

Summary
Here, we describe a protocol to culture Drosophila S2R+ cells and to extract plasmatocytes from adult flies. The modified seeding approach detailed here, in combination with coating of coverslips with concanvalin A, enables enhanced adhesion and spreading of cells. We describe the steps for confocal microscopy and a detailed quantification pipeline to evaluate changes in cortical actin cytoskeleton dynamics. The protocol can be applied to a variety of genetic or chemical perturbations.
For complete details on the use and execution of this protocol, please refer to Nath et al. (2022).
Subject areas: Cell Biology, Genetics, Immunology, Microscopy, Model Organisms
Graphical abstract
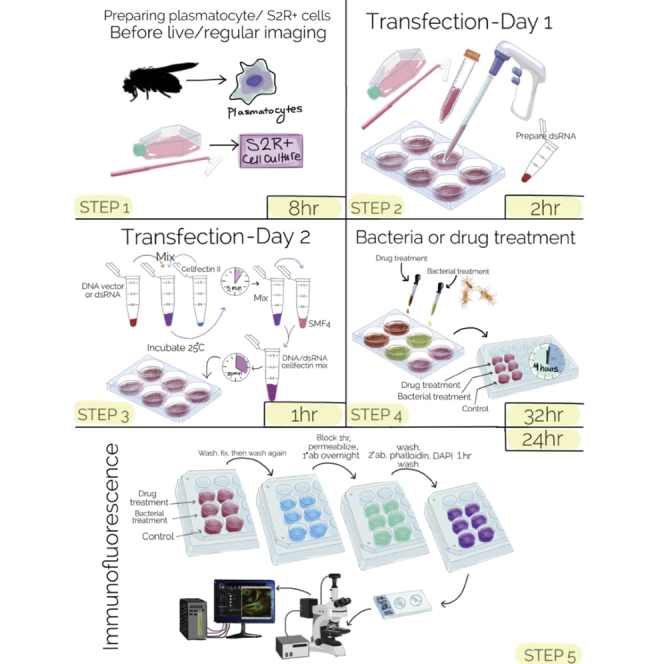
Highlights
-
•
Culturing and seeding conditions for optimal spreading of Drosophila S2R+ cells
-
•
Tools for effective genetic manipulations of Drosophila S2R+ cells
-
•
Extraction, culture, and seeding of plasmatocytes from adult Drosophila
-
•
Confocal imaging and analyses to quantify cortical cytoskeleton remodeling
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we describe a protocol to culture Drosophila S2R+ cells and to extract plasmatocytes from adult flies. The modified seeding approach detailed here, in combination with coating of coverslips with concanvalin A, enables enhanced adhesion and spreading of cells. We describe the steps for confocal microscopy and a detailed quantification pipeline to evaluate changes in cortical actin cytoskeleton dynamics. The protocol can be applied to a variety of genetic or chemical perturbations.
Before you begin
Coverslip preparation for cell seeding and immunofluorescence imaging
-
1.
Wash 12 mm diameter round glass coverslips (#2603 Ted Pella) with distilled water.
-
2.
Dry the coverslips using clean delicate task wipers (Kimtech).
-
3.
Dissolve 0.5 mg of concanavalin A (MP Biomedicals #195283) in 1 mL of distilled sterile water to prepare the concanavalin A working solution. Store at 4° for one year.
-
4.
Coat glass coverslips with 20 μL concanavalin A by submerging each coverslip in concanavalin A solution for 30 min.
-
5.
Rinse the coverslips with distilled sterile water.
-
6.
Allow to dry for 16 h before seeding cells.
CRITICAL: Coverslips need to be thoroughly dried for optimal performance. If not dry, cells may adhere unevenly to the coverslip and thus become difficult to image.
S2R+ cell culture
Note: S2R+ cell culture methods described in this protocol are adapted from Rogers and Rogers (2008).
-
7.
Thaw cell stocks by immersing the cell-containing vial in a 37°C water bath for 1 min.
-
8.
Dispense cell suspension into 10 mL room-temperature SFM4 Insect Cell Culture Medium supplemented with 50 U penicillin/mL and 50 μg streptomycin sulfate/mL in a T25 flask with a sterile pipette. Work in a level II sterile cabinet using sterile technique.
-
9.
Allow cells to attach to cell culture flasks for 1 h.
-
10.
Remove media and replace with fresh 10 mL SFM4 medium supplemented with antibiotics to remove residual chemicals from cryopreservation.
-
11.
Allow cells to grow and establish a culture for 5–6d at 25°C. Split cells and continue with routine passage.
