

Abstract
The mammalian cell cycle has been extensively studied regarding cancer etiology, progression, and therapeutic intervention. The canonical cell cycle framework is supported by a plethora of data pointing to a relatively simple linear pathway in which mitogenic signals are integrated in a stepwise fashion to allow progression through G1/S with coordinate actions of cyclin-dependent kinases (CDK)4/6 and CDK2 on the RB tumor suppressor. Recent work on adaptive mechanisms and intrinsic heterogeneous dependencies indicates that G1/S control of the cell cycle is a variable signaling pathway rather than an invariant engine that drives cell division. These alterations can limit the effectiveness of pharmaceutical agents but provide new avenues for therapeutic interventions. These findings support a dystopian view of the cell cycle in cancer where the canonical utopian cell cycle is often not observed. However, recognizing the extent of cell cycle heterogeneity likely creates new opportunities for precision therapeutic approaches specifically targeting these states.
Introduction
The mitotic cell cycle is a crucial biological process in which a cell divides into two daughter cells in a controlled manner. This general mechanism is highly conserved through evolution and was recognized as a fundamental process, termed ‘mitosis’, in 1882 by Walther Flemming. Canonically, all cellular division is the same: DNA is replicated and is then divided into two daughter cells during mitosis. Although control over the coupling of DNA replication and mitosis is crucial for limiting chromosome instability, the key rate-limiting step for the division of somatic cells is the G1 phase [1]. This is the period when a multitude of environmental and cell intrinsic cues are integrated to delineate if proliferation is appropriate. Thus, deregulated cell division, the core hallmark of cancer, is largely driven by those processes, which regulate the progression through G1 [2]. Cell cycle genes that modulate G1 progression are often mutated in human cancer, further supporting their importance in limiting inappropriate proliferation (Figure 1). Significant data have supported the conventional model of cell cycle progression through G1; however, it has emerged that the cell cycle is more pliable and heterogeneous in driving the G1/S transition and that multiple different mechanisms are at play in cancer. Although this diversity poses challenges for cancer therapy, there is a potential to utilize such information for tailoring therapeutic interventions. We discuss the evolving understanding of cell cycle control mechanisms in cancer and their relevance for precision therapeutic interventions.
Figure 1. Genetic alterations of G1/S cell cycle genes in cancer.
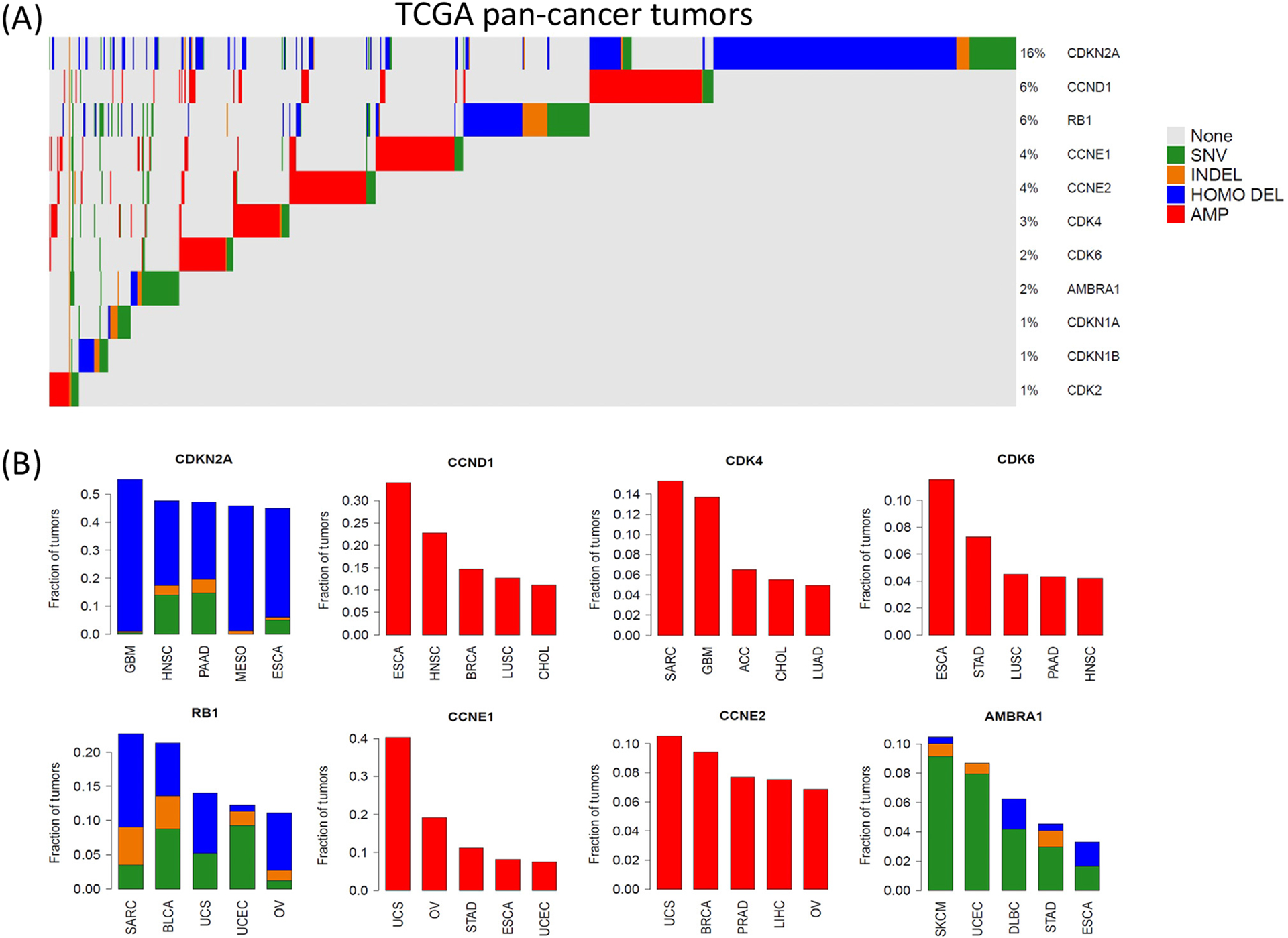
(A) Oncoprint summarizes the frequency of genetic alterations of the indicated genes in TCGA (The Cancer Genome Atlas) Pan-Cancer Cohort. (B) Barplots summarize the five histological tumor types with the most frequent disruption of a given gene, Color codes: blue, homozygous deletion; red, gene amplification; green, gene mutation; orange, insertion/deletion. Abbreviations: AMP, amplification; HOMO DEL, homozygous deletion; INDEL, insertion/deletion; SNV, single-nucleotide variant. TCGA study abbreviations in panel (B) are given in https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations.
Classical G1/S control of the cell cycle
Given the high level of conservation of the cell division process it would be expected that the fundamental mechanisms coordinating transitions through the cell cycle would be conserved. Consistent with this general notion, all eukaryotic cell cycles are controlled by cyclin-dependent kinases (CDK) and cyclins [3–5]. The active CDK/cyclin complexes phosphorylate key substrates to advance progression through all cell cycle phases. The degree of conservation is apparent in that many mammalian genes encoding cyclins and CDK were cloned by complementation of mutations in yeast models. Notably, the first identified mammalian CDK gene (CDK1) was cloned in a complementation screen for a Cdc2 mutant in the fission yeast Schizosaccharomyces pombe [6]. That CDKs and their cyclin partners are consummate drivers/modulators of cell cycle progression is irrefutable.
D-type cyclins and CDK4/6
Cyclin D1 was identified by three independent groups using different methodologies that to this day shape our understanding of G1/S control. (i) A complementation screen of G1 cyclins in Saccharomyces cerevisiae identified a human candidate G1 cyclin [7]. This approach showed that the gene promotes G1/S control in the absence of other G1 cyclins and is conserved across evolution. (ii) Analysis of genes induced as cells progress through G1 identified cyclin D1 in murine macrophages stimulated with GM-CSF [8], demonstrating that cyclin D1 is responsive to mitogenic factors. (iii) The same gene was identified at a chromosomal breakpoint in parathyroid adenomas [9]. This finding supported the concept that cyclin D1 could function as an oncogene. Over the intervening years it has become clear that cyclin D1 and the related D-type cyclins (cyclin D2 and cyclin D3) are broadly responsive to mitogenic and oncogenic signals and play important roles in driving G1/S progression [10]. Furthermore, AMBRA1 (autophagy and beclin 1 regulator 1) was recently found to be crucial in controlling the protein turnover of cyclin D1 and represents a potentially new class of tumor-suppressors whose loss leads to aberrant stabilization and accumulation of cyclin D1 [11–13] (Figure 1). The D-type cyclins preferentially bind to and activate the CDK4 and CDK6 proteins [10,14]. The CDK4 and CDK6 genes are highly related and, with the D-type cyclin genes, are largely selective to metazoans in controlling G1/S progression [15]. The regulation of CDK4/6–cyclin D is complex and involves several adapter molecules (e.g., Cdc37 and Hsp90) that contribute to the assembly and activation status of the complexes [16–18]. In addition, the protein p27KIP1 plays an important role in the catalytic activation of CDK4 which is governed by its tyrosine phosphorylation status [19,20]. Conversely, p16INK4A directly binds to and inactivates CDK4 and CDK6 [21,22]. This function of p16INK4A is believed to be crucial for blocking oncogene-induced tumor development, and instead shunts cells with deregulated oncogenes toward a senescent phenotype [23]. Concordantly, the p16INK4A gene (CDKN2A) is commonly mutated in human cancer [24,25] (Figure 1).
