

Abstract
Bacterial products can act on neurons to alter signaling and function. Here, we find that dorsal root ganglia (DRG) sensory neurons are enriched for ANTXR2, the high-affinity receptor for anthrax toxins. Anthrax toxins are composed of protective antigen (PA), which binds to ANTXR2, and the protein cargoes Edema Factor (EF) and Lethal Factor (LF). Intrathecal administration of Edema Toxin (ET; PA+EF) targeted DRG neurons and induced analgesia in mice. ET inhibited mechanical and thermal sensation, and pain caused by formalin, carrageenan, and nerve injury. Analgesia depended on ANTXR2 expressed by Nav1.8+ or Advillin+ neurons. ET modulated PKA signaling in mouse sensory and human iPS-derived sensory neurons and attenuated spinal cord neurotransmission. We further engineered anthrax toxins to introduce exogenous protein cargoes, including botulinum toxin, into DRG neurons to silence pain. Our study highlights interactions between a bacterial toxin and nociceptors which may lead to the development of novel pain therapeutics.
INTRODUCTION
Pain is an unpleasant sensation initiated by nociceptive somatosensory neurons in response to harmful thermal, mechanical and chemical stimuli1. Identifying novel strategies to selectively target and silence nociceptive neurons may contribute to the development of improved pain therapeutics. Naturally occurring toxins are a rich source of evolutionarily selected molecular agents that target neuronal function. We and others have recently found that bacterial products can act on sensory neurons to modulate pain or cough during pathogenic infection2. Here, we aimed to identify receptors for bacterial products expressed on DRG sensory neurons and determine whether the corresponding products regulate pain. By mining transcriptional data3–5, we identified ANTXR2, the high affinity receptor for anthrax toxin, to be expressed by Nav1.8+ DRG neurons.
The bacterium Bacillus anthracis is the etiologic agent of anthrax. Anthrax toxin is a major virulence factor of B. anthracis, composed of three proteins: Protective Antigen (PA), Lethal Factor (LF) and Edema Factor (EF), which form two bipartite toxins: Lethal Toxin (LT; PA + LF) and Edema Toxin (ET; PA + EF). During intoxication, PA binds to anthrax toxin receptors and forms pores in endocytic vesicles6. LF and EF bind to the PA pore through their N-terminal domains and are translocated from endosomes into the cytoplasm. LF is a zinc metalloprotease that cleaves MAP kinase kinases (MAPKKs)7, inflammasome sensor Nlrp18, and regulatory subunits of phosphoinositide-3 kinase (PI3K), p85α and p85β9. EF is a calcium- and calmodulin-dependent adenylyl cyclase that converts ATP into cAMP10.
There are two validated receptors for PA, which are structurally homologous: ANTXR1 (also known as TEM8) and ANTXR2 (also known as CMG2)11,12. PA binds to ANTXR2 with significantly higher affinity than ANTXR113, and ANTXR2-deficient mice are resistant to challenge by anthrax toxins and B. anthracis infection14. These observations indicate that ANTXR2 is the major, physiologically relevant receptor for anthrax toxin in vivo.
Beyond their roles in bacterial pathogenesis, anthrax toxins have been utilized as a delivery system for transporting functional molecular cargo into the cytoplasm of cells of interest, such as cancer cells15. Linking the N-terminal domain of LF (LFN) with exogenous molecules enables their delivery through the PA pore. Cargoes that have been delivered via this methodology include protein binders16, non-canonical polypeptides17 and nucleic acids18. The breadth of these applications supports utility of the anthrax toxin system as a tool for targeting neurons.
Here, we describe a striking pattern of ANTXR2 expression in the nervous system, where it is mostly absent in CNS neurons but enriched in Nav1.8+ DRG neurons. We found that intrathecally administered ET targets DRG neurons in vivo and silences thermal and mechanical pain modalities across multiple mouse models. This analgesia was dependent on ANTXR2 on Nav1.8+ or Advillin+ neurons, but not other non-neuronal cells. ET attenuated neurotransmission to the spinal cord dorsal horn. The anthrax toxin system delivered multiple types of non-native proteins into DRG sensory neurons, demonstrating potential as a protein delivery platform to target pain. Altogether, we identify ANTXR2 as a marker enriched in nociceptive sensory neurons and propose novel strategies for modulating sensory function based on anthrax toxin-mediated targeting of DRG neurons.
RESULTS
ANTXR2 is enriched in Nav1.8+ DRG and absent in CNS neurons
To determine novel bacterial mechanisms which may modulate pain-related signaling, we mined our mouse transcriptional dataset of FACS-sorted DRG neuron populations3 and found that Antxr2 was enriched by 5-fold in Nav1.8 lineage (Nav1.8-cre Rosa26-Tdtomato+) neurons compared to Parvalbumin lineage (Pvalb-creRosa26-Tdtomato+) proprioceptive neurons (Fig. 1a). In a single cell RNAseq dataset of mouse DRG neurons4, expression of Antxr2 was highest in those expressing nociceptor-associated transcripts such as Scn10a (Nav1.8), Trpv1, Calca (CGRP), P2rx3 and Ntrk1 (TrkA) (Fig. 1b). In a separate dataset5, Antxr2 was highly expressed in C-fiber subsets including CGRP+ neurons, non-peptidergic nociceptors and SST+ neurons (Extended Data Fig. 1a). The low-affinity anthrax toxin receptor Antxr1 was largely absent in DRG neurons (Extended Data Fig. 1b).
Figure 1. Nav1.8+ mouse DRG neurons and human DRG neurons express Antxr2.
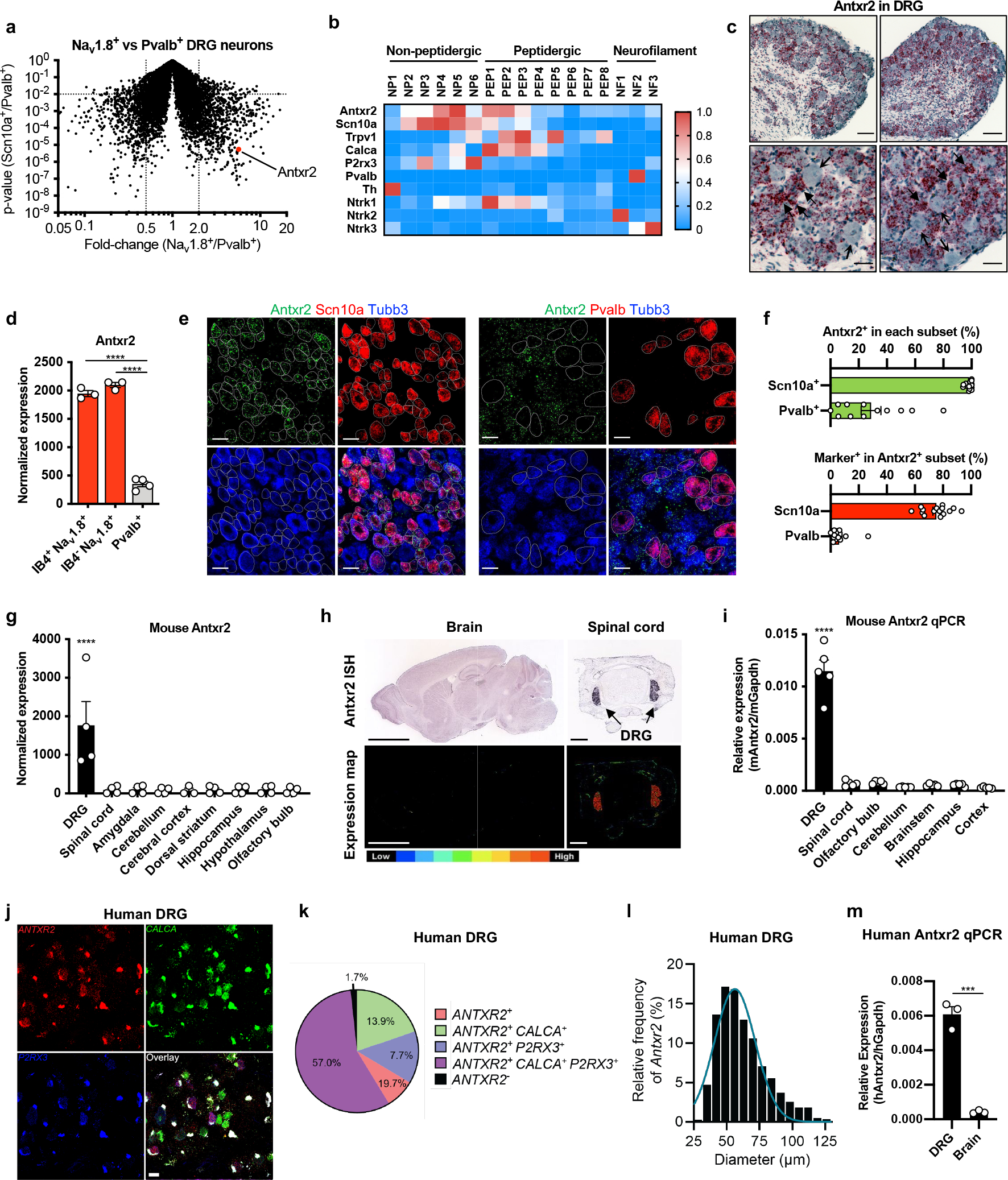
(a) Gene expression comparison of FACS-purified DRG neuron subsets3. Antxr2 is enriched in Nav1.8-lineage neurons by 5.7-fold (p=5.48×10−6).
(b) Expression of Antxr2 and subgroup markers across sensory neuron subsets from published RNA-seq data4.
(c) Representative images of in situ hybridization (ISH) analysis of Antxr2 in mouse DRG. Solid and open arrowheads point to neurons with high or low levels of Antxr2 transcripts, respectively. Scale bar, 100 μm (top row) or 35 μm (bottom row).
(d) Microarray analysis of Antxr2 expression in sorted DRG neuron subsets3 (n=3 mice for IB4+Nav1.8+ and IB4−Nav1.8+; n=4 mice for Pvalb+).
(e) Representative ISH images of Antxr2, Scn10a and Tubb3 (left) or Antxr2, Pvalb and Tubb3 (right) in mouse DRG. Scale bar, 40 μm.
(f) Expression of Antxr2, Scn10a and Pvalb were scored in all Tubb3+ neurons (n=15 fields for Scn10a analysis and n=12 fields for Pvalb analysis, both collected across 3 mice.)
(g) Antxr2 expression in mouse DRG and brain regions from public microarray data52–54 (n=4 samples).
(h) Antxr2 expression in adult (P56) brain and juvenile (P4) spinal cord from public ISH data55. Bottom row, color map of expression levels. Scale bar, 3000 μm (brain) or 400 μm (spinal cord).
(i) Quantitative PCR analysis of Antxr2 expression in mouse DRG and brain regions (n=5 mice).
(j) Representative ISH images of human DRG labeled for CALCA, P2RX3, ANTXR2 and DAPI. Lipofuscin that autofluoresced in all 3 channels and appear white in the overlay image is background signal present in all human nervous tissue.
(k) Distribution of ANTXR2 across neuronal subpopulations in human DRG (n=603 neurons from 3 donors).
(l) Size profile of all ANTXR2+ neurons in human DRG (n=594 neurons from 3 donors). Scale bar, 50 μm.
(m) Quantitative PCR analysis of ANTXR2 expression from human DRG RNA (pooled from 4 individuals) and total brain RNA (pooled from 21 individuals) (n=3 technical replicates).
Statistical significance was assessed by two-tailed t-test with unequal variances (a), one-way ANOVA with post hoc comparisons (d, g, i) or two-tailed unpaired t-test (m). ***p<0.001, ****p<0.0001. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
We next performed RNAscope analysis, which validated the presence of Antxr2 transcripts in mouse DRG. Highest levels of expression were observed in small- and medium-diameter neurons (Fig. 1c). Our transcriptional dataset of FACS-sorted DRG neuron populations3 showed that Antxr2 was evenly enriched in Isolectin B4+ and Isolectin B4− Nav1.8 lineage neurons compared to Parvalbumin lineage proprioceptors (Fig. 1d). RNAscope analysis also showed that most Scn10a+ cells express Antxr2 compared to only a small subset of Pvalb+ cells (Fig. 1e, f).
Mining whole tissue expression databases of the nervous system, we found that Antxr2 is highly expressed in the DRG but absent in CNS tissues including the spinal cord and brain regions (Fig. 1g). Public in situ hybridization data also showed that Antxr2 probe signal is detected in the DRG but mostly absent throughout the brain and spinal cord (Fig. 1h). qPCR analysis for Antxr2 expression in different mouse nervous tissues confirmed that Antxr2 expression is higher in the DRG compared to various regions of the CNS (Fig. 1i). We note that the RNAseq dataset of the mouse nervous system4 showed that Antxr2 is also expressed in enteric and sympathetic neurons in the PNS, while largely absent from most CNS neurons (Extended Data Fig. 1c).
RNAscope analysis of human DRG neurons revealed that Antxr2 is widely expressed in human somatosensory neurons, including Calca+, P2rx3+ and Calca+/P2rx3+ neurons (Fig 1j, k). We also found that Antxr2 is primarily expressed in small and medium diameter DRG neurons by size (Fig 1l). In addition, human Antxr2 was expressed at higher levels in DRG RNA compared to brain RNA (Fig. 1m). Collectively, our results demonstrated that Antxr2 is expressed in both human and mouse somatosensory neurons including nociceptors.
Lethal and Edema Toxins induce signaling in DRG cells
Given expression of ANTXR2 in sensory neurons, we next investigated whether the anthrax toxins LT and ET affect intracellular signaling in DRG neurons (Fig. 2a). We observed significant cleavage of MEK3 (Extended Data Fig. 2a) and reduced phosphorylation of p38 (Extended Data Fig. 2b) specifically with LT treatment. LF alone had no effect on either measure, indicating that PA was required for delivery of LF into cells. We also observed increases in cAMP only with ET treatment ( Extended Data Fig. 2c). Combined with a fixed concentration of PA (10 nM), EF induced cAMP with an EC50 of 46 pM (Extended Data Fig. 2d). Our results show that anthrax toxins can act on DRG cultures to perturb intracellular signaling in a PA-dependent manner.
Figure 2. Edema toxin intrathecal administration silences mechanical and thermal sensation in mice.
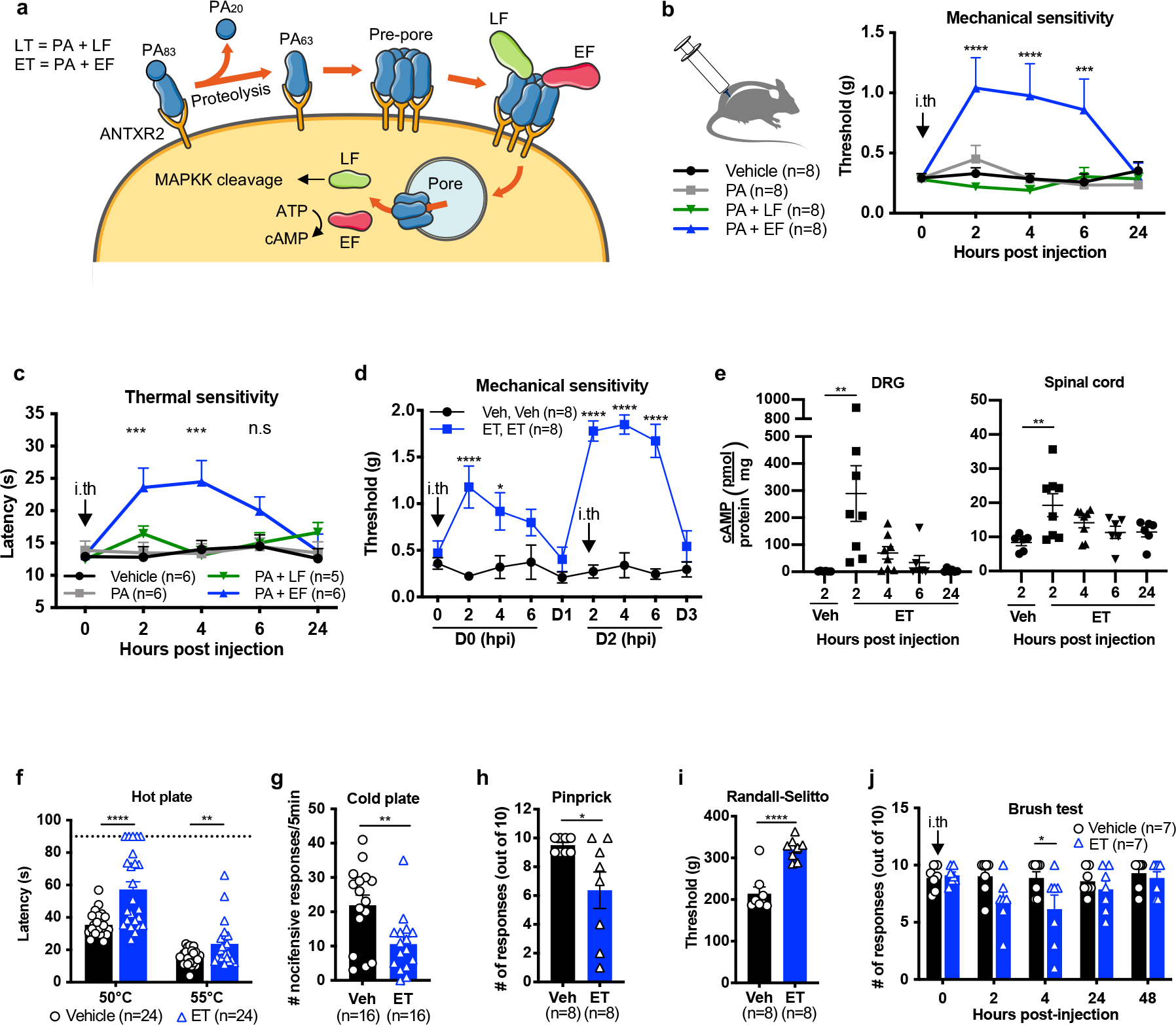
(a) Schematic depicting the intoxication mechanism of anthrax Lethal Toxin (LT) and Edema Toxin (ET).
(b) Mechanical sensitivity thresholds after intrathecal administration of vehicle (PBS), PA (2 μg), LT (2 μg PA + 2 μg LF) or ET (2 μg PA + 2 μg EF) (n=8 mice/group).
(c) Thermal sensitivity thresholds after intrathecal administration of vehicle (PBS; n=6 mice), PA (2 μg; n=6 mice), LT (2 μg PA + 2 μg LF; n=5 mice) or ET (2 μg PA + 2 μg EF; n=6 mice).
(d) Mice were treated with intrathecal vehicle (PBS) or ET (2 ug PA + 2 ug EF) at 0 and 48 hours. Mechanical sensitivity thresholds were monitored the day of and 24 hours after each injection (n=8 mice/group).
(e) cAMP levels in lumbar DRG or spinal cord after intrathecal administration of vehicle (PBS; n=6 mice) or ET (2 μg PA + 2 μg EF; n=8 mice for 2 and 4 hpi, n=6 mice for 6 and 24 hpi).
(f-j) Mice received intrathecal administration of vehicle (PBS) or ET (2 μg PA + 2 μg EF). Responses were measured at 2 hours post-injection to (f) the hot plate test (50°C, 55°C; n=24 mice), (g) cold plate test (0°C; n=16 mice), (h) pin-prick test (n=8 mice), (i) Randall-Selitto test (n=8 mice) or (j) light touch (n=7 mice).
Statistical significance was assessed by two-way RM ANOVA (b, c, d, j) with post hoc comparisons, one-way ANOVA with Dunnett’s post hoc test (e), or two tailed unpaired t-test (f-i). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
Intrathecal Edema Toxin administration blocks pain behaviors
Having determined that anthrax toxins act on DRG cultures in vitro, we next examined whether the toxins affect sensory or pain-like behaviors in mice. Given the selectivity of ANTXR2 in DRG neurons compared to CNS tissues, we reasoned that intrathecal injection of the toxins into the spinal subarachnoid space would efficiently target them to the DRG while minimizing exposure to non-neuronal cells in the periphery. We used dosages of 2 μg of PA, alone or in combination with 2 μg of LF or 2 μg of EF. In an injection volume of 5 μL, these amounts correlate to molar concentrations of 4.8 μM PA, 4.4 μM LF and 4.5 μM EF. Intrathecal administration of ET significantly increased mechanical sensitivity thresholds (Fig. 2b) and thermal latency (Fig. 2c) for several hours. The effects of ET were repeatable, where a second intrathecal injection of ET two days after the first also elevated mechanical sensitivity thresholds (Fig. 2d). The analgesic effects were stronger after the second administration, which may be due to ET-induced upregulation of ANTXR2 reported to occur in other cell types19.
We next examined whether intrathecal ET induces signaling in specific regions of the nervous system or the periphery. ET significantly increased cAMP levels in the DRG at 2 hours post-injection (hpi), which returned to baseline by 24 hours (Fig. 2e). In the spinal cord, we observed a small but significant increase at 2 hpi, potentially reflecting elevated cAMP in the central terminals of nociceptors which comprise a fraction of total spinal cord tissue (Fig. 2e). No changes were observed in the footpad (Supplementary Fig. 1a).
Intrathecal administration of ET also attenuated responses to noxious thermal and mechanical stimuli at 2 hpi, including hot plate (Fig 2f), cold plate (Fig 2g) and pinprick (Fig 2h). Responses to the Randall-Selitto test was attenuated at 2 hpi (Fig 2i) but not at 5 hpi (Supplementary Fig. 1b). We also observed a partial impairment in light touch (Fig 2j), potentially reflecting expression of ANTXR2 in some Aβ- and Aδ-LTMRs. The effects of intrathecal ET injection on thermal and mechanical sensory modalities were mostly independent of sex (Extended Data Fig. 3a–f). Overall, we found that intrathecally administered ET elevates mechanical and thermal sensitivity thresholds to both low-threshold and noxious stimuli.
Beyond sensory function, gross motor function and coordination measured by the rotarod test were not significantly affected by ET (Supplementary Fig. 1c). ET also did not affect sympathetic readouts such as heart rate or body temperature (Supplementary Fig. 2a–b), suggesting it does not produce major off-target effects on motor or sympathetic function.