-
12.
The day prior to seeding cells for treatment and/or immunofluorescence imaging, ensure plates are not confluent by splitting them 1:2. On the day of treatment seed maximum 2,000,000 cells per well in a 6-well plate or 100,000 cells in a 24-well plate.
Note: Cell growth and treatments are both performed at 25°C in a sterile cell culture incubator.
Note: We use SFM4 Insect Cell Culture Medium for all experiments. This is a defined medium that allows the cells to grow in absence of Fetal Bovine Serum (FBS). This growth media is essential to contribute to cell adherence and eliminate variability in the culture conditions that might occur when FBS is used.
Preparation of recombinant DNA or double-stranded RNA
-
13.
To perform ectopic expression of protein of interest, obtain desired DNA recombinant constructs using S2R+-appropriate destination vector. In previous work, we have successfully used the pAW series Drosophila gateway expression vectors for protein expression (Nath et al., 2022) and pAc5-sgRNA-Cas9-puro to perform Crispr/Cas9-mediated mutagenesis (Bassett et al., 2014).
Note: To obtain high transfection efficiency, use freshly extracted recombinant DNA. Use of one year old DNA samples might affect cell viability or transfection efficiency. Use an endotoxin-free Midi prep or Maxi prep extraction kit, if possible. We recommend QIAGEN Midi or Maxi prep endo-free kits.
-
14.To perform RNA interference (RNAi)-mediated depletion of a desired gene, prepare dsRNA-specific to the target gene.
-
a.Extract genomic DNA from a wild-type Drosophila strain or from S2 R+ cell pellet, using the Tissue and Blood DNA extraction kit from QIAGEN.
-
b.Using the extracted genomic DNA as template, prepare a PCR reaction to amplify the region of the gene that transcribe the mRNA sequence to target by dsRNAi. For this reaction, use gene specific primers with 5′ anchoring sequence (see key resources table reports the anchoring sequence to insert at the 5′ of each gene specific primer).
-
c.Assemble the reaction on ice in a 0.2 μL PCR tube.
Reagents Volume Primer Mix (1 μΜ each) 10 μL Template genomic DNA (10 ng/ μL) 2 μL 10× Taq PCR Buffer (NEB) 5 μL dNTP mix (25 mM each) 0.5 μL Taq Polymerase (NEB) 0.5 μL Molecular grade sterile water 32 μL Total 50 μL -
d.and run the reaction following the cycle conditions.
Step Temperature Time 1 95°C 3 min 2 95°C 30 s 3 50°C 45 s 4 72°C 1 min 5 Go to step 2, 39× more times 6 72°C 10 min 7 Pause at 4°C -
e.When the PCR run is complete, analyze the PCR product on a 1% agarose gel.
-
f.If the product is of the predicted size, use 10 μL of the amplified product to assemble PCR #2 (see table below) to amplify the anchor-tagged genomic PCR product with the T7 promoter containing primer (see key resources table).
-
g.Assemble the reaction on ice in a 0.2 μL PCR tube.
Reagents Volume PCR #1 product 10 μL T7 linker primer (10 μΜ) 2 μL dNTP (25 mM each) 0.5 μL 10× Taq PCR Buffer (NEB) 5 μL Taq 0.5 μL Molecular grade sterile water 32 μL Total 50 μL -
h.and run the reaction following the cycle conditions.
Step Temperature Time 1 95°C 3 min 2 95°C 30 s 3 42°C 30 s 4 72°C 1 min 5 Go to step 2, 4× more times 6 95°C 30 s 7 60°C 30 s 8 72°C 1 min 9 Go to step 6, 29× more times 10 72°C 10 min 11 Pause at 4°C -
i.When the run is complete, analyze the product on a 1% agarose gel.
-
j.If the product is of the expected size, use 10 μL of the amplified product as template to synthesize dsRNA.
Reagents Volume PCR #2 product 10 μL 10× Reaction 2 μL ATP (100 mM) 2 μL GTP (100 mM) 2 μL UTP (100 mM) 2 μL CTP (100 mM) 2 μL T7 RNA polymerase Mix 2 μL Total 20 μL -
k.following the cycling reported in the table below.
Step Temperature Time 1 25°C 5 min 2 37°C 7 h 3 50°C 45 s 4 85°C 5 min 5 Cool the reaction at 20°C 3 h -
l.When the run is completed, dilute the dsRNA 1:5 in deionized water and run it on a 1% agarose gel. Quantify the concentration of 2 μL of dsRNA using a Nanodrop spectrophotometer.Note: An efficient reaction should yield around 5 μg/μL of dsRNA.
