

Abstract
Preeclampsia (PE) is a high-prevalence pregnancy disease characterized by placental insufficiency, gestational hypertension, and proteinuria. Overexpression of the A isoform of the STOX1 transcription factor (STOX1A) recapitulates PE in mice, and STOX1A overexpressing trophoblasts recapitulate PE patients hallmarks in terms of gene expression and pathophysiology. STOX1 overexpression induces nitroso-redox imbalance and mitochondrial hyper-activation. Here, by a thorough analysis on cell models, we show that STOX1 overexpression in trophoblasts alters inducible nitric oxide synthase (iNOS), nitric oxide (NO) content, the nitroso-redox balance, the antioxidant defense, and mitochondrial function. This is accompanied by specific alterations of the Krebs cycle leading to reduced l-malate content. By increasing NOS coupling using the metabolite tetrahydrobiopterin (BH4) we restore this multi-step pathway in vitro. Moving in vivo on two different rodent models (STOX1 mice and RUPP rats, alike early onset and late onset preeclampsia, respectively), we show by transcriptomics that BH4 directly reverts STOX1-deregulated gene expression including glutathione metabolism, oxidative phosphorylation, cholesterol metabolism, inflammation, lipoprotein metabolism and platelet activation, successfully treating placental hypotrophy, gestational hypertension, proteinuria and heart hypertrophy. In the RUPP rats we show that the major fetal issue of preeclampsia, Intra Uterine Growth Restriction (IUGR), is efficiently corrected. Our work posits on solid bases BH4 as a novel potential therapy for preeclampsia.
Keywords: STOX1, iNOS, Malate, Nitroso-redox balance, Mitochondria, Tetrahydrobiopterin, Preeclampsia
Highlights/Significance
-
•
STOX1 overexpression drives drastic alterations of the l-malate/fumarate ratio and bioenergetic changes in trophoblasts.
-
•
Metabolic alterations upon STOX1 overexpression depend on iNOS uncoupling.
-
•
The NOS coupling agent and metabolite tetrahydrobiopterin (BH4) corrects STOX1-induced alterations including mitochondrial dysfunction in trophoblasts.
-
•
BH4 efficiently corrects the STOX1-induced preeclamptic/IUGR phenotype in mice and rats , and is a potential novel treatment.
Single sentence summary: Reduction of gestational preeclampsia/IUGR symptoms by tetrahydrobipterine is mediated by increased NOS coupling that restores l-malate levels.
1. Introduction
Preeclampsia (PE) is a major hypertensive complication of human pregnancy characterized by a generalized endothelial dysfunction, which is likely triggered by defective endovascular trophoblast invasion of maternal spiral arteries. This condition leads to gestational hypertension associated with proteinuria from mid-gestation, which are classical hallmarks of PE [1]. This syndrome affects around 5% of pregnant women worldwide, and is a leading cause of maternal mortality. Clinically it is classical divided into to presentations, early and late onset (EOPE and LOPE), according to the moment when the symptoms appear, before or after 34 weeks of amenorrhea. Moreover, PE is associated with a predisposition to strokes, chronic arterial hypertension, heart and renal dysfunction, and increased risk of vascular dementia later in life [2,3].
Placental hypoxia, oxidative stress due to reactive oxygen species (ROS) and related lipid peroxidation are well-described features of preeclampsia [[4], [5], [6]]. Although metabolomic profiles of placentas from early-onset PE show downregulation of the glutathione metabolism (i. e. antioxidant defense), to date it has not been stated whether oxidative stress is a cause or a consequence of the disease [7]. Since three decades, the reactive nitrogen species (RNS) nitric oxide (NO) is reported as an endothelium protector able to alleviate preeclampsia [8]. Nitric oxide is a physiological signaling molecule with multiple effects including on DNA synthesis, inflammation, cardiac function, and hypertension [9]. Upregulation of a specific NO synthase (NOS), i.e. inducible NOS (iNOS), linked to hypertension was observed in placentas of PE patients, leading to the suggestion of NOS inhibitors for clinical treatment of PE [10,11]. Unfortunately, treating rats with the NOS inhibitor l-NAME (L-NG-Nitro arginine methyl ester) resulted in the opposite effect, triggering preeclamptic symptoms including hypertension, thereby suggesting that iNOS up-regulation is not a cause but rather an adaptive response to this pathology. This could be due to the fact that l-NAME is a non-specific inhibitor affecting eNOS and nNOS in addition to iNOS; more spcific inhibitors such as 1400W are efficient to alleviate PE symptoms [10].
