

Abstract
Three separate classes of ribonucleotide reductases exist in nature. They differ widely in protein structure. Class I enzymes are found in aerobic bacteria and eukaryotes; class II enzymes are found in aerobic and anaerobic bacteria; class III enzymes are found in strict and facultative anaerobic bacteria. Usually, but not always, one organism contains only one or two (in facultative anaerobes) classes. Surprisingly, the genomic sequence of Pseudomonas aeruginosa contains sequences for each of the three classes. Here, we show by DNA hybridization that other species of Pseudomonas also contain the genes for three classes. Extracts from P. aeruginosa and P. stutzeri grown aerobically or microaerobically contain active class I and II enzymes, whereas we could not demonstrate class III activity. Unexpectedly, class I activity increased greatly during microaerobic conditions. The enzymes were separated, and the large proteins of the class I enzymes were obtained in close to homogeneous form. The catalytic properties of all enzymes are similar to those of other bacterial reductases. However, the Pseudomonas class I reductases required the continuous presence of oxygen during catalysis, unlike the corresponding Escherichia coli enzyme but similar to the mouse enzyme. In similarity searches, the amino acid sequence of the class I enzyme of P. aeruginosa was more related to that of eukaryotes than to that of E. coli or other proteobacteria, with the large protein showing 42% identity to that of the mouse, suggesting the possibility of a horizontal transfer of the gene. The results raise many questions concerning the physiological function and evolution of the three classes in Pseudomonas species.
Ribonucleotide reductases catalyze the reduction of ribonucleotides to the corresponding deoxyribonucleotides and thereby provide the building blocks for DNA synthesis. Three separate classes of reductases are known (20, 31, 35). Class I enzymes are aerobic enzymes, present in both bacteria and eukaryotes, with the enzyme from Escherichia coli as the prototype. They consist of two homodimers (R1 and R2) encoded by the nrdAB genes. R1 contains the catalytic site and also binds allosteric effectors. R2 contains stable tyrosyl radicals, essential for catalysis, and diferric iron centers. Radical generation requires molecular oxygen, and class I reductases therefore function only during aerobiosis. The two known subgroups of class I (class Ia and class Ib) both conform to the mentioned criteria; they differ in amino acid sequence and also slightly in their allosteric regulation. Bacteria with class Ib enzymes, encoded by the nrdEF genes, also contain a separate hydrogen donor, encoded by nrdH. Class Ib has so far been found exclusively in bacteria.
Class II reductases are widely spread among bacteria, both eubacteria and archaebacteria. They consist of a single polypeptide chain (nrdJ) whose function corresponds to that of protein R1. Adenosylcobalamin supplies the function of protein R2 and acts as radical generator in an oxygen-independent process.
Class III reductases (nrdDG) function only during anaerobiosis. The large homodimer (nrdD) contains, similar to protein R1, catalytic and allosteric sites but in addition also a glycyl radical, stable only in the absence of oxygen. The small homodimer (nrdG) carries an iron-sulfur cluster which together with S-adenosylmethionine and a reducing system generates the glycyl radical.
Why are there so many different reductases? No doubt, their already mentioned response to molecular oxygen plays a role. Conceivably, aerobic organisms could have either a class I or class II enzyme, anaerobic organisms could have a class II or class III enzyme, and facultative anaerobic organisms could have either a class II enzyme or both class I and class III enzymes. Whereas these are indeed the basic rules, many bacteria have genes for additional, apparently unnecessary enzymes. The first such example came from the facultative anaerobic enterobacteria. They contain, in addition to a class Ia and a class III enzyme, also chromosomal genes (nrdEF) for a fully active class Ib enzyme. Under usual laboratory conditions, these do not complement mutations in nrdAB, and their physiological function is not understood (21, 23, 26). Many more examples of apparently redundant reductases in a single organism have been brought to light by the recent explosion in bacterial genomics (19a). Almost any combination of reductase genes can occur. Thus, nrdEF (class Ib) and nrdJ (class II) are present simultaneously in Deinococcus radiodurans (24) and Mycobacterium tuberculosis (7), and nrdJ and nrdDG are found in Methanobacterium thermoautotrophicum (36), Pyrococcus furiosus, Pyrococcus horikoshii, and Porphyromonas gingivalis. Bacillus subtilis (28) and Streptococcus pyogenes each have several different nrdEF genes. Finally, the genomes of Clostridium acetobutylicum and Pseudomonas aeruginosa contain nrdAB, nrdJ, and nrdDG, i.e., genes encoding each class.
Such a diversity of genes is a challenge for our understanding the evolution of ribonucleotide reduction and needs exploration. For this purpose, the pseudomonads appear to be an appropriate subject. These bacteria were originally divided into five groups (I to V) and were all considered to be Pseudomonas species; more recently, only those belonging to group I have been classified as Pseudomonas species (30). Here, we also investigated some members of other groups, which we refer to as Pseudomonas-related species. Pseudomonas species are part of the gamma subdivision of proteobacteria, to which also enterobacteria belong. The Pseudomonas-related species fall into three subdivisions of proteobacteria. All species grow generally in air and are nonfermenters. They can also grow anaerobically in the presence of nitrate or arginine.
The presence of genes encoding members of each of the three classes of ribonucleotide reductases in P. aeruginosa raises two major questions. (i) Is this phenomenon limited to P. aeruginosa, or does it also occur in other Pseudomonas species? (ii) Are several of the genes expressed simultaneously to supply the deoxynucleotides required for DNA replication?
There is only scanty evidence on both points. The growth of P. aeruginosa belonging to the gamma subdivision was reported to be inhibited by hydroxyurea, a rather specific inhibitor of class I enzymes (15). On the other hand, another member of the gamma subdivision, Pseudomonas stutzeri, was shown to contain a B12-dependent class II reductase (17).