RB phosphorylation and E2F
CDK/cyclins are proline-directed kinases and have a relatively broad substrate spectrum. However, CDK4 and CDK6 complexes are particularly active toward the retinoblastoma (RB) tumor-suppressor and related proteins (i.e., p107 and p130) [26,27]. RB. the first tumor-suppressor identified, has been the subject of intense study [28–30]. Although RB can have a multitude of impacts relative to tumor biology [30–32], its function in cell cycle control is largely mediated via repressing the activity of the E2F family of transcription factors [33–37]. When unphosphorylated or hypophosphorylated, RB can assemble several protein complexes that mediate transcriptional repression of E2F-regulated genes [38–40]. Phosphorylation disrupts these complexes [27,41], and thereby leads to the expression of a highly conserved cadre of genes including DNA replication factors, genes involved in mitotic progression, and other cell cycle regulators (e.g., cyclin E and cyclin A) [42–45]. RB contains multiple phosphorylation sites, and the inactivation of RB is a multistep process [38,46,47]. RB that is only modestly phosphorylated (e.g., monophosphorylated) retains cell cycle inhibitory function, and hyperphosphorylation at multiple sites is required for the complete RB inactivation and derepression of E2F-mediated transcription.
Several key functional studies support the importance of RB phosphorylation as a downstream determinant of CDK4 and/or CDK6 function and support the canonical regulation of G1/S control. In addition to being a substrate, the requirement for cyclin D1 or CDK4/6 in driving the cell cycle is conditioned by the presence of RB. This concept was shown initially using antibody depletion of cyclin D1 function in RB-deficient tumor cell lines [48,49], and it was subsequently shown that RB is necessary for cell cycle arrest induced by ectopic expression of P16INK4A [50,51]. Cancer genetic analyses have also supported this linear pathway in which CDKN2A loss or cyclin D1 (CCND1) amplification are mutually exclusive with RB loss [52,53].
CDK2 and cyclin E
The other CDK/cyclin complex implicated in G1/S control is CDK2/cyclin E. Like cyclin D1, gene encoding cyclin E was cloned via complementation of yeast G1 cyclin deficiency [54,55]. CDK2 was identified both by complementation of Cdc28 mutants as a clone distinct from CDK1 [56], as well as by homology with human CDK1 [57]. There are two cyclin E genes, CCNE1 and CCNE2, which code for the proteins cyclin E and cyclin E2, respectively, that are both able to activate CDK2 [58]. Both genes are subject to amplification in cancer, and CCNE2 amplification often co-occurs with the MYC oncogene on chromosome 8 (Figure 1). Although cyclin E2 can contribute to G1/S progression [58], cyclin E is much more widely studied. The protein levels and activities of CDK2/cyclin E are controlled by multiple processes including the action of the CDK2 inhibitors, p27KIP1 and P21CIP1 [59,60]. The identification of cyclin E and CDK2 occurred at the same time as the identification of cyclin D1 and CDK4/6, and deciphering the respective functions of these two CDK modules has evolved over the following years. Unlike CDK4 and CDK6, CDK2 is much more permissive in substrate phosphorylation, suggesting that it could facilitate progression through latter phases of the cell cycle [61,62]. Furthermore, the induction of cyclin E is delayed relative to cyclin D1 as cells progress from G0 toward S phase [63,64]. These data and the clear mitogen-responsiveness of cyclin D1 suggested that CDK4/6 would act upstream of CDK2/cyclin E. That there is a degree of coupling or interdependencies emerged from several findings beyond temporal coordination. (i) CDK4/6 was necessary to initiate the phosphorylation of RB, and CDK2 complexes contribute to full inactivation [65,66]. (ii) RB/E2F control the expression of cyclin E and CDK2 [44]. (iii) RB in some settings was sufficient to limit the expression and/or activity of cyclin E and/or CDK2 [67,68]. (iv) CDK2 activity contributes to several downstream activities (e.g., phosphorylation of replication factors) that are not carried out by CDK4 or CDK6 [60,69].
Summary of the classical model of G1/S control
The key findings that contributed to the genesis of the classical model for G1/S control are summarized as a timeline (Figure 2A); however, the first descriptions of the ‘RB pathway’ as we know it today emerged ~25 years ago [70,71]. The overall structure is summarized in Figure 2B. (i) Oncogenic/mitogenic signals converge to activate CDK4/6, which initiates the phosphorylation of RB. (ii) The accumulation of CDK2/cyclin E complexes contributes to the hyperphosphorylation and inactivation of RB. (iii) Subsequent activation of E2F-mediated transcription allows the expression of downstream target genes. (iv) These genes are required for DNA replication and progression through mitosis, including the activation of CDK2/cyclin A and CDK1/cyclin B1 complexes. Hence, the canonical/utopian cell cycle is a linear process in which the activation of CDKs occurs in a sequential manner initiated by the action of CDK4 and/or CDK6. Moreover, RB acts as a major node to couple the activity of CDK4/6 to the rest of the cell cycle machinery. Because CDK4/6 exists as the upstream kinase for cell cycle, it could represent an ideal target in cancers.
Figure 2. The canonical G1/S pathway.
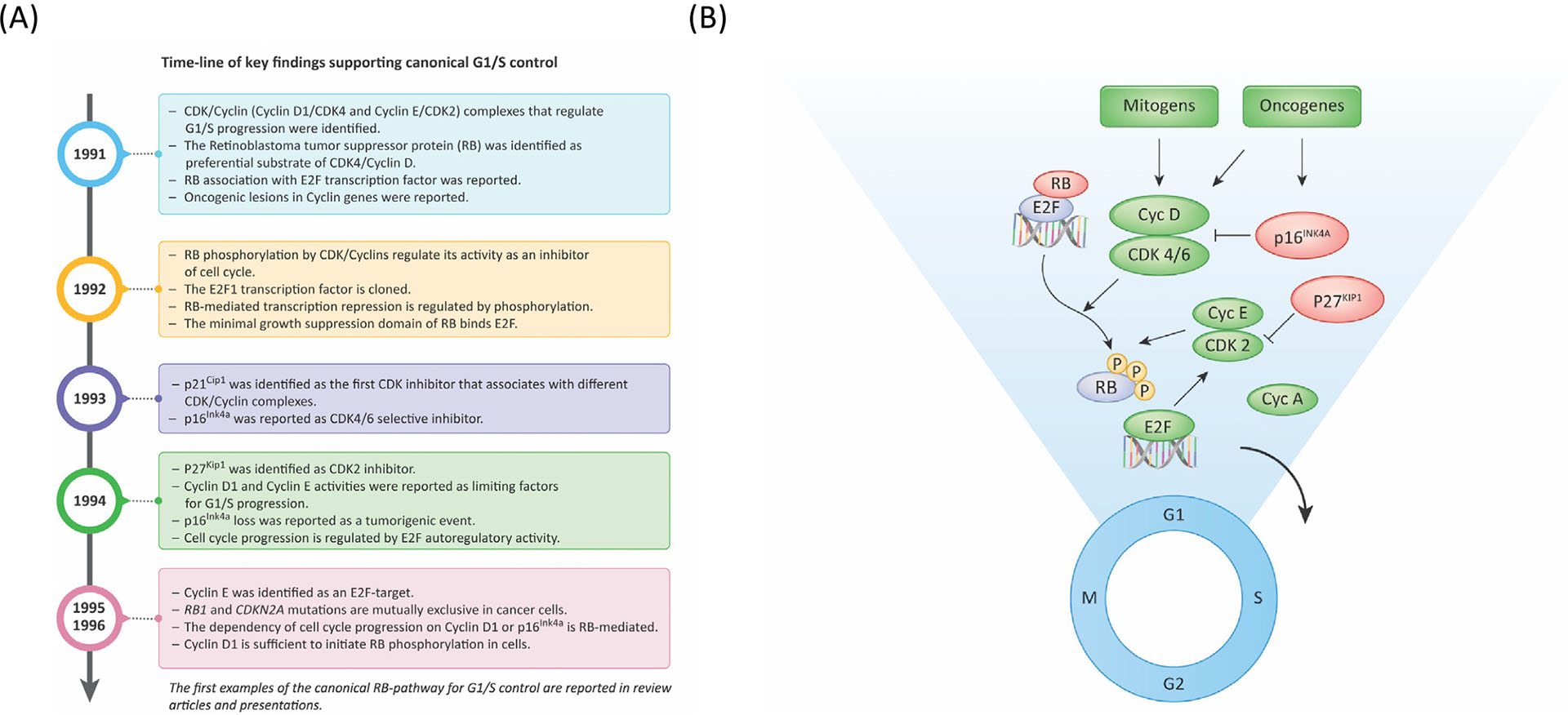
(A) Timeline summarizing discoveries which contributed to the development of the canonical regulation of G1/S progression. (B) Basic schematic of canonical G1/S control wherein mitogenic and oncogenic signals converge on CDK4/6–cyclin D activity. Although these factors support the assembly and activation of the complexes, select oncogenic signaling can induce p16INK4A to yield oncogene-induced senescence. The activation of CDK4/6 initiates the phosphorylation of RB and enables the subsequent activation of CDK2/cyclin E that contributes to the hyperphosphorylation and functional inactivation of RB. p27KIP1 plays a complex role in CDK4/6 activation state, but is a potent CDK2 inhibitor. With RB inactivation, E2F-mediated transcription elicits a program of gene expression to drive progression through the remainder of the cell cycle. Adapted from [116].
