

Abstract
Objective:
To determine cell-specific gene expression profiles that contribute to development of abdominal aortic aneurysms (AAAs).
Background:
AAAs represent the most common pathological aortic dilation leading to the fatal consequence of aortic rupture. Both immune and structural cells contribute to aortic degeneration, however, gene specific alterations in these cellular subsets are poorly understood.
Methods:
We performed single-cell RNA sequencing (scRNA-seq) analysis of AAAs and control tissues. AAA-related changes were examined by comparing gene expression profiles as well as detailed receptor-ligand interactions. An integrative analysis of scRNA-seq data with large genome wide association study (GWAS) data was conducted to identify genes critical for AAA development.
Results:
Using scRNA-seq we provide the first comprehensive characterization of the cellular landscape in human AAA tissues. Unbiased clustering analysis of transcriptional profiles identified seventeen clusters representing eight cell lineages. For immune cells, clustering analysis identified four T-cell and five monocyte/macrophage subpopulations, with distinct transcriptional profiles in AAAs compared to controls. Gene enrichment analysis on immune subsets identified multiple pathways only expressed in AAA tissue, including those involved in mitochondrial dysfunction, proliferation, and cytokine secretion. Moreover, receptor-ligand analysis defined robust interactions between vascular smooth muscle cells (SMCs) and myeloid populations in AAA tissues. Lastly, integrated analysis of scRNA-seq data with GWAS studies determined that VSMC expression of SORT1 is critical for maintaining normal aortic wall function.
Conclusions:
Here we provide the first comprehensive evaluation of single cell composition of the abdominal aortic wall and reveal how the gene expression landscape is altered in human AAAs.
MINI ABSTRACT
Using single-cell RNA sequencing we provide a comprehensive characterization of the cellular landscape in human abdominal aortic aneurysm (AAA) tissues. We demonstrate a diverse array of structural and immune cell subsets contribute to AAA pathogenesis. Integration of the scRNA-seq with GWAS data demonstrates that SORT1 within vascular smooth muscle cells exerts a critical role in maintaining normal aortic wall function.
INTRODUCTION
Abdominal aortic aneurysms (AAA) are a common vascular disease that can progress to aortic rupture which has a mortality of over 80%.1 Recent studies suggest there may be greater than 1 million people living in the United States with an AAA.2 Despite advances in the surgical and endovascular management of AAAs over the past decade, there remains no proven pharmacological intervention that slows AAA growth or prevents aortic rupture.3 Cumulative efforts to understand mechanisms that contribute to AAA development have identified smooth muscle apoptosis, elastin fragmentation and extracellular matrix degradation as a pathological hallmarks of impaired vascular remodeling resulting in weakening of the aortic wall.4,5 Over the past decade murine models of AAAs have utilized in vivo lineage tracing systems based on candidate marker genes coupled with fluorescence-activated cell sorting to examine immune and structural cells contributing to AAA development. However, these approaches require prior knowledge of in vitro cell surface markers which often do not adequately reflect in vivo functional states and transcriptional profiles.6,7 Further, although the cellular subsets that contribute to aortic wall integrity are well established8 the heterogeneity and mechanistic contribution of different immune and structural cells in control and aneurysmal human aortic tissue remains unknown.
Recently, the advent of single cell RNA sequencing (scRNAseq) allows for the dissection of cellular subpopulations within the aortic wall independent of prior assumptions on cell surface markers.9,10 scRNAseq adds unparalleled resolution to transcriptomic studies, enabling a comprehensive investigation of diverse cell types within tissues in an unbiased fashion and provides the opportunity to assess the heterogeneity of gene expression in individual cell populations during control and disease states. Recent cardiovascular studies have utilized scRNAseq to depict the cellular landscapes of endothelial cells8, vascular SMCs11, perivascular adipose tissue stem cells12, and immune cells13,14 within atherosclerotic disease. Additionally, limited investigations have employed scRNAseq in murine AAA models15 and human ascending aortic aneurysm tissues.16 However, given the pathologic development of infrarenal AAAs is distinct from ascending aortic aneurysms17–19, the dynamic gene expression profiles in abdominal aortic cell populations has not been examined in healthy and diseased tissues. Understanding the heterogeneous cellular identities, diverse functional states and subpopulations within AAAs is fundamentally important in understanding the etiology of aneurysmal disease to better define potential cell-specific targets for early intervention.
In this manuscript, we present the first comprehensive analysis of lineage heterogeneity, transcriptomic profiles, and functional states of structural and immune cells from human infrarenal AAAs and control tissues using scRNAseq. Our analysis demonstrates a marked increase in immune cell populations, particularly in T cells, in AAAs compared to controls and the interactome analysis of AAA tissues shows a profound upregulation of receptor ligand interactions between vascular SMCs and several immune cell populations. Specifically, we identify myeloid cell - smooth muscle interactions via CCL2/CCR2 and TGFB1/TGFB1R as well as T cell - smooth muscle interactions via TNFSF12/TNFRSF25 that are significantly altered in AAA tissues. Lastly, our investigation expands on prior blood-based genome-wide association studies (GWAS) findings by demonstrating cell specificity within human AAAs by integrating our scRNAseq data with publicly available GWAS data. Herein we demonstrate the importance of SORT1 in maintaining normal aortic smooth muscle cell function as well as how alterations in SORT1 signaling are present in human AAA tissue. Overall, our study provides the first unbiased evaluation of the cellular gene expression profiles of the human abdominal aorta and reveals how the gene expression landscape is altered in AAAs.
METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Frank Davis (davisfr@umich.edu) and Katherine Gallagher (kgallag@med.umich.edu).