Here, we show that the occurrence of multiple genes coding for ribonucleotide reductases is widespread among members of the Pseudomonas species. We demonstrate the presence of active class I and II enzymes in both P. aeruginosa and in P. stutzeri and describe some of their properties.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The Pseudomonas and related strains used in this work were obtained from the Colección Española de Cultivos Tipo (Spanish National Collection of Type Cultures). They are listed in Table 1 and are classified according to the three subdivisions of proteobacteria. Bacteria were grown aerobically in Luria-Bertani (LB) liquid broth at 30°C on a bacterial shaker. For microaerophilic growth, the bacteria were grown at 30°C in a nitrate reduction broth under continuous flushing with N2:CO2 (96:4). One liter of broth contained 5 g of tryptone, 5 g of NaCl, 2 g of yeast extract, and 1 g of KNO3. For DNA cloning, E. coli DH5αF′ (Clontech) was used and grown at 37°C in LB with ampicillin (50 mg/ml) when required.
TABLE 1.
Southern hybridization and ribonucleotide reductase activity of Pseudomonas and related species
| Strain | ATCC code | Southern hybridizationa
|
Ribonucleotide reductaseb (sp act)
|
||||
|---|---|---|---|---|---|---|---|
| nrdA | nrdJ | nrdD | Class I | Class II | Class III | ||
| Pseudomonas species | |||||||
| Gamma subdivision | |||||||
| P. aeruginosa | 15692 | ++ | ++ | ++ | 0.09 | 0.02 | − |
| P. stutzeri | 17588 | ++ | ++ | ++ | 0.14 | 0.12 | − |
| P. putida | 12633 | ++ | ++ | + | 0.68 | − | |
| P. fluorescens | 13525 | ++ | ++ | + | 0.28 | − | |
| Unclassified | |||||||
| P. denitrificans | 19244 | + | ++ | ++ | − | 0.051 | |
| Related species | |||||||
| Gamma subdivision | |||||||
| Stenotrophomas maltophilia | 13637 | + | ++ | − | − | − | |
| Xanthomonas campestris | 33913 | + | ++ | + | − | − | |
| Beta subdivision | |||||||
| Ralstonia pickettii | 27511 | ++ | ++ | ++ | 0.10 | − | − |
| Burkholderia cepacia | 17759 | ++ | ++ | + | 0.039 | − | |
| Comamonas testosteroni | 11996 | + | − | − | 0.096 | − | |
| C. acidovorans | 15668 | + | + | − | − | − | |
| Hydrogenophaga flava | 33667 | + | + | + | ND | ND | |
| Alpha subdivision | |||||||
| Brevundimonas diminuta | 11568 | − | + | − | − | − | |
| B. vesicularis | 11426 | − | + | − | − | − | |
Performed with the P. aeruginosa probes described in Materials and Methods. ++, positive hybridization at high stringency; +, positive hybridization at low stringency; −, no hybridization.
Specific activities of class I and II enzymes are from experiments with extracts from aerobically grown bacteria. Assays were done with CDP as the substrate, in the presence of 2 mM ATP, 20 mM DTT, and 10 mM MgCl2. Class III activity was assayed with extracts from microaerobically grown bacteria under the anaerobic conditions described for assay of the class III E. coli reductase (18). —, no activity found; ND, not determined.
DNA probes and Southern hybridization.
For PCR amplification of fragments of P. aeruginosa nrdA, nrdJ, and nrdD, the required sequence information was found in the database (30a). The fragments were used as DNA probes for Southern hybridizations. For the nrdA fragment we used primers PAO-A-up (5′-ATGCTCGATAACGTCATCGA-3′) and PAO-A-low (5′-CGACTGGGCCTGGTCGAT-3′), corresponding to peptides MLDNVID and IDQAQS, respectively; for the nrdJ fragment we used PAO-J-up (5′-ACCAACCCCTGCGGCGA-3′) and PAO-J-low (5′-GATGGTCCCGGTCGGCGCGAT-3′), corresponding to peptides TNPCGE and IAPTGTI, respectively; for the nrdD fragment we used PAO-D-up (5′-CATATCCACGACCTCGA-3′) and PAO-D-low (5′-GAGGAGTTGGTGTAGTA-3′), corresponding to peptides HIHDLD and YYTNSS, respectively.
Genomic DNA was extracted from the pseudomonads by the cetyltrimethylammonium bromide method (1). DNA from P. aeruginosa (0.2 mg) was PCR amplified by using 50 pmol of each of the three primer pairs together with 1 mM each deoxynucleoside triphosphate, 0.005 ml of 10× PCR buffer (Boehringer Mannheim), and 1.5 U of Taq polymerase in a 0.05-ml reaction mixture. The reaction was run with the following program: 3 min at 94°C; 30 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 68°C; and 7 min at 68°C. The resulting amplification products of 729 bp (nrdA fragment), 615 bp (nrdJ fragment), and 1,208 bp (nrdD fragment) were purified from an ethidium bromide–2% NuSieve-agarose gel by melting the band in 6 M NaI at 50°C and using the Wizard DNA Clean-up system (Promega) and then cloned in pGEM-T (Promega) according to the manufacturer’s protocol. The fragments were labeled by PCR amplification as described above but using a deoxynucleoside triphosphate mixture containing 0.35 mM digoxigenin-11-dUTP and 0.65 mM dTTP in place of 1 mM dTTP.
Southern hybridizations were done by standard methods (34) with a DIG DNA labeling and detection kit (Boehringer Mannheim). During high-stringency conditions, hybridization was done at 42°C in the presence of 50% formamide, with the posthybridization wash at 68°C with 0.1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% sodium dodecyl sulfate (SDS). During low-stringency conditions, hybridization was done at 30°C in formamide and the wash was at 60°C with 1× SSC–0.1% SDS. For chemiluminescence detection, the CSPD (Tropix) reagent was used.