-
m.Store the dsRNA at -80°C until it is needed.Note: For efficient gene silencing use freshly prepared dsRNA, not older than 20 days.
-
a.
Fly stocks and husbandry
-
15.
Transgenic Drosophila fly lines for this protocol were raised on standard Bloomington Drosophila Stock Center (BDSC) cornmeal medium supplemented with soy powder in insect growth chamber at 25°C. Flies were transferred to fresh vials every 2 days prior to plasmatocyte extraction.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 594 phalloidin | Invitrogen | A12381 |
| Bacterial and virus strains | ||
| Ecc15 (2141) | Dr. Edan Foley, University of Alberta | Basset et al., 2000 |
| Chemicals, peptides, and recombinant proteins | ||
| SFM4 Insect Cell Culture Medium | Hyclone | SH30913.02 |
| penicillin/ streptomycin sulfate 10× solution | Thermo Fisher Scientific | 15140122 |
| 4,6-diamidine-2-phenylindole (DAPI) | Invitrogen | D1306 |
| Pro-Gold Antifade Reagent | Invitrogen | P36930 |
| Concanavalin A | MP Biomedicals | CAT 195283 |
| Trypan Blue stain (0.4%) | Gibco | REF 15250-061 |
| Calcium–magnesium-free Phosphate Buffer Saline (PBS) | Corning | 20-031-CV |
| Normal goat serum | Sigma-Aldrich | NS02L |
| Triton X-100 | Sigma-Aldrich | CAT 9002-93-1 |
| 16% Formaldehyde | Thermo Scientific | 28906 |
| cOmpleteTM Protease Inhibitor Cocktail | Roche | 04693116001 |
| Cellfectin II reagent | Invitrogen | 10362-100 |
| Tissue and Blood DNA extraction kit | QIAGEN | 69504 |
| Experimental models: Cell lines | ||
| Drosophila melanogaster S2R+ | DGRC S2R+ (https://dgrc.bio.indiana.edu//stock/150; RRID:CVCL_Z831) | Stock Number: 150 |
| Experimental models: Organisms/strains | ||
| Drosophila melanogaster | Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/Home/Search) | #30142 |
| Oligonucleotides | ||
| Gene-specific primer with anchor for PCR#1 | 5′-GGGCGGGT (followed by gene specific sequence) | 5′-GGGCGGGT (followed by gene specific sequence) |
| T7 primer | 5′ TAATACGACTCACTATA GGGAGACCACGGGCGGGT |
5′ TAATACGACTCACTATA GGGAGACCACGGGCGGGT |
| Software and algorithms | ||
| FIJI/ImageJ software | Schindelin et al., 2012 | https://imagej.nih.gov/ij/ |
| Icy Bioimage | BioImage Analysis Lab at Institut Pasteur | http://icy.bioimageanalysis.org |
| Zeiss Zen lite Black & lite Blue | ZEISS | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html?vaURL=www.zeiss.com/zen-lite |
| Other | ||
| 12 mm diameter round glass coverslips | Ted Pella | #2603 |
| incubator for Drosophila culture | DigiTherm Tritech Research | DT2-MP-47 |
| Bench-top centrifuge | Eppendorf | 5427 R |
| Bench-top centrifuge | Eppendorf | 5810R |
| Thermocycler | Applied Biosystems | SimplyAMP |
| 24-well tissue culture-treated plates | Falcon | 38021 |
| 6-well tissue culture-treated plates | Falcon | 353046 |
| Surgical forceps | Fine Science Tools, Dumont | #55 |
| Pyrex dissecting dishes | Corning | 7220-85 |
| 8-chamber cover glass | Nunc | 155411 |
| Confocal microscope | ZEISS | LSM 880 with Airy scan |
| Stereomicroscope | ZEISS | STEMI |
| glass beads | Sigma-Aldrich | G8772 |
Materials and equipment
Fixing solution
| 4% formaldehyde | Final concentration | Amount |
|---|---|---|
| 16% Methanol-free Formaldehyde | 4% | 1 mL |
| Calcium–magnesium-free Phosphate Buffer Saline (1×) | 0.75× | 3 mL |
| Total | N/A | 4 mL |
Store at room temperature for up to one year.
PBST-working solution
| PBST 0.1% | Final concentration | Amount |
|---|---|---|
| Calcium–magnesium-free Phosphate Buffer Saline (10×) | 1× | 10 mL |
| Deionized sterile Water | – | 89.9 mL |
| Triton-X | 0.1% | 0.1 mL |
| Total | N/A | 100 mL |
Store at room temperature for up to one year.
