN, Ruhl JA, Lagerman BF, et al. Deep Neural Networks can Predict New-Onset Atrial Fibrillation from the 12-lead Electrocardiogram and Help Identify Those at Risk of AF-Related Stroke. Circulation. 2021;143:1287–1298. doi: 10.1161/CIRCULATIONAHA.120.047829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carruth ED, Beer D, Alsaid A, Schwartz MLB, McMinn M, Kelly MA, Buchanan AH, Nevius CD, Calkins H, James CA, et al. Clinical Findings and Diagnostic Yield of Arrhythmogenic Cardiomyopathy through Genomic Screening of Pathogenic or Likely Pathogenic Desmosome Gene Variants. Circ Genom Precis Med. 2021;14:e003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Firth D Bias Reduction of Maximum Likelihood Estimates. Biometrika. 1993;80:27. [Google Scholar]
- 37.Loy A, Korobova J. Bootstrapping Clustered Data in R using lmeresampler. 2021. doi: 10.48550/arxiv.2106.06568. [DOI] [Google Scholar]
- 38.Field CA, Welsh AH. Bootstrapping clustered data. J R Stat Soc Ser B (Statistical Methodol 69, 369–390 (2007). [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) vol. 57 289–300 (1995). [Google Scholar]
- 40.Rousselet GA, Pernet CR, Wilcox RR. The Percentile Bootstrap: A Primer With Step-by-Step Instructions in R: 10.1177/2515245920911881 4, (2021). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.