Extraction and partial purification of ribonucleotide reductases.
Bacteria grown to the end of the logarithmic phase were cooled on ice, and all further manipulations were carried out close to 4°C. Crude extracts were prepared as described previously (23) in a buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM KCl, 1 mM EDTA, 10 mM dithiothreitol (DTT), 1 mM phenylmethanesulfonyl fluoride, and 1.0 mg of egg white lysozyme per ml and precipitated with ammonium sulfate to 70% saturation. The dissolved precipitate was dialyzed (23) and either used directly for assay of enzyme activity or further purified by chromatography on dATP-Sepharose as described below. Extracts from bacteria grown under nitrogen were manipulated and extracted inside an anaerobic box. The ammonium sulfate step was omitted, but the extracts were dialyzed overnight inside the box before use.
Ribonucleotide reductases were purified from the bacterial extracts in buffers containing 30 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 2 mM DTT, and various amounts of KCl by chromatography on columns of dATP-Sepharose (1 ml of resin per 30 mg of protein). For the class II enzyme from P. stutzeri, no KCl was used and the reductase was eluted with 0.5 mM ATP. The class I enzyme from P. stutzeri was loaded at 0.3 M KCl, the column was washed with 0.5 M KCl, and the R1 protein was subsequently eluted with 5 mM ATP. The P. aeruginosa class I enzyme was loaded at 0.5 M KCl, the column was washed with 0.5 mM ATP, and the R1 protein was eluted with 5 mM ATP.
Ribonucleotide reductase assay.
The reduction of CDP or CTP was used as an assay (39). Incubation was at room temperature (ca. 26°C) for 20 min in a final volume of 0.05 ml containing 50 mM Tris-HCl (pH 8.0), 0.5 mM [3H]CDP or [3H]CTP (17 cpm/pmol), 10 mM MgCl2, 20 mM DTT, and 2 mM ATP or 0.3 mM dATP (as allosteric effectors). To measure class II activity, the extract was preincubated with 5 mM hydroxyurea for 30 min, the assay mixture contained 15 μM adenosylcobalamin, and the assay was done anaerobically under dimmed light.
Attempts to measure class III activity in extracts from bacteria after microaerophilic growth were made anaerobically as described earlier (18).
One unit of enzyme activity is the formation of 1 nmol dCDP or dCTP per min. Specific activity is expressed as units per milligrams of protein.
Other methods.
Protein concentrations were determined by the Bradford assay (5), with crystalline bovine serum albumin as a standard. The NH2-terminal sequence of the R1 protein from P. aeruginosa was determined in a PE-Applied Biosystems model 494 amino acid sequencer as instructed by the manufacturer. Analytical protein gel electrophoresis was done in denaturing polyacrylamide gels either with the Phastgel system (Amersham-Pharmacia Biotech) or with Mini Protean II (Bio-Rad). DNA manipulations were done by standard procedures (34).
RESULTS
Presence of genes for different ribonucleotide reductase classes in Pseudomonas species.
The genome sequence of P. aeruginosa contains the genes nrdAB, nrdJ, and nrdDG for the three classes of ribonucleotide reductases (30a). Using the sequence information, we purified and digoxigenin-labeled fragments of each gene cluster by PCR as described in Materials and Methods to provide probes for Southern hybridizations used to identify homologous genes in other Pseudomonas or Pseudomonas-related species. The genomic DNA from each species was for this purpose digested with EcoRI and doubly digested with EcoRI and BamHI.
Results of hybridization with members of various Pseudomonas and related species obtained under high- and low-stringency conditions are summarized in Table 1. Under high stringency, hybridization was found only with species from the gamma and beta subdivisions of proteobacteria; at low stringency, hybridization was found in all subdivisions. All positive results were obtained in two separate hybridizations. When the results from low-stringency hybridizations are included, we find that, in addition to P. aeruginosa, the following species contained genes for all three classes of ribonucleotide reductases: P. stutzeri, Pseudomonas putida, Pseudomonas fluorescens, Xanthomonas campestris, Ralstonia pickettii, Burkholderia cepacia, Hydrogenophaga flava, and Pseudomonas denitrificans.
Evidence for the coexistence of nrdA and nrdJ was found for Stenotrophomonas maltophilia and Comamonas acidovorans. nrdA alone was found in Comamonas testosteroni; nrdJ alone was found in Brevundimonas diminuta and B. vesicularis. No positive results were found with any species with a probe corresponding to the nrdEF genes from Corynebacterium ammoniagenes (13).
Our results demonstrate the widespread occurrence of all three classes, often all together, among Pseudomonas and related species. The apparent absence of the genes encoding certain classes from some of the species may be due to specific environmental pressures, or it may be that the genes were not detected because of limited homology to the P. aeruginosa sequence.
Ribonucleotide reductase activities in different species.
To determine the extent to which the genes for the different classes were expressed during growth, extracts from aerobically growing bacteria were prepared from all species and analyzed for class I and II activity. P. aeruginosa, P. stutzeri, and R. pickettii were also grown microaerobically under nitrogen, and their extracts were analyzed for class I, II, and III activity. Under strict anaerobic conditions, obtained by addition of 3.2 mM sodium sulfide to the media in closed bottles, growth was insufficient.
It was possible to differentiate between class I and II activity in the following way. Class II reductases show a strict requirement for adenosylcobalamin; in its absence, the assay is specific for class I. Two factors were combined to obtain an assay specific for class II reductases: (i) the extract was first treated with 5 mM hydroxyurea to inactivate the tyrosyl radical of class I; and (ii) oxygen was excluded from the assay. As demonstrated below, the Pseudomonas class I enzymes required the continuous presence of oxygen for activity. In the absence of adenosylcobalamine, anaerobic extracts treated with 5 mM hydroxyurea were completely devoid of activity. Class III enzymes finally are dependent on adenosylmethionine, K+ ions, and a specific reducing system (18) and require strict anaerobiosis. Class III enzymes are thus inactive when the assay is performed aerobically.
