

Abstract
The functional decline in T cells during their chronic stimulation, commonly referred to as T cell exhaustion, is a major limitation for current immunotherapy approaches. As modern medicine embraces therapeutic approaches that exploit the immuno-oncology interface, a primary question has become how to maintain T cell function over time in scenarios of prolonged tumor burden. Deciphering the molecular mechanisms of T cell exhaustion is now enabling the field to begin using cardinal features of T cell differentiation to develop biomarkers that can delineate responders from non-responders prior to treatment with T cell-based therapeutics. Furthermore, applying principles of basic T cell immunity toward the development of cancer treatments is laying a foundation for rational approaches to improve immunotherapy by redirecting T cells away from a dysfunctional developmental trajectory.
Keywords: cancer immunotherapy, immune checkpoint blockade, CAR T cells, epigenetics, T cell exhaustion
T cell exhaustion, a cell fate lineage
The gradual repression of a T cell’s developmental potential, now commonly referred to as T cell exhaustion, was first characterized using the murine model of chronic lymphocytic choriomeningitis viral (LCMV) infection where it was observed that virus-specific T cells eventually lost the ability to illicit effector functions after exposure to chronic viral antigen [1, 2]. Building upon the realization that the chronically stimulated cells do not simply die, efforts were made to characterize the functional changes in the cells and determine the developmental origin of T cell exhaustion. Subsequently, adoptive transfer studies were performed whereby exhausted T cells were transferred into an antigen-free environment and monitored. Resting these cells without exposure to antigen did not reinstate canonical memory features providing evidence that exhaustion is a cell fate commitment [3, 4]. Furthermore, unlike functional memory T cells that exhibit the cardinal feature of antigen-independent homeostatic proliferation, exhausted T cells do not undergo self-renewal in the presence of the homeostatic cytokines IL-7 and IL-15, rather they require antigen for maintenance [5]. To date a large body of work documenting the functional differences and unique characteristics of exhausted T cells has established exhaustion as a distinct developmental lineage.
After recognizing that exhausted T cells arise from a unique differentiation path, efforts began to determine steps in the cell fate decision process. Broadly, the studies identifying checkpoints in the exhaustion pathway have helped establish the concept that T cell exhaustion results from a distinct network of transcriptional programs. Several key transcription factors such as BATF, IRF4, c-Jun, NR4A, EOMES, and NFATC1 have been determined to play a critical role in driving T cell exhaustion [6, 7]. TOX, in particular, has been reported to be an essential transcription factor for the development and maintenance of T cell exhaustion [8–10]. In this piece, we will discuss signals that reinforce specific T cell differentiation states, examine both intrinsic and extrinsic barriers to current immunotherapy approaches and outline strategies to overcome these limitations through redirecting T cell fate decisions in order to shape the next generation of immunotherapy approaches.
Identifying immunotherapy responsive T cells
Epigenetic (see Glossary) reinforcement of T cell subset specification is thought to have evolved to tailor the function of effector and memory T cells during an immune response [11] and, in the case of T cell exhaustion, has been thought to limit immune pathology in scenarios of chronic diseases such as autoimmune disorders. However, this stable suppression of the adaptive immune response also has undesirable implications for the T cell response against cancer, thereby prompting approaches to reinvigorate dysfunctional T cells. Understanding the molecular determinants that suppress a sustained T cell effector response has allowed for strategies to redirect T cells away from an exhausted developmental trajectory and maintain cytotoxic T cell potential. One such approach, termed immune checkpoint blockade (ICB), works to reengage the endogenous immune response by interrupting T cell inhibitory signals. During the last decade of FDA approved ICB usage, many groups have investigated the cellular and molecular mechanisms driving response in the subset of patients that exhibit remission after checkpoint therapy [12, 13]. From these studies it has been concluded that ICB is most successful when applied at a time when antigen-specific T cells still possess an ability to function. Administering the checkpoint therapy itself does not reprogram T cells away from an exhausted state of differentiation [14, 15]. Instead, ICB efficacy relies on a stage of differentiation in which T cells remain poised to mount an effector response and proliferate in order to maintain anti-tumor functions. These studies illustrate the necessity for accurately defining the differentiation status of T cells as the stages of a T cell immune response can be specifically leveraged to enhance immunotherapy outcomes.