It has also been suggested to treat PE by modulating the NOS coupling [9,12,13]. Indeed, NOS coupled to its cofactors produces NO, whereas uncoupled NOS produce NO as well as the superoxide anion (O2−), a ROS. The spatial proximity of the two species strongly favors their chemical combination to form the powerful oxidant RNS peroxinitrite (ONOO−) which oxidizes and nitrates molecules and proteins [9,13]. In this context, it has been shown that increasing iNOS coupling with tetrahydrobiopterin (BH4), the essential NOS cofactor, restores iNOS activity leading to NO rather than peroxynitrite generation. Therefore, BH4 has been proposed for the treatment of pulmonary hypertension of the newborn, as well as several neurodegenerative diseases [[13], [14], [15], [16]]. Also, the addition of BH4 to human placental homogenates including from preeclamptic pregnancies stimulates NOS activity, supporting the use of BH4 in PE [17].
Hypertension is also caused by the decrease in NO and l-malate, which results from the insufficiency of fumarase (or fumarate hydratase, FH), a key enzyme in the tricarboxylic acid cycle (TCA), or Krebs cycle, that catalyzes the reversible hydration of fumarate to l-malate and is further converted to l-arginine, the substrate of NOS [18]. In a rat model of salt-sensitive hypertension due to fumarase insufficiency, l-malate supplementation increased l-arginine and NO levels, and attenuated hypertension [18]. Interestingly, although l-arginine has been shown effective for preventing PE in high-risk pregnancy, the fumarase-related l-malate pathway has not been investigated in PE [19].
The winged-helix transcription factor Storkhead box1 (STOX1) is involved in the genetic forms of preeclampsia [20]. This gene encodes two major isoforms, STOX1A and STOX1B [20]. Although both isoforms can bind the STOX1 DNA binding sites STRE1 and STRE2 (for STOX-responsive element 1 and 2) that we have recently identified, only STOX1A contains a transactivating domain, and specifically deregulates gene expression linked to the nitroso (RNS)-redox(ROS) balance [21,22]. STOX1A overexpression confers PE-like gene-expression to trophoblastic cell lines [23] and PE symptoms to pregnant mice carrying transgenic embryos [24] by deregulating more than 2000 endothelial genes [25]. STOX1A overexpression also induces nitroso-redox imbalance and mitochondrial hyper-activation both in vitro and in vivo [26]. STOX1A-overexpressing mice recapitulate long-term cardiovascular consequences such as cardiac stress with heart hypertrophy and fibrosis, consequences of human PE [27]. Overexpression of STOX1A and STOX1B has opposite effects on trophoblast function, in particular trophoblast fusion and syncytialization (acceleration and inhibition, respectively). Therefore, STOX1A/STOX1B imbalance affects gene expression possibly leading to placental abnormalities and related preeclampsia [22]. Finally, STOX1A is abundantly expressed in the brain and its expression is correlated with the severity of late-onset Alzheimer's disease (LOAD), suggesting that STOX1A controls a conserved pathway shared between placenta and brain [28].
Here, we questioned whether STOX1 overexpression affects the fumarase-related l-malate pathway, and by this way induces iNOS, which in turn alters NO levels, with subsequent nitroso-redox imbalance, metabolic dysregulation, and finally PE-symptoms. Our data reveal that STOX1A and STOX1B overexpression oppositely affect l-malate level and fumarase activity, and also alter iNOS and NO levels, the nitroso-redox balance, the antioxidant defense and bioenergetic metabolism. Treatement with either the NOS coupling agent BH4 or the NOS inhibitor l-NAME showed increase of l-malate with the former and decrease with the latter in PE trophoblasts, possibly explining the pro-preeclamptic effect of l-NAME. In vivo, we used two models of preeclampsia, the STOX1-mice [24] and the RUPP rat model, mimicking early and late onset preeclampsia, respectively. Transcriptomic analysis of mice placentas in vivo showed that BH4 rescues a high proportion of STOX1-deregulated genes including those involved in glutathione metabolism, oxidative phosphorylation, cholesterol metabolism, inflammation, lipoprotein metabolism and platelet activation. Finally, with BH4 we successfully treated gestational hypertension, proteinuria and heart hypertrophy in PE pregnant mice, demonstrating that BH4 is a potential therapeutic drug for preeclampsia. In the RUPP rat, the treatment corrected efficiently the foeto-placental phenotype.