Table 1 shows results from assays of Pseudomonas extracts done under conditions specific for either class I or class II activity. Data for P. aeruginosa and P. stutzeri are given in more detail below. Extracts from P. putida, P. fluorescens, B. cepacia, R. pickettii, and C. testosteroni contained only measurable class I activity, with specific activities ranging from 0.04 to 0.68. In contrast, in extracts prepared from P. denitrificans, only class II activity was detected. These results show that in most of the species that contained the genetic information for several enzymes, only one class had measurable activity under the growth conditions assayed. In extracts from some species we found no activity, possibly because of presence of some inhibitory activity, as addition of a small amount of crude extracts from these species was inhibitory in an assay of the P. aeruginosa class I activity (data not shown). Similar inhibitory effects may also be responsible for the absence of class II activity in some cases.
P. aeruginosa and P. stutzeri express class I and class II enzymes simultaneously.
In earlier work, P. aeruginosa was shown to contain class I activity (15) and P. stutzeri was shown to contain class II activity (17). After aerobic growth we found, instead, that extracts from each species contained both class I and class II activity (Table 2). Also, after microaerophilic growth under nitrogen, both types of activity were found (Table 2). Surprisingly, class I activity was greatly increased under these conditions. Attempts to find class III activity were unsuccessful under conditions that routinely detected class III activity in E. coli extracts.
TABLE 2.
Class I and II ribonucleotide reductase activities of P. aeruginosa and P. stutzeri extracts after aerobic and microaerobic growth
| Species | Growth | Sp act
|
|
|---|---|---|---|
| Class I + regenerated ATPa | Class II + regenerated ATPb | ||
| P. aeruginosa | Aerobic | 0.088 + 0.023 | 0.025 + 0.036 |
| Microaerobic | 1.7 + 0.35 | 0.10 + 0.11 | |
| P. stutzeri | Aerobic | 0.14 + 0.039 | 0.13 + 0.20 |
| Microaerobic | 0.54 + 0.11 | 0.15 + 0.20 | |
Assays were performed under class I conditions but in the presence of 20 mM phosphoenolpyruvate and 50 μg of pyruvate kinase per ml to regenerate ATP.
Assays were performed under class II conditions but with ATP regeneration as described in footnote a.
The assays used for the results in Table 2 were done with CDP as the substrate. In crude extracts, CDP and CTP are interconverted rapidly, and it is difficult to determine which of them serves as a substrate for the reductase. A distinction can be made, however, from the effect of added phosphoenolpyruvate and pyruvate kinase, which shifts the equilibrium between the two nucleotides toward the triphosphates. The results in Table 2 and other experiments showed that this addition inhibited both class I enzymes but slightly stimulated the class II enzymes, suggesting that the former are ribonucleoside diphosphate reductases whereas the latter are ribonucleoside triphosphate reductases.
Oxygen is continuously required for the class I enzymes.
During microaerobic growth class I enzymes were expressed to much higher levels than during aerobic growth (Table 2). As demonstrated by the results of Table 3, oxygen was continuously required during the assay of class I enzymes to generate their tyrosyl radical. In this experiment, we preincubated anaerobic extracts from microaerobically grown bacteria first for 30 min, either aerobically or anaerobically, and subsequently carried out the assay, again either aerobically or anaerobically. In repeated experiments, we found that class I activity required aerobiosis during the second step, whereas class II activity was independent of oxygen.
TABLE 3.
Oxygen dependence of class I activitya
| Preincubation | Assay | Sp act
|
|||
|---|---|---|---|---|---|
|
P. aeruginosa
|
P. stutzeri
|
||||
| Class I | Class II | Class I | Class II | ||
| Anaerobic | Anaerobic | 0.17 | 0.18 | 0.030 | ND |
| Aerobic | Aerobic | 2.2 | 0.16 | 0.82 | 0.045 |
| Anaerobic | 0.065 | 0.18 | 0.049 | 0.034 | |
Anaerobic extracts from microaerobically grown bacteria were preincubated either aerobically or anaerobically for 30 min at room temperature. The appropriate standard components required for the assay of class I or II reductases were added, and ribonucleotide reduction was determined after 20 min of aerobic or anaerobic incubation. ND, not determined.
Requirements for enzyme activity.
Class I activity from P. aeruginosa and P. stutzeri was studied in extracts from microaerobically grown bacteria, as they showed the highest activity. Class II activity from P. stutzeri was studied in extracts from aerobically grown cells, as they contained the highest ratio of class II to class I activities. Class II activity depended linearly on the concentration of protein, whereas class I activity displayed S-shaped dependence (data not shown). The latter result can be attributed to the fact that the class I enzymes consist of two loosely bound proteins (NrdA and NrdB), similar to the E. coli class Ia reductase (6).
The requirements of the two P. stutzeri enzymes for maximal activity are summarized in Table 4. Both enzymes depended on the presence of an allosteric activator, ATP for class I and dATP for class II (see also below). DTT could serve as an artificial reductant for both enzymes, presumably replacing a thioredoxin or glutaredoxin system. Mg2+ or Ca2+ stimulated both enzymes moderately. Class II activity was completely dependent on the presence of adenosylcobalamin.
TABLE 4.
Requirements for P. stutzeri class I and II ribonucleotide reductases
| Growth system | Activity (% of activity in complete system)
|
|
|---|---|---|
| Class Ia | Class IIb | |
| −Mg2+ | 29 | 41 |
| −Mg2+ + 10 mM Ca2+ | 121 | 125 |
| −DTTc | 61 | 12 |
| −ATP and dATP | 7 | 0.6 |
| −Adenosylcobalamin | 100 | 0 |
Measured in the presence of 0.5 mM CDP, 10 mM Mg2+, 20 mM DTT, and 2 mM ATP.