Tissue Samples
The protocol for collecting human tissue samples was approved by the Institutional Review Board at University of Michigan (AWD017589). The investigation conforms to the principles outlined in the Declaration of Helsinki. Written informed consent was provided by all participants or the organ donors’ legal representatives before enrollment. All experiments conducted with human tissue samples were performed in accordance with the relevant guidelines and regulations. The aneurysmal samples were taken from the midportion of the aneurysmal sac. For control samples, aortic tissue was isolated from patients with atherosclerotic occlusive disease but no history of aneurysmal disease at the time of open aorto-bifemoral bypass. Patient medical comorbidities are represented in Supplemental Table I. Patients were excluded if they had aortic dissection; a heritable form of aortopathy (eg, Marfan syndrome, Loeys-Dietz syndrome, bicuspid aortic valve); or AAA related to infection, aortitis, trauma, or isolated pseudoaneurysm.
Single Cell RNA-Seq and Bioinformatics Analysis
Generation of single cell suspensions for scRNA-seq was performed as follows: Aortic tissue from aneurysmal and non-aneurysmal controls was harvested at the time of surgical intervention. Samples were minced, digested in 0.2% Collagenase II (Life Technologies) and 0.2% Collagenase V (Sigma) in plain medium for 1 hour at 37°C, and strained through a 70μM mesh. The scRNA-seq samples were analyzed by the University of Michigan Advanced Genomics Core on the 10X Chromium system. Libraries were sequencing on the Illumina NovaSeq 6000 sequencer to generate 151-bp paired end reads. Single cell RNA sequencing data analysis
After sequencing, raw reads were aligned to the human genome (hg38) and the digital expression matrix was generated using cellranger count. Individual samples were merged to generate the digital expression matrix using cellranger aggr. To identify different cell types and find signature genes for each cell type, the R package Seurat20 was used to analyze the digital expression matrix. Cells with less than 100 genes and greater than 25% mitochondrial expression were removed from further analysis. Seurat function NormalizeData was used to normalize the raw counts. Variable genes were identified using the FindVariableGenes function. The Seurat ScaleData function was used to scale and center expression values in the dataset for dimensional reduction. Principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and uniform manifold approximation and projection (UMAP) were used to reduce the dimensions of the data, and the first two dimensions were used in plots. The FindClusters function was later used to cluster the cells. The FindAllMarkers function was used to determine the marker genes for each cluster, which were then used to define cell types. The FindMarkers function was used to determine the differentially expressed genes between two group of cells. Genes with adjusted p value < 0.05 was considered significantly differentially expressed.
Cell-cell ligand receptor interaction analysis
CellphoneDB (v2.0.0)21 was applied for ligand receptor analysis. The raw counts and cell type annotation for each cell were imputed into cellphoneDB to determine the potential ligand receptor pairs. Pairs with p value >0.05 were filtered out from further analysis. Two runs were performed on two groups of cells: cells from atherosclerotic control samples and cells from AAA samples. The results from the two runs were plotted separately.
RESULTS
scRNAseq Analysis of Human Abdominal Aortic Aneurysms
To examine the transcriptional landscape of the human abdominal aorta and determine cellular alterations during AAA pathogenesis, we obtained fresh human aortic tissue from patients with a documented AAA (aortic diameter >5.5 cm) or non-aneurysmal control aorta undergoing aortic surgery. The patients ranged from 57 to 76 years old and demonstrated similar co-morbid conditions (Supplemental Table I). The tissue was dissociated, washed, and rapidly processed via the Drop scRNA-seq platform.22 A total of 9,290 cells passed our quality controls and were then analyzed by Seurat for unsupervised clustering. Integrative unsupervised cell clustering analysis was performed, and cells were projected onto a 2-dimensional UMAP plot. We initially obtained 17 clusters (Figure 1B). After examining conserved genes in each cluster, we identified 8 major cell types in the infrarenal aortic wall. The eight major cell types included, endothelial cells, SMCs, fibroblasts, myeloid cells (including monocytes/macrophages/dendritic cells), T lymphocytes, natural killer cells, B lymphocytes, and plasma cells (Figure 1C). Distinct gene expression profiles were present within each cell type (Figure 1D and 1E). Neutrophils showed very low transcript counts and required setting low filtering thresholds to allow for their detection. PTPRC (encoding pan-hematopoietic marker CD45 [cluster of differentiation 45]) was used to distinguish immune and nonimmune cells (Figure 1D). We found that the control tissues contributed more cells than expected in the nonimmune cell group, whereas the aneurysm tissues contributed more cells than expected in the immune cell group, especially T- and B-lymphocytes (Figure 1F). Individual analysis of each sample generated a similar number of clusters and identified the same major cell types (Supplemental Figure 1). To determine if the identified transcriptomic signatures align with the cellular infiltrates in AAA, immunohistochemistry imaging was performed to identify and quantify infiltrating leukocytes. This showed positive staining for CD3+ T cells, CD20+ B cells, CD163+ monocyte/macrophages, CD11c+ dendritic cells, and CD138+ plasma cells (Figure 1F).
Figure 1. Eight major cell types identified with single-cell RNA sequencing analysis of human abdominal aortic tissue samples.

A. Experimental approach and data analysis strategy. B, C. A UMAP plot showing all 17 cell clusters with cells colored according to the 8 major cell types (n=6 patients analyzed primarily). D. Relative expression of several marker genes in all cells from all samples. Cells were projected onto a UMAP plot. E. Heatmap of top marker genes per cluster. F. The composition of each cell type within AAA vs. control tissue samples is shown in the horizontal bar plot. G. Immunofluorescence of T cells (CD3), B cells (CD20), NK cells (CD11c), monocyte/macrophage (CD163), and plasma cells (CD138) showed primary in AAA tissue sections. Scale bar equals 100 μm in distance (n = 3 patients per image with ten high powered fields viewed and representative shown).