Antigen-driven T cell differentiation can be thought of as a continuum which begins with a naïve T cell and ends with terminal exhaustion. Along this spectrum, there is a ‘point of no return’ in which T cells undergo repression of effector and memory potential mediated by various epigenetic modifications, including histone and DNA methylation, that allow for cell-division transmission of acquired gene expression programs (Figure 1 & 2) [16–19]. Recent efforts to resolve the stage of development where a T cell becomes committed to the exhaustion fate has yielded tremendous insight into the dynamic relationship between chromatin accessibility and transcriptional regulation. Reinforcement of the exhaustion fate results from the progressive acquisition of repressive DNA methylation programming mediated by the de novo DNA Methyltransferase 3 Alpha (DNMT3A). These DNA methylation programs have also been shown to limit the effectiveness of a T cell response during T cell-mediated immunotherapy efforts. In order to determine which cancers and specifically which patients will be responsive, several groups have sought to define biomarkers for identifying precursor exhausted T cells which maintain stem-like properties that mediate the proliferative burst to ICB [20–22]. For instance, a minor subset of TCF1 (express TCF7) positive precursor exhausted T cells have now been identified that exhibit the ability to self-renew and give rise to daughter cells with exhaustion properties after exposure to chronic antigen [23]. From a clinical perspective, expression of the transcription factor TCF7 among tumor infiltrating CD8+ T cells was found to be predictive of positive outcomes in patients with melanoma treated with ICB [24]. Another example of precursor-exhausted cells promoting a response to ICB was observed with the murine LCMV system. Specifically, a transitory CX3CR1+ T cell subset was identified that maintained the ability to express effector molecules, control virus, and expand after checkpoint blockade [25]. The collective body of work characterizing exhaustion suggests that ICB must be administered prior to a critical point in T cell differentiation in order to invigorate a transitory population of T cells that have not yet become fully exhausted.
Key Figure, Figure 1. T cell differentiation trajectories impacting immunotherapy efficacy.
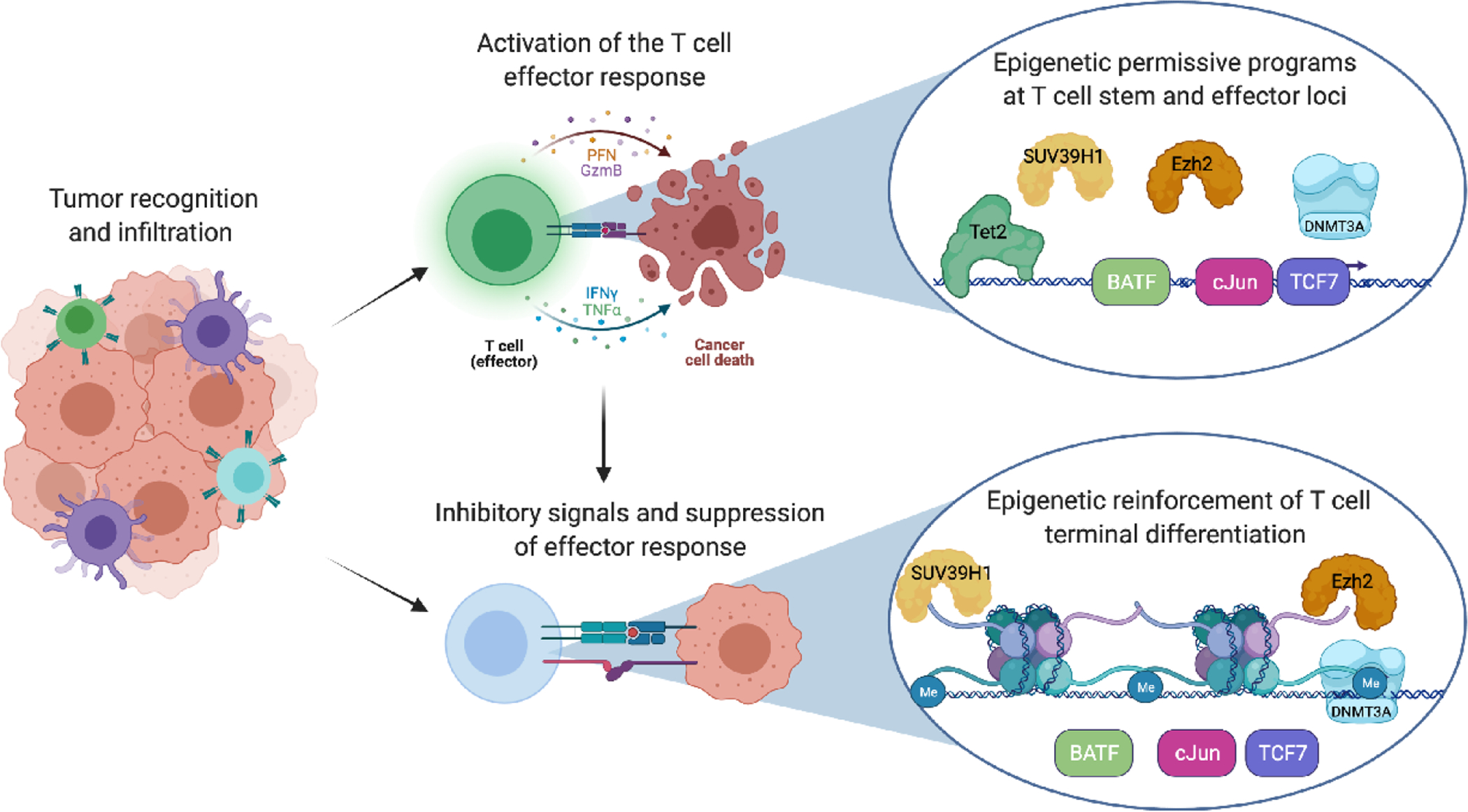
T cell function is limited by trafficking to the tumor as well as through immunosuppressive signals arising from the tumor microenvironment. Activation of the T cells requires antigen positive tumor cells as well as proper cross presentation and priming. Chronic antigen exposure results in epigenetic changes that include both DNA methylation and histone modifications consistent with T cell exhaustion.