2. Results
2.1. STOX1 regulates the hypoxia marker HIF1A and the fumarase-related l-malate pathway
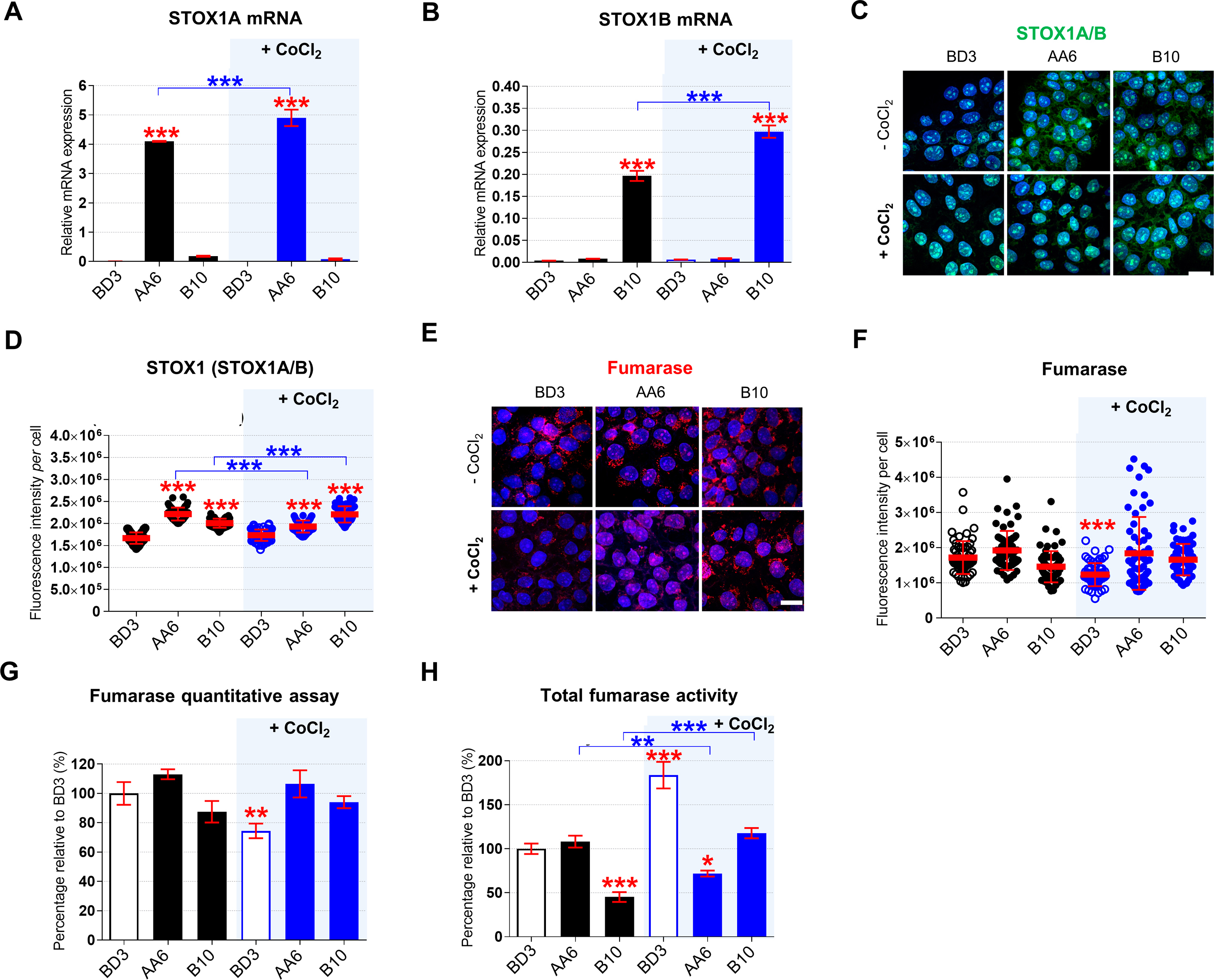
We investigated STOX1 overexpression in JEG-3 choriocarcinoma trophoblasts that have been stably transfected with: i) the empty pCMX vector containing the CMV promoter (BD3 cell line), or ii) the pCMX-STOX1A vector that overexpresses STOX1A (AA6 cell line), or iii) the pCMX-STOX1B vector that overexpress STOX1B (B10 cell line) (Fig. 1A). Following a defective invasion of the trophoblast cells in the maternal spiral arteries, the maternal vessels keep their contractile arterial vaso-active functions; it is currently admitted that this defect leads to rapid local variations in oxygen pressure (hypoxia or alternance of hypoxia-hyperoxia) at the materno-fetal interface and eventually to placental dysfunction and to preeclampsia. We simulated hypoxia in these cellular models through stabilization of the hypoxia-inducible factor 1α (HIF1α) via CoCl2 , a well-known hypoxia mimetic agent [29] (Fig. 1A). RT-qPCR confirmed STOX1A overexpression in AA6 cells (Fig. S1A), and STOX1B overexpression in B10 cells (Fig. S1B). STOX1A overexpressions did not affect STOX1B levels and vice-versa, and CoCl2 treatment further increased STOX1A and STOX1B expression (in AA6 and B10 cells, respectively) (Figs. S1A–B). STOX1A/B overexpression (in the presence and in the absence of CoCl2) was confirmed at the protein level by immunofluorescence using a polyclonal antibody that recognizes both STOX1 isoforms (Figs. S1C–D). CoCl2 treatment did not further increase the levels of STOX1A protein, possibly indicating an altered turnover of STOX1A under these conditions.
Fig. 1.