Heterogeneity of Immune and Structural Cells in the Abdominal Aorta
After identifying the cellular transcriptomic landscape and observing differences in immune and structural cell populations between the aneurysmal and control aortic tissues, we focused on specific gene expression patterns in immune and structural cells from AAA tissues. Nonimmune cells corresponding to the structural cells of the aortic wall have been shown to play critical roles in the pathophysiology of AAA development, thus we first investigated the gene expression in these cells.23 During AAA pathogenesis, phenotypic and cellular alterations in vascular SMC biology have been suggested to play an important role in murine models of AAAs.24 Within our human tissue analysis, the SMC-related cluster, high expression of contractile proteins TAGLN, ACTA2, and MYL9 was observed. We identified one cluster of endothelial cells that expressed endothelial marker genes VWF, IFI27, and CLDN5. We identified 3 types of fibroblasts, however the expression for multiple genes were slightly different between each cell cluster. Specifically, fibroblast#1 and #2 displayed an inflammatory activation with upregulation of CXCL12, CXCL14, and APOE. In contrast, fibroblast#3 contained many components of the extracellular matrix, including the collagens COL1A1, COL1A2, COL3A1, COL4A2, COL5A1, COL6A2, COL8A1, COL11A1, COL12A1, and COL21A1 as well as elastin (ELN), fibronectin (FN1), and fibrillin (FBN1) (Figure 2A). We also found overexpression of several factors involved in matrix assembly, such as MFAP2, MFAP5, SFRP2, lysyl oxidase (LOX), and FAP. To investigate whether specific functions could be assigned to the different fibroblast subpopulations, we performed gene ontology (GO) analyses using the most representative markers of each fibroblast cluster. Fibroblast#3 demonstrated strong association with extracellular matrix organization, collagen fibril organization, and protein complex subunit organization (Figure 2B)
Figure 2. Heterogeneity of nonimmune and immune cell subsets within abdominal aortic aneurysm tissue.
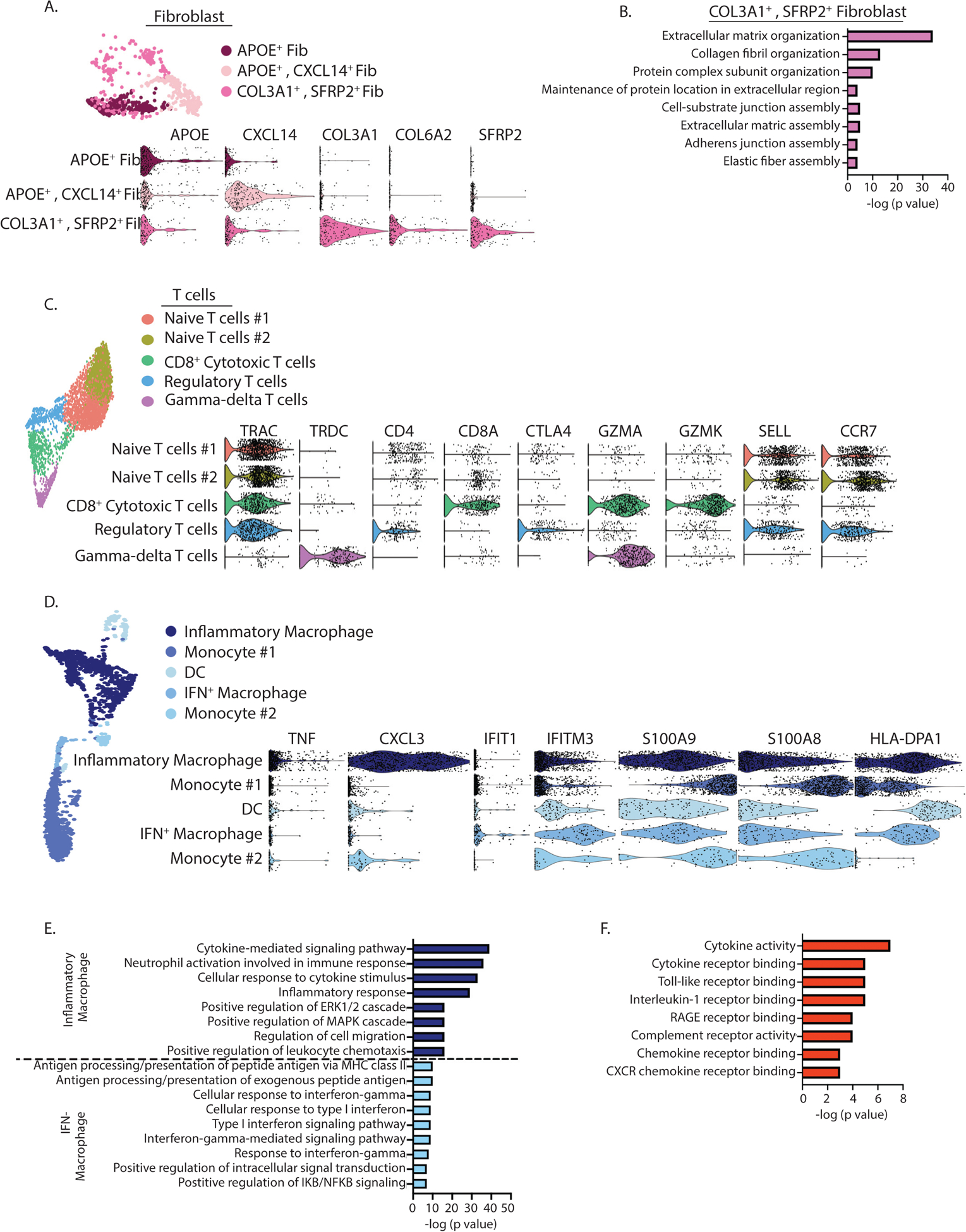
A. A t-SNE plot of fibroblast cells and accompanied violin plots of representative genes colored according to identified clusters (n=6 patients analyzed primarily). B. Gene Ontology BP enrichment analysis of differentially expressed genes in the COL3A1+, SFRP2+ fibroblast cluster. C. A t-SNE plot of T cells and accompanied violin plots of representative genes colored according to identified clusters. D. A t-SNE plot of monocytes/macrophages and accompanied violin plots of representative genes colored according to identified clusters (n=6 patients analyzed primarily). E. Gene Ontology BP enrichment analysis of differentially expressed genes in the inflammatory macrophage and IFN-macrophage clusters. F. Gene Ontology BP enrichment analysis of differentially expressed genes in the monocyte cluster #1 and #2. (n=6 patients analyzed primarily)
After investigating the transcriptomic profiles of the structural cell population, we next sought to unravel the transcriptomic heterogeneity of immune cells. T lymphocytes were the largest immune cell population identified in aortic tissue corresponding to cells with high gene expression of CD3D, CD3E, IL7R, and TRBC2. To gain further insight, we combined all of the T lymphocytes from all tissues and performed integrative unsupervised reclustering which resulted in the identification of 5 unique T lymphocyte clusters. There were two populations of naïve T cells expressing CD4 and high levels of SELL and CCR7. Regulatory CD4+ T lymphocyte (cluster) expressed Treg defining genes IL2RA, CTLA4, and TNFRSF18 (Figure 2C). In contrast we noted one population of CD8+ cytotoxic T cells expressing genes of CD8A, GZMK, and GZMA. Lastly, a gamma-delta T cell was identified with TRDC expression and absence of TRAC expression.