Blocking solution
| 5% NGS in PBST | Final concentration | Amount |
|---|---|---|
| PBST (1×) | 0.95× | 4.75 mL |
| Normal goat serum | – | 0.25 mL |
| Total | N/A | 5 mL |
Store at 4°C for maximum 24 h.
Alternatives: 0.1% Tween-20 could be used in substitution of Triton-X in PBST of S2 R+ cells.
Luria-Bertani broth
| Ingredients and procedure | Amount |
|---|---|
| Peptone | 10 g |
| Yeast Extract | 5 g |
| Sodium Chloride | 5 g |
|
Store at 20°C for up to 1 year.
Equipment
See key resources table.
Alternatives: This protocol uses a DigiTherm Tritech Research incubator for Drosophila culture but any other cell incubator that can be set at 25°C can be used.
Alternatives: This protocol uses a Class II Biological Safety Cabinet (Sterilgard III Advance, The Baker Company) but any other class II hood can be used.
Alternatives: This protocol uses bench-top centrifuges from Eppendorf but any other class II bench centrifuges be used.
Alternatives: This protocol describes the use of confocal imaging using a ZEISS LSM 880 confocal laser microscope with Airy scan. Any other confocal microscope can be used but ability to acquire z-stacks is essential.
Alternatives: This protocol uses a SimpliAMP Thermocycler (Applied Biosystems). Any other thermocycler can be used.
Alternatives: This protocol describes the use of plasticware, chamber cover glass and dissecting tools from specific brands we prefer in our laboratory. Similar tools from any other manufacturer can be used.
Step-by-step method details
Transfection of S2R+ cells with recombinant DNA or double strand (ds) RNA -day 1
Timing: 2 h
-
1.
Harvest cells from T25 flask using a disposable cell scraper (Sarstedt) and transfer to a 15 mL sterile tube (Sarstedt) using a sterile serological pipette.
-
2.
Take an aliquot of cells (100 μL) and mix it with Trypan Blue stain in a 1:1 ratio in a 1.5 mL microcentrifuge tube (Fisher Scientific).
-
3.
Count the live cells (blue cells are dead) using an inverted microscope.
-
4.
Transfer 2,000,000 cells in 2 mL of SFM4 medium supplemented with antibiotics and seed them in a well of a 6-well plate. Incubate overnight before transfection.
Note: Seed 2,000,000 cells per well in as many wells as needed. Always seed a well as an untreated control.
Transfection of S2R+ cells-day 2
-
5.
On the day of transfection/dsRNA treatment, prepare two empty 1.5 mL tubes per each DNA or dsRNA on a rack in a sterile cabinet and add 100 μL of SMF4 medium to each.
-
6.
To transfect recombinant DNA, add 2 μg of DNA to one of the prepared 1.5 mL tubes and add 18 μL of Cellfectin II transfection reagent to the other tube.
-
7.
Incubate both tubes for 5 min at room temperature.
-
8.
Transfer the 100 μL of media plus DNA to the tube containing the 100 μL of SFM4 medium plus Cellfectin II.
-
9.
Pipette up and down 4 times to mix and incubate at room temperature for 20 min.
-
10.
Add the DNA and Cellfectin II mix to one well of the 6-well plate prepared on day 1 and incubate at 25°C for 1–3 days.
Note: The time of incubation of the transfected cells should be empirically established and can vary depending on the protein that is being expressed.
Note: Cells can be used up to 20 passages without observing any reduction in cell transfection efficiency. If reduction in transfection efficiency or loss of response to stimuli is observed, fresh cells should be thawed.
Alternatives: If cells need to be treated with dsRNA, add 10 μg of dsRNA to 100 μL of SFM4 media and add to the 2,000,000 cells seeded in one 6-well plate. Swirl the plate after adding the dsRNA and incubate for 3–4 days at 25°.
Note: When performing a transfection either with dsRNA or with a recombinant DNA, always set another well of cells that will receive the vehicle and not the treatment. This well will serve as control for cell viability and for detection of phenotypes caused by the treatments.
Bacterial culture for treatment of transfected S2R+ cells-day 5
-
11.
A day before ending the treatment of the S2R+ cells, culture Erwinia carotovora carotovora (Ecc15) bacteria in 50 mL of Luria-Bertani (LB) broth at 29°C in a shaking incubator (200 RPM) for 24 h.