Hypoxia and STOX1 overexpression affect the fumarate-dependentl-malate pathway. (A) Characteristics of control, STOX1A and STOX1B over-expressing human trophoblast cell lines, and experimental plan in the presence and in the absence of the hypoxia-mimetic agent CoCl2. (B) Immunoblot of the hypoxia marker HIF1A and the internal reference GAPDH from whole-cell extracts. The immunoblot is representative of 3 independent experiments. (C) Schematic representation of the Krebs cycle where the reaction catalyzed by the enzyme fumarase from fumarate (substrate) to l-malate (product) is framed. Levels of the (D) fumarate and (E)l-malate metabolites expressed in nmol/mg of protein. (F) Fumarase specific activity (fumarase activity/fumarase protein content) expressed as percent of untreated control BD3 cell line. Dosages (fumarate, malate) and enzyme activity (fumarase), n = 3 independent experiments, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
STOX1A overexpression (AA6 cells) reduced, whereas STOX1B overexpression (B10 cells) increased, the levels of the stabilized (processed) HIF1A form [30,31] compared to control BD3 cells, as shown by Western blots (Fig. 1B). These data suggest that STOX1B overexpression mimics a hypoxic condition, whereas STOX1A overexpression does not promote the expression and activation of HIF1A. CoCl2 treatment increased both HIF1A forms in control BD3, confirming its hypoxia mimetic function, and to a lesser extent in AA6 cells, whereas it failed to do so in B10 cells (Fig. 1B). Altogether, the results indicate that STOX1 overexpression reduces the expression and stabilization of HIF1A and thereby the HIF1A-driven response in hypoxia.
Under hypoxia, the expression, processing, and stabilization of HIF1A are induced through canonical oxygen-dependent regulation [32]. HIF1A and fumarate production are possibly connected since intracellular fumarate is correlated with HIF1 up-regulation in cancer [33]. In our experimental paradigm, CoCl2 treatment decreased the levels of fumarate (Fig. 1C–D) in STOX1A (AA6) cells compared to all other conditions, and STOX1A (AA6) cells displayed reduced levels of l-malate (that is produced from fumarate) even in the absence of CoCl2 treatment (Fig. 1 E). l-malate is a crucial metabolite involved in NOS function and hypertension. To note, the levels of l-malate were slightly higher than control in STOX1B (B10) cells, and remained higher also upon CoCl2 treatment. Thus, under chemical hypoxia not only STOX1 overexpression poorly activated HIF1A, but STOX1A overexpression also reduced the levels of fumarate and l-malate, whereas these factors were moderately affected by STOX1B overexpression.
HIF1A stabilization is modulated by the activity of fumarase. The fumarase specific activity (Fig. 1F), consisting of total fumarase activity (measured with a colorimetric assay) (Fig. S1H) normalized to the amount of enzyme (assessed by immunofluorescence) (Fig. S1 E,F), was similar in STOX1A (AA6) and control cells (BD3), while it was lower in STOX1B (B10) cells. To note, the levels of fumarase assesed by IF were confirmed by a second test, fumarase quantitative assay based on sandwich ELISA assay (Fig. S1G). CoCl2 treatment increased the fumarase specific activity by about two-fold in control BD3 and STOX1B (B10) cells, thereby restoring control levels in B10 cells, but reduced this activity in STOX1A (AA6) cells (Fig. S1H). Thus, under chemical hypoxia low levels of l-malate in STOX1A overexpressing cells were correlated with low levels of both the substrate (fumarate) and the enzyme responsible for its conversion (fumarase). Upon the same principle, higher levels of l-malate in STOX1B overexpressing cells and controls under chemical hypoxia were correlated with high levels of fumarate and fumarase activity.