ANTXR2 in sensory neurons mediates ET-induced analgesia
Given broad expression of ANTXR2 in peripheral tissues outside of the nervous system20, we next investigated whether the effects of ET on sensory function are mediated through ANTXR2 expressed in DRG neurons or non-neuronal cells. We first generated conditional ANTXR2 knock-out mice lacking expression in Nav1.8+ nociceptive neurons (Nav1.8cre/+/Antxr2fl/fl) (Fig. 3a) and confirmed reduced expression of the full-length Antxr2 transcript in the DRG (Extended Data Fig. 4a). ET-induced analgesia was significantly attenuated in Nav1.8cre/+/Antxr2fl/fl mice compared to littermate controls (Fig. 3b). ET-induced induction of cAMP was also reduced in cultures established from the DRGs of Nav1.8cre/+/Antxr2fl/fl mice (Extended Data Fig. 4b).
Figure 3. Antxr2 expressed by Nav1.8+ or Advillin+ neurons mediates ET-induced analgesia.
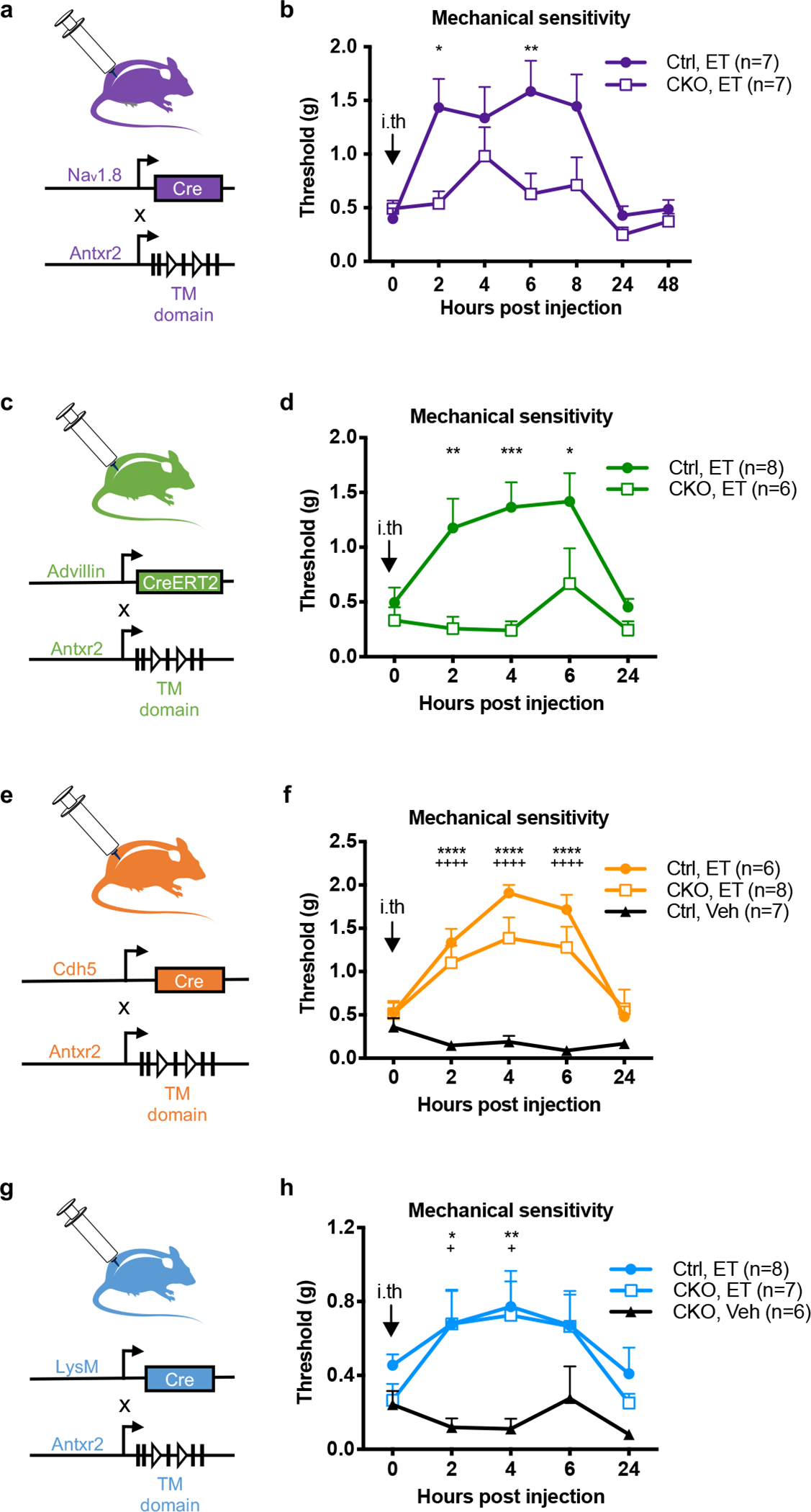
(a) Nav1.8-cre mice were bred with a conditionally targeted allele of Antxr2 in the transmembrane region (Antxr2fl/fl) to generate mice lacking Antxr2 function in Nav1.8 lineage nociceptors.
(b) Mechanical sensitivity thresholds in Nav1.8cre/+/Antxr2fl/fl mice (CKO; n=7 mice) or Nav1.8+/+/Antxr2fl/fl littermates (Ctrl; n=7 mice) injected intrathecally with ET (2 μg PA + 2 μg EF).
(c) Advillin-creERT2 mice were bred with Antxr2fl/fl mice to generate animals lacking Antxr2 function in all somatosensory neurons.
(d) Mechanical sensitivity thresholds in AdvcreERT2/+/Antxr2fl/fl mice (CKO; n=6 mice) or Adv+/+/Antxr2fl/fl littermates (Ctrl; n=8 mice) injected intrathecally with ET (2 μg PA + 2 μg EF). Antxr2 ablation was induced in adult mice by tamoxifen injection two weeks prior to the experiment.
(e) Cdh5-cre mice were bred with Antxr2fl/fl mice to generate animals lacking Antxr2 function in endothelial cells.
(f) Mechanical sensitivity thresholds in Cdh5cre/+/Antxr2fl/fl conditional KO mice (CKO) injected intrathecally with ET (2 μg PA + 2 μg EF; n=8 mice), or their Cdh5+/+/Antxr2fl/fl littermates (Ctrl) injected intrathecally with vehicle (PBS; n=7 mice) or ET (2 μg PA + 2 μg EF; n=6 mice). ‘*’ compares Ctrl, Veh vs. Ctrl, ET groups. ‘+’ compares Ctrl, Veh vs. CKO, ET groups.
(g) LysM-cre mice were bred with Antxr2fl/fl mice to generate animals lacking Antxr2 function in myeloid cells.
(h) Mechanical sensitivity thresholds in conditional KO mice (CKO; LysMcre/+/Antxr2fl/fl or LysMcre/cre/Antxr2fl/fl) injected intrathecally with vehicle (PBS; n=6 mice) or ET (2 μg PA + 2 μg EF; n=8 mice), or in control littermates (Ctrl; LysMcre/cre/Antxr2+/+, LysMcre/+/Antxr2+/+ or LysMcre/+/Antxr2fl/+) injected intrathecally with ET (2 μg PA + 2 μg EF; n=8 mice). ‘*’ compares CKO, Veh vs. Ctrl, ET groups. ‘+’ compares CKO, Veh vs. CKO, ET groups.
Statistical significance was assessed by two-way RM ANOVA with post hoc comparisons (b, d, f, h). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, +p<0.05, ++++p<0.0001. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
The endogenous function of ANTXR2 is not fully understood, with reported roles in binding to extracellular matrix proteins and mediating their internalization and degradation21. We found that absence of ANTXR2 in Nav1.8+ neurons does not affect most thermal and mechanical sensory modalities at baseline (Extended Data Fig. 4c), and produces a slight decrease in the second phase of formalin-induced pain (Extended Data Fig. 4d) with no defects in carrageenan-induced mechanical allodynia (Extended Data Fig. 4e). Overall, results indicated that ANTXR2 does not play a major role in sensory function at baseline, but is critical for ET-induced analgesia.
To further determine the role of sensory neuron-expressed ANTXR2, we generated Advillin-creERT2 driven ANTXR2 knockout mice (Advillin-creERT2/Antxr2fl/fl) (Fig. 3c) to ablate ANTXR2 expression from all peripheral somatosensory neurons in adult mice, which express Advillin22. ET-induced analgesia was nearly abolished in Advillin-specific ANTXR2 KO mice (Fig. 3d). Stronger attenuation was observed in Advillin-creERT2/Antxr2fl/fl mice compared to Nav1.8-cre/Antxr2fl/fl mice, potentially reflecting contributions from a small population of Antxr2-positive, Nav1.8-negative neurons.
We next examined whether endothelial cells or immune cells, which may be exposed to intrathecally injected ET, play any role in the analgesic mechanism. To this end, we utilized endothelial cell-specific Cdh5-cre/Antxr2fl/fl (Fig. 3e) and myeloid immune cell-specific LysM-cre/Antxr2fl/fl conditional KO mice (Fig. 3g)20. In both strains, ET-induced analgesia was not significantly affected in the conditional ANTXR2 KO mice (Fig. 3f, h). Altogether, our results showed that neuronal ANTXR2 is critical to the anti-nociceptive effects of ET in vivo, whereas endothelial cells and myeloid immune cells play a minimal role.
Intraplantar ET induces mechanical allodynia
In contrast to intrathecally administered ET, ET injected subcutaneously into the footpad induced mechanical allodynia (Extended Data Fig. 5a) accompanied by swelling of the footpad (Extended Data Fig. 5b)20. Intraplantar ET injection elevated cAMP levels in the footpad but not the DRG or spinal cord (Extended Data Fig. 5c), indicating that the activity of the toxin is locally confined. Nav1.8cre/+/Antxr2fl/fl mice developed mechanical allodynia in an identical manner to Nav1.8+/+/Antxr2fl/fl littermates (Extended Data Fig. 5d), suggesting that ET sensitizes nociceptors through an indirect mechanism in the periphery.
ET induces PKA signaling in mouse and human iPSC neurons
ET is known to signal via cAMP and PKA in target cells, which are key modulators of nociceptor signaling and pain23. We thus quantified PKA activation in DRG neurons. The type II isoforms of PKA (PKA-II) contain the regulatory subunit RIIα or RIIβ (collectively referred to as RII), of which RIIβ is highly expressed in nociceptors24. In immunological staining of fixed neurons, the phosphorylated inhibitory site of RII (pRII) becomes accessible to antibodies after cAMP triggers dissociation of the catalytic subunits25. We identified sensory neurons by ubiquitin C-terminal hydrolase L1 (UCHL1), nociceptors by RIIβ, and PKA-II activation by pRII. Positive control forskolin (Fsk) induced a long-lasting and dose-dependent increase in PKA-II activity in almost all DRG neurons (Fig. 4a). ET steadily increased PKA-II activity in a subgroup of DRG neurons to saturate by 2 hours with a picomolar EC50 value, whereas EF or PA alone had no effect (Fig. 4b). PA, LF alone or LT did not affect PKA-II activity (Extended Data Fig. 6a, b). We characterized the subset of DRG neurons responding to ET. We detected PKA activation within all nociceptive and non-nociceptive subgroups but to varying degrees (Extended Data Fig. 6c). Responses to ET positively correlated with CaMKIIα and Nav1.8 expression but inversely so with NF200 (Extended Data Fig. 6d). In addition, ET did not increase pRII levels in non-neuronal cells, in contrast to forskolin (Extended Data Fig. 6e, f). Overall, our analyses showed that ET dose-dependently activates PKA within hours of application in sensory neurons and nociceptive subsets.
Figure 4. Edema Toxin induces PKA signaling in DRG neurons but does not affect neuronal viability.
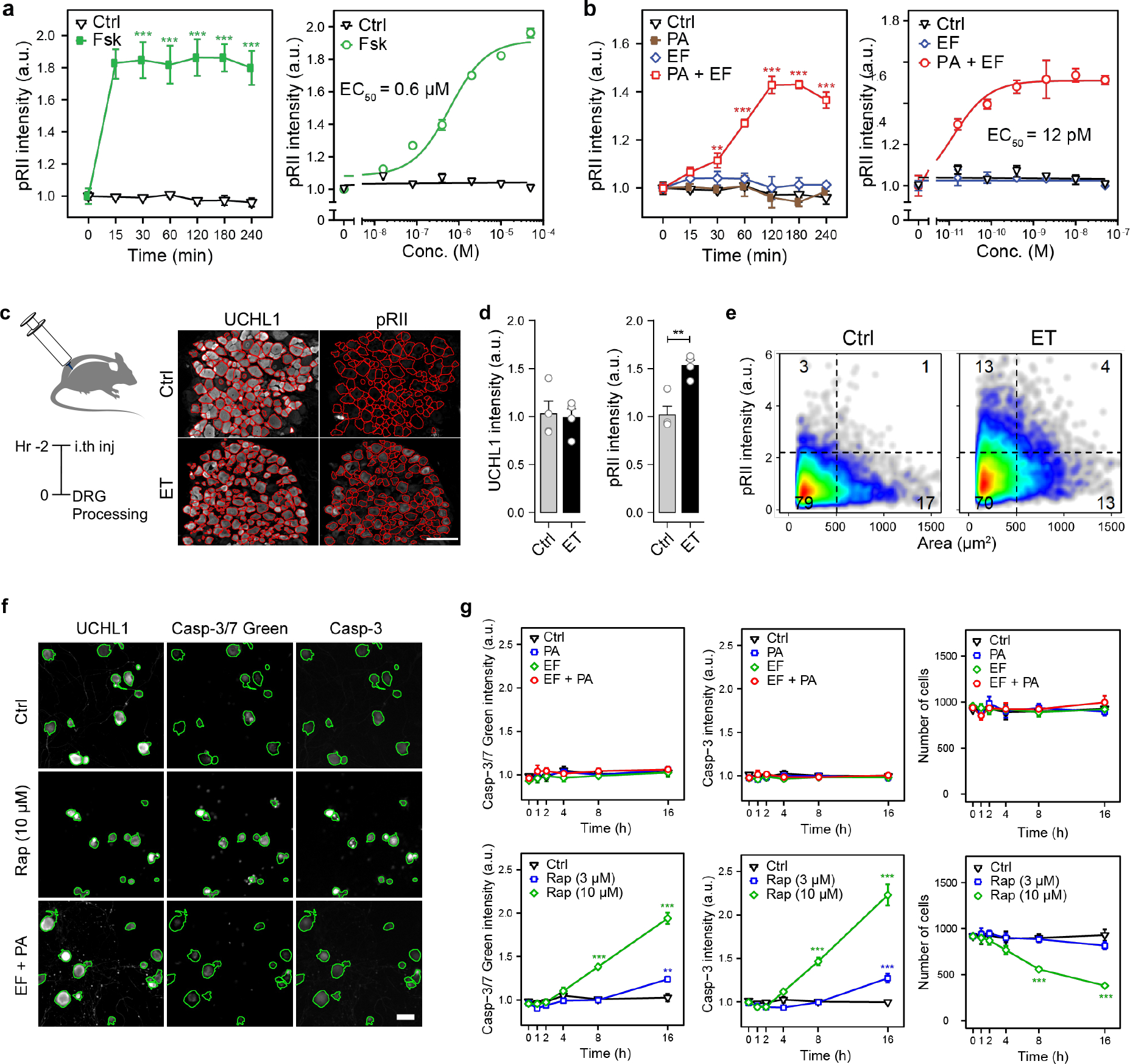
(a) Time-course (left) and dose response (right) of pRII intensity in DRG neurons stimulated with forskolin (10 μM or 2 h) (n=3 experiments, >2500 neurons/condition).
(b) (Left) Time-course of pRII intensity in DRG neurons stimulated with Ctrl (0.1% BSA), PA (10 nM), EF (10 nM) or the combination of both factors. (Right) Dose-response curve of pRII intensity in DRG neurons stimulated with EF (0 – 50 nM, 2 h) in the presence of a constant concentration of PA (10 nM) (n=3 experiments, >2500 neurons/condition).
(c) Representative images of frozen L3 - L6 DRG sections obtained from mice 2 h post intrathecal injection of vehicle (PBS) or ET (2 μg PA + 2 μg EF). The red lines indicate the mask used to quantify signal intensities in DRG neurons. Scale bar, 100 μm.
(d) Mean UCHL1 and pRII intensities quantified in DRG sections of the respective mice (n=4 mice/group, 15 – 20 images of 4 non-consecutive sections/animal, 1951 ± 279 neurons/animal).
(e) Single cell data of the quantified DRG neurons.
(f) Representative images of mouse DRG neurons stimulated with solvent control (Ctrl), raptinal (Rap; 3 or 10 μM) or ET (10 nM PA + 10 nM EF) for 16 h. Cultures were stained for UCHL1, caspase-3/7 green detection reagent, and cleaved caspase 3. The cells were fixed about 1 hour after adding the caspase-3/7 dye, stained with a standard ICC protocol, and analyzed by HCS microscopy. Green encircled neurons indicate automatically selected objects, respectively (see Methods section). Scale bar, 50 μm.
(g) Time-course of caspase-3/7 green detection reagent, cleaved caspase 3 intensity and corresponding cell numbers per analyzed well (n=4 experiments).
Statistical significance was assessed by two-way ANOVA with Bonferroni’s post hoc test (a, b, g) or two-tailed unpaired t-test (d). **p<0.01, ***p<0.001. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
We examined whether intrathecal administration of ET activates PKA in DRG neurons in vivo (Fig. 4c). Quantification of ≈2000 neurons per animal in DRG sections revealed that ET induces substantial PKA-II activity in DRG neurons in vivo (Fig. 4d, e).
As we previously found that Antxr2 is expressed in human DRG neurons, we next investigated whether ET activates PKA signaling in human iPSC-derived sensory neurons. We differentiated human iPSCs into sensory neurons of a nociceptor phenotype following a previously established small molecule-based differentiation protocol26. Stimulation with ET elevated pRII levels (Extended Data Fig. 7a, b) with a picomolar EC50 value (Extended Data Fig. 7c), suggesting that human sensory neurons may be targeted by anthrax toxin.
ET treatment does not affect the viability of DRG neurons
Systemic injection of ET has been shown to cause extensive tissue damage and necrosis in peripheral organs27. However, treatment of DRG cultures with ET for 16 hours did not induce any quantifiable caspase-3/7 activity, which was in contrast with the mitochondrial poison raptinal (Fig. 4f, g). We also failed to detect activated caspase-3 in the DRGs of ET-injected mice (Supplementary Fig. 3). Altogether, our results indicated that ET is not acutely toxic to neurons and the analgesic effects of ET are unlikely to stem from cell death.
ET modulation of neural activity and analgesia induction
Previous work has shown that cAMP and PKA activation can modify multiple conductances in nociceptors28–30, resulting in hyperexcitability and hyperalgesia23. This raised the question of how ET is affecting neuronal excitability. ET-treated DRG neurons showed enhanced excitability when tested by injection of small or moderate currents (Extended Data Fig. 8a–c), which occurred without detectable differences in resting membrane potential or input resistance of the neurons (Extended Data Fig. 8d, e).
Voltage-dependent calcium channels in nociceptive DRG neurons are regulated by many second-messenger pathways that can mediate powerful presynaptic inhibition. However, voltage-activated calcium currents recorded in ET-treated neurons did not significantly differ from control (Supplementary Fig. 4). ET treatment also did not affect KCl-induced release of CGRP in DRG culture (Supplementary Fig. 5).
We next investigated whether ET may activate nociceptive neurons to produce stress-induced analgesia mediated by endogenous opioid or cannabinoid signaling31,32. The opioid receptor antagonists naltrexone or naloxone, or the CB1 antagonist/inverse agonist rimonabant did not significantly affect the anti-nociceptive effects of ET (Supplementary Fig. 6a, b). In addition, mice that underwent chemical sympathectomy still showed significant analgesia (Supplementary Fig. 7). We alternatively hypothesized that cAMP could be secreted extracellularly and become converted to adenosine33, which then act on adenosine receptors on nociceptors to block pain34. However, systemic administration of a pan adenosine receptor antagonist did not prevent the ET-induced increase in thermal sensitivity thresholds (Supplementary Fig. 6c).
Intrathecal ET induces DRG transcriptional changes
Given that cAMP and PKA signaling is known to induce phosphorylation of CREB and other transcription factors, we hypothesized that ET may induce transcriptional changes that affect neuronal responsiveness to stimuli. Transcriptional profiling analysis of the DRG 2 hours after intrathecal administration of ET identified major transcriptional changes including upregulation of Dusp1, Fosb and Btg2 (Extended Data Fig. 9a) and alteration of several pathways (Extended Data Fig. 9b). DUSP1 is a phosphatase that blocks MAPK signaling pathways involved in pain35. Pharmacological inhibition of DUSP1 activity did not affect ET-induced analgesia (Extended Data Fig. 9c). Although we did not find a specific role for Dusp1, our results showed that ET induces intrinsic transcriptional changes in the DRG in vivo within hours of injection.
ET attenuates neurotransmission at DRG central terminals
Intrinsic changes in DRG neurons could lead to modulation of regulatory pathways localized to presynaptic DRG nerve terminals in the spinal cord in vivo, affecting central neurotransmission to second order neurons. We thus investigated the effect of ET on neurotransmission using intrathecal capsaicin injection as a stimulus for primary afferent central branches and phosphorylated ERK (pERK) as a readout for activation in dorsal horn neurons36. Intrathecal injection of vehicle followed by capsaicin increased the number of pERK-positive cells in the dorsal horn (Fig. 5a, b). Pre-injection of ET prior to capsaicin significantly reduced pERK induction (Fig. 5a, b).
Figure 5. Edema Toxin attenuates neurotransmission at nociceptor central terminals.