Figure 2. T cell exhaustion differentiation includes a point of no return after which impedes immunotherapy efforts.
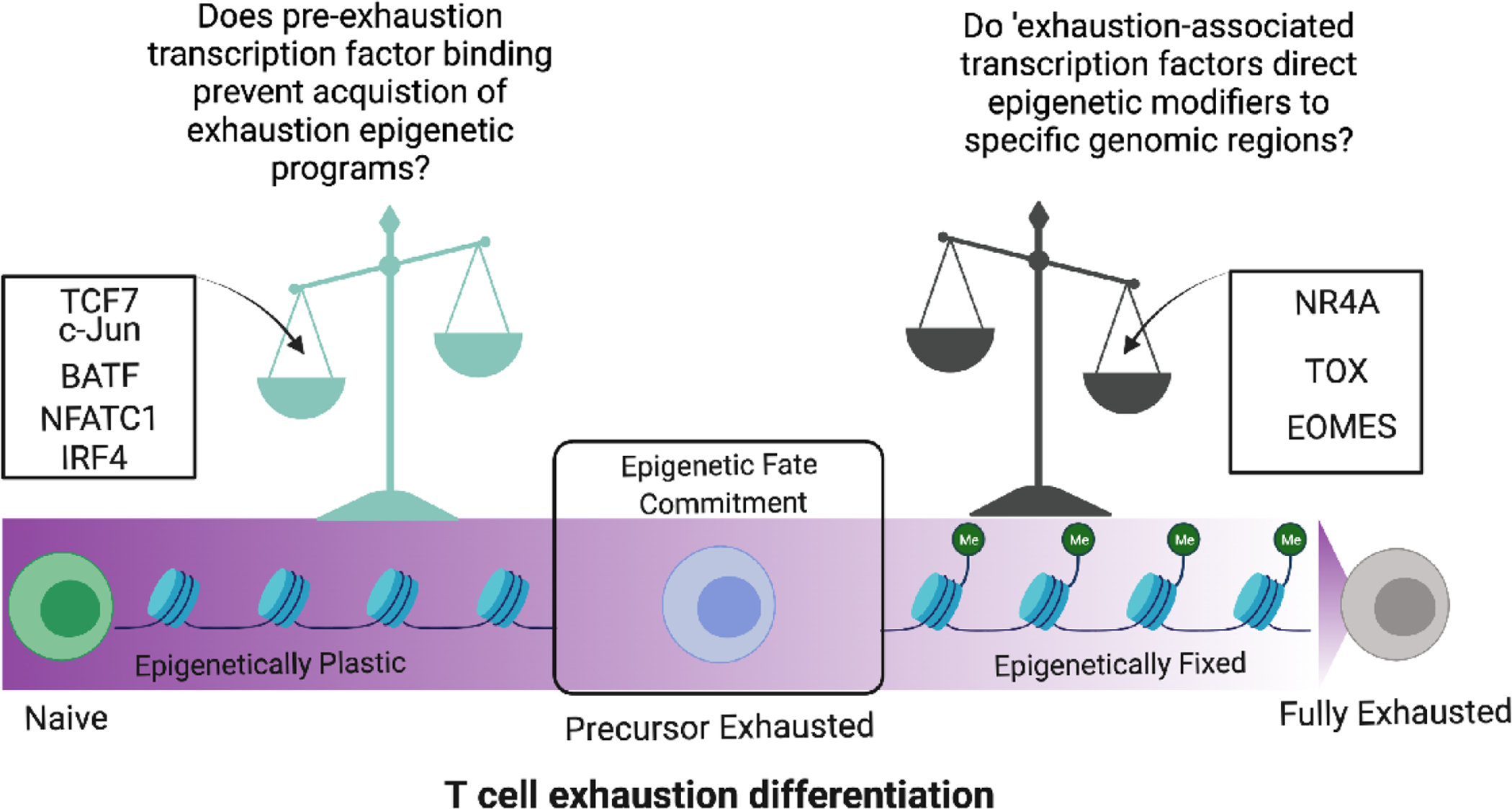
The spectrum of T cell exhaustion differentiation is influenced by the action of several key transcription factors that can either maintain T cells in an epigenetically plastic stem-like state or tip the balance toward terminal exhaustion. An epigenetic checkpoint determines the fate commitment of precursor exhausted T cells which can either become rejuvenated to maintain anti-tumor function or epigenetically fixed in a fully exhausted state.