2.2. STOX1 regulates iNOS and NO levels, the nitroso-redox balance and the antioxidant defense
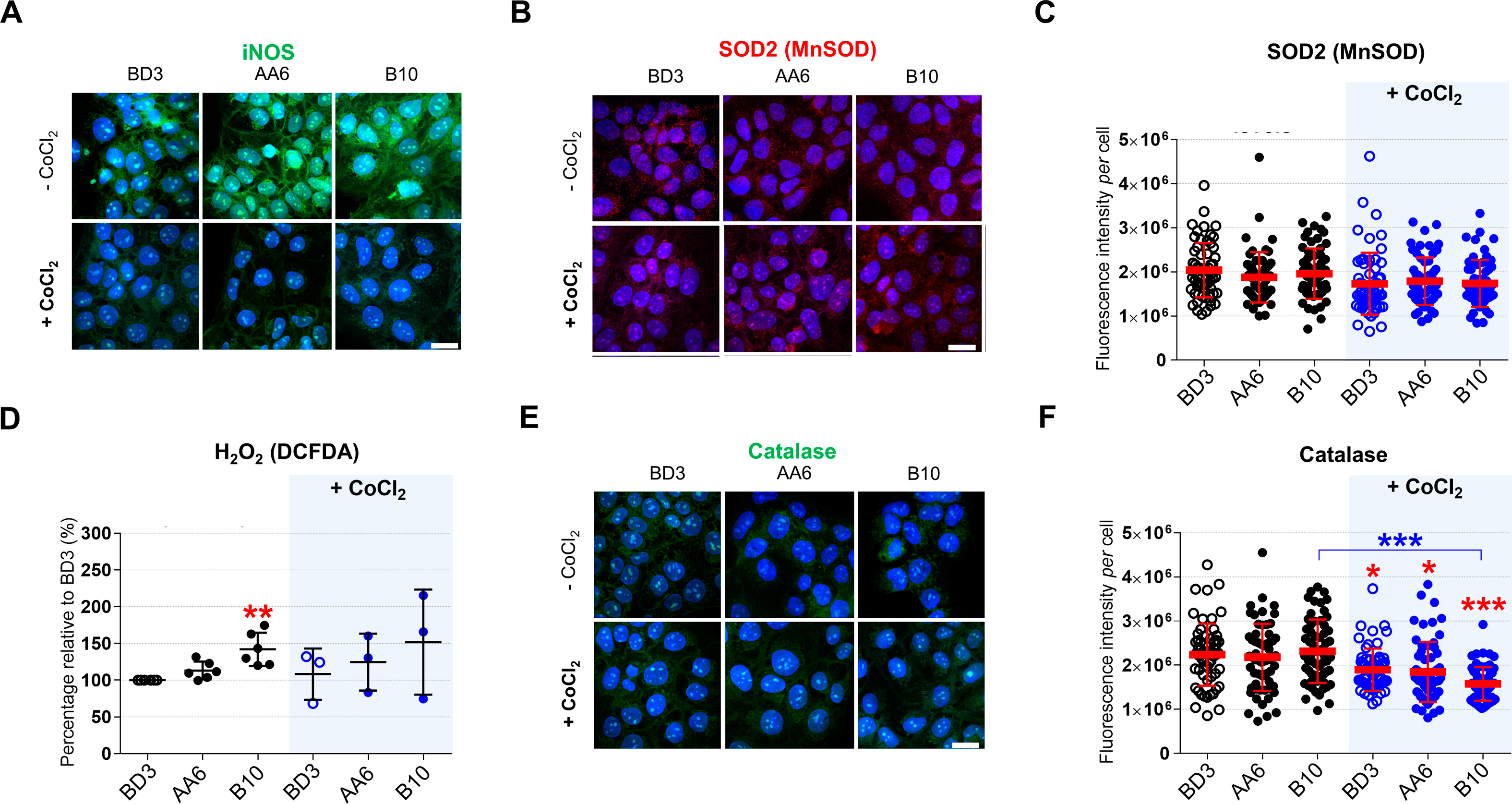
The regulator of the hypoxia response HIF1A, the levels of fumarate, l-malate, and the fumarase activity are affected by ROS (insufficient fumarase activity increases H2O2 levels [[34], [35], [36], [37]]) as well as by the NO metabolism, including iNOS [10,11,18,37,38]. iNOS (NOS2) levels measured by immunofluorescence (IF) were higher in STOX1A (AA6) and STOX1B (B10) compared to control (BD3) cells (Figs. S2A and 2A), and were reduced in all cells upon CoCl2 treatment. Accordingly, NO levels detected by the diaminofluorescein-2 diacetate (DAF-2DA) probe [39], were higher in STOX1A (AA6) and STOX1B (B10) compared to BD3 cells (Fig. 2B). The use of this technique, while possibly influenced by dehydroascorbic acid and ascorbic acid, may suffer some interferences [40], nevertheless this approach is the reference for innumerable studies for more than 20 years [[41], [42], [43], [44]]. Similarly, upon CoCl2 treatment, NO was reduced in all cells except B10, suggesting that under hypoxia STOX1B overexpression is able to maintain high levels of NO despite reduced amount of iNOS (Fig. 2B).
Fig. 2.

NO metabolism and antioxidant defense upon STOX1 overexpression and hypoxia. (A) Quantification of inducible NOS (iNOS; NOS2) fluorescence intensity per cell (images in Fig. S2A). Assessment of (B) Nitric oxide (NO) using the DAF-2 DA probe, (C) the ROS anion superoxide (O2−) using the DHE probe, and (D) the RNS peroxynitrite (ONOO−) using the DHR123 probe, expressed as percentage of the untreated control BD3 cell line. (E) Total SOD activity (measured as inhibition of the activity of SOD, percentage). (F) Catalase activity expressed in nmol/min/ml. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Dosages (NO, O2−, peroxynitrite), n = 3–6. Enzyme activities (SOD, catalase), n = 6 for untreated BD3/AA6/B10 in NO and O2− assays, n = 3 independent experiments for all other experiments, mean ± SD, *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Generation of the RNS NO, the ROS O2− (superoxide) and subsequent formation of the RNS ONOO− (peroxynitrite) are hallmarks of NOS uncoupling [9,13]. Accumulation of iNOS in STOX1A (AA6) and STOX1B (B10) cells was not associated with over-generation of O2−, detected with the dihydroethidum (DHE) probe [41], and ONOO− detected with the dihydrorhodamine 123 (DHR123) probe [42], compared to control (BD3) cells (Fig. 2C–D). No changes were observed upon CoCl2 treatment, except reduced ONOO− in controls, and to some extent in STOX1B (B10), compared to the corresponding untreated cells (Fig. 2D). Altogether, STOX1 overexpression, which we previously showed altering the nitroso-redox balance [26], induces iNOS and NO accumulation but no superoxide and peroxynitrite accumulation, suggesting altered antioxidant defence in STOX1 cells or poor NOS uncoupling.