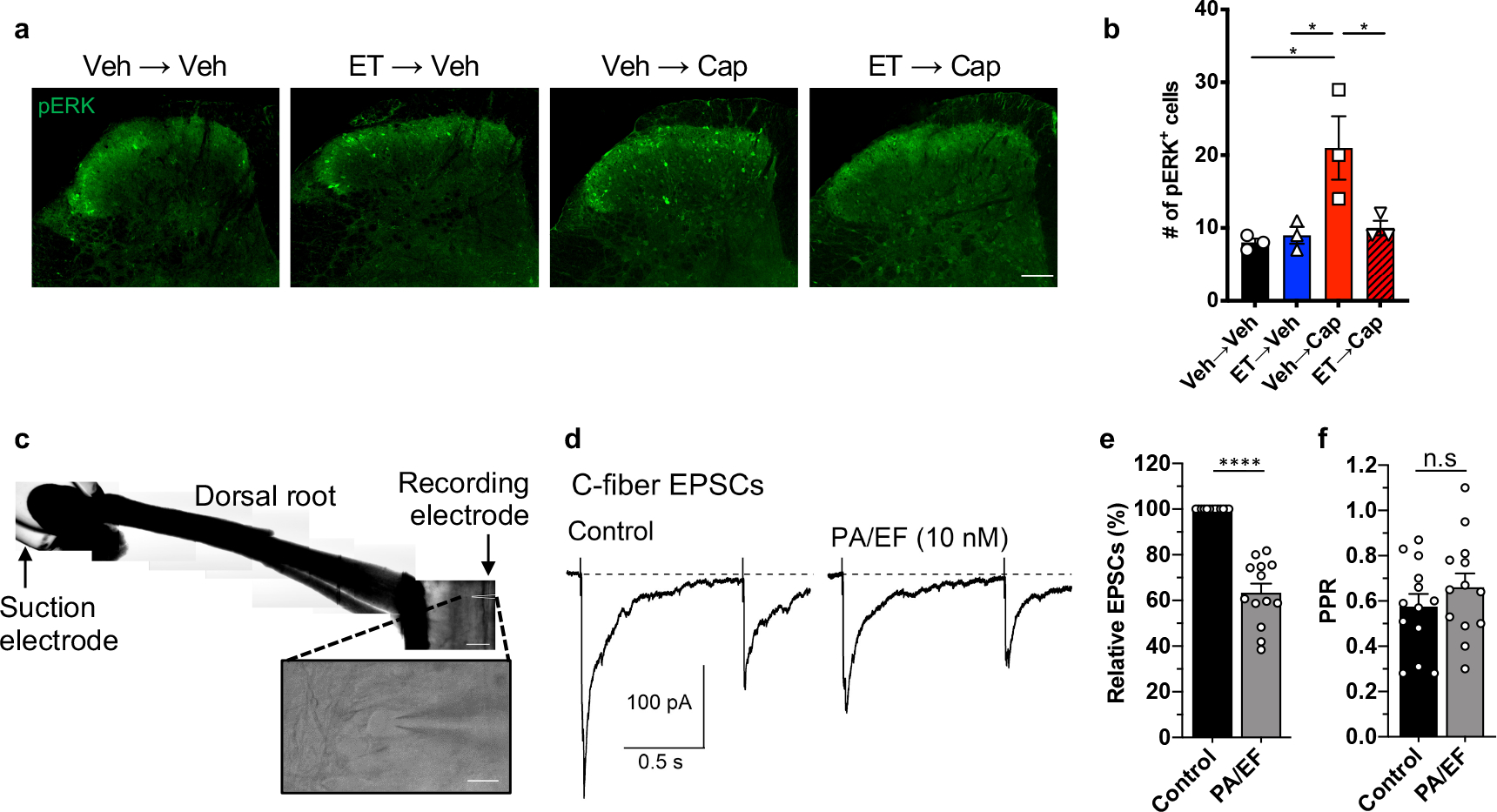
(a, b) Mice received intrathecal injection of vehicle or ET (2 μg PA + 2 μg EF), followed by intrathecal injection of vehicle or capsaicin (1 μg) after 2 hours. Spinal cords were harvested after 20 min and stained for pERK. (a) Representative images of pERK staining in the dorsal horn. Scale bar, 100 μm. (b) Quantification of the number of pERK-positive cells in the superficial laminae of the dorsal horn. 8–12 sections were quantified and averaged per animal (n=3 mice).
(c) Representative horizontal spinal cord slice preparation with the attached L4 dorsal root and a lamina I neuron (inset). Scale bar, 500 μm and 20 μm (inset).
(d) C-fiber EPSCs elicited in a lamina I neuron by stimulation of the L4 dorsal root (paired 400 μA stimuli at a 1 s interval). The measured conduction velocity was 0.7 m/s, consistent with C-fiber activation.
(e, f) Collected results (n=13 cells). (e) Application of ET (10 nM PA + 10 nM EF) reduced the first EPSC by 37±4 %. (f) No significant changes were observed in the paired pulse ratio.
Statistical significance was assessed by one-way ANOVA with Tukey’s post hoc test (b) or two-tailed paired t-test (e, f). n.s, not significant, *p<0.05, ****p<0.0001. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
We next examined the effect of ET application on excitatory post-synaptic currents (EPSCs) in lamina I neurons evoked by stimulation of dorsal roots (Fig. 5c). Application of ET slowly reduced the C-fiber-mediated EPSCs (Fig. 5d) with a reduction of 37±4 % (Fig. 5e). There was no significant change in the paired-pulse ratio (Fig. 5f). We also recorded miniature EPSCs (mEPSCs) in the absence and presence of 1 μM tetrodotoxin. In both conditions, changes in median frequency and median amplitude of mEPSCs measured in each cell before and after ET application showed no significant differences (Supplementary Fig. 8). As the majority of mEPSCs appear to originate from spinal cord interneurons with very little contribution from primary afferents37, the small effects on mEPSCs relative to the larger reduction of EPSCs are consistent with our earlier evidence that ET acts via ANTXR2 on primary afferent neurons. Overall, our results suggested that ET inhibits synaptic transmission from C-fibers to lamina I neurons and diminishes activation of the latter in the dorsal horn.
Intrathecal ET silences neuropathic and inflammatory pain
We next investigated whether ET could have therapeutic efficacy in animal models of pain. ET blocked mechanical allodynia in the spared nerve injury (SNI) model of neuropathic pain, whereas PA alone, EF alone, or LT had no effect (Fig. 6a, b). ET raised the mechanical sensitivity threshold in both ipsilateral and contralateral paws of mice (Supplementary Fig. 9) and elevated cAMP levels in both ipsilateral and contralateral DRGs (Fig. 6c). ET also attenuated the first and second phases of formalin-induced pain (Fig. 6d). In the carrageenan model of inflammatory pain, ET blocked mechanical allodynia (Fig. 6e) dependent on ANTXR2 expression on Nav1.8+ neurons (Fig. 6f). There were no differences in ET efficacy in males and females for formalin and carrageenan models (Extended Data Fig. 10).
Figure 6. Anthrax Edema Toxin silences pain in mouse models of neuropathic and inflammatory pain.
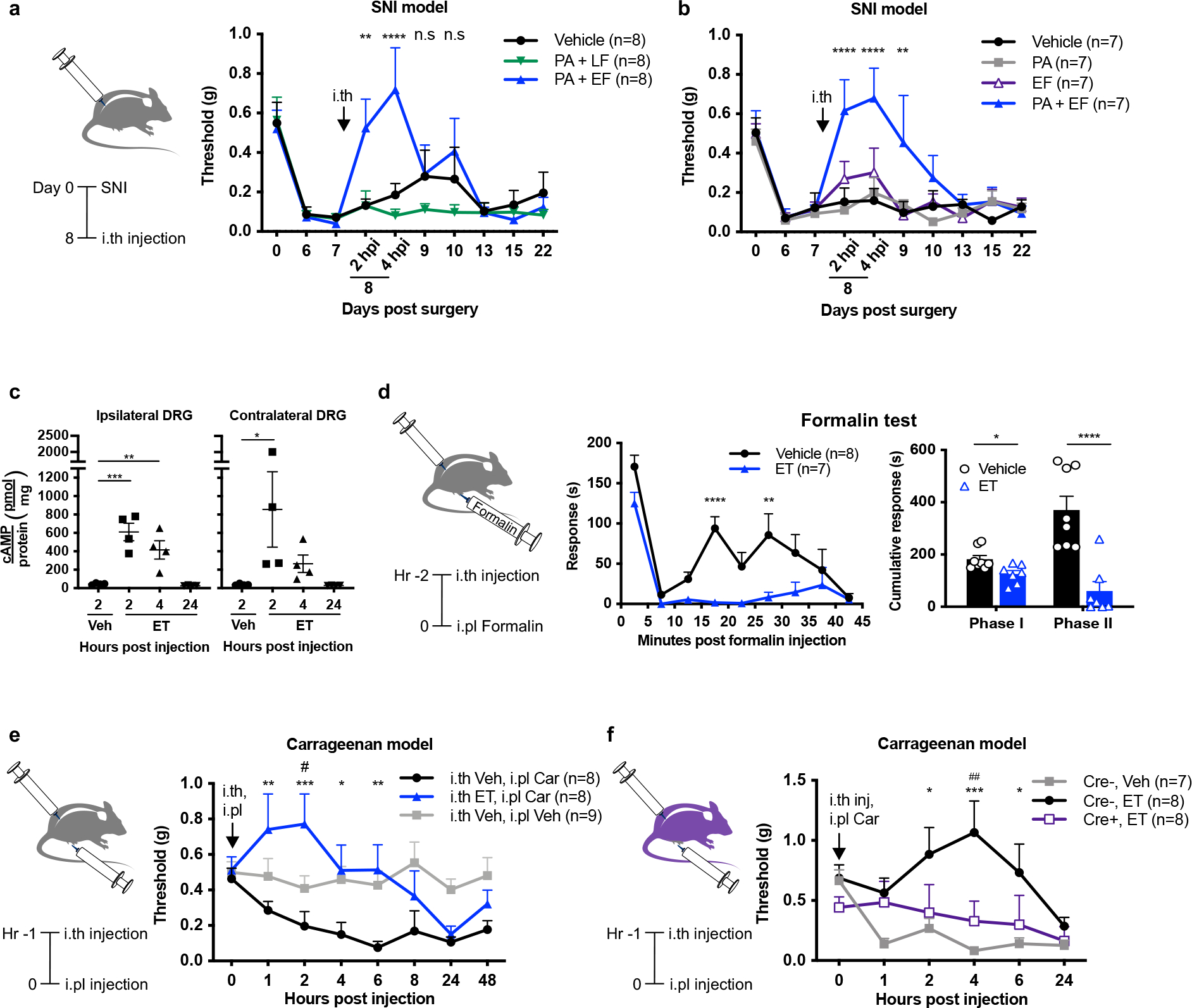
(a) Mechanical sensitivities in SNI mice injected intrathecally with vehicle (PBS), LT (2 μg PA + 2 μg LF) or ET (2 μg PA + 2 μg EF) (n=8 mice/group). ‘*’ compares vehicle and ET groups.
(b) Mechanical sensitivities in SNI mice injected intrathecally with vehicle (PBS), PA (2 μg), EF (2 μg) or ET (2 μg PA + 2 μg EF) (n=7 mice/group). ‘*’ compares vehicle and ET groups.
(c) cAMP levels in the ipsilateral or contralateral DRGs (L3 – L5) of SNI mice injected intrathecally with vehicle (PBS) or ET (2 μg PA + 2 μg EF) (n=4 mice/group).
(d) Mice were given intrathecal injection of vehicle (PBS; n=8 mice) or ET (2 μg PA + 2 μg EF; n=7 mice) 2 h prior to intraplantar injection of 5% formalin. (Left) Acute pain-like behaviors measured in 5 min intervals. (Right) Cumulative responses during Phase I (0 – 5 min) or Phase II (15 – 35 min).
(e) Mechanical sensitivities in mice injected intrathecally with vehicle (PBS) or ET (2 μg PA + 2 μg EF) 1 h prior to intraplantar injection of vehicle (0.9% saline) or 2% carrageenan (Car). (n=8 mice for i.th Veh, i.pl Car and i.th ET, i.pl Car groups; n=9 mice for i.th Veh, i.pl Veh). ‘*’ compares i.th Veh, i.pl Car vs. i.th ET, i.pl Car groups. ‘#’ compares i.th Veh, i.pl Veh vs. i.th ET, i.pl Car groups.
(f) Mechanical sensitivities in Nav1.8cre/+/Antxr2fl/fl (Cre+) mice or Nav1.8+/+/Antxr2fl/fl (Cre−) littermates injected intrathecally with vehicle (PBS) or ET (2 μg PA + 2 μg EF) 1 h prior to intraplantar injection of 2% carrageenan (Car). (n=7 mice for Cre−, Veh; n=8 mice for Cre−, ET and Cre+, ET groups). ‘*’ compares Cre−, Veh vs. Cre−, ET groups. ‘#’ compares Cre−, ET vs. Cre+, ET groups.
Statistical significance was assessed by two-way RM ANOVA with post hoc comparisons (a, b, d-left, e, f), one-way ANOVA with Dunnett’s post hoc test (c), or two-tailed unpaired t-test (d-right). n.s, not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, #p<0.05, ##p<0.01. Data represent the mean ± s.e.m. For detailed statistical information, see Supplementary Table 2.
Engineered anthrax toxins deliver cargoes into DRG neurons
We next tested whether the PA + LFN anthrax toxin system could deliver exogenous proteins into the cytoplasm of sensory neurons (Fig. 7a). First, we utilized the reagent LFN-DTA38 (Fig. 7b) where the A chain of Diphtheria Toxin (DTA) inhibits protein translation in mammalian cells. PA + LFN-DTA blocked translation (Fig. 7b) and produced cell death (Supplementary Fig. 10a) in DRG cultures. LFN-DTA alone had no effect on both measures, indicating that LFN-DTA was specifically internalized via PA.
Figure 7. Engineered anthrax toxins deliver molecular cargo into DRG sensory neurons and block pain in vivo.
(a) Schematic of exogenous cargo delivery into neurons by the PA + LFN system.
(b) (Left) Design of LFN-DTA linking the N terminal domain of LF (LFN) to the A chain of diphtheria toxin (DTA). (Right) Protein synthesis levels in DRG cultures following 6 h treatment with the indicated concentrations of LFN-DTA ± PA (10 nM) (n=3 experiments). Data represent the mean ± s.e.m.
(c) Design of LFN-LC/AC699S linking the N terminal domain of LF (LFN) to a mutated light chain (LC) of type A botulinum neurotoxin (LC/AC699S).
(d-f) DRG cultures were treated with the indicated concentrations of LFN-LC/AC699S ± PA (10 nM) for 24 h and stimulated with 80 mM KCl for 10 min (n=2 wells/condition). Dotted lines connect the means. (d) SNAP-25 cleavage in cell lysates measured by western blot. Estimated molecular weight markers are shown. (e) Percent cleavage was calculated using band intensities with the following formula: cleaved/(intact + cleaved). (f) CGRP release in the supernatant.
(g) Mechanical sensitivity thresholds in SNI mice that received three daily intrathecal injection of vehicle (PBS; n=6 mice), PA only (500 ng; n=6 mice), LFN-LC/AC699S only (200 ng; n=5 mice) or PA + LFN-LC/AC699S (500 ng + 200 ng; n=6 mice). Data represent the mean ± s.e.m.
(h, i) Mice received three daily intrathecal injection of vehicle (PBS) or PA + LFN-LC/AC699S (500 ng + 200 ng) starting on Day 0 (n=5 mice/group) and were monitored by the rotarod test (h) or wire hang test (i). Data represent the mean ± s.e.m.
(j) The effects of BoNT/A, LFN-LC/AC699S alone and PA + LFN-LC/AC699S tested on an ex vivo mouse phrenic nerve hemidiaphragm (mPNHD) preparation (n=3 experiments). Data represent the mean.
Statistical significance was assessed by two-way ANOVA with Sidak’s post hoc test (b) or two-way RM ANOVA with post hoc comparisons (g, h, i). n.s, not significant, ****p<0.0001. For detailed statistical information, see Supplementary Table 2.
Next, we generated a chimera of LF and EF where LFN is linked to the C terminal catalytic domain of EF (EFC), referred to as LFN-EFC (Supplementary Fig. 10b). PA + LFN-EFC robustly induced cAMP in DRG cultures (Supplementary Fig. 10b).
Finally, we designed a novel LFN-based construct based on botulinum neurotoxin (BoNT), which cleaves components of the SNARE complex through its enzymatic light chain (LC) to prevent neurotransmitter release from synaptic vesicles. We fused the light chain of BoNT serotype A1 (LC/A1), which targets SNAP-25, to the C terminus of LFN. In addition, a free cysteine towards the C terminus of LC/A1 was mutated to a serine (C699S) (Fig. 7c). LFN-LC/A1C699S retained enzymatic activity with an EC50 of 89.5 pM in a cell-free assay. PA + LFN-LC/A1C699S, but not LC/A1C699S alone, produced dose-dependent cleavage of SNAP-25 (Fig. 7d, e) and inhibited CGRP release (Fig. 7f) in DRG cultures.
Anthrax toxin delivery of BoNT/A light chain blocks pain
We next wished to determine whether delivery of botulinum toxin light chain by the PA + LFN system can silence pain in vivo. Three daily intrathecal administration of PA + LFN-LC/AC699S produced significant blockade of pain in the SNI model (Fig. 7g) without affecting body weight (Supplementary Fig. 10c) or motor function (Fig. 7h, i). We further investigated potential off-target effects of PA + LFN-LC/AC699S on motor neurons using the ex vivo mouse phrenic nerve hemidiaphragm (mPNHD) assay. Whereas BoNT/A produced a concentration-dependent decrease in muscle contractility, PA + LFN-LC/AC699S and LFN-LC/AC699S alone did not affect contractility at equivalent concentrations (Fig. 7j). Overall, our results demonstrate proof-of-concept for targeting sensory function and pain in vivo with high specificity using the PA + LFN anthrax toxin system.
DISCUSSION
Bacterial products targeting the nervous system may be used to modulate function and behavior. Here, we describe ANTXR2 as a receptor for anthrax toxins expressed on nociceptive sensory neurons and identify B. anthracis Edema Toxin as a modulator of neuronal signaling and pain. ET induced cAMP and PKA signaling, blockade of neurotransmission, and analgesic effects in mice in a neuronal-ANTXR2 dependent manner. This contrasts with the traditional paradigm of cAMP/PKA signaling in pain, where prior work has shown that inflammatory mediators promote nociceptor sensitization by activating adenylyl cyclases and PKA23. In addition, injection of membrane-permeable cAMP39 or forskolin40 produce mechanical hyperalgesia. A key difference between these molecules and ET is their targeting specificity and potency. ET preferentially acts on small-diameter nociceptive sensory neurons and is orders of magnitude more potent than mammalian adenylyl cyclases41, creating waves of cAMP emanating from the perinuclear region42. The nature of downstream signaling pathways induced by large quantities of unregulated cAMP in the DRG remains to be examined.
ET partially inhibited synaptic transmission from C-fibers in isolated spinal cord preparations and inhibited capsaicin-induced activation of pERK in the spinal cord dorsal horn. One possible mechanism linking inhibition of synaptic transmission to the ability of ET to increase cAMP could be a cAMP-mediated enhancement of GABAA receptor chloride channels in the nerve terminals of C-fibers. In mice, presynaptic GABAA receptors on the axons or terminals of primary nociceptors mediate spinal pain control43, most likely through a mechanism of presynaptic inhibition mediated by depolarization of the primary afferents resulting from the depolarized chloride equilibrium potential in DRGs of about −30 mV44. GABAA receptor activation on nerve terminals can produce powerful presynaptic inhibition by preventing propagation of action potentials into the terminals, as a result of depolarizing the nerve terminals and adjacent axon to inactivate sodium channels and providing a shunting conductance in the terminal44. Currents through GABAA receptor channels can be enhanced by cAMP-mediated activation of protein kinase A45.
Another well-described effect of cAMP in DRG neurons is to shift the voltage-dependence of activation of TTX-resistant sodium channels to more hyperpolarized voltages46. This effect contributes to an enhancement of excitability in DRG cell bodies by cAMP47, as we saw for ET exposure. However, in nerve terminals, the activation of a steady-state sodium current at small depolarizations could enhance the steady depolarization from GABAA channel-mediated depolarization to inactivate the transient sodium current mediating action potentials and thereby promote presynaptic inhibition. Such a mechanism might produce all-or-none loss of action potential propagation in individual branches of primary afferents, and therefore would not affect the paired pulse ratio from branches that remain functional. Although the exact mechanism by which intrathecal ET produces analgesia remains to be determined, collectively our data show that ET targets ANTXR2 on primary afferent nociceptive neurons to induce intrinsic changes at the transcriptional and signaling levels, leading to inhibition of synaptic transmission to second order neurons in the spinal cord.
Potential therapeutic and research applications
The enriched expression of ANTXR2 in DRG nociceptive neurons compared to CNS neurons offers an opportunity for selective targeting. It is important to note that the intrathecal route of injection likely restricts ET within the spinal cord and DRG, preventing potential toxicity in peripheral organs which has been observed following systemic injection of larger doses of ET27. As a cautionary note, ET has also been implicated in disrupting the integrity of the blood brain barrier during infection48 and further study will be required to characterize potential effects of intrathecal ET injection on the brain. Nonetheless, intrathecal ET or PA-based delivery systems may provide an avenue to treat pain with higher specificity compared to existing analgesics. For example, ziconotide, the synthetic version of ω-conotoxin, inhibits N-type calcium channels and is administered via intrathecal infusion devices to treat severe chronic pain. However, possibly due to channel expression in the brain, ziconotide can also cause psychiatric symptoms and neurological impairment49. Alternatively, opioids can induce addiction and dependence through acting on opioid receptors in the brain.
Beyond native anthrax toxins, the PA + LFN system delivered three separate enzymatic entities into DRG sensory neurons, demonstrating flexibility and modularity as a delivery platform. Anthrax toxin-mediated delivery of botulinum toxin light chain demonstrated proof-of-concept as a therapeutic strategy to block pain, joining an emerging field of engineered bacterial toxins that silence pain50. Potentially, multiple LFN-linked cargo could be delivered together for synergistic effects. A reported limitation of the PA + LFN system involves protein payloads that are very stable, which do not unfold efficiently enough to be translocated through the PA pore but could be successfully delivered with the introduction of destabilizing mutations51. The delivery capacity of the system, including the rate and efficiency of delivery, remains to be fully characterized in sensory neurons and optimized for in vivo applications.
As a protein-based delivery platform, the PA + LFN system may provide several advantages over genetic or viral approaches for targeting sensory neurons in vitro or in vivo, such as quicker introduction of working cargo and tighter temporal control. In addition, compared to AAV-based methods that can be limited by packaging capacity, there are no known size limitations for transportation across the PA pore. Anthrax toxin-mediated delivery of cytosolic inhibitors, effectors or sensors could complement the existing arsenal of research tools for studying somatosensory function. Altogether, we propose that anthrax toxin provides unique opportunities for targeting sensory neurons and merits further development as a platform for modulating their intracellular biology.