Parallel efforts examining the role of epigenetics and the functionality of exhausted T cells demonstrated that specific T cell differentiation states were associated with distinct epigenetic signatures. For instance, SUV39H1, LSD1, and TET2 have been identified as critical epigenetic regulators involved in restricting T cell fate potential (Figure 1) [16, 26–28]. A recent focus on resolving the crosstalk between ‘fate-defining’ transcription factors and epigenetic programs has provided evidence that several stem-associated transcription factors can serve to preserve specific chromatin states during T cell activation [29]. Progressive restriction of T cell developmental potential requires specifically localizing epigenetic modifiers to discrete regions of the genome, and while the specificity determinates for each epigenetic state are not yet defined, genetic disruption of the epigenetic modifiers have established a clear role for their involvement in restricting fate potential of the T cell. Notably, knocking out the de novo DNA methyltransferase Dnmt3a in CD8 T cells prior to exhaustion resulted in the Dnmt3a KO T cells maintaining an ability to respond to ICB despite exposure to chronic antigen. These data further confirmed that DNA methylation programming is critical for shutting down the T cell response to checkpoint blockade [14]. From these studies it was concluded that Dnmt3a-mediated DNA methylation preserved the T cells in a more multipotent developmental state conducive to ICB therapy. Moreover, understanding the association between epigenetic modifications and T cell differentiation status are contributing to identification of biomarkers that can be used to predict T cell developmental potential (T cell multipotency index [30, 31]) which is coupled to immunotherapy response. While there are nuances to murine and human T cell responses in both viral and tumor settings, the underlying commonality remains that ICB efficacy requires T cells with some developmental plasticity that precludes terminal exhaustion.
The transition from stem to exhausted T cells
In addition to the T cell intrinsic mechanisms that regulate a multipotent differentiation state that is responsive to immunotherapeutic efforts, the range of signals that contribute to the epigenetic reinforcement of T cell differentiation is another important consideration for T cell function (Figure 2). The tumor microenvironment (TME) has been shown to include a complex network of cancer cells, inflammatory cytokines, antigen-presenting cells and innate immune cells that all impact T cell function. Dendritic cells (DCs) in particular play an essential role in fighting tumor by not only priming T cells in the tumor-draining lymph node but also through regulating T cell function in the TME [32]. A discrete DC state termed DC3s was recently identified as a key player for intratumoral lymphocyte activation. Specifically, the chemokine receptor CXCR6 positions cytotoxic lymphocytes in proximity to the DC3s of the tumor stroma. Interaction between the stem-like T cells and DC3s promotes a proliferative response that mediates lymphocyte anti-tumor functions [33]. Maintaining the functional pool of transitory endogenous T cells is essential for effective homing to elicit a sustained anti-tumor response. These studies emphasize the need to not only identify the population of T cells mediating a clinical response but also the environmental factors that skew T cell differentiation toward a therapeutically effective T cell subset.
Along with informing on the efficacy of immunotherapies that utilize the endogenous population of T cells, T cell differentiation is critical for other therapeutic strategies such as chimeric antigen receptor (CAR) T cells engineered to target tumor antigen. Most CAR T cell protocols use a heterogenous starting population of bulk CD4 and CD8 T cells that span the range of T cell developmental potential. However it is well established that more plastic memory T cells, such as central memory (Tcm) and stem cell memory (Tscm) cells, possess a greater capacity for effector function and proliferation which results in heightened eradication of tumors in preclinical model systems [34–37]. Supporting these preclinical findings, a greater frequency of the less terminally differentiated T cell subsets (naive, Tscm and Tcm) found in the apheresis product used to generate CD19-CAR T cells has been associated with CAR T cell persistence beyond 6 months in clinical settings [38]. These preclinical and clinical observations suggest manufacturing CAR T cells by enriching for a desired T cell population may allow the properties of the endogenous T cell subsets to play a critical role in the clinical outcome. While this is a well-accepted goal, selecting multipotent T cell subsets for CAR T cell product generation presents with challenges in maintaining the stem-associated characteristics not only during the manufacturing process but after infusion into patients. T cell activation, transduction, proliferation, and response to antigen can all drive T cell differentiation and limit stem-associated properties. Developing the most efficacious CAR T cell product begins prior to the engineering process by considering the starting population of T cells and continues after generation of the CAR T cells with strategies to maintain CAR T cell function.
While selecting a desired starting population of T cells to generate CAR T cells is ideal, this scenario is not always an option when caring for patients. For instance, patients who are eligible for CAR T cell therapy typically have relapsed/refractory disease. With an extensive cancer history, the patient’s T cells have been exposed to chronic tumor antigen as well as a multitude of chemotherapeutic agents. Indeed, leukemia induced T-cell dysfunction has been reported to result in suboptimal autologous CAR T cells that are less effective in clearing tumor as compared to CAR T cells generated from healthy individuals [39, 40]. Additionally, chemotherapy itself has been shown to diminish T cell function and ultimately skew the T cell subsets toward the more terminally differentiated effector memory (Tem) population [41]. These inherent limitations to the starting pool of T cells impose potential limitations in CAR T cell function even before infusing the cells into patients. The current barriers described above in using patient-derived T cells further emphasizes the need to focus on approaches to prevent or revert T cell terminal differentiation prior to CAR T cell generation.