CDK4/6 inhibitors: mechanisms of action, the question of senescence, and an evolving cell cycle
The development of pharmacological CDK4/6 inhibitors emerged from the longstanding concept that targeting CDK activity would halt cell proliferation and thus limit the pathogenesis of cancer [72,73]. Based on utopian G1/S control, one would predict that CDK4/6 inhibitors should elicit G1 arrest via RB activation. Consistent with this concept, multiple preclinical studies have demonstrated that CDK4/6 inhibitors can block RB phosphorylation and contribute to the suppression of E2F target genes [72–74].
Despite these findings, monotherapy with CDK4/6 inhibitors appears to be effective in only a subset of RB-positive tumor types [75–78]. One tumor in which CDK4/6 inhibitors have had substantial success as monotherapy and in combination with endocrine therapy is HR+/HER2− breast cancer. The combination of CDK4/6 inhibitors with endocrine therapy and monotherapy with abemaciclib is FDA-approved for HR+/HER2− metastatic breast cancer [79]. In this setting, the mechanism of action of the CDK4/6 inhibitor is consistent with expectation – profound cell cycle inhibition as determined from neoadjuvant studies in which the combination elicits ‘complete cell cycle arrest’ [80–82]. Despite the success of CDK4/6 inhibitors in the context of HR+/HER2− breast cancer, several questions remain. (i) Do CDK4/6 inhibitors elicit an irreversible senescent-like cell cycle arrest for their mechanism of action? (ii) Why do CDK4/6 inhibitors only have efficacy in a narrow range of tumors if G1/S control is relatively invariant? (iii) How do HR+/HER2− breast cancers ultimately evade CDK4/6 inhibition during disease progression?
CDK4/6 inhibition and senescence
Although one could argue that the ‘gold-standard’ for senescence is telomere erosion, as defined by Hayflick and colleagues [83], it is clear that multiple cancer drugs can induce features of cellular senescence. Senescence is characterized as irreversible G1 cell cycle arrest. RB activation induced by CDK4/6 inhibitors in non-malignant cells causes visible changes in nuclear organization that are related to stable cell cycle exit [84]. Indeed, pharmacological inhibition of CDK4/6 induces a senescence-associated secretory phenotype (SASP) that is enriched in p53 targets and less in proinflammatory components [85]. In preclinical models it has been shown that CDK4/6 inhibitors, alone or in combination with other therapies (e.g., MEK inhibition), can induce at least some features of senescence, including SASP components and the expression of senescence-associated β-galactosidase [86–89]. Despite fairly robust induction of this phenotype, under most circumstances tumor cells escape cell cycle arrest upon cessation of CDK4/6 inhibitor treatment. It remains unclear whether this represents a reversal of senescence or the existence of small pools of tumor cells that did not become senescent.
Although senescence is challenging to assess in clinical specimens, there is evidence for and against a senescence-like arrest. Arguing against senescence is the finding from neoadjuvant studies with endocrine therapy and CDK4/6 inhibition that, during a break from CDK4/6 inhibition, Ki67 and other markers of proliferation resume relatively rapidly in most cases [80]. This phenomenon has also been recently observed in clinical cases using a circulating thymidine kinase assay (TKa). The expression of thymidine kinase is highly cell cycle-regulated and is under the control of RB/E2F. TKa is significantly suppressed in on-treatment samples in most clinical cases [90]. However, because palbociclib is given for 21 days of a 28 day cycle, during the break from treatment the TKa rebounds in the majority of cases [90]. Such findings suggest that, under standard-of-care conditions, most HR+/HER2− breast cancers do not undergo irreversible growth arrest. However, in support of a senescence-like state, CDK4/6 inhibitor-based treatment regimens can induce an interferon-like response in the neoadjuvant setting; although this is not a direct indication of senescence, it resembles the senescence-associated secretory phenotype [81,86]. In addition, a subgroup of tumors do not exhibit a rise in Ki67 or TKa during treatment cessation [90]. Whether this fraction of cancers represents senescence will need to be more rigorously evaluated.
Limitations of CDK4/6 inhibitors
This question remains challenging to address because several clinical studies based on robust preclinical data or suspected genetic vulnerabilities have failed. For example, although esophageal cancers frequently harbor CCND1 amplification and would be expected to be sensitive to CDK4/6 inhibitors, clinical studies demonstrated no significant activity [91]. The same lack of clinical efficacy has been observed in the context of combination studies; for example, the use of CDK4/6 inhibition with androgen blockade in metastatic prostate cancer and the combination of CDK4/6 inhibitors with traztuzumab in HER2+ breast cancer [92,93]. In these cases, the preclinical data supported a benefit of including the CDK4/6 inhibitor, and clear cooperation with the standard standard-of-care therapy [94,95]. The lack of antitumor activity in these studies, as well as others, suggests that the requirement for CDK4/6 could be much narrower than expected through the canonical cell cycle. Additional limitations in the efficacy of CDK4/6 inhibitors include sequestration of these drugs into tumor cells lysosomes, as demonstrated in triple-negative breast cancers resistant to CDK4/6 inhibition [96,97].
Resistance mechanisms
Given the prevalence of patients with HR+/HER2− metastatic breast cancer, the emergence of resistance to CDK4/6 inhibitors has been the subject of multiple studies, as recently systematically reviewed [98]. Although RB loss can drive resistance, most of the resistance is not associated with RB loss [98,99]. Instead, it appears that the ability of the inhibitor to completely block the activity of CDK4/6 is compromised, or that rewiring of the cell cycle drives a state of CDK4/6-independence (Figure 3A). These findings are rapidly reshaping the overarching concepts of G1/S control.
Figure 3. Mechanisms of resistance to CDK4/6 inhibitors.
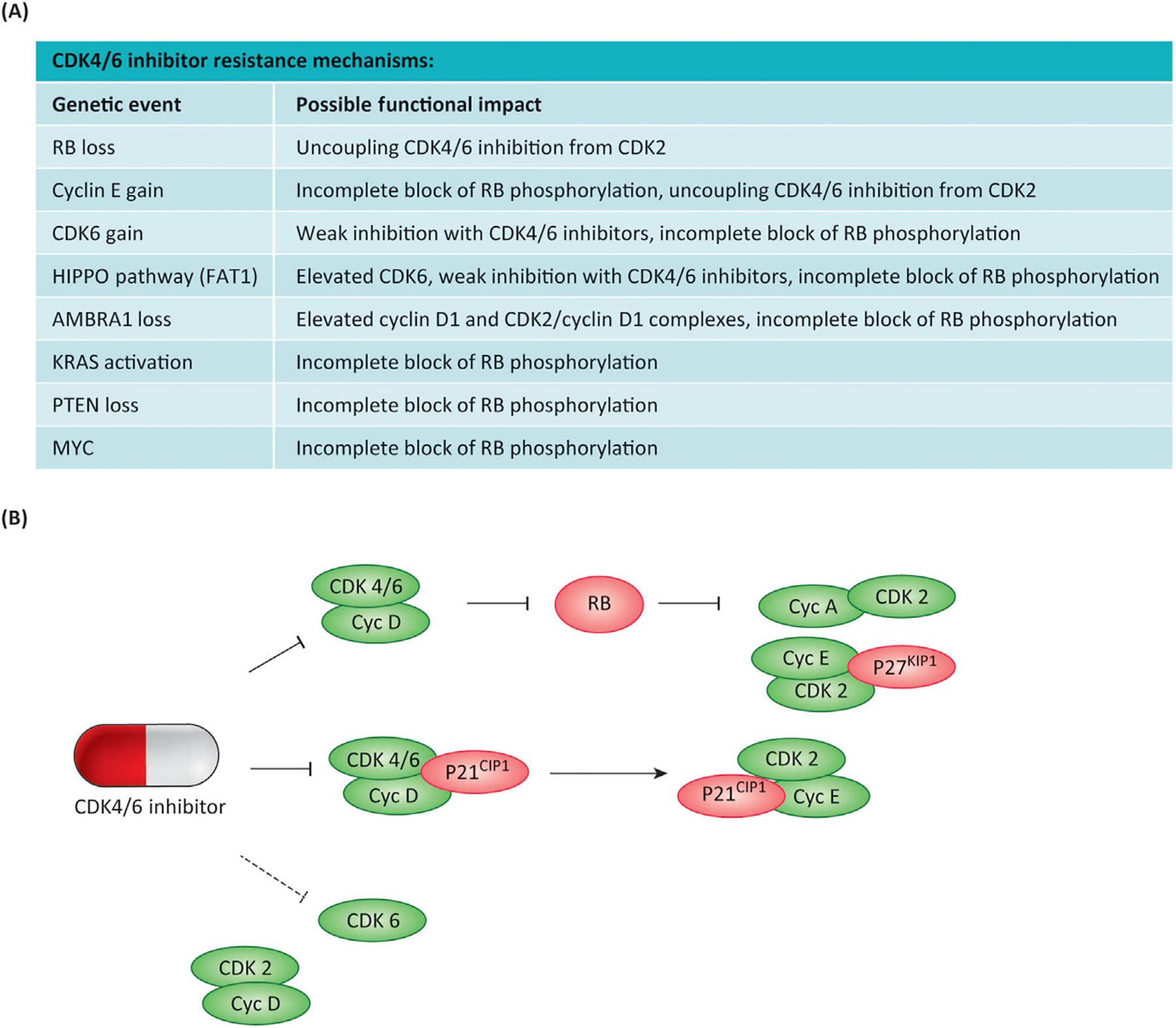
(A) The table summarizes different genetic events that are associated with resistance to CDK4/6 inhibition. (B) The presumed mechanistic consequences for the cell cycle are summarized. The schematic represents different nodes through which CDK4/6 inhibition is coupled to suppression of CDK2 activity, whereas select CDK6 complexes and CDK2/cyclin D1 complexes are resistant to the action of the FDA-approved inhibitors.