METHODS
Animals
C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and bred at Harvard Medical School. For high content screening (HCS) microscopy experiments, male C57Bl/6N mice were obtained from Charles River. For the mPNHD assay, male CD1 mice were purchased from Janvier Labs. For histological analysis of pERK and biotelemetry experiments, C57BL/6 mice were obtained from the Central Animal Care Facility of Ribeirao Preto Medical School, University of Sao Paulo (USP). For spinal cord electrophysiology experiments, Sprague-Dawley rats were purchased from Envigo. Nav1.8-Cre mice were provided by John Wood (University College London)56 on the C57BL/6 background. Antxr2fl/fl mice in which the transmembrane domain of Antxr2 is flanked by loxP sites were obtained from Jackson Laboratory (#027703) on the C57BL/6 background. Mice lacking functional ANTXR2 in Nav1.8-lineage neurons were generated at Harvard Medical School by crossing Nav1.8-Cre mice with Antxr2fl/fl mice to obtain Nav1.8cre/+/Antxr2fl/fl mice and control Nav1.8+/+/Antxr2fl/fl littermates. Genotyping of this strain was performed as previously described20. Advillin-CreERT2 mice on a mixed C57Bl6J and CD1 background were provided by David Ginty (Harvard Medical School). Mice lacking functional ANTXR2 in somatosensory neurons were generated at Harvard Medical School by crossing Advillin-CreERT2 mice with Antxr2fl/fl mice to obtain AdvillincreERT2/+/Antxr2fl/fl mice and control Advillin+/+/Antxr2fl/fl littermates. Endothelial cell-specific Cdh5cre/+/Antxr2fl/fl conditional ANTXR2 KO mice and their Cdh5+/+/Antxr2fl/fl control littermates; myeloid cell-specific LysMcre/+/Antxr2fl/fl or LysMcre/cre/Antxr2fl/fl conditional ANTXR2 KO mice and their control littermates (LysMcre/cre/Antxr2+/+, LysMcre/+/Antxr2+/+ or LysMcre/+/Antxr2fl/+) were generated and bred by Stephen Leppla (NIH) and transported to Harvard Medical School for testing. Both endothelial- and myeloid-specific specific ANTXR2 KO mouse strains were on the C57BL/6 background.
Expression analyses of Antxr2 (Figures 1c, 1e–f, 1i and Extended Data Fig. 4a), analyses involving primary cells or tissue (Figures 6c, 7b and 7d–f; Extended Data Figures 2a–d, 4b, 5c and 9c; Supplementary Figures 1a, 5 and 10a–b), transcriptional profiling analysis of the DRG (Extended Data Fig. 9c) and all behavior experiments were performed with age-matched mice between 6–14 weeks of age. Male and female mice were used at equivalent ratios for behavior experiments, except for the following which were performed with male mice only: Fig. 2j, 6a and 6b. For high content screening (HCS) microscopy experiments, male C57Bl/6N mice were used between 8–10 weeks of age and >24 g in weight. For the mPNHD assay, male CD1 mice were used between 18–28 g in weight at the time of experiment. For histological analysis of pERK and biotelemetry experiments, male C57BL/6 mice were used between 6–10 weeks of age. For spinal cord electrophysiology experiments, Sprague-Dawley rats were used between P18 - P27 of age at equivalent ratios of males and females.
Animal Care and Ethics
Animal experiments were approved by the Harvard Medical School Institutional Animal Care and Use Committee, Stony Brook University Institutional Animal Care and Use Committee, the Committee for Ethics in Animal Research of the Ribeirao Preto Medical School (Process no 16/2021), or the State Office for Nature, Environment and Consumer Protection North Rhine-Westphalia (LANUV) in Germany in compliance with German animal welfare law. The mPNHD assay was performed in accordance with Council Directive No. 2010/63/UE of September 22nd, 2010, on the protection of animals used for scientific purpose in France. Animals were housed in temperature (22 ± 2°C) and humidity (55 ± 5%) controlled care facilities at the respective institutions on a 12 h light/dark cycle and provided with food and water ad libitum.
Reagents
Forskolin (10 mM in DMSO) was purchased from Tocris (Bristol, UK). Lethal factor (LF) was purchased from List Biological Laboratories (#169, recombinant from B. anthracis). Protective antigen (PA) was obtained through BEI Resources, NIAID, NIH (#NR-140, recombinant from B. anthracis).
Antibodies
The following antibodies were used in this study: Rabbit polyclonal anti-MEK-3 (1:500, Santa Cruz Biotechnology, #sc-961), rabbit polyclonal anti-p38 (1:1000, Cell Signaling Technology, #9212), rabbit monoclonal anti-phospho-p38 (Thr180/Tyr182) (clone D3F9, Cell Signaling Technology, #4511, 1:1000), rabbit polyclonal anti-SNAP-25 (1:1000, Millipore Sigma #S9684), rabbit polyclonal anti-GAPDH (1:30,000, Millipore Sigma #G9545), goat anti-rabbit IgG (1:1000, Cell Signaling Technology, #7074), chicken polyclonal anti-UCHL1 (1:2000, Novus, Cambridge, UK, #NB110-58872), rabbit monoclonal anti-RIIα (phospho-Ser96) (1:1000, clone 151, Abcam, Cambridge, UK, #ab32390), mouse monoclonal anti-RIIβ (1:2000, BD Transduction Laboratories, #610625), mouse monoclonal anti-NF200 (clone N52, Sigma, #N0142, 1:1000), mouse monoclonal anti-CaMKII alpha subunit (clone 6G9, Thermo Fisher Scientific, #MA1-048, 1:1000), mouse monoclonal anti-NaV1.8 (clone N134/12 Neuromab Facility, #75–166, 1:500), mouse monoclonal anti-CGRP (clone 4901, biorbyt, #orb319478, 1:500), goat polyclonal anti-TrkA, (R&D Systems, #AF1056, 1:500), goat polyclonal anti-TRPV1 (R&D Systems, #AF3066, 1:500), rabbit monoclonal anti-Cleaved Caspase 3 (Asp175, clone 5A1E, Cell Signaling Technology, #9664, 1:500), rabbit anti-p-ERK (Thr202/Tyr204) (D13.14.4E) (Cell Signaling Technology, #4370S, 1:200), highly cross-adsorbed Alexa 647, 555, and 488 conjugated secondary antibodies (Thermo Fisher Scientific).
Recombinant protein expression and purification
Sequences for all recombinant proteins are reported in Fig. S22. The Edema Factor (EF) clone employed in this study contains an extra alanine at the N terminus compared to the native sequence, which has been shown to have a stabilizing effect on activity57. EF and LFN-DTA were expressed using the Champion pET SUMO expression system (ThermoFisher Scientific) in BL21(DE3) E. coli and purified using a HisTrap FF Ni-NTA column (GE Healthcare Life Sciences). The SUMO tag was cleaved by incubation with SUMO protease (Thermo Fisher Scientific) for 1 h at RT and removed by size-exclusion chromatography (SEC). EF underwent additional endotoxin removal by anion exchange chromatography (AEX) using a HiTrap Q HP anion exchange column (GE Healthcare Life Sciences). Endotoxin levels in the final product were measured using the Pierce LAL Chromogenic Endotoxin Quantitation Kit (ThermoFisher Scientific) to be 0.29 EU/mg.
LFN-EFC linking the N terminal domain of LF (residues 1 – 255 of native LF) and C terminal catalytic domain of EF (residues 258 – 766 of native EF) was cloned into the expression vector pSJ115 using standard molecular techniques. LFN-EFC and EF were expressed and purified from the avirulent B. anthracis strain BH460 as previously described58,59.
For the LFN-LC/AC699S construct, the N terminal domain of LF (residues 1 – 262 of native LF) and the catalytic domain of Botulinum neurotoxin type A1 (BoNT/A1) (residues 1 – 448 of native BoNT/A1) separated by a (GGS)2 linker was codon-optimized and cloned into the pK8 expression vector with a cleavable C-terminal His-tag. This was expressed in E. coli strain NiCo21 (DE3) using conditions previously described60 and purified by IMAC using NiHP (GE Healthcare Life Sciences) and AEC using QHP (GE Healthcare Life Sciences) columns. The His-tag was removed by O/N incubation with TEV protease (Millipore Sigma) at 4 °C followed by negative INAC, and the final product was desalted into PBS pH 7.2 and stored at −80 °C.
Analysis of published microarray, single-cell RNAseq, tissue expression and in situ hybridization data
Microarray data of sorted mouse DRG neuron populations were obtained from Chiu et al3. The dataset is deposited at the NCBI GEO database under accession number GSE55114. Affymetrix CEL files were normalized using the Robust Multi-array Average (RMA) algorithm with quantile normalization, background correction, and median scaling. Microsoft excel was used to calculate p-values by two-tailed, unequal variance t-tests.
Single-cell RNAseq data of mouse DRG neurons were obtained from Zeisel et al.4 and Sharma et al.5, respectively available from the SRA database (accession code SRP135960) or GEO database (accession code GSE139088). Average transcript levels of Antxr2, Scn10a, Trpv1, Calca, Ntrk1, Ntrk2 and Ntrk3 across DRG neuron clusters were obtained from mousebrain.org4 and plotted as a relative heat map using GraphPad Prism. The clusters were originally designated as PSPEP1-8, PSNF1-3 and PSNP1-6 by Zeisel et al. Expression of Antxr2 and Antxr1 in DRG neurons across development5 were plotted as a force-directed layout through https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/Sharma2019/all and filtered for adult neurons only.
Microarray data of Antxr2 expression in the DRG and brain regions were obtained from BioGPS.org52 (http://biogps.org/#goto=genereport&id=71914) for the datasets GeneAtlas GNF1M53 and MOE43054. In situ hybridization data of Antxr2 in the adult brain (P56) and juvenile spinal cord (P4) were obtained from the Allen Mouse Brain Atlas55 (2004) (http://mouse.brain-map.org/experiment/show/69526659) and Allen Spinal Cord Atlas (2008) (http://mousespinal.brain-map.org/imageseries/detail/100019979.html) from the Allen Institute.
qPCR
For analysis of mouse tissue, animals were anesthetized with Avertin solution (500 mg/kg, Millipore Sigma) and perfused with 10 mL of cold PBS prior to harvest. RNA was isolated using the RNeasy mini kit (Qiagen). For analysis of human tissue, total brain or DRG RNA was obtained from Clontech (#636530 and #636150). Brain RNA was pooled from 4 Asian males, aged 21 – 29, with an unknown cause of death. DRG RNA was pooled from 21 Caucasian males and females, aged 16 – 65, who underwent sudden death. Per the supplier, RNA was isolated by a modified guanidium thiocyanate method and integrity and purity was confirmed using an Agilent 2100 Bioanalyzer. Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative real-time PCR was performed using the Power SYBR Green PCR Master Mix (ThermoFisher Scientific) on a StepOnePlus RT PCR system (Applied Biosystems) or a LightCycler 96 (Roche). Expression relative to Gapdh was calculated using the comparative CT method.
mAntxr1 F: CTCGCCCATCAAGGGAAAACT
mAntxr1 R: TACTTGGCTGGCTGACTGTTC
mAntxr2 F: CAGTGAGCATTCAGCCAAGTTC
mAntxr2 R: CTGCAATCCCATTGGTACATTCTG
Primers to measure Antxr2 expression in Nav1.8/Antxr2 mice were designed to span the deleted region:
mAntxr2 F (for conditional KO): ATTGCAGCCATCGTAGCTATTT
mAntxr2 R (for conditional KO): GCCAAAACCACCACATCAAG
mGapdh F: GGGTGTGAACCACGAGAAATATG
mGapdh R: TGTGAGGGAGATGCTCAGTGTTG
hAntxr2 F: TGTGTGGGGGAGGAATTTCAG
hAntxr2 R: AGGATAGGTGCAGGACAAAGC
hGapdh F: TGGCATTGCCCTCAACGA
hGapdh R: TGTGAGGAGGGGAGATTCAGT
In situ hybridization of mouse DRG neurons
For chromogenic detection of Antxr2, freshly dissected lumbar DRGs were post-fixed in 10% NBF (Fisher Scientific) for 2 hours at 4°C, dehydrated in 30% sucrose in PBS overnight at 4°C, embedded in OCT medium (Tissue-Tek) and frozen in a dry ice/isopentane bath. For fluorescent detection of Antxr2, Scn10a, Pvalb and Tubb3, DRGs were embedded and frozen immediately after dissection. DRGs were cryo-sectioned to 12 μm and mounted onto Superfrost Plus slides (Fisher Scientific). In situ hybridization was performed using the RNAscope system (Advanced Cell Diagnostics) following manufacturer’s protocol. For chromogenic detection, sections were digested with Protease Plus for 5 min at RT and processed with the RNAscope 2.5HD RED detection kit using the probe Mm-Antxr2-C1 (#46851). The manufacturer’s Positive Control Probe against mouse PPIB (#313911) and Negative Control Probe against the bacterial DapB gene (#310043) were used as controls. For fluorescent detection, sections were digested with Protease IV for 30 min at RT and processed with the RNAscope Multiplex Fluorescent detection kit v2 using the following probe combinations: (a) Mm-Antxr2-C1 (#46851), Mm-Scn10a-C2 (#426011-C2) and Mm-Tubb3-C3 (#423391-C3) or (b) Mm-Antxr2-C1 (#46851), Mm-Tubb3-C2 (#423391-C2) and Mm-Pvalb-C3 (#421931-C3). The manufacturer’s 3-plex Negative Control Probe against DapB (#320871) was used as a negative control. Widefield images were acquired at 20x magnification on an Eclipse TE2000-E Inverted Fluorescence Microscope (Nikon) using NIS-Elements software (Nikon). The raw image files were brightened and contrasted using ImageJ software. Aside from adjusting brightness and contrast, we did not perform digital image processing to subtract background. For quantification, cell boundaries were drawn manually in ImageJ based on the Tubb3 signal. Each cell was scored manually by a blinded observer as positive or negative for Antxr2, Scn10a or Pvalb expression. Results from 12 – 15 fields (4 – 5 fields from 3 mice) were combined for analysis.
In situ hybridization of human DRG neurons
Tissue preparation
All human tissue procurement procedures were approved by the Institutional Review Boards at the University of Texas at Dallas. Samples from medically cleared donors were provided by the Southwest Transplant Alliance with written familial consent. All samples were deidentified prior to use in the study. The sex, age and cause of death of the donors are provided in Supplementary Table 1. Human dorsal root ganglion (L5) were collected, frozen on dry ice and stored in a −80°C freezer. The human DRGs were gradually embedded with OCT in a cryomold by adding small volumes of OCT over dry ice to avoid thawing. All tissues were cryostat sectioned at 20 μm onto SuperFrost Plus charged slides. Sections were only briefly thawed in order to adhere to the slide but were immediately returned to the −20°C cryostat chamber until completion of sectioning. The slides were then immediately utilized for histology.
RNAscope in situ hybridization
RNAscope in situ hybridization multiplex version 1 was performed as instructed by Advanced Cell Diagnostics (ACD). Slides were removed from the cryostat and immediately transferred to cold (4°C) 10% formalin for 15 minutes. The tissues were then dehydrated in 50% ethanol (5 min), 70% ethanol (5 min) and 100% ethanol (10 min) at room temperature. The slides were air dried briefly and then boundaries were drawn around each section using a hydrophobic pen (ImmEdge PAP pen; Vector Labs). When hydrophobic boundaries had dried, protease IV reagent was added to each section until fully covered and incubated for 2–5 minutes at room temperature. The protease IV incubation period was optimized for the specific lot of Protease IV reagent and for each DRG as recommended by ACD. Slides were washed briefly in 1X phosphate buffered saline (PBS, pH 7.4) at room temperature. Each slide was then placed in a prewarmed humidity control tray (ACD) containing dampened filter paper and a 50:1:1 dilution (as directed by ACD due to stock concentrations) of ANTXR2 (ACD Cat # 855501; Channel 1), CALCA (ACD Cat # 605551; Channel 2), P2RX3 (ACD Cat # 406301; Channel 3) was pipetted onto each section until fully submerged. This was performed one slide at a time to avoid liquid evaporation and section drying. The humidity control tray was placed in a HybEZ oven (ACD) for 2 hours at 40°C. Following probe incubation, the slides were washed two times in 1X RNAscope wash buffer and returned to the oven for 30 minutes after submersion in AMP-1 reagent. Washes and amplification were repeated using AMP-2, AMP-3 and AMP-4 reagents with a 15-min, 30-min, and 15-min incubation period, respectively. AMP-4 ALT C (Channel 1 = Atto 550, Channel 2 = Atto 647, Channel 3 = Alexa 488) was used for all experiments. Slides were washed two times in 0.1M phosphate buffer (PB, pH7.4) and then incubated in DAPI (1/5000) in 0.1M PB for 1 min before being washed, air dried, and cover-slipped with Prolong Gold Antifade mounting medium.
Tissue Quality Check
All human DRGs were checked for RNA quality by using a positive control probe cocktail (ACD) which contains probes for high, medium and low-expressing mRNAs that are present in all cells (ubiquitin C > Peptidyl-prolyl cis-trans isomerase B > DNA-directed RNA polymerase II subunit RPB1). DRGs that showed signal for all 3 positive control probes were used to generate experimental data. A negative control probe against the bacterial DapB gene (ACD) was used to check for non-specific/background label.
Image Analysis
DRG sections were imaged on an Olympus FV3000 confocal microscope at 20X magnification. 3 20X images were acquired of each human DRG section, and 3–4 sections were imaged per human donor. The acquisition parameters were set based on guidelines for the FV3000 provided by Olympus. In particular, the gain was kept at the default setting 1, HV ≤ 600, offset = 4, and laser power ≤ 15%. The raw image files were brightened and contrasted in Olympus CellSens software (v1.18), and then analyzed manually one cell at a time for expression of each gene target. Cell diameters were measured using the polyline tool. Total neuron counts for human samples were acquired by counting all of the probe-labeled neurons and all neurons that were clearly outlined by DAPI (satellite cell) signal and contained lipofuscin in the overlay image.
Large globular structures and/or signal that auto-fluoresced in all 3 channels (488, 550, and 647; appears white in the overlay images) was considered to be background lipofuscin and was not analyzed. Aside from adjusting brightness/contrast, we performed no digital image processing to subtract background. We attempted to optimize automated imaging analysis tools for our purposes, but these tools were designed to work with fresh, low background rodent tissues, not human samples taken from older organ donors. As such, we chose to implement a manual approach in our imaging analysis in which we used our own judgement of the negative/positive controls and target images to assess mRNA label. Images were not analyzed in a blinded fashion.
Data Analysis and Statistics
Graphs were generated using GraphPad Prism version 8.2.0 – 8.4.3 (GraphPad Software, Inc. San Diego, CA USA). All pie-charts were generated by averaging the population distributions from each human DRG (n=3). The total number of neurons assessed between all subjects is indicated on the pie-chart; however, the pie-charts were generated based on population averages as noted above. Relative frequency distribution histograms with a Gaussian distribution curve were generated using the diameters of all target-positive neurons.
Dorsal root ganglia neuron dissection and culture
Dorsal root ganglia cultures were prepared as previously described61 with minor modifications. Briefly, adult mice 6 – 12 weeks of age were euthanized by CO2 asphyxiation and dorsal root ganglia (DRG) were harvested from all segments of the spinal cord. Following enzymatic dissociation in Collagenase A and Dispase II for 40 min at 37 °C, DRGs were triturated with decreasing diameters of syringe needles (18G, 22G, 25G) and purified through a layer of 15% BSA in neurobasal media (NBM). The resulting pellet was filtered through a 70 μm strainer and resuspended in NBM supplemented with B27 (Thermo Fisher) and L-glutamine (Thermo Fisher). Cells were then seeded in laminin-coated tissue culture plates and cultured in the presence of 50 ng/μL nerve growth factor (NGF) (Thermo Fisher) unless otherwise noted.
cAMP detection from DRG cultures
DRG neurons were prepared as described in Dorsal root ganglia neuron dissection and culture and seeded 4000 cells per well in 96 well plates. Following overnight culture, cells were treated with the indicated toxin components in NBM containing 50 ng/μL NBM for 2 h at 37 °C, lysed by manual scrapping in 0.1M HCl containing 0.5% triton X-100, and clarified by centrifugation. cAMP levels in clarified lysates were measured using the Direct cAMP ELISA kit from Enzo Life Sciences (#ADI-900-066) following manufacturer’s protocol. Absorbance was measured on a Synergy Mx multi-mode microplate reader (BioTek) and fitted using GraphPad Prism.
cAMP detection from tissue
Mice were given intrathecal or intraplantar administration of PBS or ET (2 μg PA + 2 μg EF). At the indicated timepoint, animals were euthanized by CO2 asphyxiation and blood was drained by cardiac puncture. Lumbar DRGs, lumbar spinal cord or the glabrous skin of the footpad were harvested and homogenized in 0.1M HCl + 0.5% triton X-100 using glass beads and a TissueLyser II (Qiagen). Lysate was centrifuged at 18,000 ×g and 4 °C for 40 – 60 min to pellet cell debris. cAMP levels in clarified lysates were measured using the Direct cAMP ELISA kit from Enzo Life Sciences (#ADI-900-066) following manufacturer’s protocol. The protein concentration in clarified lysates were measured using the Pierce BCA Protein Assay kit (Thermo Fisher Scientific) following manufacturer’s protocol. Absorbances were measured on a Synergy Mx multi-mode microplate reader (BioTek) and fitted using GraphPad Prism. The concentration of cAMP in each sample was normalized by corresponding protein concentration.