Rational approaches to epigenetically reprogram T cells for immunotherapy
CAR T cell therapy is an attractive treatment modality for relapsed or refractory B cell malignancies yet less than half of the patients maintain long-term disease control and often require hematopoietic stem cell transplantation [42, 43]. Of the patients that relapse after CD19-CAR T cell therapy, over half recur with CD19+ leukemia suggesting a decline in CAR T cell function over time [31, 43]. While CAR T cell exhaustion in a clinical setting has not been extensively defined, the mechanisms that contribute to CAR T cell dysfunction are thought to overlap with the process of exhaustion that occurs in endogenous T cells. Therefore, recent efforts from our group and others have attempted to characterize and prevent CAR T cell exhaustion based on our understanding of endogenous T cell differentiation.
Given the link between the strength of T cell stimulation and exhaustion observed among endogenous T cell responses, one approach to examine CAR T cell dysfunction involved interrogation of a tonic signaling CAR T cell model system to induce exhaustion. In this hyper-stimulating model, a functional deficiency in the AP-1 factor c-Jun was found to mediate CAR T cell dysfunction thus prompting efforts to overexpress c-Jun in order to prevent exhaustion [44, 45] (Figure 2). To more broadly assess mechanisms controlling T cell ‘stemness’ several groups have performed CRISPR-based screens looking for novel hits that preserve T cell proliferation and effector function. One such example of this screen was the discovery of Regnase, an RNA helicase critical for promoting terminal differentiation of murine T cells during an in vivo tumor challenge [46]. A complementary approach was applied to primary human T cells by using a library of >75,000 guide RNAs to screen for genes that impact on T cell proliferation in an ex vivo assay. Broadly, the authors compiled a list of established and novel genes involved in TCR signaling including SOCS1, CBLB, and Rasa2. Inhibition of these genes involved in T cell activation can be targeted to improve T cell anti-tumor functions, [47].
Additional strategies to impede exhaustion involve reversal and prevention of repressive epigenetic programs. For instance, disruption of Regnase, Tet2, and LSD1 have been targeted to enhance immunotherapies [16, 46, 48]. Given that DNA methylation programs are causal in establishing T cell exhaustion among endogenous T cell responses, a logical approach to preserve CAR T cell function involves preventing the acquisition of DNMT3A-mediated methylated programming. As compared to the controls, healthy donor-derived DNMT3A KO CAR T cells exhibited a preserved functional capacity against chronic tumor in both in vitro chronic stimulation assays as well as in vivo NSG murine tumor models involving rechallenge experiments in which the CAR T cells were reintroduced to tumor antigen after >100 days [17]. The ability of DNMT3A KO CAR T cells to control a secondary tumor challenge after a long period of rest following the primary tumor challenge provides evidence that deletion of DNMT3A in CAR T cells preserves memory potential and allows the CAR T cells to protect against tumor relapse. These results involving suppression of DNMT3A function in preclinical systems are also relevant in a clinical setting. Examination of publicly available DNMT3A gene expression from CD19-CAR T cells prior to infusion into patients with chronic lymphocytic leukemia (CLL) revealed that patients who responded to therapy had higher DNMT3A target gene expression as defined by the genes identified in the preclinical studies. The ability to detect differences in the expression of DNMT3A target genes in the CAR T cell product prior to infusion suggests these tools can be applied to predict which patients will undergo clinical response to therapy and perhaps could guide efforts to intervene for patients who are less likely to respond. In addition to knocking out DNMT3A to prevent acquisition of DNA methylation, other groups have sought to use DNA demethylating agents such as decitabine to revert exhaustion-associated DNA methylation programs in CAR T cells. Decitabine treated CAR T cells were found to have improved proliferation, cytokine production, and anti-tumor ability in preclinical settings [49]. Taken together, these studies apply the fundamental principles of T cell exhaustion to describe a molecular framework for CAR T cell exhaustion and provide epigenetic-based strategies to preserve CAR T cell function.
The characterization of CAR T cell exhaustion identified in preclinical model systems has now been extended to clinical settings. CAR T cells collected from leukemia patients were analyzed in order to identify the dysfunctional programs that become established during an in vivo anti-tumor response. Specifically, CD8+ CD19-CAR T cells were subjected to DNA methylation profiling both prior to infusion and after being isolated longitudinally from patients [31]. Post-infusion, the CAR T cells underwent global DNA methylation reprogramming, which was coupled to gene regulatory elements that broadly repressed memory potential and demonstrate a progression toward exhaustion-progenitor T cells. Further, the development of CAR T cell exhaustion was associated with acquisition of DNA methylation at sites previously shown to be targeted by DNMT3A. Upon reemergence of antigen positive cells within the patient, the remaining CD19-CAR T cells were unable to mount a recall response consistent with an exhausted fate. Collectively these studies support a gene editing approach to knock out DNMT3A in order to preserve CAR T cell function.