CDK4/6–CDK2 coupling as a key element in G1/S control
In searching for common threads related to different mechanisms of resistance, it appears that dependence on CDK4/6 largely involves inhibition of CDK2 [13,100,101]. Although CDK4/6 inhibitors are clearly specific and do not directly inhibit CDK2, there is coupling between CDK4/6 inhibition and the suppression of CDK2 activity. This can occur through several levels of interactions (Figure 3B). First, full RB activation can limit CDK2 activity; this is presumed to involves downregulation of cyclin E and/or A and CDK2 proteins [100]. Second, p21CIP1 and p27KIP1 can shuttle between CDK4/6 complexes and CDK2 [100,102,103]. These proteins serve as CDK2 inhibitors and are likely related to the threshold of sensitivity to CDK4/6 inhibitors. Non-canonical CDK2/cyclin D1 complexes that are not blocked by CDK4/6 inhibition have been reported [13,101], and some CDK6 complexes appear to be resistant to pharmacological inhibition with existing FDA-approved inhibitors [104]. Presumably, these varied mechanisms may co-occur in different settings, underscoring the multifactorial mechanism of resistance. However, CDK4/6–CDK2 coupling suggests that resistance to CDK4/6 inhibitors could be attenuated by combination with CDK2-selective inhibitors. Such inhibitors have been a challenge to develop because of the similarity in the kinase domains of CDK2 and CDK1; however, newly developed CDK2 inhibitors are being tested in clinical studies. Furthermore, a CDK4/6 and CDK2 inhibitor has been shown to suppress the proliferation of multiple tumor models that exhibit resistance to CDK4/6 inhibitors [100,105,106]. These data are driving a resurgence in considering CDK inhibitors in tumor types that are unresponsive to the FDA-approved CDK4/6 inhibitors.
The dystopian cell cycle model
Several studies have shown that classical G1/S cell cycle control and the requirement for specific CDK and cyclins can be subverted by genetic manipulation. However, the extent to which intrinsic heterogeneity and adaptation occur in the context of human cancer continues to evolve, particularly in relation to therapeutic interest in targeting the cell cycle.
Mouse models
Seminal work carried out in multiple laboratories has demonstrated that mouse models can adapt to the loss of multiple CDK and cyclins which would be expected to be essential for G1/S progression. Complete blockade of cell cycle progression in mice would be expected to yield early embryonic lethality due to failure of somatic cells to divide. Because there are multiple D-type cyclins and a degree of redundancy between CDK4 and CDK6, composite knockouts needed to be generated before it was possible to conclude that ‘mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6’, and ‘mouse development and cell proliferation occurs in the absence of D-cyclins’ [107,108]. These models are not without phenotypes, but cells derived from the models have the capacity to transition from G1 to S-phase. These findings have led to doubts that CDK4/6 inhibitors would represent effective therapeutic agents. However, these cyclins appear to be essential for the proliferation of specific oncogene-driven tumors, and CDK4/6 inhibition could be a viable strategy in some cancer settings [109]. Similarly, deletions of genes encoding cyclin E or CDK2 can be tolerated; although there are some deficits in G1/S control, adaptive developmental mechanisms enable the cell cycle to progress, even though depletion suppressed tumor development in some settings [110–112]. This work was carried to its apex when it was shown that, in essence, CDK1 could be sufficient for cell division when genes encoding CDK4, CDK6, and CDK2 were deleted [113]. Whether this occurs physiologically and/or in specific cancer models remains unclear to date. However, these data clearly illustrate that there is intrinsic plasticity in coordinating G1/S in mouse models with the elimination of CDK and cyclin genes.
Intrinsic cell cycle heterogeneity in tumor cells and the importance of context
Human tumor models have shown that the classical model for G1/S control can be broken. Foremost, the RB-deficient cell cycle is CDK4/6-independent. Furthermore, analyses of RB-deficient tumors demonstrated that they often express high-levels of p16INK4A and minimal levels of cyclin D1 [114]. The ensemble of these data suggests that RB-deficient tumors are not only resistant to CDK4/6 inhibition, but to a first approximation these tumors are already partially deficient in CDK4/6 activity [114]. These data have raised the question of how RB-deficient cells divide, particularly because the RB-related proteins, p107 and p130, mediate similar control over E2F-mediated transcription, are regulated by CDK-mediated phosphorylation, and can function as redundant mediators of cell cycle control/tumor suppression [29,115]. Recent studies suggest that there are two forms of RB-deficient cell cycles [116], one driven by cyclin E that is required for the phosphorylation and inactivation of p130, and another driven by currently unidentified kinases (Figure 4).
Figure 4. Differential mechanisms for the control of RB-deficient cell cycles.
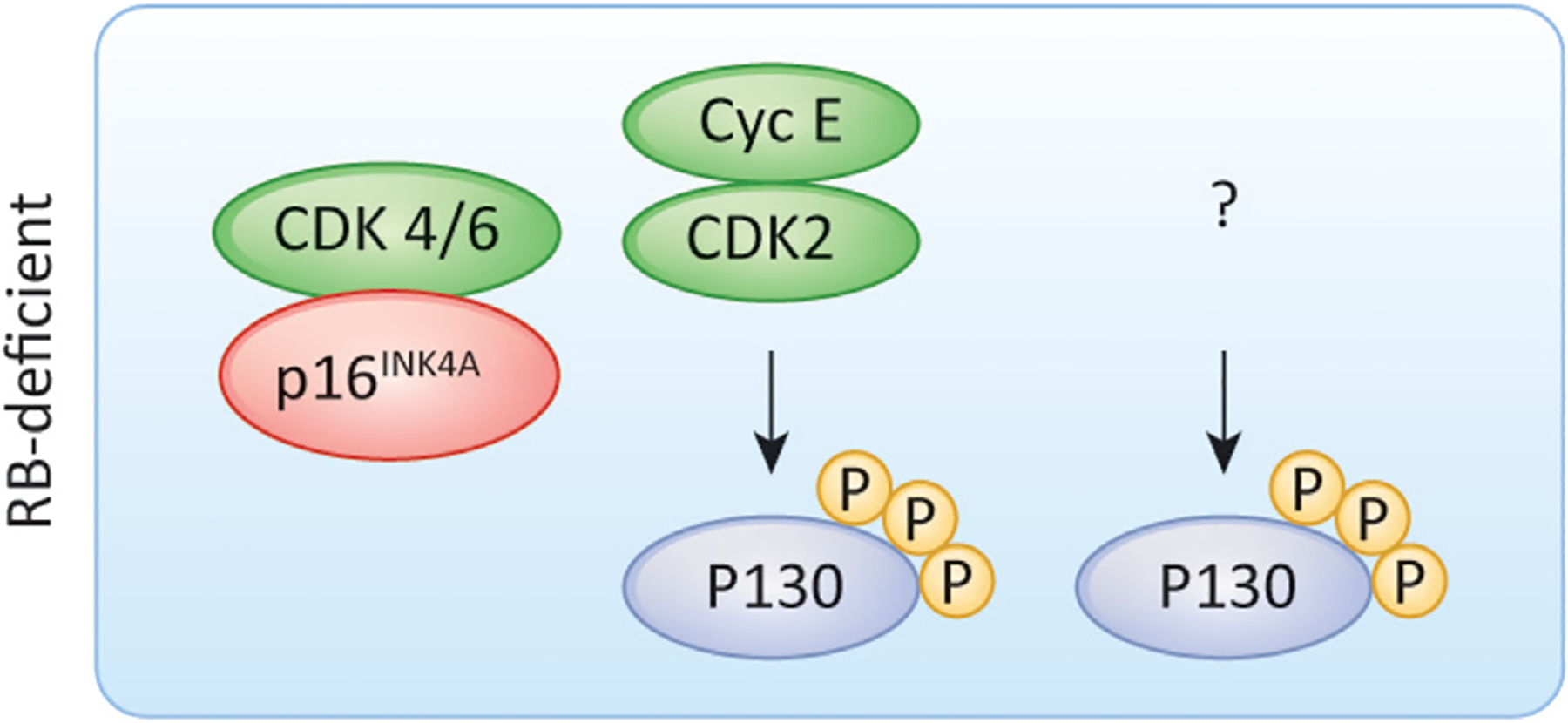
There appear to be at least two different RB-deficient cell cycles. One is cyclin E-dependent, and controls the phosphorylation of p130. In the second the mechanisms controlling p130 phosphorylation to drive G1/S progression are unknown.