Western Blot Analysis
DRG neurons were prepared as described in Dorsal root ganglia neuron dissection and culture and seeded 15,000 cells per well in 96 well plates. Following overnight culture, cells were treated with combinations of 10 nM PA, 10 nM LF and 10 nM EF in NBM containing 50 ng/μL for 24 h at 37 °C. Cells were then lysed by manual scrapping in cold NP40 lysis buffer (Thermo Fisher Scientific) containing Halt protease and phosphatase inhibitors (Thermo Fisher Scientific). Samples were separated by SDS-PAGE and transferred to a PVDF membrane, which was blocked in TBST + 5% non-fat milk for 1 h at RT and washed with TBST. The membrane was incubated with the following primary antibodies in TBST + 5% BSA at 4°C overnight: rabbit polyclonal anti-MEK-3 (1:500, Santa Cruz Biotechnology, #sc-961), rabbit polyclonal anti-p38 (1:1000, Cell Signaling Technology, #9212) or rabbit monoclonal anti-phospho-p38 (Thr180/Tyr182) (clone D3F9, Cell Signaling Technology, #4511, 1:1000). Incubation with HRP-linked goat anti-rabbit IgG (1:2000, Cell Signaling Technology, #7074) in TBST + 2.5% BSA was performed for 1 h at RT. Signal was developed using the SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific) and imaged on an Amersham Imager 600 (GE Healthcare Life Sciences). The membrane was then stripped with Restore western blot stripping buffer (Thermo Fisher Scientific) for 15 min at 37°C, and re-probed with rabbit polyclonal anti-GAPDH (1:30,000, Millipore Sigma #G9545) in TBST + 5% BSA. Band intensities were quantified using ImageJ and each target was normalized to its respective loading control.
Protein synthesis assay
DRG neurons were prepared as described in Dorsal root ganglia neuron dissection and culture and seeded 15,000 cells per well in 96 well plates. Following overnight culture, cells were treated with 0.1 pM to 10 nM LFN-DTA, with or without 10 nM PA, in NBM containing 50 ng/μL NGF for 6 h at 37 °C. Cells were then washed with leucine-free F12K media and incubated with 20 μCi/mL 3H-leucine in the same media for 1 h at 37 °C. Cells were washed three times with PBS and lysed in MicroScint-20 (PerkinElmer). Scintillation counting was performed on a MicroBeta TriLux (PerkinElmer).
Botulinum toxin light chain activity assay
Activity of botulinum toxin light chains were measured using the BoTest A/E BoNT detection kit (BioSentinel) following manufacturer’s instructions. Fluorescence was measured on a Synergy Mx multi-mode microplate reader (BioTek).
CGRP release and SNAP-25 cleavage assays
DRG neurons were prepared as described in Dorsal root ganglia neuron dissection and culture and seeded 7500 cells per well in 96 well plates. Cells were cultured for a week in NBM + 50 ng/μL NGF and pulsed with an additional 10 mM cytosine arabinoside for three days during day 3 – 5 of culture. Cells were then incubated with 0.1 pM to 1 nM LFN-LC/AC699S, with or without 10 nM PA, for 24 h at 37°C. Cells were stimulated with Krebs-Ringer buffer containing 80 mM KCl for 10 min at 37°C. The concentration of CGRP in the supernatant was determined using a CGRP ELISA kit (Cayman Chemical) following manufacturer’s protocol. Remaining cells were lysed in Bolt LDS sample buffer (Thermo Fisher Scientific) containing 0.1M DTT (Thermo Fisher Scientific) and 0.125 units/μL Benzonase (Millipore Sigma). Samples were then separated by SDS-PAGE and transferred to a PVDF membrane, which was blocked with TBST + 5% non-fat milk (NFM) for 1 h at RT. The membrane was blotted with rabbit polyclonal anti-SNAP-25 (1:1000, Millipore Sigma #S9684) in TBST + 5% NFM and HRP-linked goat anti-rabbit IgG (1:1000, Cell Signaling Technology, #7074) in TBST + 5% NFM. Signal was developed using the SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific) and imaged on an Amersham Imager 600 (GE Healthcare Life Sciences). Band intensities were quantified using ImageJ and percent cleavage was calculated using the following formula: Cleaved SNAP-25 / (Uncleaved SNAP-25 + Cleaved SNAP-25).
Mouse DRG recordings
Cell preparation
DRG neurons were prepared as described in Dorsal root ganglia neuron dissection and culture and cultured overnight in NBM containing 50 ng/μL NGF and 2 ng/μL GDNF. Cells were plated on laminin-coated glass coverslips and incubated at 37 °C (95% O2, 5% CO2) overnight. Cells were treated with 10 nM PA + 10 nM EF in NBM + 50 ng/μL NGF + 2 ng/μL GDNF or vehicle for 2–10 h at 37 °C. Recordings were made within 1 h after removal from treatment.
Whole-cell current clamp and calcium current recordings
Small DRG neurons (membrane capacitance: 6.6 ± 0.5 pF) were recorded at room temperature. Whole-cell recordings were made using an Axon Instruments Multiclamp 700B Amplifier (Molecular Devices) and a Digidata 1322A data acquisition interface using pClamp 9.2 software (Molecular Devices). Membrane voltage and current signals were filtered at 10 kHz and digitized at 100 kHz. Analysis was performed with Igor Pro (Wavemetrics, Lake Oswego, OR) using DataAccess (Bruxton Software) to import pClamp data. Recordings were made using using electrodes with resistances of 5–9 MOhm (current clamp) or xx-xx MOhm (voltage clamp) with tips wrapped by strips of Parafilm to reduce pipette capacitance. Current clamp recordings were made using an internal solution containing 140 mM K aspartate, 13.5 mM NaCl, 1.6 mM MgCl2, 0.09 mM EGTA, 9 mM HEPES, 14 mM creatine phosphate (Tris salt), 4 mM MgATP, 0.3 mM Tris-GTP, pH 7.2 adjusted with KOH and an external Tyrode’s solution containing 155 mM NaCl, 3.5 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4 adjusted with NaOH. Reported membrane potentials were corrected for a liquid junction potential of −10 mV between the internal and extracellular solutions65. After breaking into the cells, pipette capacitance was compensated (50% - 60%) and bridge balance was set to compensate for series resistance. Action potential firing was recorded with injection of a series of 1-s depolarizing current pulses ranging from 1 pA to 150 pA. Cell input resistance was measured using a 30-pA hyperpolarizing current injection. Voltage clamp recordings of currents through voltage-dependent calcium channels were made using an internal solution containing 140 mM CsCl, 13.5 mM NaCl, 1.8 mM MgCl2, 0.09 mM EGTA, 9 mM HEPES, 14 mM creatine phosphate (Tris salt), 4 mM MgATP, 0.3 mM Tris-GTP, pH 7.2 adjusted with CsOH and an external solution containing 5 mM BaCl2, 160 mM TEACl, 10 mM HEPES, 10 mM glucose, pH 7.4 adjusted with TEAOH, with 5 μM TTX. Calcium channel currents were evoked using a series of 100-ms voltage steps from −60 to +70 mV, delivered from a holding voltage of −70 mV. Collected data are presented as mean ± SEM.
Spinal cord slice recordings
Horizontal spinal cord slices
Horizontal spinal cord slices were made from P18-P27 Sprague-Dawley rats. Rats were deeply anesthetized with isoflurane prior to decapitation. After decapitation, the ventral aspect of the vertebral column was exposed and immersed in ice-cold dissecting solution (in mM): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4.H2O, 26 NaHCO3, 6 MgCl2, 0.5 CaCl2, 20 Glucose, 77 sucrose, 1 kynurenic acid, oxygenated with 95/5% O2/CO2. Using a bilateral ventral laminectomy, the lumbar part of the spinal cord was exposed and carefully removed with L4 and L5 dorsal roots attached. Horizontal slices were made manually. First, the spinal cord was cut in half with a micro ophthalmic scissor through the parasagittal plane to produce a hemisected spinal cord. With a second cut, ~ 30–45-degree angle with respect to the parasagittal plane, the ventral part of the hemisected cord was removed such that the result was a horizontal slice (400–500 μm thick) with the L4-L5 dorsal roots (7–10 mm length) attached66. Spinal cord slices were then immersed in oxygenated recovery solution (same as dissecting solution, but without kynurenic acid) at 35 °C and allowed to recover for 1 hour. After 1 hour the slices were transferred to a storage solution (same as recovery solution, at room temperature) and kept for the next 4–5 hours. In horizontal slices, lamina I neurons were visualized through the white matter by using an infrared light-emitting diode (IR-LED) illumination67–69, a CCD video camera (Oly 150, Olympus), and a 40X water immersion objective mounted on an upright microscope (BX51WI, Olympus). To restrict the analysis mainly to lamina I projection neurons, large neurons with soma cross-sectional area larger than 250 μm2 and cell capacitance larger than 75 pF, located in the outer portion of lamina I, just below the white matter, were selected for recording66,70,71. Neurons had a cross sectional area of 313±90 μm2 and capacitance of 98±31 pF (n=13).
Patch clamp electrophysiology
After recovery, slices were transferred to a submersion recording chamber and mounted on the stage of an upright microscope (BX51WI, Olympus). Slices were then perfused with artificial cerebrospinal fluid (aCSF) (in mM: 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 20 glucose), oxygenated with 95/5% O2/CO2. Whole-cell voltage clamp recordings were made with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Patch pipettes were pulled from borosilicate glass (WPI, Sarasota, FL) using a Sutter P97 puller (Sutter Instrument, Novato, CA). The resistance of the patch pipette was 1.3–1.8 MΩ when filled with the standard internal Cs-methanesulfonate-based solution. The shank of the patch pipette was wrapped with parafilm to reduce pipette capacitance. In whole-cell mode, the capacity current was reduced by using the amplifier circuitry. To reduce voltage errors, 40–50% of series resistance compensation was applied. For recording EPSCs in voltage clamp, the external solution was the oxygenated aCSF; the internal solution was (in mM): 125 Cs-methanesulfonate, 10 NaCl, 2 MgCl2, 14 phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP, 10 EGTA, 10 HEPES, 5 mM QX-314, pH 7.2 with CsOH. EPSCs in voltage clamp were recorded at −70 mV. Drugs were dissolved in the aCSF and applied to the spinal cord slice through the perfusion system at a speed of 2 ml/min. Recordings were made at 35 ± 1 °C by heating the solutions with a temperature controller (Warner TC-344B, Warner Instruments, Hamden, CT).
Dorsal root stimulation
EPSCs were evoked by stimulating the dorsal root with a suction electrode connected to an isolated current stimulator (ISO-Flex Stimulus Isolator; A.M.P.I., Jerusalem, Israel). The dorsal root was stimulated at 0.016 Hz (duration 0.1 ms). The stimulus intensity was gradually increased to recruit fibers with increasing threshold. Stimulation of the dorsal root started with 25 μA of current to test for synaptic inputs from Aα/β-fibers72–74. The stimulus intensity was then gradually increased to 80, 200, 300, 400, and 500 μA of current to recruit Aδ- and C-fibers and until the maximum EPSC was elicited. Once the maximum EPSC was elicited, then the stimulus intensity was set at 30 % higher (usually between 400 and 500 μA). Only monosynaptic responses were included in the analysis. Monosynaptic responses elicited by stimulation of Aδ- and C-fibers were identified based on the absence of synaptic failures and low variability (less than 15%) in synaptic delay during 15 consecutive stimuli: 80 μA, 2 Hz (for Aδ-fibers), and 500 μA, 1 Hz (for C-fibers)66,74–77. Isolation of Aδ- and C-fiber components were confirmed with the measured conduction velocity (CV): Aδ-fibers (0.5–2 m/s), C-fibers (<0.5 m/s)72,74.
Data acquisition and analysis
Currents and voltages were controlled and sampled using a Digidata 1440A interface and pCLAMP 10.3 software (Molecular Devices, Sunnyvale, CA). In voltage-clamp, currents were filtered at 2 kHz (3 dB, 4-pole Bessel) and digitized at 50 kHz. Analysis was performed using Clampfit 10.3 and IGOR Pro (version 6.2; WaveMetrics, Lake Oswego, OR) using DataAccess (Bruxton, Seattle, WA) to import pCLAMP files into IGOR. Analyses of evoked synaptic currents and PPR were carried out by measuring the peak current of three averaged consecutive traces in each condition. For analysis of mEPSCs, the threshold was set at twice the average noise. Frequency and peak of mEPSCs were determined during a 2 min period in each condition. Reported voltages were corrected for −8 mV junction potential (pipette relative to bath, for the methanesulfonate-based internal solution) which was measured using a flowing 3M KCl reference electrode65.
DRG neurons for High Content Screening (HCS) microcopy
Mice were euthanized between 9 – 12 AM by CO2 intoxication, and cervical, lumbar and thoracic DRGs were removed within 30 min per animal. DRGs were incubated in Neurobasal A/B27 medium (Invitrogen, Carlsbad, CA) containing collagenase P (Roche, Penzberg, DE) (0.2 U/mL, 1 h, 37°C, 5% CO2). DRGs were dissociated by trituration with fire-polished Pasteur pipettes. Axon stumps and disrupted cells were removed by bovine serum albumin (BSA) gradient centrifugation (15% BSA, 120 g, 8 min). Viable cells were resuspended in Neurobasal A/B27 medium, plated in 0.1 mg/mL poly-L-ornithine / 5 μg/mL laminin-precoated 96-well imaging plates (Greiner, Kremsmünster, AU) and incubated overnight (37°C, 5% CO2). Neuron density was 1500 neurons/cm2. DRG neurons were stimulated 24 h after isolation in 96-well imaging plates.
Generation of iPSC-derived sensory neurons
This study employed the iPSC line UKBi013-A (https://hpscreg.eu/cell-line/UKBi013-A). The study was approved by the Ethics Committee of the Medical Faculty of the University of Bonn (approval number 275/08), and informed consent was obtained from the patient. Differentiation of iPSC into sensory neurons was performed as previously reported26 with slight modifications. Briefly, single cell iPSCs were seeded at 3×105 cells/cm2 in StemMAC iPS-Brew (Miltenyi Biotec, Bergisch Gladbach, DE) in presence of 10 μM Rock-Inhibitor Y-27632 (Cell Guidance Systems, Cambridge, UK) on Geltrex (Thermo Fisher Scientific, Waltham, USA) coated T175-flasks at day −1. After 24 h the medium was change to differentiation medium. Neural differentiation was initiated by dual-SMAD inhibition using 100 nM LDN 193189 (Axon Medchem BV, Groningen, NL) and 10 μM SB 431542 (Biozol, Eching, DE) from day 0 to day 6. To specify the differentiating cells into sensory neurons 3 μM CHIR 99021 (Miltenyi Biotec, Bergisch Gladbach, DE), 10 μM SU5402 (Sigma Aldrich, St. Louis, USA) and 10 μM DAPT (Axon Medchem BV, Groningen, NL) were added to the culture from day 3 to day 14. Two basal media were used during differentiation: Media1 consists of Knock out DMEM (Thermo Fisher Scientific, Waltham, USA) with 20% Knock Out Serum Replacement (Thermo Fisher Scientific, Waltham, USA), 2 mM (1x) GlutaMAX (Thermo Fisher Scientific, Waltham, USA), 100 μM (1x) NEAA (Thermo Fisher Scientific, Waltham, USA) and 0.02 mM 2-Mercaptoethanol (Thermo Fisher Scientific, Waltham, USA). Media2 consists of Neurobasal Media (Thermo Fisher Scientific, Waltham, USA) supplemented with 1% N2 supplement (Thermo Fisher Scientific, Waltham, USA), 2% B27 supplement (Thermo Fisher Scientific, Waltham, USA), 2 mM (1x) GlutaMAX (Thermo Fisher Scientific, Waltham, USA) and 0.02 mM 2-Mercaptoethanol (Thermo Fisher Scientific, Waltham, USA). Between d0 and d3 cells were kept in Media1, from day 4 to 5 cells were kept in 75% Media1 and 25% Media2, for day 6 cells were kept in 50% Media1 and 50% Media2, from day 7 to 9 cells were kept in 25% Media1 and 75% Media2 and from day 10 to 14 differentiating cells were kept in 100% Media2. At day 14 of differentiated cells were dissociated with Accutase (Thermo Fisher Scientific, Waltham, USA) and frozen in cold CryoStor CS10 freezing medium (Sigma Aldrich, St. Louis, USA) at −80 °C. After 24 h frozen cells were transferred to a liquid nitrogen tank for long-term storage.
Cells were plated after thawing in Geltrex coated 96-well imaging plates at a density of 40.000 cells per well in maturation medium. Maturation medium consisting of Neurobasal A (Thermo Fisher Scientific, Waltham, USA) supplemented with 1% N2 supplement (Thermo Fisher Scientific, Waltham, USA), 2% B27 supplement (Thermo Fisher Scientific, Waltham, USA), 2 mM (1x) GlutaMAX (Thermo Fisher Scientific, Waltham, USA), 0.02 mM 2-Mercaptoethanol (Thermo Fisher Scientific, Waltham, USA), 12 μg/mL Gentamycin (Thermo Fisher Scientific, Waltham, USA), 200 μM Ascorbic acid (Sigma Aldrich, St. Louis, USA), 0.1 μg/mL human recombinant Laminin LN521 (BioLamina, Sundyberg, SWE), 10 ng/mL GDNF (Cell Guidance Systems, Cambridge, UK), 10 ng/mL BDNF (Cell Guidance Systems, Cambridge, UK), 10 ng/mL NGF (Peprotech, Hamburg, DE) and 10 ng/mL NT3 (Peprotech, Hamburg, DE). Four days after plating cultures were treated with 1μg/mL Mitomyin C (Sigma Aldrich, St. Louis, USA) for 2 h at 37 °C to inactivate proliferative cells. Medium was changes twice per week. Cells were matured for three weeks prior to stimulation.
Cell culture stimulation and fixation for HCS microscopy
DRG neurons and human IPSC-derived sensory neurons were stimulated in 96-well imaging plates. Compounds were dissolved in 12.5 μL PBS in 96-well V-bottom plates, mixed with 50 μL medium from the culture wells, and added back to the same wells. Stimulations were performed with automated 8 channel pipettes (Eppendorf, Hamburg, DE) at low dispense speed on heated blocks, and stimulated cells were placed back in the incubator. The cells were fixed for 10 minutes at RT by adding 100 μL of 8% paraformaldehyde resulting in a final concentration of 4%.
Quantitative HCS microscopy
Fixed DRG cells were treated with goat or donkey serum blocking (2% serum, 1% BSA, 0.1% Triton X-100, 0.05% Tween 20, 1 h, RT) and incubated with respective primary antibodies diluted in 1% BSA in PBS at 4 °C overnight. After three washes with PBS (30 min, RT), cells were incubated with secondary Alexa dye-coupled antibodies (1:1000, 1 h, RT). After three final washes (30 min, RT), wells of 96-well plates were filled with PBS, sealed, and stored at 4 °C until scanning. For IPSC-derived cultures the Nissl staining was performed after primary and secondary staining. Wells were rinse with 0.1% Triton X-100 (5 min, RT) in PBS followed by 20 min incubation with Nissl solution (1:500, 20 min, RT). Three rinsing steps were performed as follow: 5 min PBS, 5 min 0.1% Triton X-100, 30 min three final washes with PBS. Latest, wells were filled with PBS, sealed, and stored at 4 °C.
We used a Cellomics ArrayScan XTI microscope equipped with an X1 CCD camera and LED light source to scan stained cultures of DRG neurons in 96-well imaging plates. 2×2 binned images (1104 × 1104 pixels) were acquired with 10× (NA = 0.3) EC Plan Neo-Fluor objective (Zeiss) for DRG and 20x objective for IPSC-derived nociceptors. Images were analyzed using the Cellomics software package. Briefly, images of UCHL1/Nissl staining were background corrected (low pass filtration), converted to binary image masks (fixed threshold), segmented (geometric method), and neurons were identified by the object selection parameters: size of 80–7500 μm2 (DRG), 10–600 μm2 (IPSC-SN), circularity (perimeter2 / 4π area) of 1–3; length-to-width ratio of 1–2 (DRG), 1–3 (IPSC-SN); average intensity of 800–12000 (DRG), 300–5000 (IPSC-SN), and total intensity of 2×105−5×107 (DRG), 500–109 (IPSC-SN). The image masks were then used to quantify signal in other channels. To calculate spill-over between fluorescence channels, three respective controls were prepared for each triple staining: (i) UCHL1/Nissl alone, (ii) UCHL1/Nissl + antibody 1, and (iii) UCHL1/Nissl + antibody 2. Raw fluorescence data of the controls were used to calculate the slope of best fit straight lines by linear regression, which was then used to compensate spill-over. Compensated data were scaled to a mean value of 1 for the unstimulated cells to adjust for variability between experimental days. One and two-dimensional probability density plots were generated using R packages. Gating of subpopulations was performed by setting thresholds at local minima of probability density plots. Statistical analyses were performed with Students t-tests, one-, or two-way ANOVA with respective post hoc tests as indicated in the figure legends. P < 0.05 was considered statistically significant. Dose-response curves from HCS microscopy were generated using non-linear regression curve-fitting (three parameter, standard Hill slope) using Prism software (GraphPad, La Jolla, CA). The parameters of the model (top, bottom, or pEC50/pIC50 values) were compared using the extra-sum-of-squares F test. High content screening kinetic experiments were analyzed with R using ordinary two-way ANOVA. Bonferroni’s post hoc analysis was applied to determine p values of selected pairs defined in a contrast matrix using the R library multcomp. Error bars represent the standard error of the mean (SEM) of three independent replicate experiments using cells of different animals.
Analysis of DRG sections for PKA activation
Mice received intrathecal injection of PBS or ET (2 μg PA + 2 μg EF). At 2 h post-injection, L3 – L6 DRGs or lumbar spinal cord were harvested and fixed with 4% PFA (Electron Microscopy Sciences) in PBS for 2 h on ice and dehydrated in 30% sucrose in PBS at 4 °C overnight. The DRGs were then embedded in OCT medium (Tissue-Tek) and snap frozen in an isopentane/dry ice bath. Frozen blocks were cut into 12 μm sections, mounted on slides, dried for 30 minutes at RT, and stored at −80 °C. Thawed sections were post-fixed in 2% PFA for 10 minutes at 4 °C, rinsed in PBS for 30 minutes, incubated in goat serum blocking (2% serum, 1% BSA, 0.1% Triton X-100, 0.05% Tween 20, 1h, RT), and incubated with respective primary antibodies diluted in 1% BSA in PBS at 4 °C overnight. After three washes with PBS (30 min, RT), sections were incubated with secondary Alexa dye-coupled antibodies (1:1000, 1h, RT). After three final washes (30 min, RT), the sections were mounted with Fluoromount-G (Southern Biotech) containing DAPI (0.5 μg/mL).