Myeloid cells represented the second largest cluster of immune cells and were a heterogenous group. Analysis of the subclusters within this population demonstrated two macrophage clusters. Macrophage cluster #1 demonstrated upregulation of well-defined proinflammatory (M1-like) marker genes: TNF, IL1B, and NFKB1. This cluster also expressed several cytokine genes, including CCL3L, CXCL3, CXCL2, and TNF consistent with an inflammatory function. The second macrophage cluster (macrophage#2) had upregulation of a predominance of genes involved in interferon signaling, including IFI44L, ISG15, IFIT1, and IFITM3 (Figure 2D). GO analysis of these macrophage clusters demonstrated marked inflammatory pathway activation in macrophage cluster #1 while macrophage cluster #2 exhibited significant upregulation of antigen presenting capabilities and type I interferon signaling (Figure 2E). The two monocyte clusters expressed CD14, NAMPT, S100A9, S100A8, STAB1 and an absence of CD16 expression, consistent with a classical monocyte population25,26 and demonstrated several cytokine receptor genes, including CCR1 and CSF1R. GO pathway analysis of these clusters demonstrates activation of cytokine receptor activity and inflammatory signaling pathways suggestive of monocyte differentiation into macrophages (Figure 2F). Lastly, the DC cluster expressed IRF4 and IRF8 as well as high levels of major histocompatibility complex genes consistent with the antigen-presentation function of DCs. Finally, aortic tissue samples had two clusters of naïve B cells with marker genes MS4A1, CD79, and CD74 as well as one cluster of each NK cell and plasma cells with marker genes of NKG7, GNLY, and FGFBP227 compared IGHG1–4 genes respectively.
Compositional differences and receptor-ligand interactions in AAA tissues compared with in controls
To identify key changes in AAA tissues compared to control aortic tissue, we compared data between AAA and control tissues and performed differential analysis between conditions based on cell type. Of the eight different cell types, five demonstrated >300 differentially expressed genes (DEGs). To evaluate the synthetic properties and functions of the major cellular populations, DEGs (false-discovery rate (FDR) <0.05) distinguishing each AAA cell cluster from the corresponding control cell cluster were identified and evaluated for enriched gene ontology biological processes. In vascular SMCs, 1,695 genes were significant upregulated and enriched processes for the upregulated genes reflect regulation of cell migration/proliferation as well as TGFβ receptor signaling (Figure 3A). In the myeloid cells from AAA tissue, expression of multiple inflammatory cytokine genes (CXCL8, CCL3, IL1B, CCL4, and CXCL2) were among the 455 upregulated genes in comparted to control. As such enriched processes for AAA myeloid cells paralleled a significant pro-inflammatory component with activation of TNF response, NFκB signaling, and the Type I IFN pathway. Within AAA fibroblasts, 189 upregulated genes reflected pathways involved in response to reactive oxygen species, IFNγ signaling and cytokine stimulus. Lastly, across multiple cell populations within AAA tissue, there was upregulation of cellular pathways involved in oxidative phosphorylation that has recently been shown to be aberrant in ascending aortic aneurysm tissue (Figure 3B).16
Figure 3. Comparison of molecular and cellular interactions in AAA vs. control tissue.