There also appears to be heterogeneity in RB-proficient cancer cells. For example, although overexpression of p16INK4A arrests RB-proficient cells in G1 by suppressing CDK4/6 activity, this event can be overcome by ectopic expression of cyclin E [67,117]. Similarly, RB expression arrests the cell cycle of some RB-deficient cells in G1 (e.g., the osteosarcoma cell line SAOS-2). In this setting multiple cyclins, in addition to D-type cyclins, can drive the phosphorylation and inactivation of RB [118]. In retrospect, this work suggested that many CDKs or cyclins can initiate RB phosphorylation/inactivation and G1/S progression under specific conditions, although the extent to which these atypical cell cycles normally operate was unclear. For added complexity, the p38 family of protein kinases have been reported to phosphorylate RB at CDK sites in response to cellular stress [119,120]. Interestingly, these kinases can trigger both RB activation and inactivation in a CDK-independent manner, and their activity is cellular context- and stress condition-specific.
A conundrum in the field has been the requirement for different CDK and cyclins in the canonical cell cycle and how these dependencies are maintained. For example, tumors with genetic alterations driving cyclin D1 activity were associated with a requirement for CDK4/6 and sensitivity to CDK4/6 inhibitors [121]. However, cyclin D1 overexpression driven by AMBRA1 loss is associated with resistance to CDK4/6 inhibitors [12,13]. Similarly, disruption of the HIPPO signaling pathway that drives CDK6 expression is associated with resistance [122]. These findings are also consistent with the observation that depletion of cyclin D1 cooperates with CDK4/6 inhibitors in some contexts, and that ectopic overexpression of cyclin D1 can attenuate the impact of those inhibitors on cell cycle [100,123]. The same point is relevant for the requirement for CDK2 and cyclin E, which have been considered to be either ideal drug targets or irrelevant for cancer cell cycles [60,124]. Interestingly, although the canonical model would suggest a correlation or coupling between the expression of cyclins D1 and E, it has been recently found that the expression of these genes is reciprocal in many cancer cell lines and tumors, suggesting that there are distinct modes through which the cells transition G1/S [116]. It is likely the composite protein stoichiometry of different G1/S CDKs, cyclins, and CDK inhibitors will delineate dependencies.
Given these complexities, a key question is the extent to which there are intrinsic differences in the cell cycle in cancer. The differential dependencies on cell cycle regulatory elements can be probed using large-scale functional genomic screens [116,125]. An example of this analysis is shown in Figure 5A where data from the Cancer Dependency Map (DepMap; https://depmap.org/portal/) CRISPR screening are interrogated in an unbiased fashion [116]. All tumor cells are dependent on CDK1 and cyclin A, as illustrated by the consistent negative CERES scores (CRISPR/Cas essentiality screen: a measure of gene essentiality) across >700 cell lines. However, there is significant heterogeneity in the requirements for the G1/S CDKs and cyclins. Using clustering algorithms, it is possible to define tumors that appear to have intrinsically different requirements for CDKs and/or cyclins (Figure 5B). Some of these distinctions are consistent with genetic analyses; for example, cells in which the cyclin E gene is amplified are more often dependent on the cyclin E. Others track with the known roles of the gene in a particular lineage; for example, hematological tumors are more dependent on CDK6 [116]. Despite these features, there can be multiple distinct requirements within a given histological tumor type. A key determinant of these dependencies is the relative expression of select CDK or cyclin genes [116,125]. Functional and biochemical studies carried out recently have defined multiple different G1/S transitions – those solely driven by cyclin E, which controls RB phosphorylation status; cyclin D1-driven cell cycles, which appear to be CDK4/6-independent; and those that are dependent on CDK4 or 6, but are cyclin E-independent (Figure 5C). In addition to these pathways, non-canonical CDK/cyclin complexes need to be considered; for example, CDK2/cyclin D1 or cyclin D1/P27KIP1 complexes add a further level of complexity [11–13,101]. In essence, it appears that any single CDK activity and perhaps other kinases are capable of driving the G1/S transition in a particular context (Figure 5C).
Figure 5. Distinct G1/S CDK and cyclin dependencies in cancer cells.
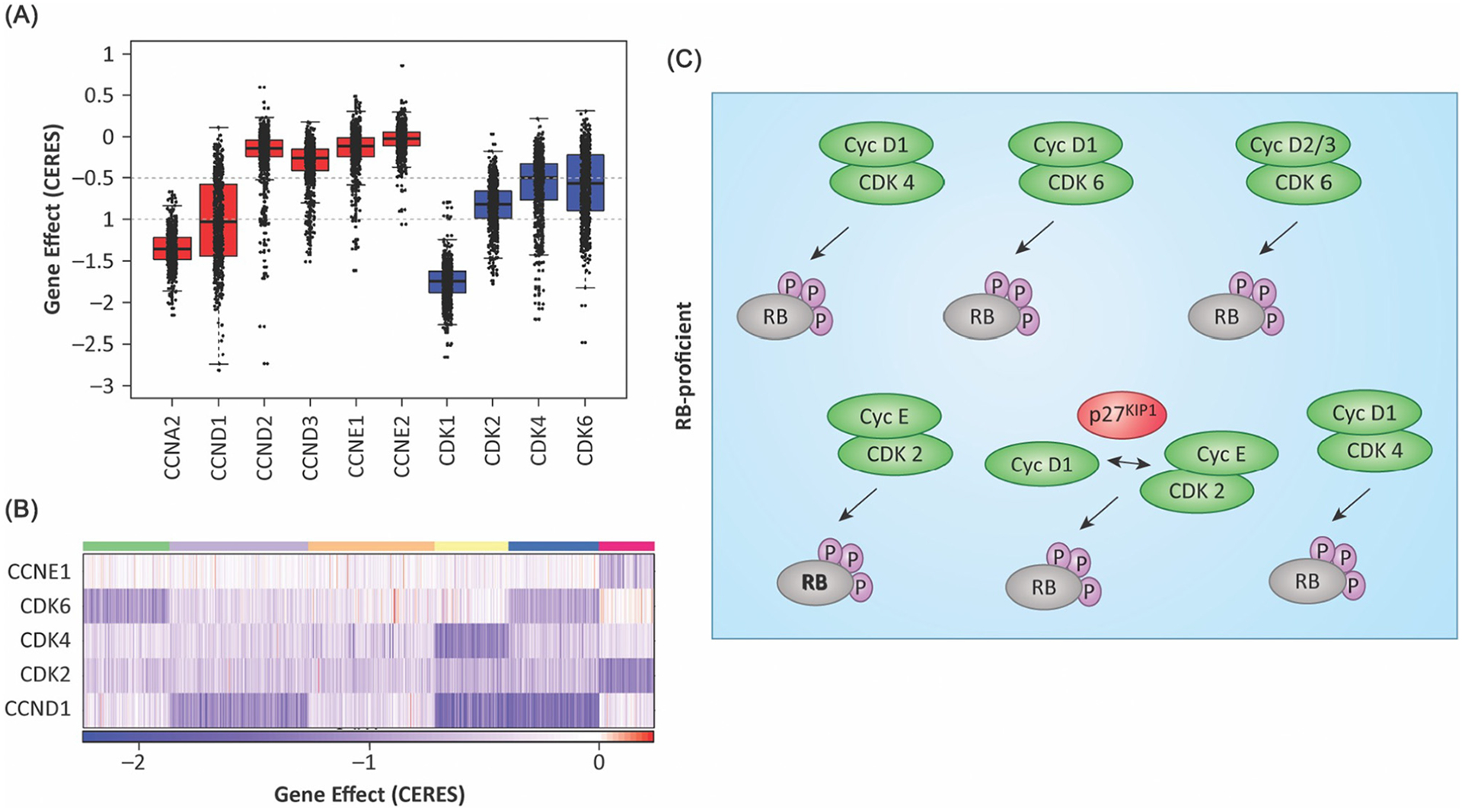
(A) The boxplot summarizes the requirement for the indicated CDK or cyclin as determined from DepMAP data. The CERES (CRISPR/Cas essentiality screen) score is an indication of essentiality, but there is substantial heterogeneity in the requirements for different CDK and cyclin gene in different CRISPR screens. (B) The heatmap illustrates the differential requirement for select CDK or cyclin genes as determined by clustering based on the CERES score. Darker blue indicates a larger magnitude of effect, as summarized in the color bar. (C) Schematic representation of different RB-proficient cell cycles that have been uncovered through screens and functional analyses.