Images of sections were acquired with a Leica DM6000B epifluorescence microscope controlled by Leica MetaMorph (version 1.4.0) using a 20x objective (Leica HC PLAN APO, 0.7). The images were analyzed using a custom Fiji Image J plugin. Briefly, UCHL1 images (8 bit) were background corrected (rolling ball radius of 500 pixel), blurred (Gaussian with 1 pixel), and contrast enhanced (Enhance contrast value of 0.7). Then a fixed threshold was applied (40–250) followed by watershedding and particle analysis (size of 1000–20000, circularity of 0.3–1). The obtained image mask was transferred to the background corrected pRII images.
Cell viability assay
To measure the induction of apoptosis, isolated DRG neurons were cultured for 2 days and stained alive with Molecular Probes CellEvent™ Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, Cat# C10723, 4 μM) for 1 h prior to paraformaldehyde fixation. Fixed cells were stained for UCHL1 and cleaved caspase 3 followed by HCS microcopy analysis.
Transcriptional profiling analysis of the DRG
Mice were given intrathecal injection of vehicle (PBS) or ET (2 μg PA + 2 μg EF). After 2 hours, mice were given terminal injection of Avertin (500 mg/kg i.p, Millipore Sigma) and perfused with 10 mL of cold PBS. Lumbar DRGs (L3 – L6) were harvested, immediately homogenized in Trizol (Thermo Fisher Scientific) and frozen on dry ice, then stored at −80°C until further processing. Samples from 2 male and 2 female mice were collected each day over 3 days for n=6 total per group. RNA isolation was performed using the RNeasy Micro kit (Qiagen). Total-RNA in each of the samples was quantified using an Agilent 4200 TapeStation instrument and normalized to 200 ng of input in 50 μL (4 ng/μL). The mRNA was captured using oligo-dt beads as part of the KAPA mRNA HyperPrep workflow. cDNA synthesis, adapter ligation, and amplification were conducted subsequently as part of the same workflow. Following amplification, residual primers were eluted away using KAPA Pure Beads in a 0.63× SPRI-based cleanup. qPCR with the KAPA Library Quantification kit was run to confirm the functional concentration. Molarity values obtained from this assay were used to normalize all samples in equimolar ratio for one final pool. The pool was denatured and loaded onto an Illumina NextSeq 500 instrument, with a High-Output 75-cycle kit to obtain Single-End 75bp reads. The pool was loaded at 1.9 pM, with 5% PhiX spiked in as a sequencing control. The basecall files were demultiplexed through the Harvard BPF Genomics Core’s pipeline (bcl2fastq v2.20.0.422), and the resulting fastq files were used in subsequent analysis. All fastq files passed the read quality checks performed using FastQC (v0.11.5). The sequencing reads from each sample were aligned to the GRCm38 or mm10 genome builds from Ensembl (Genome Reference Consortium Mouse Build 38) and quantified using Salmon (v1.5.2) and tximport (v1.18.0) in RStudio (v4.0). Differential gene expression analysis and variance-stabilizing transformation (to obtain normalized read counts) were performed using R/Bioconductor package DESeq2 (v1.30.1). GO pathway enrichment analysis was performed using Bioconductor/R package ClusterProfiler (v3.18.1). We used the encrichGO function in the ClusterProfiler package (with parameters key type=“ENSEMBL”, pAdjustMethod = “BH”, ont = “BP” and qvalueCutoff = 0.05) to compare and plot the enriched KEGG pathways. We visualized this pathway analysis using the dotplot function from ClusterProfiler (v3.18.1).
Immunohistochemistry for pERK in the spinal cord
Mice (male C57BL/6) were pre-treated with vehicle or ET (2 μg/5 μL, i.th.) followed (2 h after) by vehicle or Capsaicin (1 μg/5 μL, i.th. – Cat. n. Sigma M2028). 20 minutes later, mice were deeply anesthetized with ketamine and xylazine and perfused through the ascending aorta with PBS, followed by 4% paraformaldehyde (20 mL/min for 3 minutes). L4–L5 spinal cord segments were dissected and post-fixed for 4 hours. Transverse spinal cord sections (free floating, 40 μm) were cut and processed for immunofluorescence. Briefly, sections were blocked with 1% BSA, 22.52 mg/mL glycine in PBS for 1 hour at room temperature (RT) and incubated overnight at 4°C with primary antibody against p-ERK (1:200; Cell Signaling cat. n. 4370S). After incubation with the primary antibody, the sections were washed in PBS-T 3 × 5 min and incubated at room temperature for 2 hours with Alexa fluor 488®goat anti-rabbit (1:400 Invitrogen cat. n A11008). The sections were washed with PBS-T as described earlier, and mounted on glass slides, covered with cover slips with Vectashield with DAPI (Vector cat. n. H1200). Eight to twelve nonadjacent sections from the L4–L5 lumbar spinal cord of each mice analysed, and the numbers of immunoreactive neuronal profiles in the dorsal superficial laminae of the dorsal horn in each section were counted (under a 10x object field). The values from sections (per side of the dorsal horn of the spinal cord) were averaged for each animal.
Treatment with drugs and chemicals
Rimonabant (Tocris) was dissolved in 4% DMSO and 1% Tween-80 in saline and administered i.p at 5 mg/kg (10 mL/kg). Naltrexone (Tocris) was dissolved in saline and administered i.p at 10 mg/kg (10 mL/kg). Naloxone (Sigma-Aldrich) was dissolved in saline and administered i.p at 2 mg/kg (10 mL/kg). CGS15943 was dissolved in 5% DMSO, 0.3% Tween-80 in saline and administered i.p at 10 mg/kg (1 mL/kg). For induction of chemical sympathectomy, 6-OHDA free base (Tocris) was dissolved in 0.1% ascorbic acid in saline and administered i.p at 100 mg/kg (10 mL/kg) one week prior to ET injection. BCI (EMD Millipore) was dissolved in 1% DMSO in saline and administered intrathecally (1.6 μg in 5 μL).
Behavioral tests
Brush test
Mice were placed in individual test compartments separated by an opaque divider on an elevated mesh-bottomed platform. The left hind paw was gently stroked in the direction of heel to toe with a soft paintbrush (Raphaël Kaerell fan brush, size 2). The number of fast paw withdrawals out of 10 stimulations was recorded, with a 2 min interval between applications.
Measurement of mechanical and thermal sensitivity thresholds
Mechanical and thermal sensitivity thresholds were measured as previously described61 with minor modifications. Briefly, mice were habituated on the behavior apparatus for 2 consecutive days for 1 hour each. After habituation, 3 baseline measurements were obtained on separate days prior to treatment. Mechanical sensitivity thresholds were measured using von Frey filaments and the up/down method64. Thermal sensitivity thresholds were measured using the Hargreaves apparatus (Model 390G, IITC Life Science) with a 40 s cut-off.
Pinprick test
Mice were placed in individual test compartments separated by an opaque divider on an elevated mesh-bottomed platform. An Austerlitz insect pin (size 000) (FST) was applied gently to stimulate the plantar surface of the left paw without penetrating the skin. The number of fast paw withdrawals out of 10 stimulations was recorded, with a 2 min interval between applications.
Randall-Selitto test
Mice were gently restrained with a cloth and allowed to acclimate for 5 min. The base of the tail was stimulated using calibrated digital forceps (Rodent Pincher Algometer, BioSeb, France) and the pressure necessary to evoke a response was recorded. Clear signs of discomfort or attempts to escape were considered as responses. 3 trials were performed with a 3 min rest period between each stimulation, and the results were averaged.
Cold plate and Hot plate tests
For cold plate tests, mice were placed on a temperature-controlled metal plate (IITC Life Science) at 0°C and the number of nocifensive responses (jumping or hind paw licking, lifting or flinching) were recorded within a 5 min window. For hot plate tests, temperatures were set to 50°C or 55°C and the latency to response (jumping or hind paw licking) was recorded. A 90 s cut-off was imposed to avoid tissue injury.
Formalin model
Prior to formalin injection, mice were habituated for 45 min in individual chambers in an infrared Behavior Observation Box (iBOB) (Harvard Apparatus), which allows recording of mice in the dark independent of an observer. Mice were then given intraplantar injection of 10 μL of 5% neutral-buffered formalin (NBF) (Fisher Scientific) in 0.9% saline (Millipore Sigma). Injections were performed under light restraint without anesthesia. Mice were immediately placed in iBOB and recorded for 45 min. Videos were scored by a blinded observer to quantify the total time spent licking and shaking the injected paw in 5 min intervals. The first phase of the response was defined as 0 – 5 min and the second phase as 15 – 35 min post formalin injection.
Carrageenan model
Carrageenan (Millipore Sigma) was dissolved in 0.9% saline (Millipore Sigma) to a 2% concentration and autoclaved. Mice were given intraplantar injection of 20 μL of 2% carrageenan and mechanical sensitivity thresholds were monitored as described above.
Spared Nerve Injury (SNI) model
Mice were anesthetized with 100 mg/kg ketamine (Patterson Veterinary) and 10 mg/kg xylazine (Patterson Veterinary) given via intraperitoneal injection. 1 mL of sterile saline (Patterson Veterinary) was injected subcutaneously between the shoulder blades, and petrolatum ophthalmic ointment (Patterson Veterinary) was applied. The surgical area was shaved and disinfected with betadine (Patterson Veterinary) and ethanol (VWR). The skin and muscle layer were opened and separated using blunt dissection to reveal the sciatic nerve. Under a stereomicroscope, the tibial and peroneal nerves were ligated with suture (Silk, 3–0, non-absorbable) and a short segment below the ligation was cut and removed. The muscle layer was closed and skin was sutured (Ethilon, 4–0, absorbable). Mice were placed on a heating pad to recover. Sham surgery was performed identically to the regular procedure except the sciatic nerve was exposed and the incision closed without carrying out the final lesion. Mechanical sensitivity thresholds were monitored as described above.
Wire hang test
Mice were placed on an elevated wire mesh and inverted for 5 min. The latency to fall were recorded. Each session consisted of 3 trials, of which the maximum value was used.
Rotarod
Motor function and coordination were measured using a Rotarod apparatus (Med Associates). Mice were habituated on 3 consecutive days at constant speed (5 rpm for 60 s per trial, 3 trials per day). Following the indicated treatment, mice were placed on an accelerating rotarod (4 to 40 rpm in 5 min) and the time until the first passive rotation or fall was recorded. Each measurement consisted of 3 trials which were performed 15 min apart and averaged.
Biotelemetry
Under inhalatory anesthesia (isoflurane 2%) and sterile conditions, mice received the implant HDX-11 (ADInstruments) under the lateral abdomen skin, with electrodes positioned for ECG measures in the chest. After 5 days of recovery, animals were followed by 24 hours for a baseline evaluation of heart rate (bpm) and body temperature (°C). Two days after (7 days after implant), animals received vehicle (aCSF) or ET (2 μg/5 μL), intrathecally, and followed by 24 hours. One hour after the end of measures, mice were treated with adrenaline (0.2 mg/kg, i.v.) and target parameters evaluated for 60 minutes. Data were collected and analyzed by LabChart software (ADInstruments).
Phrenic nerve-hemidiaphragm assay (PNHD)
The phrenic nerve-hemidiaphragm assay (PNHD) has been the gold standard for assessing the potency of BoNTs at the neuromuscular junction, but also the inhibitory effect of neutralizing antibodies62,63. It mimics the blocking of respiratory function as it happens in nature when BoNTs penetrate the body. For the preparation of the hemidiaphragm, after deep anesthesia, the left nerve-hemidiaphragm of male CD1 mice (Janvier Labs) was removed from the thoracic cage, after cutting the phrenic nerve above the heart atria and the diaphragm all around the thoracic cavity, while carefully saving the lowest ribs of the ribcage. After cleaning of the tissue and installation onto the holder via the lowest rib, the phrenic nerve was inserted into the stimulation rings of the holder, while a hook was secured into the ligament of the diaphragm and connected to the transducer. The muscles were suspended (1g passive tension) in organ baths containing 10 mL Krebs-Henseleit buffer (KHB) containing 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 11 mM glucose, pH 7.4) at 37°C and gassed with carbogen. The phrenic nerve was stimulated with 20 μsec pulses delivered at 1 Hz. Toxins were added to the bath at a final concentration of 10 to 100 pM. Experimental data were recorded with software IOX v2.9 from Emka Technologies. After application of the toxin into the bath, the contraction force of the organ was recorded every 30 seconds for 3 hours. The half paralysis time (t50, min), i.e. the time necessary to inhibit 50% of the initial contraction, was calculated as a measure for the potency of each toxin/concentration. Calculations were done by fitting a four-parameter dose-response logistic curve model (Y=Bottom + (Top-Bottom)/(1+10^((LogIC50-X) * Hill Slope)) onto the experimental data. Natural botulinum neurotoxins A from List Biologicals Laboratories, Campbell, CA (LIST/A) was used a positive control, and LHN/A as a negative control. PA was added when required at 10 nM working concentration. All toxins were tested to n=3 independent experiments.
Statistics and Reproducibility
Statistical analyses were performed using GraphPad Prism. For detailed statistical information, please see Supplementary Table 2. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications61,78 and standard practices in the field. Data distribution was assumed to be normal, but this was not formally tested. Instead, graphs display individual data points and s.e.m. No animals or data points were excluded from the analyses. Behavioral tests were performed by observers blinded to the treatment groups or genotypes. Treatment groups were randomized and evenly distributed across cages and sex. Image analyses were repeated independently with similar results across 3 mice (Fig. 1c, 1e, 5a and Supplementary Fig. 3), 3 donors (Fig. 1j), 4 mice (Fig. 4c) or 4 experiments (Fig. 4f and Extended Data Fig. 6e). Image analysis for Extended Data Fig. 7a was replicated over 4 experiments from one culture of differentiated neurons. Images in Fig. 1h were obtained from online databases at the Allen Mouse Brain Atlas (2004) (http://mouse.brain-map.org) and Allen Spinal Cord Atlas (2008) (http://mousespinal.brain-map.org). To the best of our knowledge, each were obtained from single mice using an ISH platform with validated reproducibility across ISH runs and independent riboprobe syntheses55.
Data availability
Transcriptional profiling data of the DRG after ET administration is available from the Gene Expression Omnibus (GEO) database under the accession code GSE184619. Microarray data of sorted mouse DRG neuron populations3 is available from the GEO database under the accession code GSE55114. Single-cell RNAseq data of the mouse nervous system4 is available from the Sequence Read Archive (SRA) under the accession code SRP135960 and at http://mousebrain.org. Single-cell RNAseq data of mouse DRG neurons across development5 is available from the GEO database under the accession code GSE139088. Microarray data from BioGPS.org55 is available at http://biogps.org. ISH data of mouse brain and spinal cord are available from the Allen Mouse Brain Atlas55 (2004) (http://mouse.brain-map.org) and Allen Spinal Cord Atlas (2008) (http://mousespinal.brain-map.org). Datasets generated during this study are presented in the figures and available from the corresponding author upon reasonable request. Source data are provided with this paper for Fig. 7d and Extended Data Fig. 2a–b.
Materials availability
The LFN-LC/AC699S construct is available from Ipsen and require a material transfer agreement.
Code availability
Custom code or algorithms were not used to generate results in this study. High content screening (HCS) analysis of DRG neurons were performed using the Cellomics software package as described in Methods. Analysis of DRG sections were performed using Fiji Image J plugins as described in Methods. Transcriptional profiling data of the DRG was analyzed using RStudio as described in Methods.
Extended Data
Extended Data Figure 1. Antxr2 but not Antxr1 is enriched in DRG sensory neurons.
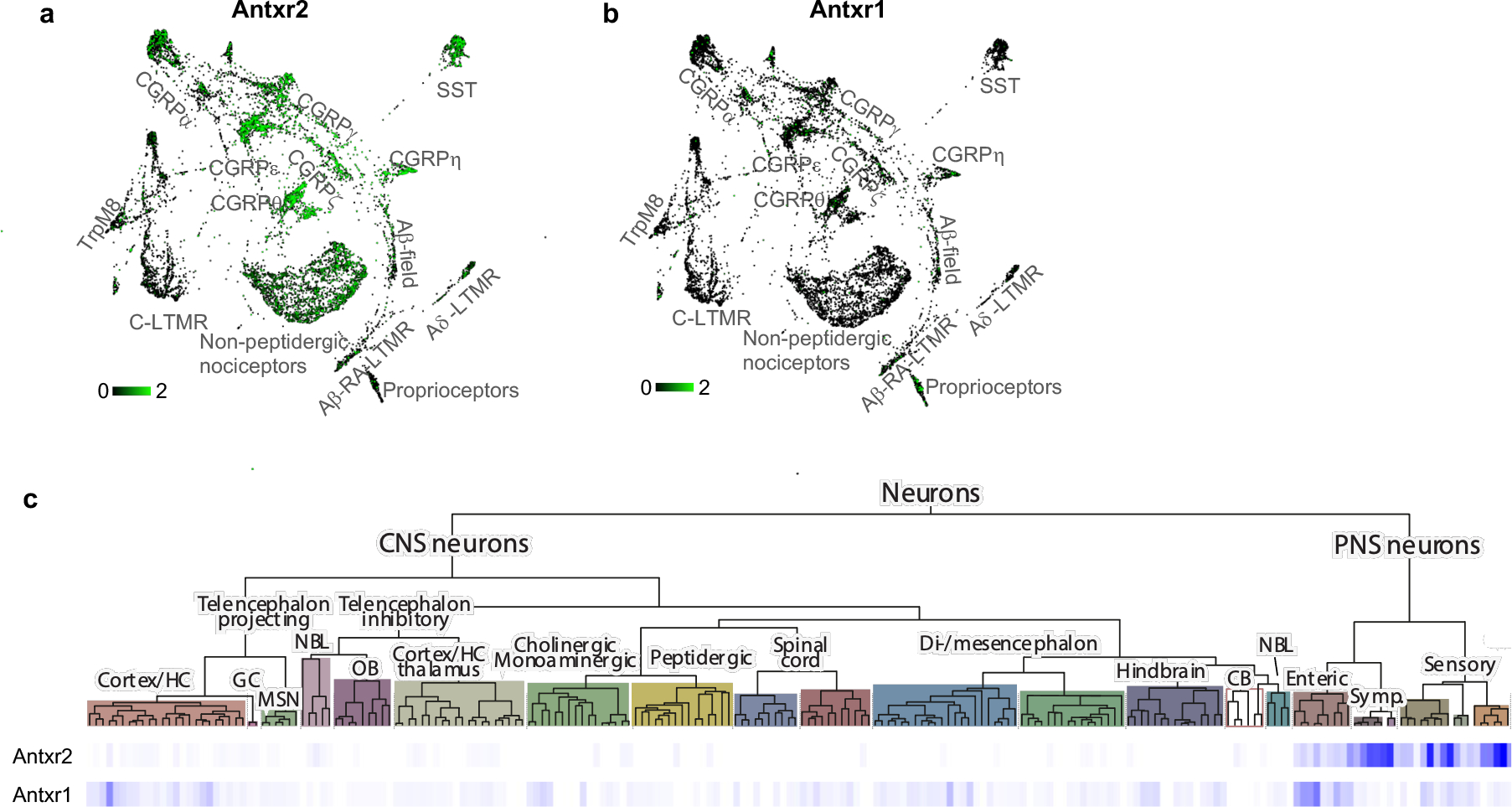
Extended Data Figure 2. Anthrax toxins modulate MAPK and cAMP signaling in DRG sensory neurons.
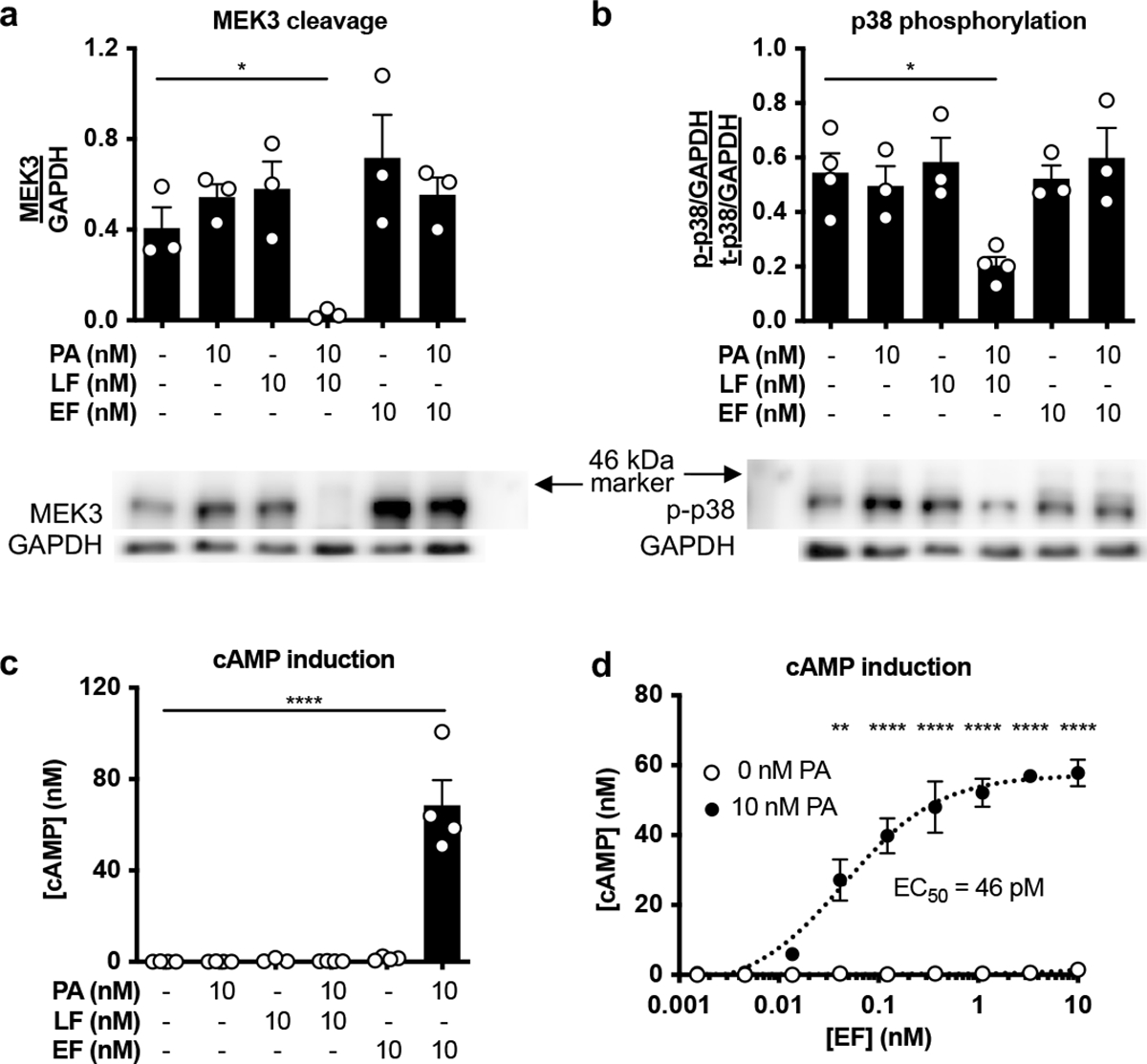
Extended Data Figure 3. ET-induced analgesia does not show significant sex-dependent effects.