Safety considerations for gene editing T cells
While all therapeutically-directed gene editing approaches should be performed with caution, a specific focus on DNMT3A warrants further assessment given that DNMT3A mutations have been associated with hematologic malignancies [50]. However, recent work suggests that DNMT3A-mutations alone are not drivers of malignancy. This has been examined in murine models, where only a cooperative knockout of DNMT3A with activating mutations of NOTCH1 in hematopoietic cells was able to induce leukemogenesis [51]. Furthermore, these mutations in mature T cells were not sufficient to drive leukemia development [51]. Additionally, multiple reports describing DNMT3A mutations that result in enrichment of DNMT3A mutant cells in hematopoietic lineages have shown no signs of malignancy [52, 53].
As opposed to driving malignant transformation, DNMT3A mutations may actually be beneficial in certain instances. For example, mutations in DNMT3A were identified as candidate drivers of age-related clonal hematopoiesis in a healthy supercentenarian with unexpectedly functional T cell immunity and no evidence of malignancy [53]. Another study evaluated donor DNMT3A clonal hematopoiesis (CH) transmitted to recipients during allogeneic hematopoietic stem cell transplantation. Sequencing was performed on over 1700 donors who were 40 years of age or older and CH was present in 22.5% with DNMT3A and TET2 accounting for the most common mutations [54]. The presence of DNMT3A CH was shown to be associated with a significant improvement in survival due to reduced risk of relapse. Interestingly, the improvements in relapse, progression free survival, and overall survival associated with DNMT3A clonal hematopoiesis were lost if the patient received post-transplant cyclophosphamide. Given that cyclophosphamide is administered to deplete donor-derived T cells, the benefits of DNMT3A clonal hematopoiesis in this allogeneic transplant setting are seemingly related to T cell function. To evaluate whether malignant transformation evolved directly from donor CH, this study also tracked development of donor cell leukemia (DCL). Samples from six patients who developed DCL were sequenced and three patients with DCL were identified to have mutations in myelodysplastic syndrome-associated splicing factors, two had TP53 mutations, and one had a mutation in a gene previously identified as a leukemia predisposition gene, DDX41. No recipients of DNMT3A- or TET2-mutated donor CH developed DCL without additional genetic mutations. Taken together, these results suggest that DNMT3A deletion does not result in malignant transformation and further argues that individuals should not be excluded as transplant donors based solely on the presence of DNMT3A CH.
CONCLUDING REMARKS
Cancer treatment is shifting from a protocol-centered one-size-fits-all approach to a patient-centered, individually tailored therapy. Incorporating a patient’s immune system into the treatment schema is a promising tactic to not only fight disease but also lessen the life-long side effects associated with harsh chemotherapeutic agents. Therefore, characterizing the barriers that restrict a T cell response is critical in order to implement strategies to overcome such limitations. Insights gained from studying basic mechanisms regulating the cell fate commitment processes of exhaustion have served as an example for translational science. Future efforts must work to incorporate our understanding of T cell exhaustion into cellular therapy approaches in order to limit T cell dysfunction and establish a durable anti-tumor response [55] (see Outstanding Questions).
Outstanding questions box.
Do CAR T cells need survival signals to remain functional?
Can epigenetically engineered T cells overcome the suppressive TME and maintain function against solid tumors?
Will preventing CAR T cell exhaustion prevent tumor relapse and potentially lead to CAR T cell therapy as a curative treatment without the necessity for subsequent hematopoietic stem cell transplant?
Can multipotent transcription factor overexpression reverse exhaustion in epigenetically fate committed cells?
Box 1: Heterogeneity along the path to T cell exhaustion.
The developmental pathway to T cell exhaustion is composed of heterogenous population of T cells. Historically TCF1 has been identified as a critical transcription factor regulating the T cell transition from precursor exhausted to fully exhausted. The self-renewing capacity of precursor-exhausted cells, which are TCF1 positive, plays an essential role in preserving T cells in an environment of continued antigen exposure that will ultimately become fully exhausted. TCF1 positive T cell populations are found in secondary lymphoid tissues while the TCF1 negative cells are found in peripheral tissues at the site of antigen [20].
Box 2: Strategies to rationally reprogram T cells for immunotherapy.