Peroxynitrite and NO accumulation have been associated with low superoxide dismutase (SOD) activity [45]. Therefore, we tested SOD that metabolizes O2− into the ROS hydrogen peroxide (H2O2) [[46], [47], [48]] and is the first line of defence against oxidative stress. The levels of mitochondrial SOD2 (MnSOD), tested by IF, were not affected by STOX1 overexpression nor by CoCl2 treatment (Figs. S2B–C), and total SOD activity was not altered beyond a 5% range (Fig. 2E). However, upon CoCl2 treatment SOD activity decreased in STOX1B cells (Fig. 2E). The product of SOD activity, the ROS H2O2, detected with the 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) probe [42], was essentially not affected, and increased upon STOX1B overexpression in normoxic conditions, in parallel with a mild increase of SOD activity (Fig. S2D). Hence, we reasoned that catalase activity may have been altered instead. Catalase is the antioxidant enzyme that metabolizes H2O2 into H2O and O2 [48,49] and also catalytically scavenges peroxynitrite. Catalase can be inactivated by NO accumulation and by single oxygen generation through interaction of peroxynitrite with H2O2 [50,51]. The levels of catalase, tested by IF, were not affected by STOX1 overexpression (Figs. S2E–F), but strikingly its activity increased by 11-fold and 9.6-fold in STOX1A (AA6) and STOX1B (B10) cells, respectively, compared to control (BD3) cells (Fig. 2F). CoCl2 treatment further increased catalase activity in control and STOX1B cells (in this last case despite reduced levels of the protein), but dramatically decreased it in STOX1A cells (Fig. 2F, S2E-F). Altogether, in hypoxia-mimetic conditions STOX1A overexpression resulted in a slight decrease of SOD activity and a dramatic drop of catalase activity, finally evoking a strongly impaired antioxidant defence. Conversely, STOX1B overexpression reduced SOD activity in chemical hypoxia, but maintained a strong increase of catalase activity, thereby displaying antioxidant defence.
2.3. STOX1 regulates the bioenergetic metabolism
We then assessed whether the observed alterations were associated with mitochondrial stress in STOX1 overexpressing cells. Indeed, insufficient fumarase activity increases H2O2 levels by altering the expression of mitochondrial OXPHOS complexes [[34], [35], [36], [37]]. Decreased fumarase and increased H2O2 levels were indeed observed upon STOX1B overexpression in normoxia (see above, Fig. S1H and Fig. S2D). Moreover, HIF1A stabilization is associated with severe damage of the mitochondrial oxidative phosphorylation (OXPHOS) complexes [32]. The amount of the mitochondrial ROS superoxide anion (mtO2-) [52], detected by MitoSOX Red staining, was slightly higher in STOX1B (B10) than STOX1A (AA6) and control (BD3) cells, and returned back to control levels under CoCl2 treatment (Fig. 3A and B). Intact mitochondria are classically detected by the mitochondrial marker citrate synthase (CS) that catalyzes the condensation of oxaloacetate with the glycolytic end-product acetyl-coenzyme A (acetyl-CoA) to produce citrate and CoA in the Krebs cycle [[53], [54], [55], [56]] (Fig. 3C). Both STOX1A (AA6) and STOX1B (B10) reduced CS activity (−37% and −54%, respectively) compared to control (BD3) cells, suggesting a reduction of active mitochondria (Fig. 3D). Conversely, CoCl2 treatment strongly activated CS activity in STOX1 overexpressing cells beyond the levels of controls, suggesting an increase of active mitochondria in these cells (Fig. 3D). However, the levels of the CS substrate acetyl-CoA were lower in STOX1B (B10), and upon CoCl2 treatment also in STOX1A (AA6), compared to control (BD3) cells (Fig. 3E), indicating that CS activity in STOX1 overexpressing cells is possibly limited by the amount of its reactants, and therefore may not correspond to reduced mitochondrial activity.
Fig. 3.