Opportunities for precision medicine
Because the cell cycle is highly variable and is not hard-wired, this has considerable implications for cancer therapy.
Biomarkers
The variability of the cell cycle reinforces the need for biomarkers related to the requirement for a given CDK. Surprisingly, although CDK4/6 inhibitors are widely used in the treatment of HR+/HER2− breast cancer, specific biomarkers to guide treatment have not emerged for clinical utilization [126]. From existing analyses, RB loss and high expression of cyclin E would appear to denote patients with a shorter response to CDK4/6 inhibitor [98,127,128], but RB loss is rare in HR+/HER2− breast cancer and there are clearly other genetic events which limit the effectiveness of CDK4/6 inhibitors [98,99]. Defining the spectrum of these genetic alterations prospectively and delineating intrinsic versus adaptive resistance could represent not only a clinical challenge but also an important opportunity for precision medicine.
In addition to CDK inhibitors, cell cycle regulators are downstream targets of multiple signaling pathways that are targets for therapeutic intervention. For example, the response to KRAS inhibition (e.g., MRTX849) or MEK inhibition is associated with cytostatic cell cycle arrest mediated at least in part by downregulation of cyclin D1 and upregulation of P27KIP1 [100,129]. Such a negative impact on cell cycle could in principle be overcome by RB loss or cyclin E overexpression. Similarly, high levels of cyclin E can drive resistance to a variety of cancer therapeutics (e.g., trastuzumab) [130]. In a similar fashion, endocrine therapy used in prostate and breast cancer therapy also acts through the cell cycle, and RB loss has been associated with resistance to these therapies [131–133]. In the context of breast cancer, gene expression signatures that evaluate E2F activity are widely used in determining benefit from chemotherapy or the requirement for extended adjuvant endocrine treatment to limit disease recurrence [134]. Thus, monitoring the cell cycle could be used to make treatment decisions related to specific targeted therapies, although this approach is currently not widely employed.
Holistically inhibiting the cell cycle
One means to build upon the activity of CDK4/6 inhibitors would be to combine them with other targeted agents (Table 1). This is being interrogated in multiple clinical trials supported by preclinical data which have been the subject of several comprehensive reviews [73,78]. In general, these strategies employ a pathway-selective inhibitor (e.g., MEK inhibition in KRAS mutant cancer) to limit cell cycle plasticity and force more dependence on CDK4/6 [89,100,135]. Whether these combinations will prove to be effective is still being evaluated. An alternative strategy would be to combine a CDK4/6 inhibitor with a CDK2 inhibitor. In this context one would expect that bypass of the G1/S regulatory mechanisms would be ‘impossible’. Although toxicity could be a challenge in such therapies, recently developed CDK4/6/2 and CDK2 inhibitors are under clinical investigation in multiple disease settings [106] (Table 1).
Table 1.
Selectively exploiting the cell cycle for cancer
| Holistic cell cycle suppression | Representative clinical trials | Clinical trial number |
|---|---|---|
| CDK4/6 + endocrine therapy | Multiple | Multiple |
| CDK4/6 + MEK inhibition | Study of the CDK4/6 inhibitor palbociclib (PD-0332991) in combination with the MEK inhibitor binimetinib (MEK162) for patients with advanced KRAS mutant non-small-cell lung cancer | NCT03170206 |
| Palbociclib and binimetinib in advanced triple-negative breast cancer (PALBOBIN) | NCT04494958 | |
| Binimetinib and palbociclib or TAS-102 in treating patients With KRAS and NRAS mutant metastatic or unresectable colorectal cancer | NCT03981614 | |
| CDK4/6 + KRAS (G12C)inhibition | Adagrasib in combination with palbociclib in patients with advanced solid tumors (KRYSTAL-16) | NCT05178888 |
| Sotorasib activity in subjects with advanced solid tumors with KRAS p. G12C mutation (CodeBreak 101) | NCT04185883 | |
| CDK4/6 + ERK inhibition | A basket trial of an ERK1/2 inhibitor (LY3214996) in combination with abemaciclib | NCT04534283 |
| CDK4/6 + PI3K inhibition | Testing the addition of copanlisib to usual treatment (fulvestrant and abemaciclib) in metastatic breast cancer | NCT03939897 |
| A study evaluating the efficacy and safety of inavolisib + palbociclib + fulvestrant versus placebo + palbociclib + fulvestrant in patients with PIK3CA-mutant, hormone receptor-positive, Her2-negative, locally advanced or metastatic breast cancer | NCT04191499 | |
| CDK4/6 + EGFR inhibition | Osimertinib and abemaciclib in EGFR mutant non-small-cell lung cancer after osimertinib resistance | NCT04545710 |
| CDK4/6/2 inhibition | A safety, pharmacokinetic, pharmacodynamic, and antitumor study of PF-06873600 as a single agent and in combination with endocrine therapy | NCT03519178 |
| Study of NUV-422 in combination with enzalutamide in patients with metastatic castrate-resistant prostate cancer | NCT05191017 | |
| Study of NUV-422 in combination with fulvestrant in patients with HR+HER2−aBC | NCT05191004 | |
| CDK2 inhibition | PF-07104091 as a single agent and in combination therapy | NCT04553133 |
| Collateral vulnerabilities | ||
| CHK1 inhibition | A study of LY2606368 (prexasertib) in patients with solid tumors with replicative stress or homologous repair deficiency | NCT02873975 |
| A study of prexasertib (LY2606368) in participants with extensive stage disease small cell lung cancer | NCT02735980 | |
| WEE1 + PARP inhibition | A study of ZN-c3 and niraparib in subjects with platinum-resistant ovarian cancer | NCT05198804 |
| WEE1 inhibition | Phase II, single-arm study of AZD1775 monotherapy in relapsed small-cell lung cancer patients | NCT02593019 |
| AURK inhibition | Alisertib and pembrolizumab for the treatment of patients with RB-deficient head and neck squamous cell cancer | NCT04555837 |
Collateral vulnerabilities
Because many targeted therapies have cytostatic mechanisms of action, alternative cell cycles (i.e., driven by cyclin E or by RB loss) are associated with resistance. However, these alternative cell cycles are associated with replication stress and have compromised cell cycle checkpoints [136–138]. In the case of RB loss, it has been possible to define multiple therapeutic vulnerabilities. Notably, RB loss can impart sensitivity to several drugs that exploit the loss of different cell cycle checkpoints (Figure 5). These include Aurora A kinase inhibitors, Polo-like kinase 1PLK1 inhibitors, WEE1 inhibitors, and PARP inhibitors [116,132,139–142]. It appears that there is some context-selectivity to these vulnerabilities because RB loss is sufficient to yield sensitivity to PARP inhibitors in prostate adenocarcinoma and osteosarcoma models [142]. However, in vivo, concurrent inhibition of PARP and ATR was more effective in prostate cancer models [132]. These findings and ongoing studies suggest that combining agents that are individually effective against RB-deficient tumors will be more broadly effective and increase the therapeutic index [116]. Importantly, in recent clinical studies in small-cell lung cancer, which frequently loses RB, the Aurora A kinase inhibitor (Alisertib) in combination with paclitaxel was more effective selectively in tumors that harbor cell cycle alterations [143]. Cyclin E overexpression has been associated with sensitivity to single-agent WEE1 and PARP inhibition, with the combination being synergistic [136]. In addition, elevated cyclin E expression is associated with sensitivity to the checkpoint kinase 1 (CHK1) inhibitor, prexasertib, in ovarian cancer [144]. Using multiple markers of replication stress, including RB loss and cyclin E gain, another study illustrated improved response to gemcitabine in patients with replication stress-high tumors [145]. Basket trials interrogating tumors with cyclin E gain and RB loss denoting ‘replicative stressed’ cancers are ongoing with CHK inhibitors. However, more studies will clearly be necessary to determine whether aberrant cell cycles can be universally targeted and whether additional vulnerabilities are associated with non-standard G1/S control.
Concluding remarks and future perspectives
The genesis of the common understanding of G1/S regulation was based upon multiple ground-breaking discoveries. Together, these findings supported a straightforward model which today is taught in introductory biology courses. However, a growing list of exceptions to that model in cancer suggest that G1/S control is similar to other cancer signaling pathways where redundancies, complementary mechanisms, and adaptations force a re-evaluation of the diversity of cell cycle in cancers. How these different cell cycles states emerge and whether they are represented in normal cells or specific cellular lineages is an interesting but underexplored concept in human cells. In many contexts, depletion of CDK or cyclins does have a negative impact on proliferation; however, this dependency is rapidly overcome. This has been differentially ascribed to adaptation or selection; single-cell analyses could yield insights into the underlying mechanisms of cell cycle rewiring and the genetic/epigenetic processes that underlie distinct mechanisms of G1/S control. Although this complexity represents a challenge in clinical translation and targeting the cell cycle therapeutically, understanding G1/S control more holistically could be employed to utilize target therapies more efficaciously. Notably, biomarkers and a full understanding of collateral vulnerabilities related to the cell cycle could ultimately be leveraged to advance more precise approaches to cancer treatment (see Outstanding questions).