Treatment of transfected S2R+ cells with microbes to induce cytoskeleton rearrangement-day 6
-
12.
Transfer 1 mL of the overnight Ecc15 culture in a plastic cuvette and measure the Optical Density (OD) in a spectrophotometer at 600 nm to calculate bacterial cell number per mL. Based on the reading of the spectrophotometer at OD of 600 nm, calculate the concentration of bacteria following the following formula for Gram-negative bacteria:
OD600 of 1= 8 × 108 cells/mL.
-
13.
Pellet and resuspend the culture in an appropriate volume to obtain a concentration of 20 × 106 cells/mL in PBS 1×.
-
14.
Take the 6-well plate from the incubator, scrape cells off and transfer the suspension to a 15 mL tube.
-
15.
Take an aliquot of S2R+ cells (100 μL) and mix it in a 1.5 mL microcentrifuge tube (Fisher Scientific) with Trypan Blue stain in a 1:1 ratio.
-
16.
Count the live cells (blue cells are dead) using an inverted microscope.
-
17.
Take the coverslips coated with concanavalin A prepared on day 1 and place them in a 24- well plate.
-
18.
Seed 100,000 cells on the coverslip per well in SFM4 medium supplemented with antibiotics. Seed two coverslips for each treatment and control group. Incubate for 4 h at 25°C.
Alternatives: When performing live imaging, seed 100,000 cells in an 8-chamber coverglass (Thermo Fischer Scientific) rather than on a slide. Seed two coverslips for cells from each treatment and control groups. Incubate for 4 h at 25°C and add the bacteria to the chamber as described above.
Alternatives: Studies of cytoskeleton dynamics in response to mutations and/or external stimuli can also be performed in Drosophila plasmatocytes to validate the effect of specific genetic manipulations in primary cells. The following procedure is a quick and reproducible method to extract plasmatocytes from adult Drosophila.
Extraction of plasmatocytes from adult flies
-
19.
For extraction of plasmatocytes from adults, anaesthetize groups of 20 flies (10 males and 10 females) from control group or mutant using gaseous CO2.
-
20.
Transfer the anaesthetized flies onto a 1.5 mL centrifuge tube.
-
21.
Cover the flies with glass beads (Sigma-Aldrich, G8772) and vortex at high intensity for 60 s to wound the flies and mobilize plasmatocytes.
-
22.
Transfer the flies to a glass dissecting dish and cut each fly into head, thorax and abdomen with surgical forceps and allow to bleed for 1 min into a glass dish that is filled with 100 mL of SFM4 Insect Cell Culture Medium containing cOmpleteTM Protease Inhibitor Cocktail to avoid coagulation.
-
23.
Filter the cell suspension on 70-μm microcentrifuge filters and collect the liquid containing plasmatocytes into a 1.5 mL tube.
-
24.
Repeat steps 19–23 5 times, each time using 20 flies.
-
25.
Take and aliquot of cells (20 μL) and mix it in a 1.5 mL tube (Fisher Scientific) with Trypan Blue stain in a 1:1 ratio.
-
26.
Count the live cells using an inverted microscope.
-
27.
Move the cell suspension (we get around 50,000 cells) in an 8-chamber cover glass coated with concanavalin A. Seed plasmatocytes from each group of flies in duplicate.
-
28.
Incubate for 2 h at 25°C.
-
29.
Transfer 100 μL of the bacterial suspension prepared in step 15 to one of the two wells. Mark the well where bacteria were added. This represents the stimulated population. Incubate for 2 h at 25°C.
Note: To visualize cortical cytoskeleton dynamics in live imaging, the Drosophila strains used for the study should express a fluorescent marker for F-actin (e.g Lifeact-GFP, an actin-binding peptide fused to a fluorescent protein) in plasmatocytes. Likewise, S2R+ cells used in live imaging should express an actin-fluorescent fusion protein such as LifeAct (Ibidi). To perform live imaging on plasmatocytes or S2R+ cells, nuclei can be stained by adding 4,6-diamidine-2-phenylindole (DAPI) to the media (dilution 1:500).
-
30.
Transfer 100 μL of the bacterial suspension prepared in step 15 to one of the 24-well plates prepared for each treatment in step 18. Mark the well where bacteria were added. This represents the stimulated population. Incubate for 2 h at 25°C.
Indirect immunofluorescence and confocal imaging of phalloidin-stained cells
-
31.
Remove the media from cells seeded on coverslips.
-
32.
With a pipette, rinse cells gently with 500 μL of PBS 1×.
-
33.