Mitochondrial ROS and mitochondrial bioenergetic upon STOX1 overexpression and hypoxia. (A) 3D reconstructions of cells stained with MitoSOX (red) to detect mitochondrial ROS (superoxide anion, O2−), counterstained with Hoechst (blue). Scale bar 10 μm, and (B) Quantification of MitoSOX fluorescence intensity per cell. (C) Schematic representation of Krebs cycle including the reaction catalyzed by citrate synthase from oxaloacetate (substrate) to citrate (product). (D) Citrate synthase activity expressed in mU/mg of protein. (E) Acetyl-CoA expressed in nmol/g protein. (F)Total ATP expressed as percentage of the untreated control BD3 cell line with non-OXPHOS and OXPHOS fractions. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Dosage (ATP, Acetyl-CoA), enzyme activity (citrate synthase), n = 3 independent experiments, mean ± SD, *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001; based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To meet bioenergetic demands, ATP is produced by glycolysis as well as mitochondrial OXPHOS [55,56]. Total ATP levels increased by 2.5-fold in STOX1A (AA6) and 5.5-fold in STOX1B (B10) compared to control (BD3) cells (Fig. 3F), indicating that either their mitochondria generate more ATP or, alternatively, that this ATP is largely produced by glycolysis. Compatibly with the second hypothesis, the fraction of ATP in the presence of the OXPHOS inhibitor oligomycin (i.e. the OXPHOS fraction) remained low in all samples (less than 15%) and ATP changes were mostly not dependent on OXPHOS (Fig. 3F). Glycolytic ATP appeared also responsible for the inversed situation upon CoCl2 treatment, with control and STOX1A (AA6) overexpressing cells displaying high, and STOX1B (B10) overexpressing cells low, ATP levels (Fig. 3F). Nevertheless, CoCl2 treatment strongly enhanced the OXPHOS fraction in STOX1A (AA6) cells. Thus, in hypoxia mimetic condition STOX1A overexpression restored, at least in part, mitochondrial respiration and activated CS activity without affecting the ROS O2−.
2.4. The BH4 metabolite controls STOX1 effects through iNOS regulation
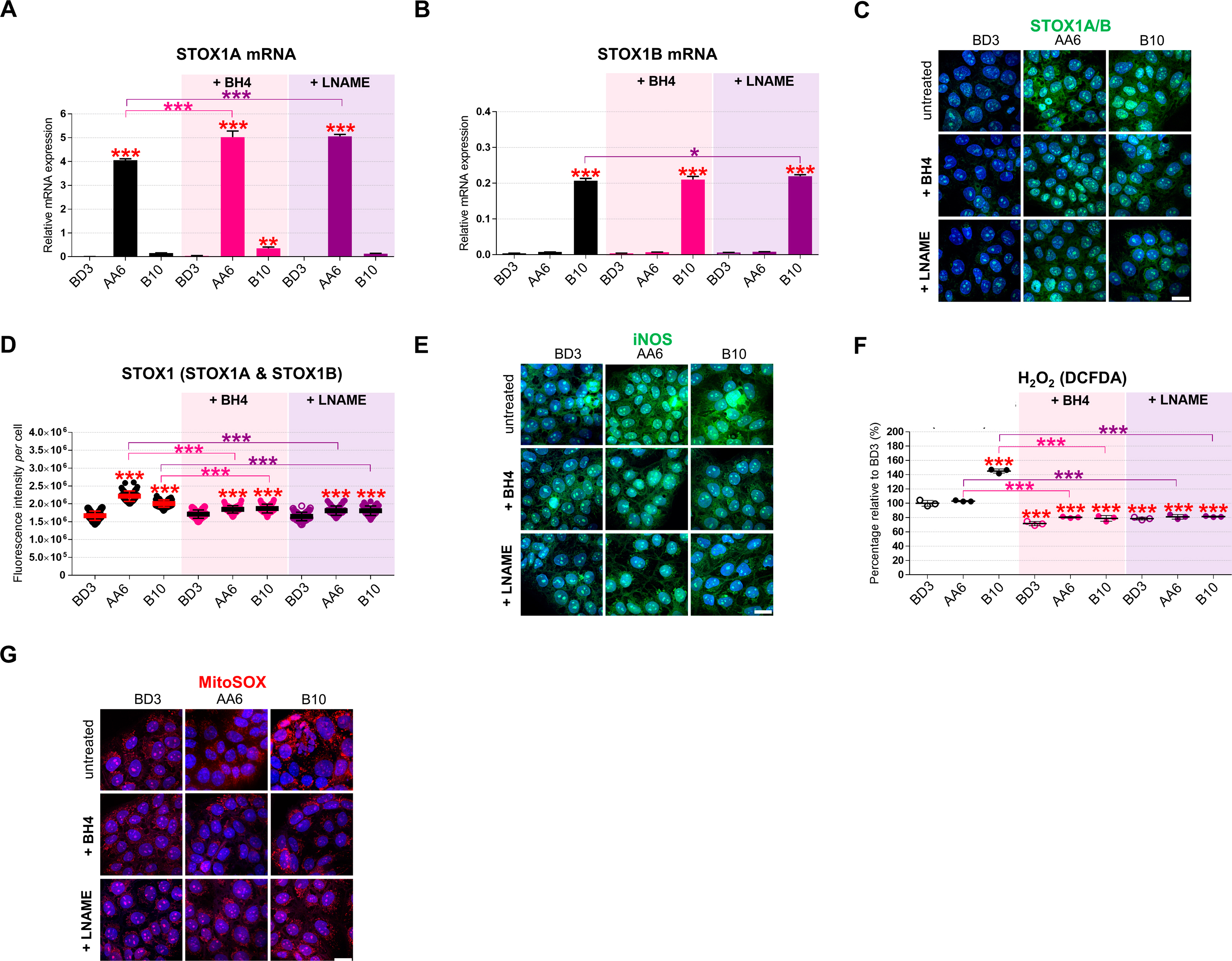
The nitric oxide signalling pathway modulates mitochondrial respiration in particular in hypoxia. Moreover, iNOS expression is involved in metabolic syndromes and chronic metabolic inflammation [57,58]. We reasoned that increasing the iNOS coupling via the BH4 cofactor [14,17] or, alternatively, inhibiting iNOS with l-NAME [10,11,59] are potential therapeutic strategies to counteract dysfunction due to STOX1 overexpression in preeclampsia. Either BH4 or l-NAME treatment maintained STOX1 isoforms accumulation at the mRNA level (Figs. S3A–B), albeit slightly reduced the STOX1 protein, compared to untreated cells (Figs. S3C–D). Either treatment reduced iNOS in STOX1A and STOX1B to the levels of untreated cells (Fig. 4A; S3E), and dramatically reduced the ROS H2O2 in all conditions (Fig. S3F). Either treatment also reduced the high mitochondrial ROS in STOX1B (B10) cells (Figs. 4B and S3G). The RNS peroxynitrite was reduced in STOX1A and control cells by either treatment, but not in STOX1B cells with l-NAME (Fig. 4C). Thus, the two treatments appear equivalent in rescuing key ROS/RNS alterations due to STOX1 overexpression, with the exception of l-NAME that failed to reduce RNS in STOX1B (B10) cells.