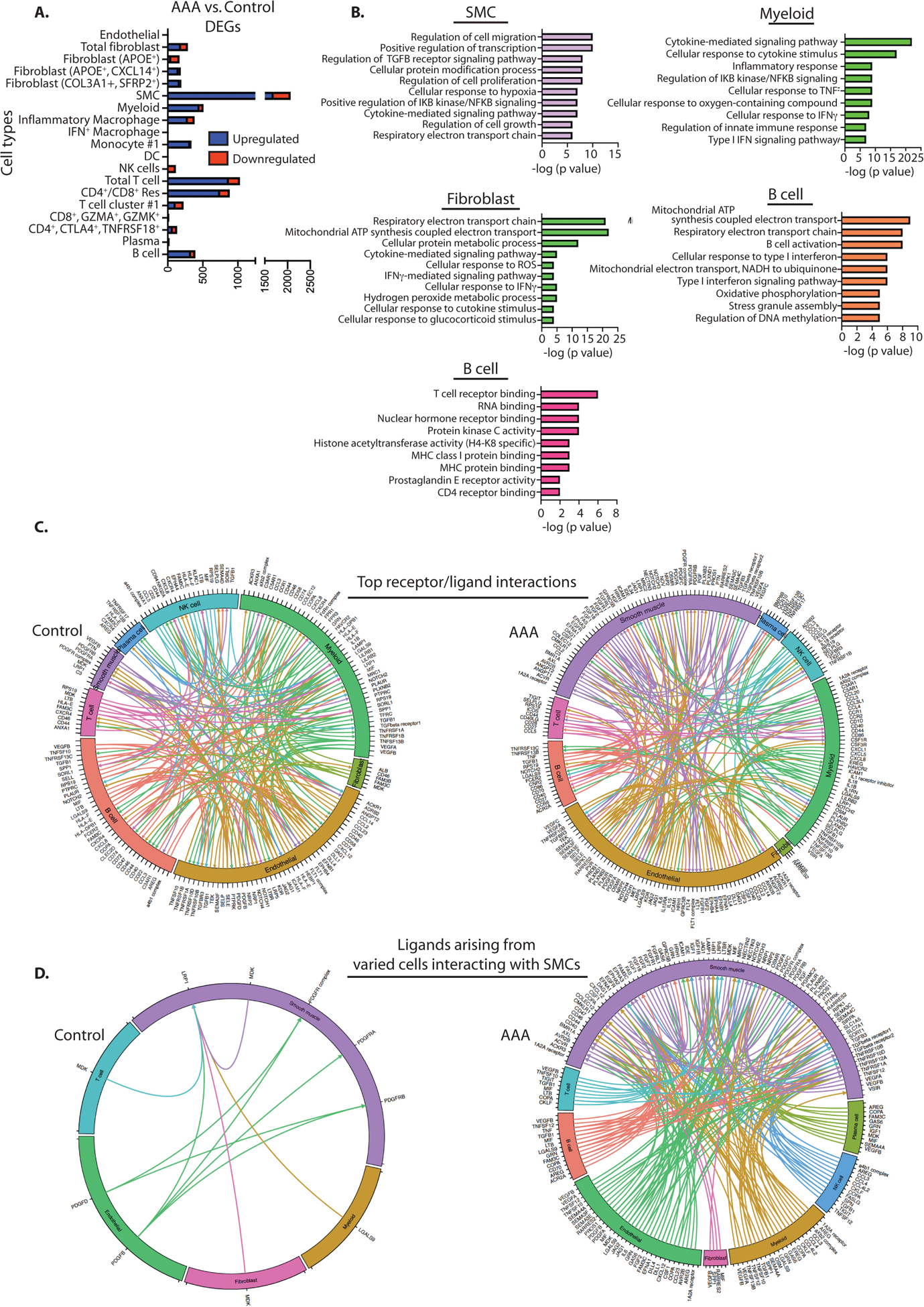
A. Comparison between AAA and control tissues according to cluster. The x axis of the left column represents differentially expressed gene (DEG) counts between the AAA and control groups. Downregulated genes are shown in red; upregulated genes are shown in blue. B. Gene Ontology BP enrichment analysis of differentially expressed genes in each cell cluster in AAA vs. control tissues. C. Cell-cell receptor-ligand communication between inflammatory infiltrate and stromal tissues for the top 200 receptor-ligand pairs in AAA vs control tissues. D. Data from single-cell sequencing were used to map receptor-ligand interactions from varying cells interacting with smooth muscle cells in control and AAA tissues (n=6 patients analyzed primarily).
scRNA-seq also provides novel opportunities to identify communicating pairs of cells based on the expression of cell-surface receptors and their interacting ligands.28 To learn cell-cell communication between the cell types, we performed CellPhoneDB ligand-receptor analysis and plotted the top ranked 200 pairs in a Circos plot (Figure 3C). The ligand-receptor analysis demonstrated extensive interactions between all the major cell subsets in AAA, including interactions of nonimmune and immune cell components. Within the AAA tissue, there was a marked increase in receptor-ligand interactions arising from and binding to the SMC population include members of the Notch pathway with endothelial and myeloid cell populations, VEGF signaling, and interactions of CXCL12 and TGFβ signaling pathways with multiple immune and nonimmune cells (Figure 3D and Supplemental Figure 2). Prior investigations have demonstrated that interactions between infiltrating immune cells and SMCs play an instrumental role in AAA development and progression.29 Within the current investigation, both T cells and myeloid cells were shown to have a significant increase in receptor-ligand interactions with SMCs in AAA in comparison to control tissue. Instrumental receptor/ligand pathways between SMCs and myeloid cells that were found to be altered included CSF1 or IL34/CSFR, CCL2/CCR2, JAG/Notch2, TGFB1/TGFB1R and CXCL12/CXCR4. Likewise, receptor ligand pathways between SMCs and T cells suggestive to play a role in aneurysmal dilation included: FGF2/CD44, LTB/LTBR, FAM3c/LAMP1, and TNFSF12/TNFRSF25. Taken together, this identifies key receptor-ligand interactions based on cell types that require further exploration and may identify key interactions for therapeutic manipulation.