Remove the PBS 1× and add 500 μL of 4% formaldehyde to fix the cells.
-
34.
Incubate for 30 min at room temperature.
-
35.
Remove formaldehyde by aspiration and wash the cells three times with PBST to fully remove formaldehyde and permeabilize cells.
-
36.
Incubate cells in 1× PBS with 5% normal goat serum for 1 h at room temperature (20°C).
-
37.
Dilute primary antibody in blocking buffer and apply primary antibody at 1:100 dilution in 1× PBS with 5% normal goat serum for 16 h at 4°C.
Note: Steps are required only if a primary antibody is used to visualize a specific protein. If only cortical cytoskeleton needs to be visualized, phalloidin staining is sufficient (step 30).
Note: The incubation time with primary antibody might vary depending on the antibody and should be adjusted accordingly.
-
38.
The following day, remove primary antibody and wash the cells three times with PBST.
-
39.
Incubate cells for 1 h at room temperature with appropriate secondary Alexa Fluor antibody at 1:1000 in 1× PBS with 5% normal goat serum, DAPI (1:1000 dilution) and fluorescently labeled phalloidin (1:500 dilution) to visualize the cytoskeleton.
-
40.
Wash cells four times, each for 10 min with PBST.
-
41.
Mount cells in Pro-Gold Antifade Reagent. Remove excess mounting medium by aspiration.
-
42.
Allow slides to dry completely.
-
43.
Image slides at ZEISS LSM 880 using a 100× oil immersion objective, NA = 1.4.
-
44.
Analyze the images following the protocol we adopted in ImageJ.
Note: Use an appropriate number of replicates. Since transient transfections of recombinant vectors or RNA interference-mediated depletion of targeted genes are transient, the chances of variation arising among cells in the same well and between wells are substantial. We recommend to always use at least three biological replicates, and to image at least 30 cells per replicate, if possible.
Expected outcomes
Upon imaging, the phalloidin-stained F-actin should be clearly visible. S2R+ cells should spread evenly, without crowding or occlusion by stacking. If applicable, reporter-labeled structures or antibody detection of cytoskeleton-interacting proteins should be visible, and relevant morphological changes to cellular edge structure upon experimental conditions should be detectable.
Cortical cytoskeletal remodeling is an essential process that occurs in different cells in response to environmental stimuli. Our protocol is an easy and robust assay to follow and quantify changes in cytoskeleton remodeling in response to genetic mutations or stimuli such as immune elicitors.
S2R+ cells are an excellent system to quickly perform genetic manipulations and to study their effects on the ability of the cortical cytoskeleton to rearrangement in response to perturbation of specific pathways and/or in response to stimuli. The simple analysis platform adopted in this protocol can be used to quantify actin remodeling in cells in response to genetic mutations and treatment with immune elicitors by calculating the mean cell area of phalloidin-stained cells. We adapted the method used by Aflaki et al. (2011) because it is more accurate than simply measuring phalloidin intensity. Phalloidin intensity may not change during actin remodeling at the cell cortex linked to changes in cell shape. Moreover, phalloidin intensity has been observed to be reduced during actin remodeling when a cell spreads (Albertinazzi et al., 1999). There appears to be general agreement that in quantifying cellular actin reorganization in microscopy, both the intensity of phalloidin staining and the cell area need to be considered (Liu et al., 2018) (Figure 1). Transfected S2R+ cells expressing LifeAct or fluorescently-labeled proteins that localize at the cortical cytoskeleton in response to stimuli can be analyzed in live imaging. In this protocol, we reported how to analyze the live imaging to generate heatmaps of localization for the protein of interest over time (Methods Video S1).
Figure 1.

Quantification of phalloidin-defined area using ImageJ
(A) Example of images acquired at the confocal microscope. Left image represents a spread cell with complex cortical phalloidin staining (red). The right image shows cells that do not spread and have defects in cortical cytoskeleton structuring. Red is polymerized actin stained with phalloidin; Blue is nucleus stained with Dapi. Scale bar 10 μm.
(B) Screenshot of the final step of the ImageJ-based analysis used to quantify the area defined by the red channel staining and the intensity of the staining.
Extracting and analyzing Drosophila plasmatocytes via immunofluorescence provides insights into responses to infection primary immune cells. As Drosophila analogs of macrophages, plasmatocytes have an important role in innate immune responses to infection. Drosophila plasmatocytes can then be used to monitor changes in actin cytoskeletal remodeling under different experimental conditions and can be combined with easily produced transgenic insertions or mutant alleles to tease out individual components of the machinery in response to stimuli such as immune responses.