Measured in the presence of 0.5 mM CTP, 10 mM Mg2+, 20 mM DTT, 0.3 mM dATP, and 15 μM adenosylcobalamin.
A small amount of DTT (2 mM for class I; 0.25 mM for class II) remained from the extract.
The effects of the allosteric modulators ATP and dATP on class I and II activity are shown in Fig. 1 and 2, respectively. In extracts of both P. aeruginosa and P. stutzeri, the reduction of CDP by the class I enzyme was strongly stimulated by ATP (Fig. 1A). The stimulatory effect of ATP on the activity of the P. stutzeri enzyme decreased above concentrations of 1 mM. No such decrease was observed for the P. aeruginosa enzyme. At high concentration of ATP, increasing amounts of CDP were removed by phosphorylation to CTP, thus depleting the substrate concentration (data not shown). The Km values for CDP were 0.15 mM for the P. stutzeri reductase and 0.55 mM for the P. aeruginosa enzyme. The latter is therefore less affected by the lowered CDP concentration. The decrease in enzyme activity of the P. stutzeri enzyme above 1 mM ATP was not seen when the initial concentration of CDP was increased from 0.5 to 2 mM (data not shown).
FIG. 1.

Effect of ATP (A) or dATP (B) on CDP reduction by Pseudomonas class I reductase. Extracts from microaerobically grown P. aeruginosa (0.14 mg of protein; ●) or P. stutzeri (0.20 mg; □) were assayed under class I conditions except for the concentrations of ATP or dATP indicated on the abscissa.
FIG. 2.

Effect of ATP (●) or dATP (□) on CTP reduction by P. stutzeri class II reductase. An extract (0.17 mg of protein) from aerobically grown P. stutzeri was assayed under class II conditions except for the concentrations of ATP and dATP indicated on the abscissa.
dATP partially replaced ATP as positive effector for both the P. aeruginosa and the P. stutzeri class I reductases (Fig. 1B). With the P. aeruginosa enzyme, a strong stimulation was found at all concentrations of the effector. With the P. stutzeri enzyme, maximal stimulation occurred at 10 μM dATP but then decreased, and the nucleotide became inhibitory. This dual effect of low concentrations of dATP is similar to the effect of dATP on the E. coli class Ia reductase and suggests that the nucleotide acts as both an allosteric stimulator and inhibitor. For the class II activity of the P. stutzeri extract (Fig. 2), dATP was a better positive effector for CTP reduction than ATP, with no inhibitory effects at high effector concentrations. These allosteric effects are discussed further in Discussion.
Separation and purification of class I and II reductases.
Affinity chromatography on dATP-Sepharose has enabled the rapid purification of many ribonucleotide reductases (3, 10, 11, 23). As both the NrdA (class I) and NrdJ (class II) proteins of the Pseudomonas reductases contain allosteric dATP-binding sites, we used this method for their purification and separation as described in Materials and Methods. When a bacterial extract at low salt concentration was chromatographed on a dATP-Sepharose column, NrdB (class I) was recovered in the flowthrough fraction, whereas NrdA (class I) and NrdJ (class II) were retained. NrdJ could be eluted with 0.5 mM ATP; NrdA required 5 mM ATP. With this procedure, NrdA from both P. aeruginosa (Fig. 3, lane 1) and P. stutzeri (lane 3) were obtained in a close to homogeneous form with subunit molecular masses of approximately 100 kDa. NrdJ from P. stutzeri was purified 31-fold but on gels showed several bands in addition to one band at close to 100 kDa that may correspond to the reductase (lane 4).
FIG. 3.
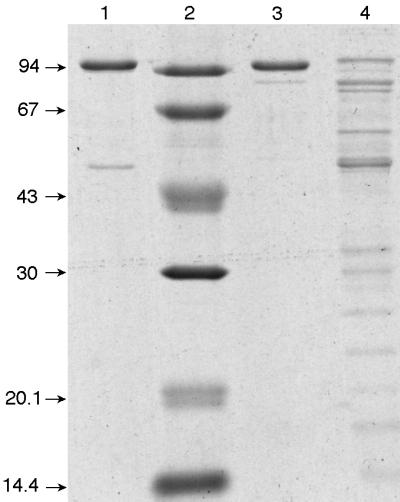
Separation by 12% polyacrylamide denaturing gel electrophoresis of the enzymes purified by dATP-Sepharose chromatography. Lane 1, P. aeruginosa NrdA; lane 2, molecular weight markers; lane 3, P. stutzeri NrdA; lane 3, P. stutzeri NrdJ. Sizes are indicated in kilodaltons.
For enzyme activity, the NrdA proteins absolutely required complementation with NrdB present in the flowthrough fraction, whereas the NrdJ protein was active by itself but was stimulated slightly by the flowthrough fraction (data not shown). This effect could be due to the presence of a physiological electron donor protein, such as thioredoxin. The purified NrdJ protein was characterized further with respect to allosteric effects, with results identical to those shown in Fig. 2, i.e., with dATP being the preferred effector for CTP reduction (data not shown).
The P. aeruginosa NrdA protein was of sufficient purity to permit the determination of its NH2-terminal sequence, resulting in the sequence MHTDTTRENPQA. A comparison with the nrdA genome sequence (30a) establishes the identity of the purified protein and unequivocally determines the translation starting codon, giving a calculated molecular mass of 107 kDa for the polypeptide chain, in reasonable agreement with the apparent mobility on the denaturing gel (Fig. 3).