Association of DEGs in AAA Tissues with GWAS Data from Peripheral Blood of AAA patients
Given that several large, recent GWAS datasets30–33 that have analyzed peripheral blood from AAA patients, we sought to identify if the genes identified from AAA patient blood samples in these GWAS studies is relevant in human aortic tissues. Although there is often a correlation between biomarkers identified in peripheral blood and tissues, it is unknown whether these genes identified in the blood are relevant in AAA tissues. Further, cell-specificity of the relevant GWAS genes is unknown. Utilizing the results of recent studies, by Klarin et al. and Jones et al.31,34 we extracted all aneurysm-related single nucleotide polymorphisms (SNPs), as well as the chromatin regions of those SNPs. From these data, we extracted putative target genes of aneurysm-associated SNPs and overlapped them with our DEGs. From these putative target genes of aneurysm associated SNPs we identified eight genes (APOE, MM9, LIPA, SORT1, LRP1, CDKN1A, CRISPLD2, and LDLR) that were differentially expressed in four cell types between control and AAA tissues (Figure 4A). Specifically, the expression of LDLR was found to be significantly decreased in clusters of T-cell (CD8+,CD247+,CD3D+) and fibroblast (APOE+) while APOE was found to be significantly decreased in clusters of inflammatory macrophages and T cells (CD8+,CD247+,CD3D+) (Figure 4B). Lastly, the SMC cluster had the highest level of putative GWAS genes that were differentially expressed and found to have significant upregulation of SORT1, LRP1, CDKN1A, and CRISPLD2 in comparison to the control tissue smooth muscle cluster. Of these genes, SORT1 was identified as a putative target gene of the aneurysm-associated SNP rs599839. To further identify potential interactions genes regulated by SORT1 we examined known SORT1 related pathway genes35 identified in other studies with DEGs by cluster. We identified IL6, STAT3, JAK1, and JAK3 genes that interact with SORT1 (Figure 4C). Together, these findings indicate that SORT1 in SMCs may be critical for the normal physiology of the aortic wall and play a critical role in altering SMC function associated with AAA development.
Figure 4. Potential role of AAA associated SNPs in cell specific alterations.

A. DEGs that were identified as the targets of aneurysm associated SNPs based on GWAS results according to cell cluster. B. Expression of SORT1 in control and abdominal aortic aneurysm (AAA) tissues in cell clusters. C. Fold-change (AAA/control) of potential interacting genes of SORT 1 in smooth muscle cells.
DISCUSSION
Herein, using scRNAseq of human AAA tissue samples, we performed integrative and differential analyses of the cellular composition of the human abdominal aortic wall and provided novel insight into how the gene expression landscape is altered in immune and structural AAA cells. Our analysis demonstrates, a marked increase in immune cell populations, especially T cells, in AAA compared to control tissues. Additionally, AAA tissues demonstrate a profound upregulation of receptor-ligand interactions between vascular SMCs and immune cell populations. We further demonstrate based on integration of GWAS data from AAA patient blood and DEGs from our human AAA tissues that aberrant SORT1 expression in vascular SMCs may play an important role in pathological aneurysm dilation.
Unsupervised clustering demonstrated both immune and non-immune cell populations with the abdominal aorta. For the immune cell populations, T lymphocytes were the largest population identified. T lymphocytes are known for having many subtypes and prior investigations have shown that both CD4+ T cells and CD8+ T cells promote AAA formation.36,37 Within the current analysis, we did not detect all the typical T lymphocyte subtypes in our analysis but did identify one CD4+ population, two populations of CD8+ cells and a unique CD4+/CD8+ resting T cell population. Of the CD4+ T cells, there was moderate expression of Treg-defining genes (IL2RA, CTLA4, TNFRSF18 and TNFRSF9) which are known to play an instrumental role in AAA development.38 The contribution of monocyte/macrophages to AAA development is well established4 with these cells exhibiting diverse functions, including amplification of the local inflammatory response by secreting pro-inflammatory cytokines, chemokines and producing proteases and reactive oxygen.39 A number of lineage tracing experiments and scRNA-seq data have demonstrated the heterogeneity of monocytes/macrophages within the atherosclerotic lesion and arteries.14 Based on the scRNA-seq data herein, we now characterized the monocyte/macrophage transcription profiles and identified their functional heterogeneity. We identified two macrophage and two monocyte populations within the abdominal aortic wall. Within AAA tissue these cell types displayed upregulation of multiple inflammatory genes with one macrophage population demonstrating significant upregulation genes involved in IFN signaling (IFI44L, ISG15, IFIT1, and IFITM3) confirming that IFN pathways have a detrimental role in AAA pathogenesis.