Quantification and statistical analysis
Quantification of cell area defined by phalloidin staining
Software used: Fiji/ImageJ software (https://imagej.nih.gov/ij/).
We measured the cell area defined by phalloidin staining for each cell in a region of interest. Cells falling in the defined pixel threshold were counted as positive. To determine the intensity of the defined cells within the image:
-
1.
Open image.
File -> Open.
-
2.
Select image type.
Image -> type -> 8-bit.
-
3.
Analysis.
Analyze -> Set Measurements -> Check the boxes: area, perimeter and intensity.
-
4.
Draw a region of interest:
Select rectangle -> select a region of interest (ROI) around a cell.
-
5.
Analysis.
Image -> Adjust -> Brightness/Contrast -> Auto.
Analyze -> Measure.
-
6.
Repeat 4 and 5 for every cell in a frame in each biological replicate for each genotype and under each condition. Infected S2 cells with cortical cytoskeleton defects presented an area value between 20,000 and 50,000 pixels, while control S2 cells presented an area value between 60,000 and 200,000 pixels.
Quantification of signaling molecule location
Software used: Icy Bioimage Analyzer (http://icy.bioimageanalysis.org/), ZEISS ZEN lite Black & ZEISS ZEN lite Blue (https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html?vaURL=www.zeiss.com/zen-lite).
This protocol allows the creation of an intensity gradient for fluorescently-labeled proteins or reporters in a cell during live imaging analyses. This method can be used to follow the cellular localization of the protein or reporter of interest over time and determine where it localizes and accumulates in the cell. Methods Video S1 shows the gradient of fluorescent intensity generated with this method. In the video, the phosphatidylinositol-3,4,5-triphosphate-binding protein fused to green fluorescent protein dynamic upon bacterial treatment can be followed. The fusion protein gradient moves from cytoplasm to cell edge over time in response to the bacterial treatment.
Heatmaps can be generated by applying the following steps to each video:
-
7.
Launch Icy and Open video.
Image/Sequence -> Open.
-
8.
Choose the scene of interest.
-
9.
Filter to remove noise.
Processing -> Filtering -> Median filter (Input: Scene name, Type: 2D) -> Apply.
-
10.
Export video for Zen formatting.
Image/Sequence -> Rendering -> ARGB image.
Image/Sequence -> Save as (File type: AVI sequence).
-
11.
Launch Zen black and Zen blue for Image Processing.
Drag and drop exported video -> File -> Send to Zen- blue edition.
-
12.
Adjust histogram intensity to the required preference for the channels.
-
13.
Dimension -> Drop down for Channel of interest -> LUT -> Rainbow.
-
14.
Export video with preferred dimension and frames/second.
Processing ->Method -> Create Image Subset -> Parameters -> Select Channels, Time & Region of interest -> Apply.
New window generates with applied settings -> Processing ->Method -> Movie Export -> Parameters -> Format (AVI uncompressed) -> Mapping (2.5fps) -> Apply.
Example of the result obtained from this analysis can be observed in Methods Video S1.
Limitations
S2R+ cells are poorly adherent to surfaces but are a useful tool to perform targeted gene manipulation. This protocol describes the methods by which they can be suitably induced to attach to surfaces for imaging. If cells are too closely spaced or are confluent, the described malleability and ease with which the cytoskeletal components are visualized is lost. Therefore, the protocol requires constant monitoring and adjusting of the cell number. This does not allow live imaging for multiple days. Plasmatocytes are primary cells and can die a few hours after extraction. This also limits the possibility of performing live imaging over a time course. S2R+ and plasmatocytes are very small cells; therefore, precise colocalization of proteins or reporters at the cell cortex can be challenging.
Troubleshooting
Problem 1
Cells too closely packed.
Upon imaging, S2R+ cells are closely packed, and cytoskeletal changes are not visible or are difficult to see (steps 43 and 44).
Potential solution
Carefully monitor cell counts before beginning immunofluorescence protocol and ensure cells are growing at normal rates. Ensure all treatments or experimental conditions have been discontinued for some time before imaging preparation. Make sure to split cells before imaging and ensure cell counts are adequate for the size of the tissue culture plates.
Problem 2
Cells not adhering to coverslip.
Upon immunofluorescence visualization, cells are not exhibiting the spreading/adhering phenomenon characteristic of concanavalin A coating. Cells are still markedly round and cytoskeletal changes are not immediately visible in experimental condition (steps 18, 43 and 44).