DISCUSSION
Our data demonstrate that several Pseudomonas species and Pseudomonas-related species contain the genes for all three classes of ribonucleotide reductases and that two separate reductases are simultaneously active in at least two of these species. To date, when an organism was found to contain two separate active ribonucleotide reductases, the proteins were expressed under different circumstances, e.g., during aerobiosis or anaerobiosis (14, 32). The present finding of two active enzymes that are expressed in parallel poses new questions concerning the physiology, gene regulation, and evolution of ribonucleotide reduction.
How reliable are our results? Positive DNA hybridization data are probably good indicators for the existence of a gene, whereas negative data may simply indicate that the DNA sequence is too divergent to give a positive signal. It is, however, safe to conclude that many species of Pseudomonas contain the genetic information for several different ribonucleotide reductases. With respect to the expression of enzyme activity, negative results may be due to technical problems, as ribonucleotide reduction is notoriously difficult to assay in extracts from slowly growing cells; therefore, our data may underestimate the generality of simultaneous enzyme expression.
As described in the introduction, class I enzymes are divided into two subclasses, Ia and Ib, coded by nrdAB and nrdEF genes, respectively. The class I reductases of Pseudomonas belong to class Ia. For P. aeruginosa, this is apparent from the nucleotide sequences of the two genes as well as from the absence of nrdHI genes in the genome (21, 25). An additional distinctive criterion for class Ia enzymes is their inhibition by dATP, which depends on an allosteric dATP-binding site (activity site) present at the NH2 terminus of the NrdA protein but absent from NrdE of class Ib enzymes (9, 12). The P. stutzeri enzyme is inhibited by dATP and thus conforms to this pattern. The P. aeruginosa reductase was not inhibited by dATP. Its amino acid sequence at the NH2 terminus does, however, present many of the structural features typical for a dATP-binding site (12). Lack of inhibition by dATP despite the presence of a binding site was found previously for some other class Ia enzymes (2, 19) and also some class II enzymes (10). This observation suggests the presence of a degenerate site that during evolution has lost its function (10).
A comparison of the complete amino acid sequence of the P. aeruginosa NrdAB enzyme with that of other reductases gives surprising results. Both NrdA and NrdB show higher homologies with enzymes from eukaryotes (42% identity with mouse NrdA; 28% for NrdB) than with enzymes from closely related bacteria (27% identity with E. coli NrdA; 24% for NrdB). An exception occurs for the enzyme of an intracellular pathogenic bacterium, Chlamydia trachomatis (37), to which P. aeruginosa is 52% identical for NrdA and 57% for NrdB. Possibly related to this is our finding that the Pseudomonas enzyme required the continuous regeneration of its tyrosyl radical by oxygen during catalysis, similar to the mouse enzyme. For the E. coli NrdAB (16) and the Lactococcus lactis NrdEF (22) class I reductases, it was shown that once this radical was generated, molecular oxygen was no longer required.
The two class II reductases are allosterically regulated ribonucleoside triphosphate reductases, similar to some other microbial class II enzymes. The sequence of the P. aeruginosa enzyme contains the catalytically active cysteines, first found in the Lactobacillus leishmanii reductase (4), as well as residues for the allosteric substrate specificity site identified from the crystallographic investigation of enzyme-effector complexes of the E. coli class I reductase (12) and present also in other class II reductases (24). The NH2 terminus does not contain the amino acids required for a dATP-binding site (12, 24), and consequently the enzymes were not inhibited by dATP.
The deduced amino acid sequence of the P. aeruginosa class III reductase includes appropriately positioned residues for the characteristic glycyl radical (38), the putative catalytically active cysteines (29, 35), and the effector-binding site (24). The DNA thus has the hallmarks of a gene coding for an active enzyme. However, we could not demonstrate a class III activity in bacterial extracts after microaerophilic growth, conditions that induce this activity in E. coli. We cannot exclude the possibility that this was the result of problems with the enzyme assay. However, microaerophilic growth induced a large increase in class I activity, similar to what was observed in an E. coli nrdD mutant strain, i.e., in a strain that lacks class III activity (16). Moreover, the finding that the assayed Pseudomonas strains did not grow under complete anaerobiosis is consistent with the lack of a catalytically active class III enzyme.
How does Pseudomonas employ its class I and II reductases? Why is it that the oxygen-requiring class I reductase is primarily increased during microaerobiosis and not the class II enzyme that works equally well in the absence of oxygen? The two enzymes use different substrates. The class I enzyme is a diphosphate reductase, the class II enzyme a triphosphate reductase. In the case of P. stutzeri, the class I enzyme is allosterically inhibited by dATP and the class II enzyme is stimulated. These different requirements make it difficult to understand how the reductases are regulated in vivo and how enzyme activity is attuned to the requirements of DNA replication. Further work with genetically modified bacteria is clearly required to answer some of these questions.
An additional, equally puzzling problem is how this situation has arisen during evolution. We suggested earlier that ribonucleotide reduction arose early during the evolution of life under anaerobic conditions during the transition of an RNA to a DNA world (20, 31). Of the enzymes present today, a class III-like reductase appeared to be the best candidate for an early enzyme. Class II and class I enzymes, in that order, evolved later, when oxygen appeared in the atmosphere. Oxygen became a major driving force for the diversification of ribonucleotide reductases, since the glycyl radical of class III enzymes is oxygen sensitive and new methods of radical generation were required during aerobiosis.
How do P. aeruginosa and other Pseudomonas species fit into this a model? The occurrence of all three classes in one bacterium could mean that each class, once it had evolved, was maintained. But why should an organism with a functioning class II enzyme evolve a class I reductase? One conceivable reason would be that the bacterium had lost the ability to synthesize B12. However, P. denitrificans can make its own B12 (8, 33), and also P. aeruginosa contains the necessary genomic information. A possible explanation may derive from the finding that the class I enzyme of P. aeruginosa is closely related to eukaryotic class I reductases and also possesses a high degree of sequence similarity to the enzyme of C. trachomatis, a bacterium containing many genes presumably transferred from its eukaryotic host (37). This finding suggests horizontal gene transfer as the source for the class I enzymes of pseudomonads. The question of why two active enzymes are maintained by the bacteria thus remains.