For nonimmune cells, our cellular composition data demonstrated that there are multiple subtypes of both fibroblasts and SMCs within the aortic wall. Within the fibroblast populations, there was inflammatory activation with upregulation of CXCL12, CXCL14, and APOE in multiple subtypes. Further, AAA fibroblast clusters exhibited increased IFNγ-mediated signaling and mitochondrial dysfunction. Mitochondrial dysfunction has been associated with aging and chronic diseases,40 supporting the notion that the long-term presence of AAA may impair mitochondrial function. Separately, SMCs displayed the highest level of DEGs in AAA compared to control tissue and had upregulation of multiple pathways involved with cell migration/proliferation as well as TGFβ receptor signaling. Prior investigations have demonstrated that SMC function and phenotypic transition is instrumental in AAA pathogenesis.41 Beyond SMC specific pathways, we were also able to analyze cell-cell interactions present within maximally dilated aortic aneurysm tissue. Most interestingly, SMCs from AAA displayed marked differences in receptor-ligand interactions in comparison to control samples. Of note these SMC receptor ligand interactions involved interactions with both immune and non-immune cells with cell signaling pathways of CCL2/CCR2, TGFB1/TGFB1R, and Notch signaling demonstrating marked upregulation in AAA tissue. Indeed, cellular crosstalk has been suggested to be instrumental in the development of aortic degeneration in both murine experimental studies and human tissue investigations.29,42
Finally, one of the significant post-GWAS challenges is the identification of candidate genes and cell-specific pathways with clinical potential.43 Using scRNAseq, we identified genes that were differentially expressed in the aorta of patients with AAA. We then integrated genes that were differentially expressed in AAA tissues with GWAS results (aneurysm-associated SNPs) identified in peripheral blood from patients with AAA disease. Our analysis showed altered expression of AAA-associated genes in myeloid, SMCs, T cells, and fibroblasts. Specifically, we found that SORT1 was markedly altered in SMCs from AAA compared to control. Previous studies have suggested that SORT1 has a pathological role in SMC vascular remodeling via the recruitment of extracellular vesicles.44 Further, SORT1 has also been demonstrated to regulate proinflammatory cytokine exocytosis, most notably IL6, as well as induce JAK/STAT signaling pathways both of which are known to be is pathological in AAA development.45 Indeed, previous studies showed that the levels of IL-6 and downstream IL-6 receptor signaling, such as STAT3, are significantly increased in human AAA aortic tissue and plasma compared with those of normal controls.46 Together, our findings suggest that SORT1 may play a pathological role in SMC function during AAA progression and positively regulate proinflammatory signaling.
Although this study provides valuable insight into the cellular heterogeneity and transcription during AAA development, some limitations must be addressed. In this study, we integrated scRNAseq data sets from a finite number of AAA and control tissues. The smaller sample size of the control group than the AAA group may limit the statistical power of this comparison. Additionally, to obtain a single-cell suspension for sequencing, we used an enzyme cocktail to digest the aortic wall and as such, the recovery rate of each cell type was not equal. Although neutrophils have been suggested to play a minor role in AAA development, we did not recover neutrophils during our aortic tissue digestion.47 This recovery bias may be attributed to cell properties or distribution (eg, the media may be harder to digest than the adventitia). Alternatively, some types of cells may be more vulnerable to the digestion process, resulting in increased cell loss. Eliminating the influence of tissue processing is difficult, but it is possible to use the same duration of time for processing control and aneurysmal tissues to minimize the influence of tissue processing. Indeed, there is a fine line between increasing digestion time to isolate more cells and generating a pure sample containing a high number of viable cells.
In conclusion, our study provides the first comprehensive evaluation of the cellular composition of the human abdominal aorta and reveals how the gene expression landscape is altered within unique cell populations during AAA development. Our analysis demonstrates, a marked increase in immune cell populations, especially T cells, in AAA compared to control and that AAA tissues show a profound upregulation of receptor-ligand interactions between vascular SMCs and immune cell populations. Further, the results of our comparative analyses suggest SORT1 plays an important role in maintaining smooth muscle aortic wall function. These findings expand our understanding of AAA pathogenesis and may contribute to the development of new treatments.
Supplementary Material
Supplementary Figure 1. Single-cell RNA sequencing of aortic tissue from AAA and control populations identifies 17 clusters. A. UMAP plots showing the contribution of each sample to the distinct cell populations identified in the integrated analysis. B. Overlaid UMAP projection of AAA and control samples demonstrating consistent distribution within non-linear reduction between samples.
Supplemental Figure 2. Interactions of SMCs with the microenvironment in AAAs. Data from single-cell sequencing were used to map receptor-ligand interactions from and to smooth.
Acknowledgements:
We thank Robin Kunkel for her assistance with the graphical illustrations.
Funding Sources:
This work is supported in part by National Institutes of Health [R01-HL137919 to KG, F32-DK117545 to FD, P30 AR075043 to JG and LT, R01-AR069071 to JG]; American College of Surgeons Resident Fellowship to FD; Vascular and Endovascular Surgery Society Resident Research Award to FD; and the Doris Duke Foundation to KG.
Footnotes
This work was presented at the 2022 American Surgical Association meeting in Chicago, IL.
Conflicts of Interest: The authors have no conflicts of interest to report.