Outstanding questions.
What is the extent of divergent G1/S cell cycle control in normal tissues/cells?
Are non-canonical cell cycles hardwired into the cell of origin of different cancers?
Are different tumor lineages related to their dependency on a given G1/S regulatory mechanism?
Is the emergence of resistance to CDK4/6 inhibitors due to adaptation or selection?
Does epigenetic plasticity enable switching between different G1/S control mechanisms?
Can cell cycle dependencies be accurately predicted and utilized to direct therapeutic agents?
Do different G1/S control mechanisms impart vulnerabilities beyond existing therapies that target the cell cycle?
Highlights.
The classical model for G1/S cell cycle control is widely accepted as an invariant construct through which mitogenic and oncogenic signals promote cellular division.
Emerging work indicates that the cancer cell cycle varies widely and does not conform to a single canonical model of cell cycle control.
Altered cell cycle states represent one means through which the response to cyclin-dependent kinases (CDK)4/6 inhibitors is increased, and has driven new research into the dystopian plasticity of G1/S control.
New strategies to target the cell cycle either by holistic blockade of G1/S progression or by exploiting aberrant regulatory processes are making their way to clinical application.
Acknowledgments
The authors would like to thank their colleagues for thought-provoking conversations and editorial suggestions. Any oversight of work in this area was unintended, and we have tried to provide a balanced and rigorously cited review article while conforming to Trends in Cancer editorial guidelines. Dr Jianxin Wang and Ms Paris Vail assisted with the data presentation in the figures. This work was supported by grants from the National Institutes of Health to A.K.W. and E.S.K. (CA24736, CA130995, CA211878).
Footnotes
Declaration of interests
The authors declare no conflicts of interest.
References
- 1.Matson JP and Cook JG (2017) Cell cycle proliferation decisions: the impact of single cell analyses. FEBS J. 284, 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3.Harashima H et al. (2013) Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 23, 345–356 [DOI] [PubMed] [Google Scholar]
- 4.Malumbres M et al. (2009) Cyclin-dependent kinases: a family portrait. Nat. Cell Biol 11, 1275–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cross FR et al. (2011) Evolution of networks and sequences in eukaryotic cell cycle control. Philos. Trans. R. Soc. Lond. B Biol. Sci 366, 3532–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee MG and Nurse P (1987) Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327, 31–35 [DOI] [PubMed] [Google Scholar]
- 7.Xiong Y et al. (1991) Human D-type cyclin. Cell 65, 691–699 [DOI] [PubMed] [Google Scholar]
- 8.Matsushime H et al. (1991) Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65, 701–713 [DOI] [PubMed] [Google Scholar]
- 9.Motokura T et al. (1991) A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature 350, 512–515 [DOI] [PubMed] [Google Scholar]
- 10.Sherr CJ (1995) D-type cyclins. Trends Biochem. Sci 20, 187–190 [DOI] [PubMed] [Google Scholar]
- 11.Simoneschi D et al. (2021) CRL4 (AMBRA1) is a master regulator of D-type cyclins. Nature 592, 789–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaikovsky AC et al. (2021) The long-lost ligase: CRL4 (AMBRA1) regulates the stability of D-type cyclins. DNA Cell Biol. 40, 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaikovsky AC et al. (2021) The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 592, 794–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl JA (2002) Cycling to cancer with cyclin D1. Cancer Biol. Ther 1, 226–231 [DOI] [PubMed] [Google Scholar]
- 15.Cao L et al. (2014) Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bockstaele L et al. (2006) Regulation of CDK4. Cell Div 1, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallett ST et al. (2017) Differential regulation of G1 CDK complexes by the Hsp90–Cdc37 chaperone system. Cell Rep. 21, 1386–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stepanova L et al. (1996) Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 [DOI] [PubMed] [Google Scholar]
- 19.Guiley KZ et al. (2019) p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366, eaaw2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel P et al. (2018) Dual inhibition of CDK4 and CDK2 via targeting p27 tyrosine phosphorylation induces a potent and durable response in breast cancer cells. Mol. Cancer Res 16, 361–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong Y et al. (1993) Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 7, 1572–1583 [DOI] [PubMed] [Google Scholar]
- 22.Serrano M et al. (1993) A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 [DOI] [PubMed] [Google Scholar]
- 23.Serrano M et al. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 24.Nobori T et al. (1994) Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368, 753–756 [DOI] [PubMed] [Google Scholar]
- 25.Liggett WH Jr. and Sidransky D (1998) Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol 16, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 26.Matsushime H et al. (1992) Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 71, 323–334 [DOI] [PubMed] [Google Scholar]
- 27.Kato J et al. (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 7, 331–342 [DOI] [PubMed] [Google Scholar]
- 28.Dick FA et al. (2018) Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 18, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burkhart DL and Sage J (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knudsen ES et al. (2019) Cell cycle and beyond: exploiting new RB1 controlled mechanisms for cancer therapy. Trends Cancer 5, 308–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein ME et al. (2018) CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell 34, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodrich DW (2006) The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 25, 5233–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 34.Nevins JR (2001) The Rb/E2F pathway and cancer. Hum. Mol. Genet 10, 699–703 [DOI] [PubMed] [Google Scholar]
- 35.Kent LN and Leone G (2019) The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 19, 326–338 [DOI] [PubMed] [Google Scholar]
- 36.Sellers WR and Kaelin WG (1996) RB as a modulator of transcription. Biochim. Biophys. Acta 1288, M1–M5 [DOI] [PubMed] [Google Scholar]
- 37.Harbour JW and Dean DC (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14, 2393–2409 [DOI] [PubMed] [Google Scholar]
- 38.Sanidas I et al. (2019) A code of mono-phosphorylation modulates the function of RB. Mol. Cell 73, 985–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brehm A and Kouzarides T (1999) Retinoblastoma protein meets chromatin. Trends Biochem. Sci 24, 142–145 [DOI] [PubMed] [Google Scholar]
- 40.Brehm A et al. (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391, 597–601 [DOI] [PubMed] [Google Scholar]
- 41.Knudsen ES and Wang JY (1996) Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J. Biol. Chem 271, 8313–8320 [DOI] [PubMed] [Google Scholar]
- 42.Ren B et al. (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markey MP et al. (2002) Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res. 62, 6587–6597 [PubMed] [Google Scholar]
- 44.DeGregori J et al. (1995) Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol 15, 4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishida S et al. (2001) Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol 21, 4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narasimha AM et al. (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3, e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubin SM (2013) Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci 38, 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lukas J et al. (1995) Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol. Cell. Biol 15, 2600–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukas J et al. (1994) Cyclin D1 protein oscillates and is essential for cell cycle progression in human tumour cell lines. Oncogene 9, 707–718 [PubMed] [Google Scholar]
- 50.Lukas J et al. (1995) Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 375, 503–506 [DOI] [PubMed] [Google Scholar]
- 51.Medema RH et al. (1995) Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc. Natl. Acad. Sci. U. S. A 92, 6289–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aagaard L et al. (1995) Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct sub-sets of human cancer cell lines. Int. J. Cancer 61, 115–120 [DOI] [PubMed] [Google Scholar]
- 53.Knudsen ES et al. (2020) Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun. Biol 3, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koff A et al. (1991) Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 66, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 55.Lew DJ et al. (1991) Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 66, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 56.Ninomiya-Tsuji J et al. (1991) Cloning of a human cDNA encoding a CDC2-related kinase by complementation of a budding yeast cdc28 mutation. Proc. Natl. Acad. Sci. U. S. A 88, 9006–9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai LH et al. (1991) Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature 353, 174–177 [DOI] [PubMed] [Google Scholar]
- 58.Gudas JM et al. (1999) Cyclin E2, a novel G1 cyclin that binds Cdk2 and is aberrantly expressed in human cancers. Mol. Cell. Biol 19, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tadesse S et al. (2020) Targeting CDK2 in cancer: challenges and opportunities for therapy. Drug Discov. Today 25, 406–413 [DOI] [PubMed] [Google Scholar]
- 60.Hwang HC and Clurman BE (2005) Cyclin E in normal and neoplastic cell cycles. Oncogene 24, 2776–2786 [DOI] [PubMed] [Google Scholar]
- 61.Chi Y et al. (2020) A novel landscape of nuclear human CDK2 substrates revealed by in situ phosphorylation. Sci. Adv 6, eaaz9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anders L et al. (2011) A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bertoli C et al. (2013) Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol 14, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohtani K et al. (1995) Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. U. S. A 92, 12146–12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lundberg AS and Weinberg RA (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol 18, 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harbour JW et al. (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869 [DOI] [PubMed] [Google Scholar]
- 67.Knudsen ES et al. (1998) Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 12, 2278–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang HS et al. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC–Rb–hSWI/SNF and Rb–hSWI/SNF. Cell 101, 79–89 [DOI] [PubMed] [Google Scholar]
- 69.Fagundes R and Teixeira LK (2021) Cyclin E/CDK2: DNA replication, replication stress and genomic instability. Front. Cell Dev. Biol 9, 774845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherr CJ (1996) Cancer cell cycles. Science 274, 1672–1677 [DOI] [PubMed] [Google Scholar]