Before we can reach an understanding, more work is required, in particular concerning the division of labor between the two enzymes of P. aeruginosa during aerobic and microaerophilic growth. We also need to investigate reductases from other Pseudomonas species, particularly those that appear to contain only one of the two classes (Table 1).
ACKNOWLEDGMENTS
We acknowledge the Pseudomonas Genome Project for making sequence data available prior to publication.
Our work was supported by grants from the Karolinska Institutet and the Wallenberg foundation (to P.R.) and from the Spanish Dirección General de Enseñanza Superior e Investigación Científica (PB97-0196) and the Universitat Autònoma de Barcelona-CIRIT (Acciones Especiales 148020) (to I.G.). A.J. was supported by a fellowship from the Wenner-Gren Foundation, and E.T. was supported by a predoctoral grant from the Dirección General d’Universitats de la Generalitat de Catalunya and a short-term fellowship from Margit and Folke Pehrzon Foundation.
REFERENCES
- 1.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1995. [Google Scholar]
- 2.Berglund O. Ribonucleoside diphosphate reductase induced by bacteriophage T4. II. Allosteric regulation of substrate specificity and catalytic activity. J Biol Chem. 1972;247:7276–7281. [PubMed] [Google Scholar]
- 3.Berglund O, Eckstein F. Synthesis of ATP- and dATP-substituted sepharoses and their application in the purification of phage-T4-induced ribonucleotide reductase. Eur J Biochem. 1972;28:492–496. doi: 10.1111/j.1432-1033.1972.tb01936.x. [DOI] [PubMed] [Google Scholar]
- 4.Booker S, Licht S, Broderick J, Stubbe J. Coenzyme B12-dependent ribonucleotide reductase: evidence for the participation of five cysteine residues in ribonucleotide reduction. Biochemistry. 1994;33:12676–12685. doi: 10.1021/bi00208a019. [DOI] [PubMed] [Google Scholar]
- 5.Bradford M M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Brown M C, Reichard P. Ribonucleoside diphosphate reductase. Formation of active and inactive complexes of proteins B1 and B2. J Mol Biol. 1969;46:25–38. doi: 10.1016/0022-2836(69)90055-2. [DOI] [PubMed] [Google Scholar]
- 7.Cole S T, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 8.Crouzet J, Levy-Schil S, Cameron B, Cauchois L, Rigault S, Rouyez M-C, Blanche F, Debussche L, Thibaut D. Nucleotide sequence and genetic analysis of a 13.1-kilobase-pair Pseudomonas denitrificans DNA fragment containing five cob genes and identification of structural genes encoding cob(I)alamin adenosyltransferase, cobyric acid synthase, and bifunctional cobamide kinase-cobinamide phosphate guanylyltransferase. J Bacteriol. 1991;173:6074–6087. doi: 10.1128/jb.173.19.6074-6087.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eliasson R, Pontis E, Jordan A, Reichard P. Allosteric regulation of the third ribonucleotide reductase (NrdEF enzyme) from Enterobacteriaceae. J Biol Chem. 1996;271:26582–26587. doi: 10.1074/jbc.271.43.26582. [DOI] [PubMed] [Google Scholar]
- 10.Eliasson R, Pontis E, Jordan A, Reichard P. Allosteric control of three B12-dependent (class II) ribonucleotide reductases. Implications for the evolution of ribonucleotide reduction. J Biol Chem. 1999;274:7182–7189. doi: 10.1074/jbc.274.11.7182. [DOI] [PubMed] [Google Scholar]
- 11.Eliasson R, Pontis E, Fontecave M, Gerez C, Harder J, Jörnvall H, Krook M, Reichard P. Characterization of components of the anaerobic ribonucleotide reductase system from Escherichia coli. J Biol Chem. 1992;267:25541–25547. [PubMed] [Google Scholar]
- 12.Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg B-M, Eklund H. Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure. 1997;5:1077–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 13.Fieschi F, Torrents E, Toulokhonova L, Jordan A, Hellman U, Barbé J, Gibert I, Karlsson M, Sjöberg B-M. The manganese-containing ribonucleotide reductase of Corynebacterium ammoniagenes is a class Ib enzyme. J Biol Chem. 1998;273:4329–4337. doi: 10.1074/jbc.273.8.4329. [DOI] [PubMed] [Google Scholar]
- 14.Fontecave M, Eliasson R, Reichard P. Oxygen-sensitive ribonucleoside triphosphate reductase is present in anaerobic Escherichia coli. Proc Natl Acad Sci USA. 1989;86:2147–2151. doi: 10.1073/pnas.86.7.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gale G R, Kendall S M, McLain H H, Dubois S. Effect of hydroxyurea on Pseudomonas aeruginosa. Cancer Res. 1964;24:1012–1019. [PubMed] [Google Scholar]
- 16.Garriga X, Eliasson R, Torrents E, Jordan A, Barbé J, Gibert I, Reichard P. nrdD and nrdG genes are essential for strict anaerobic growth of Escherichia coli. Biochem Biophys Res Commun. 1996;229:189–192. doi: 10.1006/bbrc.1996.1778. [DOI] [PubMed] [Google Scholar]
- 17.Gleason F, Hogenkamp H P C. 5′-Deoxyadenosylcobalamin-dependent ribonucleotide reductase: a survey of its distribution. Biochim Biophys Acta. 1972;277:466–470. doi: 10.1016/0005-2787(72)90089-5. [DOI] [PubMed] [Google Scholar]
- 18.Harder J, Eliasson R, Pontis E, Ballinger M D, Reichard P. Activation of the anaerobic ribonucleotide reductase from Escherichia coli by S-adenosylmethionine. J Biol Chem. 1992;267:25548–25552. [PubMed] [Google Scholar]
- 19.Hofer A, Schmidt P P, Gräslund Å, Thelander L. Cloning and characterization of the R1 and R2 subunits of ribonucleotide reductase from Trypanosoma brucei. Proc Natl Acad Sci USA. 1997;94:6959–6964. doi: 10.1073/pnas.94.13.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Institute for Genomic Research. TIGR database. [Online.] http://www.tigr.org. The Institute for Genomic Research, Rockville, Md. [28 December 1998, last date accessed.]