Therapeutic strategies for epigenetic reprogramming of T cells to improve immunotherapy, elaborated on in greater detail by others [56], is now beginning to leverage the wealth of data obtained from preclinical studies. Over the last several years many clinical trials have been initiated that have combined epigenetic-targeted therapeutics with immune checkpoint blockade approaches. The rationale for such approaches has been to broadly increase the immunogenicity of the tumors and enhance/recover anti-tumor properties the T cells. Such combination therapy approaches have had some success but there remains significant room for improvement. As opposed to broadly reversing DNA methylation using demethylating agents such as decitabine, targeted approaches using small molecules can be used to specifically modify genes identified to be associated with response to ICB and CAR T cell therapy. Another potential option involves cytokines which have been shown to alter T cells at an epigenetic level [57]. Addition of specific cytokines during the CAR T cell manufacturing process to could be used to induce desired properties. Collectively, correlates of therapeutic response from combination therapies will be able to feedback into the preclinical model systems to better define the mechanisms enabling improved efficacy.
Highlights.
T cell exhaustion is a cell fate decision that limits the efficacy of immunotherapy
T cell-based immunotherapy approaches need to be applied prior to terminal exhaustion
Strategies to epigenetically reprogram T cells can redirect their developmental fate resulting in sustained anti-tumor ability
Acknowledgments:
This work was supported by the National Institutes of Health (1R01AI114442 to BY and LRP to CCZ), the National Comprehensive Cancer Network Young Investigator Award (to CZ), Alex’s Lemonade Stand Foundation Young Investigator Grant (to CZ), Stand Up to Cancer- SU2C (to BY), the American Lebanese Syrian Associated Charities (ALSAC) to BY, and Assisi foundation to BY. Illustrations in the figures were generated using Biorender. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. BY and CCZ have patents related to preserving stemness of CAR T cells.
Glossary
- Immunotherapy
Therapeutic modalities that utilize the endogenous immune system or adoptive transfer of immune cells into a host with the goal of eliminating diseased cells or pathogens
- Immune checkpoint blockade
Therapeutic procedure involving the infusion of antibodies capable of blocking ligand-receptor interactions of inhibitory receptors such as CTLA-4 and PD-1 on T cells
- Chimeric antigen receptor (CAR) T-cell therapy
A form of immunotherapy whereby T cells are engineered to express an antibody-derived surface fragment specific for a particular antigen associated with a tumor or pathogen, and then subsequently transferred into the afflicted individual to treat the disease
- Precursor exhausted T cells (TPEX)
TCF1+ precursor T cells that maintain the ability to self-renew and can replenish exhausted T cells
- T cell exhaustion
A differentiation state of T cells induced by persistent antigen stimulation that results in a functional impairment of the cell, which can be reinforced by epigenetic modifications
- Epigenetic
Covalent modifications to the DNA and/or histones that can impact on chromatin accessibility and ultimately reinforce cell type-specific gene expression programs
- DNA methylation
An epigenetic modification whereby a methyl group is added to a DNA base, most commonly observed at cytosines in mammalian cells. This modification can result in changes in transcriptional activity when occurring at promoter of enhancer elements
- T cell multipotency index
A novel index based on the DNA methylation status of the T cells that assigns a score ranging from 0–1 that is predictive of the general differentiation status of the T cell
- TCF1
Among a group of transcription factors associated with the Wnt signaling pathway that contribute to preservation of the multipotent developmental potential among T cells
- TOX
HMG-box transcription factor involved in driving the cell fate commitment of exhausted T cells
- c-Jun
transcription factor that is part of the AP-1 C-FOS-C-Jun heterodimer which has been shown to prevent T cell exhaustion
- BATF
Leucine zipper transcription factor involved in regulating effector T cell differentiation
- NFATC1
Nuclear Factor of Activated T cells 1, transcription factor involved in T cell differentiation which promotes T cell exhaustion
- IRF4
Interferon Regulatory Factor, transcription factor that promotes T cell exhaustion while limiting memory T cell development
- NR4A
Family of orphan nuclear receptors including the transcription factors NR4A1, NR4A2, and NR4A3 that are involved in the development of T cell exhaustion
- EOMES (Eomesodermin)
T-box transcription factor induced in effector CD8 T cells that mediates CD8 T cell exhaustion
- Naïve T cell
A T cell that has exited thymic selection but has not yet undergone antigen-driven differentiation
- Central memory T cell (Tcm)
A subset of memory T cells expressing CD62L and CCR7 that are able to recirculate between the blood and secondary lymphoid organs
- Effector memory T cell (Tem)
A subset of memory T cells lacking CD62L and CCR7, generally excluded from secondary lymphoid organs and able to circulate between the blood and peripheral tissues
- Stem cell memory T cell (Tscm)
A subset of memory T cells that has an ability to undergo antigen independent self-renewal and develop into other effector and memory T cell subsets
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zajac AJ et al. (1998) Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188 (12), 2205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zinkernagel RM et al. (1993) Effector T-cell induction and T-cell memory versus peripheral deletion of T cells. Immunol Rev 133, 199–223. [DOI] [PubMed] [Google Scholar]
- 3.Wherry EJ et al. (2004) Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A 101 (45), 16004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Utzschneider DT et al. (2013) T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol 14 (6), 603–10. [DOI] [PubMed] [Google Scholar]
- 5.Shin H and Wherry EJ (2007) CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol 19 (4), 408–15. [DOI] [PubMed] [Google Scholar]
- 6.Man K et al. (2017) Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47 (6), 1129–1141 e5. [DOI] [PubMed] [Google Scholar]
- 7.Martinez GJ et al. (2015) The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42 (2), 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfei F et al. (2019) TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571 (7764), 265–269. [DOI] [PubMed] [Google Scholar]