Potential solution
Ensure coverslips have recommended concentration of concanavalin A and are fully dried before beginning the immunofluorescence protocol.
Problem 3
Cell growth problems.
Cells are growing faster or slower than in described protocol (steps 13–18).
Potential solution
Carefully monitor incubation temperatures, dilution of culture media, antibiotic concentrations, and initial seeding cell volume. Ensure adequate washing to remove residual chemicals from cryopreservation and ensure utilization of sterile technique.
Problem 4
Low transfection efficiency.
The number of transfected cells is lower than 30% per well (steps 43 and 44).
Potential solution
The recombinant DNA used in the transfection could be low quality or too old. Make a fresh DNA mini or maxi prep. Check also the expiration date on the transfection reagent and ensure that no white precipitates are visible in the solution. If precipitates are observed, purchase a new batch of transfection reagent.
Problem 5
High mortality observed upon transfection with recombinant DNA or dsRNA treatment.
Overexpression or depletion of certain proteins can lead to cell death. Likewise, the use of DNA or dsRNA preparations older than one year and 20 days, respectively will affect cells viability (step 15).
Potential solution
Perform a titration experiment to empirically establish the mount of DNA and/or dsRNA that does not affect cell survival. Perform a time course experiment to determine whether shorter treatment time can improve cells survival. Ensure that the DNA and/or dsRNA used in your experiment are not from old preparations and were properly stored.
Problem 6
Quantification of molecule location cannot be determined.
The protein is highly expressed in the cells and calculation of a gradient of localization in response to stimuli over time cannot be established (step 43).
Potential solution
Reduce the expression time of the protein or the amount of DNA transfected. Alternatively, consider expressing it using a vector that has a weaker or inducible promoter.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Francesca Di Cara (dicara@dal.ca).
Materials availability
This protocol does not generate any new materials or cell lines and can be performed with standard Drosophila cell culture techniques and readily available antibodies and reporter systems.
Acknowledgments
This work was funded by a Project Grant from the Canadian Institutes of Health Research to F.D., a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to F.D., a Canada Foundation for Innovation JELF equipment grant to F.D., and a Dalhousie Medical Research Foundation start-up fund to F.D.
Author contributions
C.M.W. wrote the first draft of the manuscript and assisted with figure creation. S.M. made the graphical abstract and edited the manuscript. F.D.C. conceived the project, originated methodology, created figures, and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101588.
Data and code availability
This protocol does not provide original datasets for use in other studies. Immunofluorescence image analysis can be performed using the generalized steps analyzed here.
References
- Aflaki E., Balenga N.A.B., Luschnig-Schratl P., Wolinski H., Povoden S., Chandak P.G., Bogner-Strauss J.G., Eder S., Konya V., Kohlwein S.D., et al. Impaired Rho GTPase activation abrogates cell polarization and migration in macrophages with defective lipolysis. Cell. Mol. Life Sci. 2011;68:3933–3947. doi: 10.1007/s00018-011-0688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertinazzi C., Cattelino A., de Curtis I. Rac GTPases localize at sites of actin reorganization during dynamic remodeling of the cytoskeleton of normal embryonic fibroblasts. J. Cell Sci. 1999;112:3821–3831. doi: 10.1242/jcs.112.21.3821. [DOI] [PubMed] [Google Scholar]
- Basset A., Khush R.S., Braun A., Gardan L., Boccard F., Hoffmann J.A., Lemaitre B. The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc. Natl. Acad. Sci. USA. 2000;97:3376–3381. doi: 10.1073/pnas.97.7.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett A.R., Tibbit C., Ponting C.P., Liu J.L. Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol. Open. 2014;3:42–49. doi: 10.1242/bio.20137120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Mollaeian K., Ren J. An image recognition-based approach to actin cytoskeleton quantification. Electronics. 2018;7:443. doi: 10.3390/electronics7120443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A.S., Parsons B.D., Makdissi S., Chilvers R.L., Mu Y., Weaver C.M., Euodia I., Fitze K.A., Long J., Scur M., et al. Modulation of the cell membrane lipid milieu by peroxisomal β-oxidation induces Rho1 signaling to trigger inflammatory responses. Cell Rep. 2022;38:110433. doi: 10.1016/j.celrep.2022.110433. [DOI] [PubMed] [Google Scholar]
- Rogers S.L., Rogers G.C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat. Protoc. 2008;3:606–611. doi: 10.1038/nprot.2008.18. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Cardona A. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This protocol does not provide original datasets for use in other studies. Immunofluorescence image analysis can be performed using the generalized steps analyzed here.