REFERENCES
- 1.Nordon IM, Hinchliffe RJ, Loftus IM, et al. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nature Reviews Cardiology. 2011;8:92–102. [DOI] [PubMed] [Google Scholar]
- 2.Kent KC, Zwolak RM, Egorova NN, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg. 2010;52:539–548. [DOI] [PubMed] [Google Scholar]
- 3.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raffort J, Lareyre F, Clément M, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. [DOI] [PubMed] [Google Scholar]
- 5.Rabkin SW. The Role Matrix Metalloproteinases in the Production of Aortic Aneurysm. In: Progress in Molecular Biology and Translational Science. Elsevier B.V.:239–265. [DOI] [PubMed] [Google Scholar]
- 6.Bjornson ZB, Nolan GP, Fantl WJ. Single-cell mass cytometry for analysis of immune system functional states. Current Opinion in Immunology. 2013;25:484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ginhoux F, Schultze JL, Murray PJ, et al. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nature Immunology. 2016;17:34–40. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri AS, Vellarikkal SK, Edelman ER, et al. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation. 2019;140:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z, Solomonidis EG, Meloni M, et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur Heart J. 2019;40:2507–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCracken IR, Taylor RS, Kok FO, et al. Transcriptional dynamics of pluripotent stemcell-derived endothelial cell differentiation revealed by single-cell RNA sequencing. Eur Heart J. 2020;41:1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobnikar L, Taylor AL, Chappell J, et al. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun.;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu W, Nowak WN, Xie Y, et al. Single-cell RNA-Sequencing and metabolomics analyses reveal the contribution of perivascular adipose tissue stem cells to vascular remodeling. Arterioscler Thromb Vasc Biol. 2019;39:2049–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkels H, Ehinger E, Vassallo M, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. 2018;122:1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cochain C, Vafadarnejad E, Arampatzi P, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674. [DOI] [PubMed] [Google Scholar]
- 15.Zhao G, Lu H, Chang Z, et al. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Ren P, Dawson A, et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation. 2020;142:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milewicz DM, Trybus KM, Guo D-C, et al. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler Thromb Vasc Biol. 2017;37:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda N, Komuro I. Genetic basis of hereditary thoracic aortic aneurysms and dissections. Journal of Cardiology. 2019;74:136–143. [DOI] [PubMed] [Google Scholar]
- 19.Pinard A, Jones GT, Milewicz DM. Genetics of Thoracic and Abdominal Aortic Diseases: Aneurysms, Dissections, and Ruptures. Circulation Research. 2019;124:588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Efremova M, Vento-Tormo M, Teichmann SA, et al. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc. 2020;15:1484–1506. [DOI] [PubMed] [Google Scholar]
- 22.Zilionis R, Nainys J, Veres A, et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc. 2017;12:44–73. [DOI] [PubMed] [Google Scholar]
- 23.Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: Translating preclinical studies into clinical therapies. Heart. 2014;100:1498–1505. [DOI] [PubMed] [Google Scholar]
- 24.Petsophonsakul P, Furmanik M, Forsythe R, et al. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation involvement of Vitamin K-dependent processes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39:1351–1368. [DOI] [PubMed] [Google Scholar]
- 25.Villani AC, Satija R, Reynolds G, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (80-).;356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutertre CA, Becht E, Irac SE, et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity. 2019;51:573–589.e8. [DOI] [PubMed] [Google Scholar]
- 27.Crinier A, Milpied P, Escalière B, et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity. 2018;49:971–986.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vento-Tormo R, Efremova M, Botting RA, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018;563:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Kong W. Cellular signaling in Abdominal Aortic Aneurysm. Cellular Signalling.;70. Epub ahead of print June 1, 2020. DOI: 10.1016/j.cellsig.2020.109575. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Pan C, Zhang S, et al. Decoding the Genomics of Abdominal Aortic Aneurysm. Cell. 2018;174:1361–1372.e10. [DOI] [PubMed] [Google Scholar]
- 31.Jones GT, Tromp G, Kuivaniemi H, et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ Res. 2017;120:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golledge J, Kuivaniemi H. Genetics of abdominal aortic aneurysm. Current Opinion in Cardiology. 2013;28:290–296. [DOI] [PubMed] [Google Scholar]
- 33.Klarin D, Verma SS, Judy R, et al. Genetic Architecture of Abdominal Aortic Aneurysm in the Million Veteran Program. Circulation. 2020;142:1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klarin D, Verma SS, Judy R, et al. Genetic architecture of abdominal aortic aneurysm in the million veteran program. Circulation. 2020;142:1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kjolby M, Nielsen MS, Petersen CM. Sortilin, Encoded by the Cardiovascular Risk Gene SORT1, and Its Suggested Functions in Cardiovascular Disease. Current Atherosclerosis Reports.;17. Epub ahead of print April 1, 2015. DOI: 10.1007/s11883-015-0496-7. [DOI] [PubMed] [Google Scholar]
- 36.Galle C, Schandené L, Stordeur P, et al. Predominance of type 1 CD4+ T cells in human abdominal aortic aneurysm. Clin Exp Immunol. 2005;142:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sagan A, Mikolajczyk TP, Mrowiecki W, et al. T Cells Are Dominant Population in Human Abdominal Aortic Aneurysms and Their Infiltration in the Perivascular Tissue Correlates With Disease Severity. Front Immunol.;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng X, Yang J, Dong M, et al. Regulatory T cells in cardiovascular diseases. Nature Reviews Cardiology. 2016;13:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizas KD, Ippagunta N, Tilson MD. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiology in Review. 2009;17:201–210. [DOI] [PubMed] [Google Scholar]
- 40.Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. 2007;83:84–92. [DOI] [PubMed] [Google Scholar]
- 41.Clément M, Chappell J, Raffort J, et al. Vascular Smooth Muscle Cell Plasticity and Autophagy in Dissecting Aortic Aneurysms. Arterioscler Thromb Vasc Biol. 2019;39:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hadi T, Boytard L, Silvestro M, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat Commun.;9. Epub ahead of print December 1, 2018. DOI: 10.1038/s41467-018-07495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goettsch C, Hutcheson JD, Aikawa M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talbot H, Saada S, Naves T, et al. Regulatory roles of sortilin and sorla in immune-related processes. Frontiers in Pharmacology.;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawson J, Cockerill GW, Choke E, et al. Aortic aneurysms secrete interleukin-6 into the circulation. J Vasc Surg. 2007;45:350–356. [DOI] [PubMed] [Google Scholar]
- 47.Eliason JL, Hannawa KK, Ailawadi G, et al. Neutrophil Depletion Inhibits Experimental Abdominal Aortic Aneurysm Formation. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Single-cell RNA sequencing of aortic tissue from AAA and control populations identifies 17 clusters. A. UMAP plots showing the contribution of each sample to the distinct cell populations identified in the integrated analysis. B. Overlaid UMAP projection of AAA and control samples demonstrating consistent distribution within non-linear reduction between samples.
Supplemental Figure 2. Interactions of SMCs with the microenvironment in AAAs. Data from single-cell sequencing were used to map receptor-ligand interactions from and to smooth.