- 71.Bartek J et al. (1997) The retinoblastoma protein pathway in cell cycle control and cancer. Exp. Cell Res 237, 1–6 [DOI] [PubMed] [Google Scholar]
- 72.Asghar U et al. (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov 14, 130–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suski JM et al. (2021) Targeting cell-cycle machinery in cancer. Cancer Cell 39, 759–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Knudsen ES and Witkiewicz AK (2017) The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patnaik A et al. (2016) Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 [DOI] [PubMed] [Google Scholar]
- 76.DeMichele A et al. (2015) CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res 21, 995–1001 [DOI] [PubMed] [Google Scholar]
- 77.Infante JR et al. (2016) A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin. Cancer Res 22, 5696–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alvarez-Fernandez M and Malumbres M (2020) Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 37, 514–529 [DOI] [PubMed] [Google Scholar]
- 79.Spring LM et al. (2020) Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet 395, 817–827 [DOI] [PubMed] [Google Scholar]
- 80.Ma CX et al. (2017) NeoPalAna: neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor positive breast cancer. Clin. Cancer Res 23, 4055–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hurvitz SA et al. (2020) Potent cell-cycle inhibition and upregulation of immune response with abemaciclib and anastrozole in neoMONARCH, phase II neoadjuvant study in HR+/HER2− breast cancer. Clin. Cancer Res 26, 566–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnston S et al. (2019) Randomized phase II study evaluating palbociclib in addition to letrozole as neoadjuvant therapy in estrogen receptor-positive early breast cancer: PALLET trial. J. Clin. Oncol 37, 178–189 [DOI] [PubMed] [Google Scholar]
- 83.Linskens MH et al. (1995) Replicative senescence and cell death. Science 267, 17. [DOI] [PubMed] [Google Scholar]
- 84.Krishnan B et al. (2022) Active RB causes visible changes in nuclear organization. J. Cell Biol 221, e202102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang B et al. (2022) Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 41, e108946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoshida A et al. (2016) Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76, 2990–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruscetti M et al. (2020) Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181, 424–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruscetti M et al. (2018) NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McCartney A et al. (2020) Plasma thymidine kinase activity as a biomarker in patients with luminal metastatic breast cancer treated with palbociclib within the TREnd trial. Clin. Cancer Res 26, 2131–2139 [DOI] [PubMed] [Google Scholar]
- 91.Karasic TB et al. (2020) Phase II trial of palbociclib in patients with advanced esophageal or gastric cancer. Oncologist 25, e1864–e1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Palmbos PL et al. (2021) A randomized phase II study of androgen deprivation therapy with or without palbociclib in rb-positive metastatic hormone-sensitive prostate cancer. Clin. Cancer Res 27, 3017–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goel S et al. (2019) Ribociclib plus trastuzumab in advanced HER2-positive breast cancer: results of a phase 1b/2 trial. Clin. Breast Cancer 19, 399–404 [DOI] [PubMed] [Google Scholar]
- 94.Comstock CE et al. (2013) Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 32, 5481–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goel S et al. (2016) Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fassl A et al. (2020) Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci. Adv 6, eabb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Llanos S et al. (2019) Lysosomal trapping of palbociclib and its functional implications. Oncogene 38, 3886–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Asghar US et al. (2022) Systematic review of molecular biomarkers predictive of resistance to CDK4/6 inhibition in metastatic breast cancer. JCO Precis Oncol. 6, e2100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wander SA et al. (2020) The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor positive metastatic breast cancer. Cancer Discov. 10, 1174–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kumarasamy V et al. (2021) Functional determinants of cell-cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res. 81, 1347–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herrera-Abreu MT et al. (2016) Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pack LR et al. (2021) Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D–CDK4 to inhibit CDK2. Nat. Commun 12, 3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rubin SM et al. (2020) Integrating old and new paradigms of G1/S control. Mol. Cell 80, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li Q et al. (2022) INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer Discov. 12, 356–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pandey K et al. (2019) Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Cancer 145, 1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Freeman-Cook K et al. (2021) Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 39, 1404–1421 [DOI] [PubMed] [Google Scholar]
- 107.Malumbres M et al. (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504 [DOI] [PubMed] [Google Scholar]
- 108.Kozar K et al. (2004) Mouse development and cell proliferation in the absence of D-cyclins. Cell 118, 477–491 [DOI] [PubMed] [Google Scholar]
- 109.Malumbres M and Barbacid M (2006) Is cyclin D1–CDK4 kinase a bona fide cancer target? Cancer Cell 9, 2–4 [DOI] [PubMed] [Google Scholar]
- 110.Gopinathan L et al. (2014) Loss of Cdk2 and cyclin A2 impairs cell proliferation and tumorigenesis. Cancer Res. 74, 3870–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aleem E et al. (2004) Cdk2 as a master of S phase entry: fact or fake? Cell Cycle 3, 35–37 [PubMed] [Google Scholar]
- 112.Berthet C et al. (2003) Cdk2 knockout mice are viable. Curr. Biol 13, 1775–1785 [DOI] [PubMed] [Google Scholar]
- 113.Santamaria D et al. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 [DOI] [PubMed] [Google Scholar]
- 114.Witkiewicz AK et al. (2011) The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 10, 2497–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sage J et al. (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14, 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Knudsen ES et al. (2022) CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep. 38, 110448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lukas J et al. (1997) Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 11, 1479–1792 [DOI] [PubMed] [Google Scholar]
- 118.Hinds PW et al. (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993–1006 [DOI] [PubMed] [Google Scholar]
- 119.Tomas-Loba A et al. (2019) p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature 568, 557–560 [DOI] [PubMed] [Google Scholar]
- 120.Gubern A et al. (2016) The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol. Cell 64, 25–36 [DOI] [PubMed] [Google Scholar]
- 121.Gong X et al. (2017) Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 32, 761–776 [DOI] [PubMed] [Google Scholar]
- 122.Li Z et al. (2018) Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 34, 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Knudsen ES et al. (2019) Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 38, 3355–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tetsu O and McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3, 233–245 [DOI] [PubMed] [Google Scholar]
- 125.Zhang Z et al. (2022) Functional genomic analysis of CDK4 and CDK6 gene dependency across human cancer cell lines. Cancer Res. Published online April 8, 2022. 10.1158/0008-5472.CAN-21-2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anurag M et al. (2020) CDK4/6 inhibitor biomarker research: are we barking up the wrong tree? Clin. Cancer Res 26, 3–5 [DOI] [PubMed] [Google Scholar]
- 127.Turner NC et al. (2019) Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol 37, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bertucci F et al. (2019) Genomic characterization of metastatic breast cancers. Nature 569, 560–564 [DOI] [PubMed] [Google Scholar]
- 129.Xue JY et al. (2020) Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Scaltriti M et al. (2011) Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. U. S. A 108, 3761–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bosco EE and Knudsen ES (2007) RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell Cycle 6, 667–671 [DOI] [PubMed] [Google Scholar]
- 132.Nyquist MD et al. (2020) Combined TP53 and RB1 loss promotes prostate cancer resistance to a spectrum of therapeutics and confers vulnerability to replication stress. Cell Rep. 31, 107669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Beltran H et al. (2019) The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res 25, 6916–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sotiriou C et al. (2006) Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst 98, 262–272 [DOI] [PubMed] [Google Scholar]
- 135.Knudsen ES et al. (2021) Targeting dual signalling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut 70, 127–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen X et al. (2018) Cyclin E overexpression sensitizes triple-negative breast cancer to wee1 kinase inhibition. Clin. Cancer Res 24, 6594–6610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tort F et al. (2006) Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 66, 10258–10263 [DOI] [PubMed] [Google Scholar]
- 138.Guerrero Llobet S et al. (2020) Cyclin E expression is associated with high levels of replication stress in triple-negative breast cancer. NPJ Breast Cancer 6, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Witkiewicz AK et al. (2018) Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell Rep. 22, 1185–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gong X et al. (2019) Aurora-A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 9, 248–263 [DOI] [PubMed] [Google Scholar]
- 141.Oser MG et al. (2019) Cells lacking the RB1 tumor suppressor gene are hyperdependent on Aurora B kinase for survival. Cancer Discov. 9, 230–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zoumpoulidou G et al. (2021) Therapeutic vulnerability to PARP1,2 inhibition in RB1-mutant osteosarcoma. Nat. Commun 12, 7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Owonikoko TK et al. (2020) Randomized phase II study of paclitaxel plus alisertib versus paclitaxel plus placebo as second-line therapy for SCLC: primary and correlative biomarker analyses. J. Thorac. Oncol 15, 274–287 [DOI] [PubMed] [Google Scholar]
- 144.Lee JM et al. (2018) Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. 19, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Konstantinopoulos PA et al. (2021) A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat. Commun 12, 5574. [DOI] [PMC free article] [PubMed] [Google Scholar]