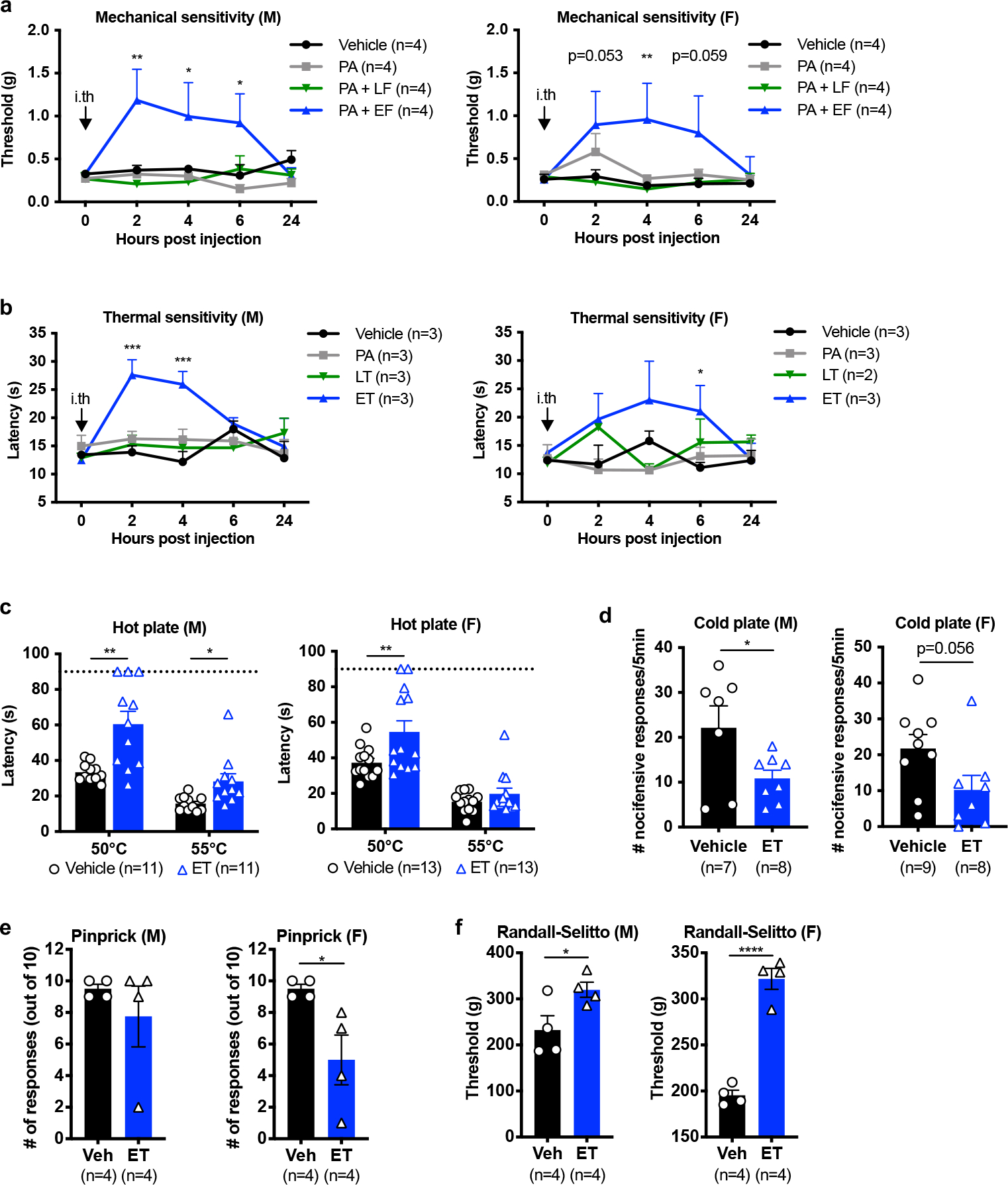
Extended Data Figure 4. ANTXR2 ablation from Nav1.8+ neurons attenuates cAMP signaling in DRG culture but causes minimal effects on baseline sensory function.
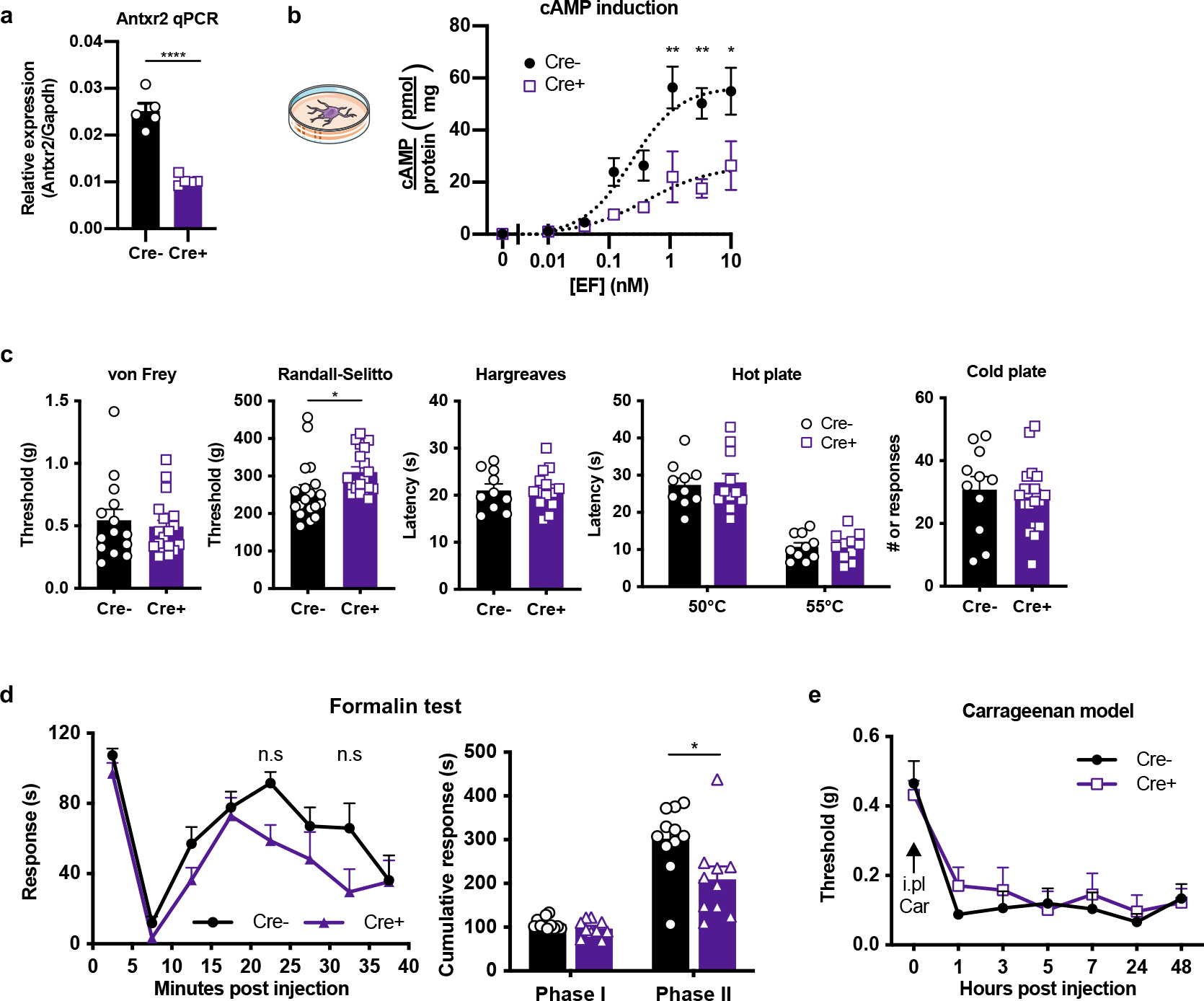
Extended Data Figure 5. Intraplantar administration of Edema Toxin induces mechanical allodynia and edema.
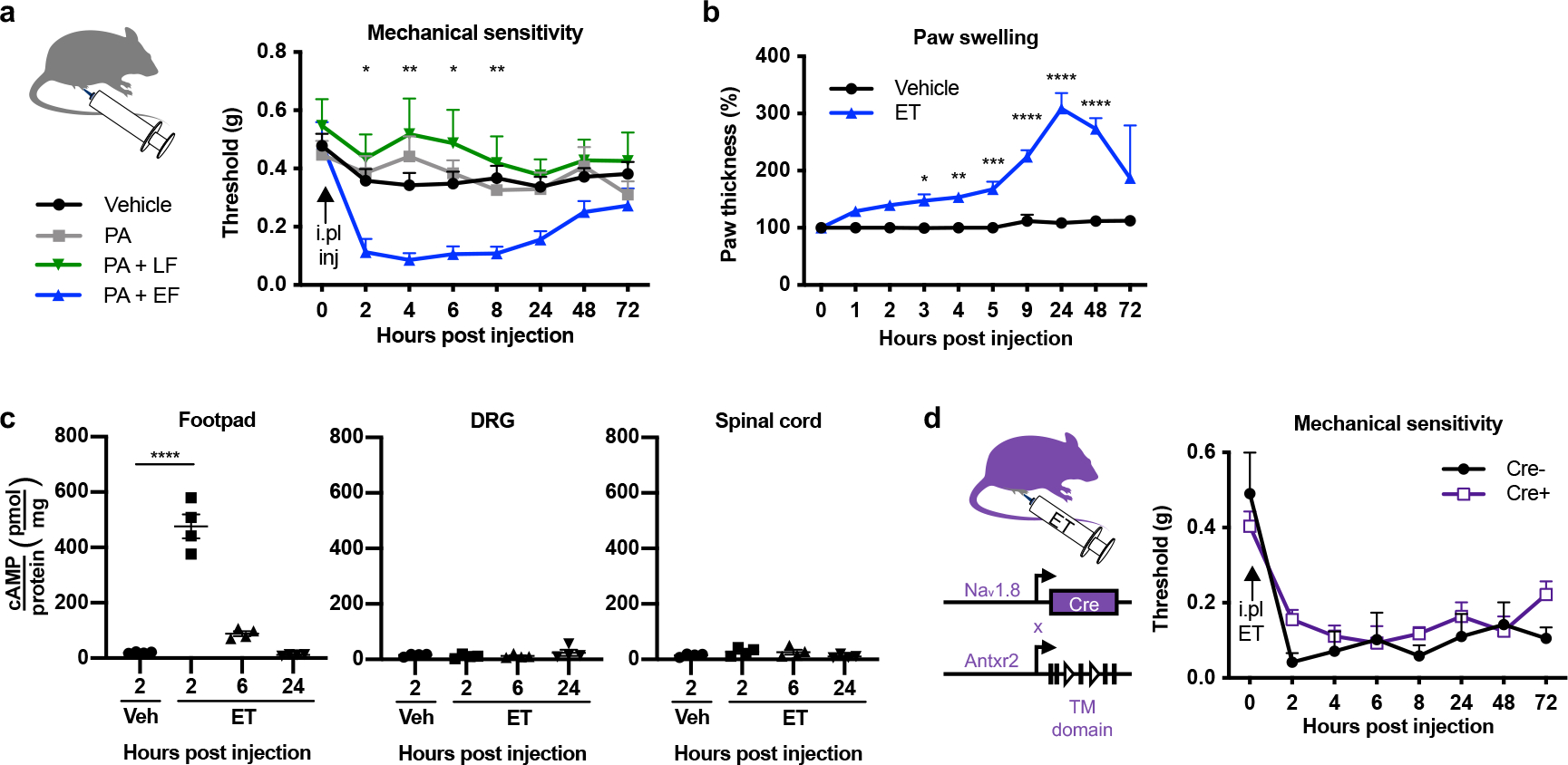
Extended Data Figure 6. Edema Toxin induces PKA signaling in DRG neurons but not non-neuronal cells.
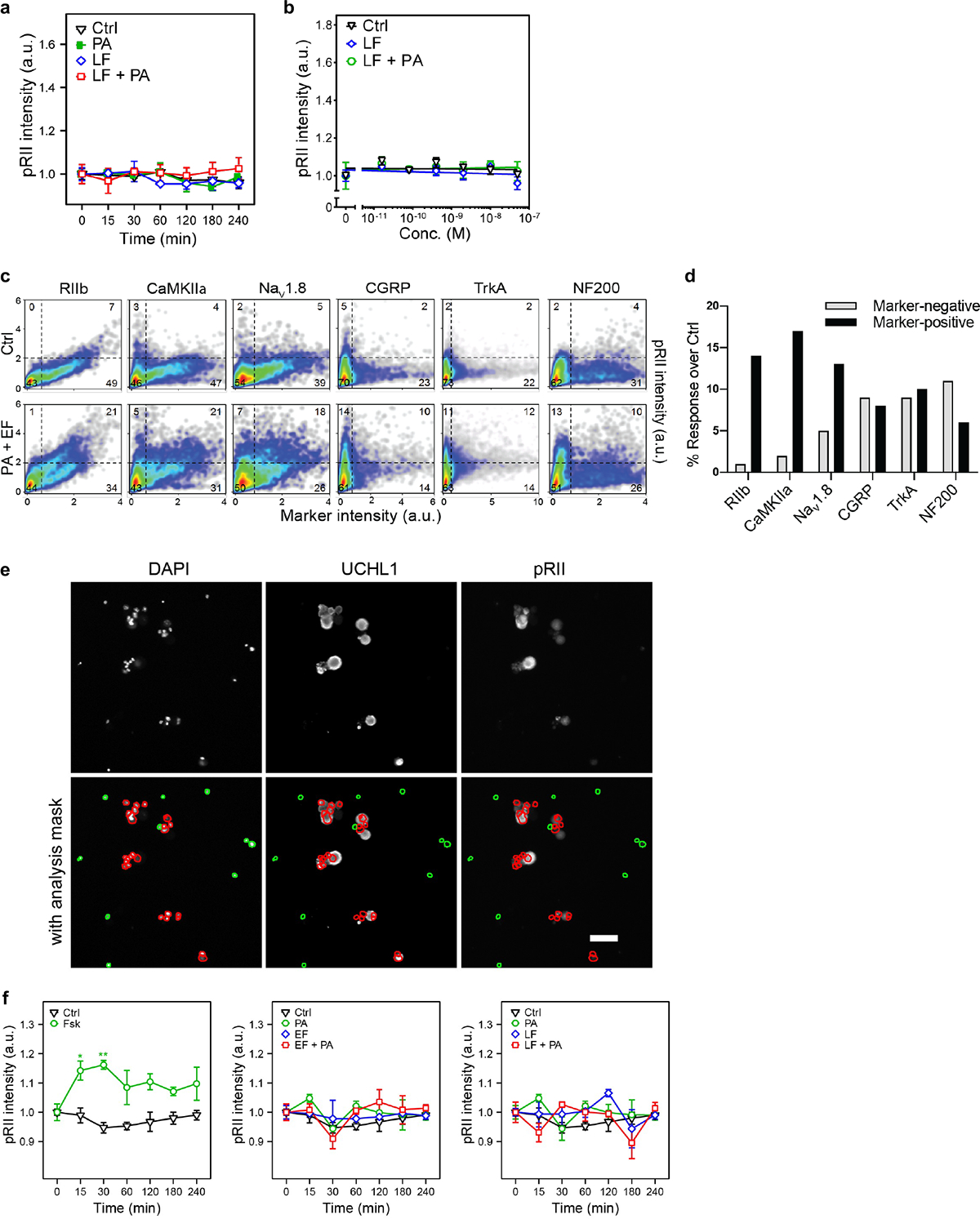
Extended Data Figure 7. Edema Toxin induces PKA signaling in human iPSC-derived sensory neurons.
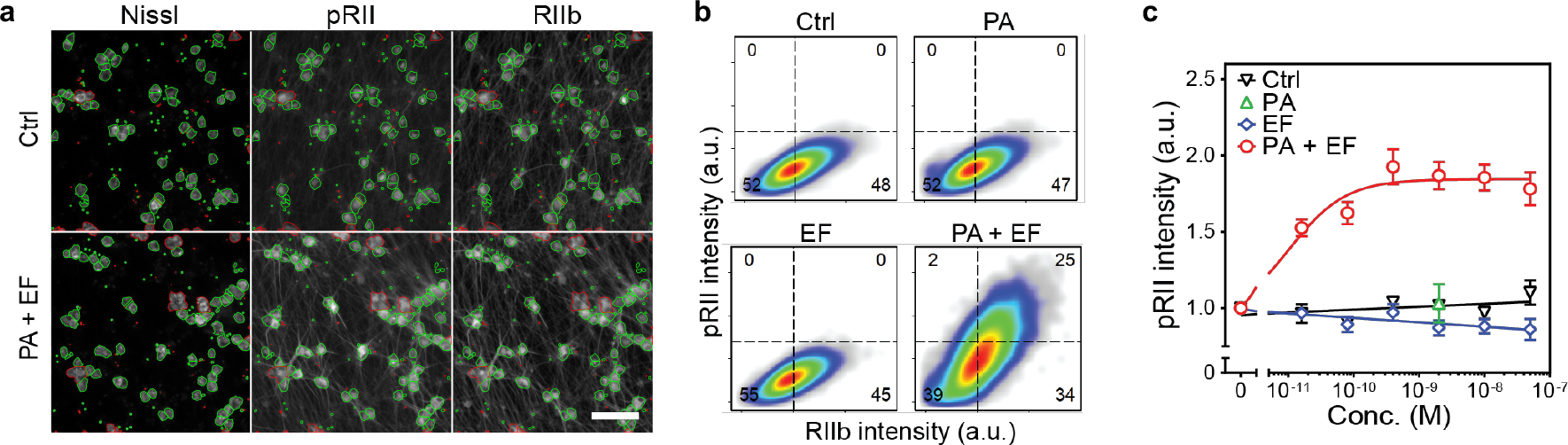
Extended Data Figure 8. Edema Toxin treatment enhances excitability of small-diameter DRG neurons.
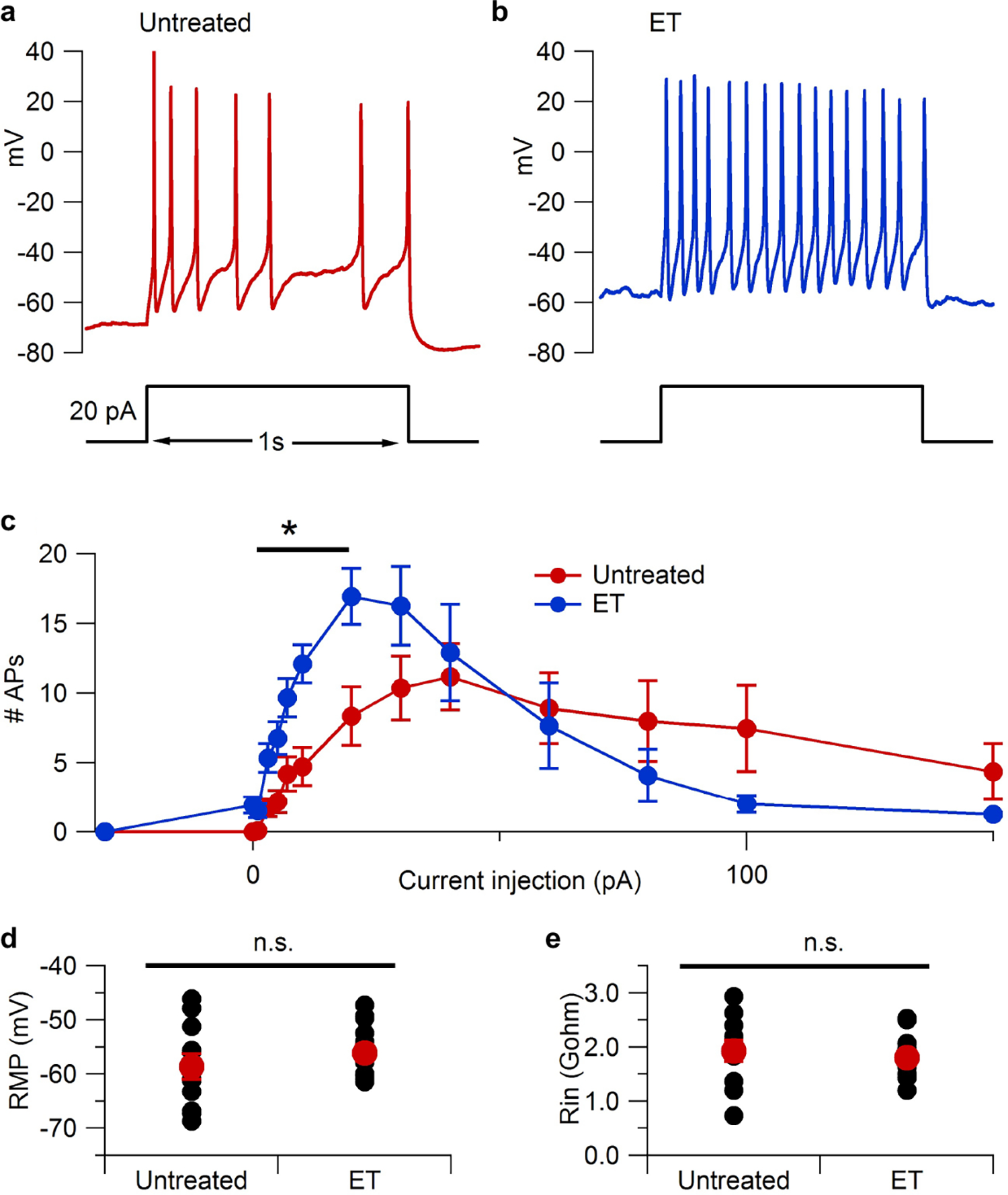
Extended Data Figure 9. Intrathecal administration of ET induces transcriptional changes in the DRG.