- 20.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 21.Jordan A, Aragall E, Gibert I, Barbe J. Promoter identification and expression analysis of Salmonella typhimurium and Escherichia coli nrdEF operons encoding one of two class I ribonucleotide reductases present in both bacteria. Mol Microbiol. 1996;19:777–790. doi: 10.1046/j.1365-2958.1996.424950.x. [DOI] [PubMed] [Google Scholar]
- 22.Jordan A, Pontis E, Åslund F, Hellman U, Gibert I, Reichard P. The ribonucleotide reductase system of Lactococcus lactis. Characterization of an NrdEF-enzyme and a new electron transport protein. J Biol Chem. 1996;271:8779–8785. doi: 10.1074/jbc.271.15.8779. [DOI] [PubMed] [Google Scholar]
- 23.Jordan A, Pontis E, Atta M, Krook M, Gibert I, Barbé J, Reichard P. A second class I ribonucleotide reductase in Enterobacteriaceae: characterization of the Salmonella typhimurium enzyme. Proc Natl Acad Sci USA. 1994;91:12892–12896. doi: 10.1073/pnas.91.26.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan A, Torrents E, Jeanthon C, Eliasson R, Hellman U, Wernstedt C, Barbé J, Gibert I, Reichard P. B12-dependent ribonucleotide reductases from deeply rooted eubacteria are structurally related to the aerobic enzyme from E. coli. Proc Natl Acad Sci USA. 1997;94:13487–13492. doi: 10.1073/pnas.94.25.13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan A, Åslund F, Pontis E, Reichard P, Holmgren A. Characterization of E. coli NrdH: a glutaredoxin-like protein with thioredoxin-like activity profile. J Biol Chem. 1997;272:18044–18050. doi: 10.1074/jbc.272.29.18044. [DOI] [PubMed] [Google Scholar]
- 26.Jordan A, Gibert I, Barbé J. Cloning and sequencing of the genes from Salmonella typhimurium encoding a new bacterial ribonucleotide reductase. J Bacteriol. 1994;176:3420–3427. doi: 10.1128/jb.176.11.3420-3427.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kauppi B, Nielsen B B, Ramaswamy S, Larsen I K, Thelander M, Thelander L, Eklund H. The three-dimensional structure of mammalian ribonucleotide reductase protein R2 reveals a more-accessible iron-radical site than Escherichia coli. J Mol Biol. 1996;262:706–720. doi: 10.1006/jmbi.1996.0546. [DOI] [PubMed] [Google Scholar]
- 28.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell S C, Bron S, Brouillet S, Bruschi C V, Caldwell B, Capuano V, Carter N M, Choi S K, Codani J J, Connerton I F, Danchin A, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 29.Logan D T, Andersson J, Sjöberg B-M, Nordlund P. A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science. 1999;283:1499–1504. doi: 10.1126/science.283.5407.1499. [DOI] [PubMed] [Google Scholar]
- 30.Palleroni N J. Pseudomonas classification. A new case history in the taxonomy of gram-negative bacteria. Antonie Leeuwenhoek. 1993;64:231–251. doi: 10.1007/BF00873084. [DOI] [PubMed] [Google Scholar]
- 30a.Pseudomonas Genome Project. [Online.] http://www.pseudomonas.com. [28 December 1998, last date accessed.]
- 31.Reichard P. From RNA to DNA, why so many ribonucleotide reductases? Science. 1993;260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 32.Reichard P. The anaerobic ribonucleotide reductase from Escherichia coli. J Biol Chem. 1993;268:8383–8386. [PubMed] [Google Scholar]
- 33.Roth J R, Lawrence J G, Bobik T A. Cobalamin (coenzyme B12): synthesis and biological significance. Annu Rev Microbiol. 1996;50:137–181. doi: 10.1146/annurev.micro.50.1.137. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 35.Sjöberg B-M. Ribonucleotide reductases—a group of enzymes with different metallosites and a similar reaction mechanism. Struct Bonding. 1997;8:139–173. [Google Scholar]
- 36.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, Harrison D, Hoang L, Keagle P, Lumm W, Pothier B, Qiu D, Spadafora R, Vicaire R, Wang Y, Wierzbowski J, Gibson R, Jiwani N, Caruso A, Bush D, Reeve J N. Complete genome sequence of Methanobacterium thermoautotrophicum DH: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephens R S, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov R L, Zhao Q, Koonin E V, Davis R W. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Ollagnier S, Schmidt P P, Atta M, Mulliez E, Lepape L, Eliasson R, Gräslund A, Fontecave M, Reichard P, Sjöberg B-M. The free radical of the anaerobic ribonucleotide reductase from Escherichia coli is at glycine 681. J Biol Chem. 1996;271:6827–6831. [PubMed] [Google Scholar]
- 39.Thelander L, Sjöberg B-M, Eriksson S. Ribonucleotide diphosphate reductase (Escherichia coli) Methods Enzymol. 1978;51:227–237. doi: 10.1016/s0076-6879(78)51032-x. [DOI] [PubMed] [Google Scholar]