- 9.Khan O et al. (2019) TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571 (7764), 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott AC et al. (2019) TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571 (7764), 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frias AB et al. (2021) Epigenetic regulation of T cell adaptive immunity. Immunol Rev 300 (1), 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shields BD et al. (2017) Indicators of responsiveness to immune checkpoint inhibitors. Sci Rep 7 (1), 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morad G et al. (2021) Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184 (21), 5309–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghoneim HE et al. (2017) De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170 (1), 142–157 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pauken KE et al. (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354 (6316), 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y et al. (2021) LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat Commun 12 (1), 6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prinzing B et al. (2021) Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med 13 (620), eabh0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdel-Hakeem MS et al. (2021) Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol 22 (8), 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yates KB et al. (2021) Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol 22 (8), 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Im SJ et al. (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537 (7620), 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brummelman J et al. (2018) High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 215 (10), 2520–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gettinger SN et al. (2018) A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun 9 (1), 3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Utzschneider DT et al. (2020) Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol 21 (10), 1256–1266. [DOI] [PubMed] [Google Scholar]
- 24.Sade-Feldman M et al. (2018) Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175 (4), 998–1013 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudson WH et al. (2019) Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity 51 (6), 1043–1058 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace L et al. (2018) The epigenetic control of stemness in CD8(+) T cell fate commitment. Science 359 (6372), 177–186. [DOI] [PubMed] [Google Scholar]
- 27.Lee M et al. (2021) Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating Lymphocytes. Cancer Res 81 (8), 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henning AN et al. (2018) Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 18 (5), 340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong Y et al. (2022) Hierarchical regulation of the resting and activated T cell epigenome by major transcription factor families. Nat Immunol 23 (1), 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelsamed HA et al. (2020) Beta cell-specific CD8(+) T cells maintain stem cell memory-associated epigenetic programs during type 1 diabetes. Nat Immunol 21 (5), 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zebley CC et al. (2021) CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep 37 (9), 110079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerhard GM et al. (2021) Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J Exp Med 218 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Pilato M et al. (2021) CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184 (17), 4512–4530 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flynn JK and Gorry PR (2014) Stem memory T cells (TSCM)-their role in cancer and HIV immunotherapies. Clin Transl Immunology 3 (7), e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gattinoni L et al. (2011) A human memory T cell subset with stem cell-like properties. Nat Med 17 (10), 1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabatino M et al. (2016) Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 128 (4), 519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klebanoff CA et al. (2012) Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother 35 (9), 651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen GM et al. (2021) Integrative bulk and single-cell profiling of pre-manufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin H et al. (2018) Murine pre-B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood 132 (18), 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palma M et al. (2017) T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 102 (3), 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sommermeyer D et al. (2016) Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30 (2), 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraietta JA et al. (2018) Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24 (5), 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Summers C et al. (2021) Hematopoietic Cell Transplantation after CD19 Chimeric Antigen Receptor T Cell-Induced Acute Lymphoblastic Lymphoma Remission Confers a Leukemia-Free Survival Advantage. Transplant Cell Ther. [DOI] [PubMed] [Google Scholar]
- 44.Lynn RC et al. (2019) c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576 (7786), 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seo H et al. (2021) BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol 22 (8), 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei J et al. (2019) Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576 (7787), 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shifrut E et al. (2018) Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175 (7), 1958–1971 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fraietta JA et al. (2018) Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558 (7709), 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y et al. (2021) Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun 12 (1), 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ley TJ et al. (2010) DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25), 2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kramer AC et al. (2017) Dnmt3a regulates T-cell development and suppresses T-ALL transformation. Leukemia 31 (11), 2479–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tovy A et al. (2020) Tissue-Biased Expansion of DNMT3A-Mutant Clones in a Mosaic Individual Is Associated with Conserved Epigenetic Erosion. Cell Stem Cell 27 (2), 326–335 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van den Akker EB et al. (2020) Dynamic clonal hematopoiesis and functional T-cell immunity in a supercentenarian. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gibson CJ et al. (2021) Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol, JCO2102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zebley CC et al. (2020) Rewriting History: Epigenetic Reprogramming of CD8(+) T Cell Differentiation to Enhance Immunotherapy. Trends Immunol 41 (8), 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Topper MJ et al. (2020) The emerging role of epigenetic therapeutics in immune-oncology. Nat Rev Clin Oncol 17 (2), 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zebley CC et al. (2021) Proinflammatory cytokines promote TET2-mediated DNA demethylation during CD8 T cell effector differentiation. Cell Rep 37 (2), 109796. [DOI] [PMC free article] [PubMed] [Google Scholar]