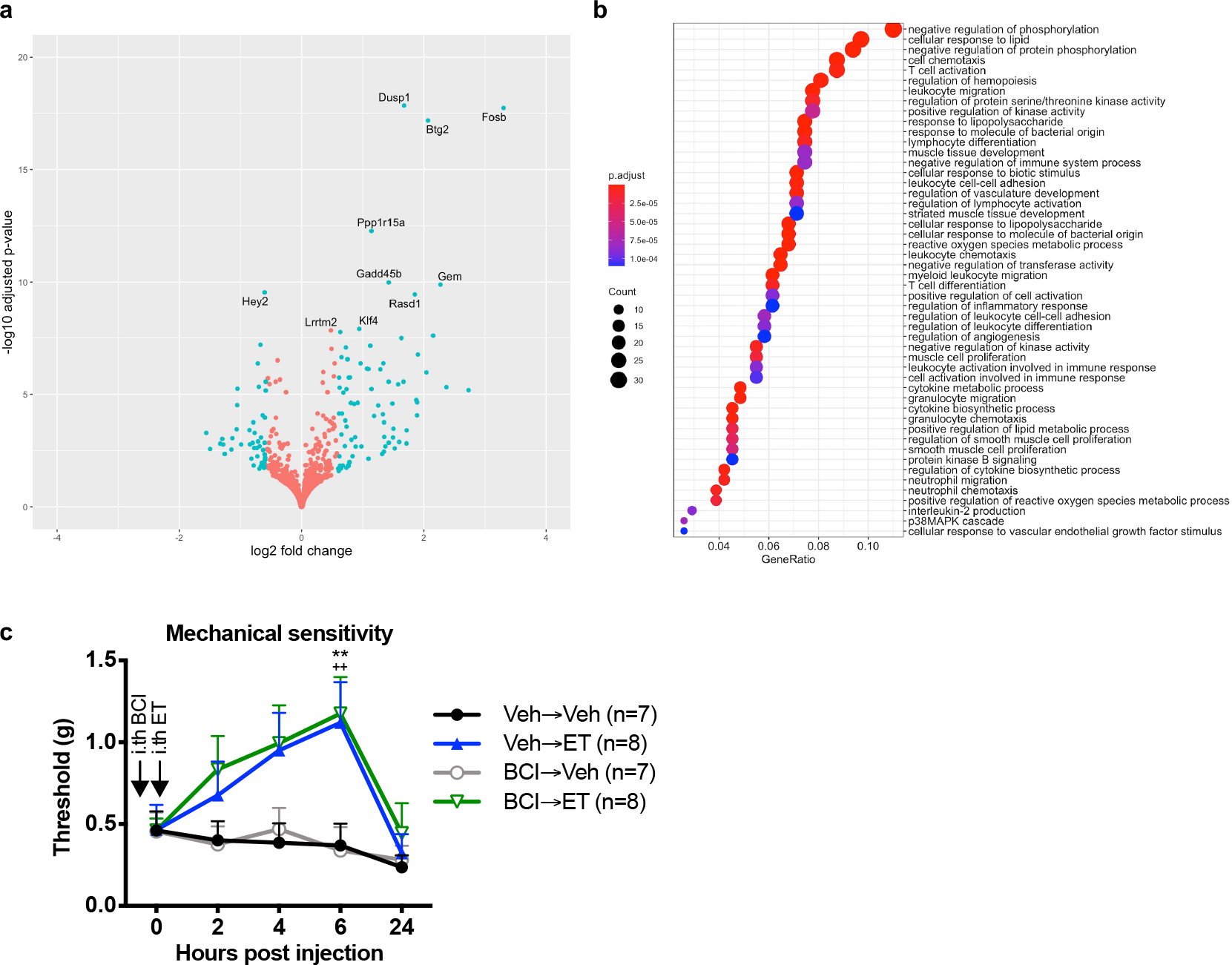
Extended Data Figure 10. ET-induced pain blockade does not show sex-dependent effects in models of pain.
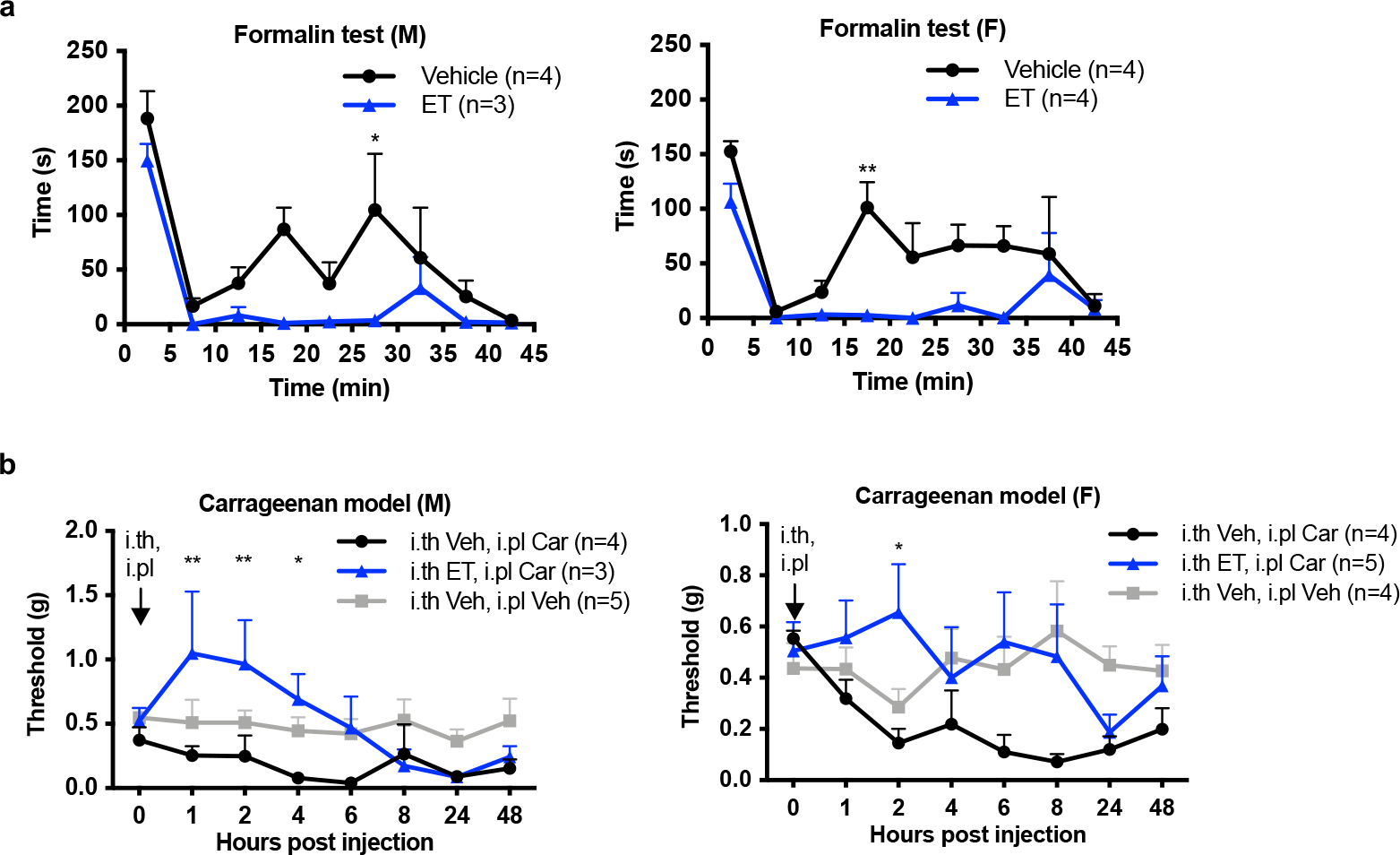
Supplementary Material
ACKNOWLEDGEMENTS
We thank Kaitlin Goldstein, Sebastien Sannajust, Michelle Heyang, Nicholas LoRocco, Ahmed Albakri for excellent technical assistance, and Tiphaine Voisin for helpful discussions. Research was supported by Burroughs Wellcome Fund, Chan-Zuckerberg Initiative, NIH DP2AT009499, R01AI130019 to I.M.C.; NIH NIA 5T32AG000222 fellowship to N.J.Y.; NIH NIGMS T32GM007753 fellowship to D.V.N.; the Intramural Program of the National Institute of Allergy and Infectious Diseases, NIH to S.H.L.; Internal funds from Department of Anesthesiology, Stony Brook Medicine to M.P.; NIH R01NS036855 to B.P.B.; the European Regional Development Fund (NeuRoWeg; EFRE-0800407 and EFRE-0800408) and the Innovative Medicines Initiative 2 Joint Undertaking (116072-NGN-PET) to O.B.; NIH NINDS NS111929 to T.J.P; and the São Paulo Research Foundation (FAPESP) (2013/08216-2 (Center for Research in Inflammatory Diseases - CRID)) to T.M.C; Deutsche Forschungsgemeinschaft (271522021 and 413120531), EFRE-0800384, and Leitmarktagentur.NRW LS-1-1-020d to T.H. This study was supported in part by a sponsored research grant from Ipsen Pharmaceuticals to I.M.C.
Footnotes
COMPETING INTERESTS
S.M.L., S.P., S.M., J.M., V.T. and K.A.F. are employees of Ipsen. I.M.C. has received sponsored research support from Ipsen, GSK and Allergan Pharmaceuticals, and is a member of scientific advisory boards for GSK and Kintai Pharmaceuticals. This work is related to patent applications PCT/US16/49099 and PCT/US16/49106, “Compositions and methods for treatment of pain,” of which R.J.C., I.M.C., B.L.P., K.A.F., S.P. and S.M.L. are co-inventors. O.B. is a co-founder and shareholder of LIFE & BRAIN GmbH. The remaining authors declare no competing interests.
REFERENCES
- 1.Basbaum AI, Bautista DM, Scherrer G & Julius D Cellular and Molecular Mechanisms of Pain. Cell 139, 267–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng L & Chiu IM Microbes and pain. PLoS Pathog. 17, e1009398 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu IM et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. eLife 3, e04660 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeisel A et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma N et al. The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young JAT & Collier RJ Anthrax Toxin: Receptor Binding, Internalization, Pore Formation, and Translocation. Annu. Rev. Biochem. 76, 243–265 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Duesbery NS et al. Proteolytic Inactivation of MAP-Kinase-Kinase by Anthrax Lethal Factor. Science 280, 734–737 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Hellmich KA et al. Anthrax Lethal Factor Cleaves Mouse Nlrp1b in Both Toxin-Sensitive and Toxin-Resistant Macrophages. PLoS ONE 7, e49741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendenhall MA et al. Anthrax lethal factor cleaves regulatory subunits of phosphoinositide-3 kinase to contribute to toxin lethality. Nat. Microbiol. 5, 1464–1471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leppla SH Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. 79, 3162–3166 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley KA, Mogridge J, Mourez M, Collier RJ & Young JAT Identification of the cellular receptor for anthrax toxin. Nature 414, 225–229 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Scobie HM, Rainey GJA, Bradley KA & Young JAT Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. 100, 5170–5174 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wigelsworth DJ et al. Binding Stoichiometry and Kinetics of the Interaction of a Human Anthrax Toxin Receptor, CMG2, with Protective Antigen. J. Biol. Chem. 279, 23349–23356 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Liu S et al. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. 106, 12424–12429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachran C & Leppla SH Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao X, Rabideau AE & Pentelute BL Delivery of Antibody Mimics into Mammalian Cells via Anthrax Toxin Protective Antigen. ChemBioChem 15, 2458–2466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabideau AE, Liao X, Akçay G & Pentelute BL Translocation of Non-Canonical Polypeptides into Cells Using Protective Antigen. Sci. Rep. 5, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dyer PDR et al. Disarmed anthrax toxin delivers antisense oligonucleotides and siRNA with high efficiency and low toxicity. J. Controlled Release 220, Part A, 316–328 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Maldonado-Arocho FJ, Fulcher JA, Lee B & Bradley KA Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol. Microbiol. 61, 324–337 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Liu S et al. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 501, 63–68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bürgi J et al. CMG2/ANTXR2 regulates extracellular collagen VI which accumulates in hyaline fibromatosis syndrome. Nat. Commun. 8, 15861 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau J et al. Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible Advillin-Cre-ERT2 recombinase mouse. Mol. Pain 7, 100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aley KO & Levine JD Role of Protein Kinase A in the Maintenance of Inflammatory Pain. J. Neurosci. 19, 2181–2186 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isensee J et al. Pain modulators regulate the dynamics of PKA-RII phosphorylation in subgroups of sensory neurons. J. Cell Sci. 127, 216–229 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Isensee J et al. PKA-RII subunit phosphorylation precedes activation by cAMP and regulates activity termination. J. Cell Biol. 217, 2167–2184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambers SM et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 30, 715–720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Firoved AM et al. Bacillus anthracis Edema Toxin Causes Extensive Tissue Lesions and Rapid Lethality in Mice. Am. J. Pathol. 167, 1309–1320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzgerald EM, Okuse K, Wood JN, Dolphin AC & Moss SJ cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J. Physiol. 516, 433–446 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emery EC, Young GT, Berrocoso EM, Chen L & McNaughton PA HCN2 Ion Channels Play a Central Role in Inflammatory and Neuropathic Pain. Science 333, 1462–1466 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Nuwer MO, Picchione KE & Bhattacharjee A PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J. Neurosci. Off. J. Soc. Neurosci. 30, 14165–14172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis JW, Cannon JT & Liebeskind JC Opioid and nonopioid mechanisms of stress analgesia. Science 208, 623–625 (1980). [DOI] [PubMed] [Google Scholar]
- 32.Hohmann AG et al. An endocannabinoid mechanism for stress-induced analgesia. Nature 435, 1108–1112 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Brundege JM, Diao L, Proctor WR & Dunwiddie TV The Role of Cyclic AMP as a Precursor of Extracellular Adenosine in the Rat Hippocampus. Neuropharmacology 36, 1201–1210 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Post C Antinociceptive effects in mice after intrathecal injection of 5′-N-ethylcarboxamide adenosine. Neurosci. Lett. 51, 325–330 (1984). [DOI] [PubMed] [Google Scholar]
- 35.Ndong C, Landry RP, DeLeo JA & Romero-Sandoval EA Mitogen activated protein kinase phosphatase-1 prevents the development of tactile sensitivity in a rodent model of neuropathic pain. Mol. Pain 8, 34 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji R-R, Baba H, Brenner GJ & Woolf CJ Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci. 2, 1114–1119 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Yang K & Li Y-Q Origins of spontaneous and noxious stimuli-evoked miniature EPSCs in substantia gelatinosa. NeuroReport 12, 39–42 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Arora N & Leppla SH Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun. 62, 4955–4961 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sluka KA Stimulation of Deep Somatic Tissue with Capsaicin Produces Long-Lasting Mechanical Allodynia and Heat Hypoalgesia that Depends on Early Activation of the cAMP Pathway. J. Neurosci. 22, 5687–5693 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H-B et al. cAMP-dependent protein kinase activated Fyn in spinal dorsal horn to regulate NMDA receptor function during inflammatory pain. J. Neurochem. 116, 93–104 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Zhukovskaya NL, Guo Q, Florián J & Tang W-J Calcium-independent calmodulin binding and two-metal–ion catalytic mechanism of anthrax edema factor. EMBO J. 24, 929–941 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dal Molin F et al. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J. 25, 5405–5413 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Witschi R et al. Presynaptic alpha2-GABAA receptors in primary afferent depolarization and spinal pain control. J. Neurosci. Off. J. Soc. Neurosci. 31, 8134–8142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willis WD Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp. Brain Res. 124, 395–421 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Celotto L, Eroli F, Nistri A & Vilotti S Long-term application of cannabinoids leads to dissociation between changes in cAMP and modulation of GABAA receptors of mouse trigeminal sensory neurons. Neurochem. Int. 126, 74–85 (2019). [DOI] [PubMed] [Google Scholar]
- 46.England S, Bevan S & Docherty RJ PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J. Physiol. 495 (Pt 2), 429–440 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gold MS, Levine JD & Correa AM Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J. Neurosci. Off. J. Soc. Neurosci. 18, 10345–10355 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebrahimi CM, Sheen TR, Renken CW, Gottlieb RA & Doran KS Contribution of Lethal Toxin and Edema Toxin to the Pathogenesis of Anthrax Meningitis. Infect. Immun. 79, 2510–2518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidtko A, Lötsch J, Freynhagen R & Geisslinger G Ziconotide for treatment of severe chronic pain. The Lancet 375, 1569–1577 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Maiarù M et al. Selective neuronal silencing using synthetic botulinum molecules alleviates chronic pain in mice. Sci. Transl. Med. 10, (2018). [DOI] [PubMed] [Google Scholar]
- 51.Verdurmen WPR, Luginbühl M, Honegger A & Plückthun A Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J. Controlled Release 200, 13–22 (2015). [DOI] [PubMed] [Google Scholar]
REFERENCES FOR METHODS
- 52.Wu C, Macleod I & Su AI BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 41, D561–565 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su AI et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. 101, 6062–6067 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lattin JE et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 4, 5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lein ES et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Abrahamsen B et al. The Cell and Molecular Basis of Mechanical, Cold, and Inflammatory Pain. Science 321, 702–705 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Leysath CE et al. Anthrax Edema Factor Toxicity Is Strongly Mediated by the N-end Rule. PLoS ONE 8, e74474 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park S & Leppla SH Optimized Production and Purification of Bacillus anthracis Lethal Factor. Protein Expr. Purif. 18, 293–302 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Pomerantsev AP et al. A Bacillus anthracis strain deleted for six proteases serves as an effective host for production of recombinant proteins. Protein Expr. Purif. 80, 80–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tao L et al. Engineered botulinum neurotoxin B with improved efficacy for targeting human receptors. Nat. Commun. 8, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinho-Ribeiro FA et al. Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell (2018) doi: 10.1016/j.cell.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simpson LL Studies on the binding of botulinum toxin type A to the rat phrenic nerve-hemidiaphragm preparation. Neuropharmacology 13, 683–691 (1974). [DOI] [PubMed] [Google Scholar]
- 63.Pellett S Progress in cell based assays for botulinum neurotoxin detection. Curr. Top. Microbiol. Immunol. 364, 257–285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chaplan SR, Bach FW, Pogrel JW, Chung JM & Yaksh TL Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 (1994). [DOI] [PubMed] [Google Scholar]
- 65.Neher E Correction for liquid junction potentials in patch clamp experiments. in Methods in Enzymology vol. 207 123–131 (Academic Press, 1992). [DOI] [PubMed] [Google Scholar]
- 66.Lu Y et al. Presynaptic Inhibition of Primary Nociceptive Signals to Dorsal Horn Lamina I Neurons by Dopamine. J. Neurosci. Off. J. Soc. Neurosci. 38, 8809–8821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Safronov BV, Pinto V & Derkach VA High-resolution single-cell imaging for functional studies in the whole brain and spinal cord and thick tissue blocks using light-emitting diode illumination. J. Neurosci. Methods 164, 292–298 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Szucs P, Pinto V & Safronov BV Advanced technique of infrared LED imaging of unstained cells and intracellular structures in isolated spinal cord, brainstem, ganglia and cerebellum. J. Neurosci. Methods 177, 369–380 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Li J, Kritzer E, Ford NC, Arbabi S & Baccei ML Connectivity of pacemaker neurons in the neonatal rat superficial dorsal horn. J. Comp. Neurol. 523, 1038–1053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ikeda H, Heinke B, Ruscheweyh R & Sandkühler J Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 299, 1237–1240 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Al Ghamdi KS, Polgár E & Todd AJ Soma size distinguishes projection neurons from neurokinin 1 receptor-expressing interneurons in lamina I of the rat lumbar spinal dorsal horn. Neuroscience 164, 1794–1804 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dickie AC, McCormick B, Lukito V, Wilson KL & Torsney C Inflammatory Pain Reduces C Fiber Activity-Dependent Slowing in a Sex-Dependent Manner, Amplifying Nociceptive Input to the Spinal Cord. J. Neurosci. Off. J. Soc. Neurosci. 37, 6488–6502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Betelli C, MacDermott AB & Bardoni R Transient, activity dependent inhibition of transmitter release from low threshold afferents mediated by GABAA receptors in spinal cord lamina III/IV. Mol. Pain 11, 64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J, Kritzer E, Craig PE & Baccei ML Aberrant synaptic integration in adult lamina I projection neurons following neonatal tissue damage. J. Neurosci. Off. J. Soc. Neurosci. 35, 2438–2451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nakatsuka T, Ataka T, Kumamoto E, Tamaki T & Yoshimura M Alteration in synaptic inputs through C-afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience 99, 549–556 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Torsney C & MacDermott AB Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J. Neurosci. Off. J. Soc. Neurosci. 26, 1833–1843 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clark AK et al. Selective activation of microglia facilitates synaptic strength. J. Neurosci. Off. J. Soc. Neurosci. 35, 4552–4570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chiu IM et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptional profiling data of the DRG after ET administration is available from the Gene Expression Omnibus (GEO) database under the accession code GSE184619. Microarray data of sorted mouse DRG neuron populations3 is available from the GEO database under the accession code GSE55114. Single-cell RNAseq data of the mouse nervous system4 is available from the Sequence Read Archive (SRA) under the accession code SRP135960 and at http://mousebrain.org. Single-cell RNAseq data of mouse DRG neurons across development5 is available from the GEO database under the accession code GSE139088. Microarray data from BioGPS.org55 is available at http://biogps.org. ISH data of mouse brain and spinal cord are available from the Allen Mouse Brain Atlas55 (2004) (http://mouse.brain-map.org) and Allen Spinal Cord Atlas (2008) (http://mousespinal.brain-map.org). Datasets generated during this study are presented in the figures and available from the corresponding author upon reasonable request. Source data are provided with this paper for Fig. 7d and Extended Data Fig. 2a–b.