Fig. 4.

The BH4 metabolite counteracts STOX1 effects through iNOS regulation and rescue ofl-malate levels. (A) Quantification of iNOS/NOS2 fluorescence intensity per cell. (B) Quantification of mitochondrial ROS fluorescence intensity per cell. (C) Percentage of peroxynitrite RNS relative to the untreated control BD3 cell line. (D) Fumarase specific activity (ratio of fumarase activity/fumarase protein content) expressed as percent of untreated control BD3 cell line. Levels of l-malate (E) and fumarate (F) expressed in nmol/mg of protein. Data of untreated cells are from the corresponding panels in Fig. 2A (iNOS), Fig. 3B (mitochondrial ROS), Fig. 2D (RNS), Fig. 1F (fumarase specific activity), Fig. 1D (fumarate), Fig. 1E (l-malate). Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Dosages (peroxynitrite, fumarate, malate) and enzyme activity (fumarase), n = 3 independent experiments, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (pink or purple stars). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
These two treatments have then been assessed for regulation of the antioxidant response. The total antioxidant SOD activity and the mitochondrial component of this activity (SOD2) were not remarkably different in STOX1 overexpressing compared to control cells, and either BH4 or l-NAME had little effect on these parameters, except slightly reduced SOD activity and SOD2 levels in STOX1B (B10) cells upon BH4 treatment (Figs. S4A–C). Conversely, the activity of the other major antioxidant, catalase, which was elevated in STOX1 cells, was largely reverted by either treatment, and in particular BH4, despite roughly comparable levels of the protein in all cell lines (Figs. S4D–F).
We then assessed whether these treatments affected the bioenergetic metabolism. Either treatment increased CS activity in all cells, essentially rescuing low CS levels in STOX1 cells, except BH4 in STOX1A cells (Fig. S5A). In addition, both treatments strongly decreased the central bioenergetic metabolite acetyl-CoA levels in all the cell types, and more severely upon STOX1A overexpression (Fig. S5B). Finally, BH4 slightly increased and l-NAME dramatically reduced ATP levels in STOX1A overexpressing cells, whereas l-NAME strongly increased ATP levels in STOX1B overexpressing cells, this being essentially due to the non OXPHOS-related ATP production (Figs. S5C–D). Despite both treatments increased the fraction of mitochondrial ATP when STOX1A and STOX1B are overexpressed, in particular STOX1A cells treated with BH4 and to a larger extent STOX1B cells treated with l-NAME (Fig. S5C), in absolute values the contribution of mitochondrial ATP remained quite modest as most ATP was not produced through OXPHOS. These observations suggest that in all cells, acetyl-CoA does not systematically feed the Krebs cycle to produce ATP, despite BH4 or l-NAME treatments tend to some extent to tilt the balance toward the consumption of acetyl-CoA and mitochondrial ATP production.
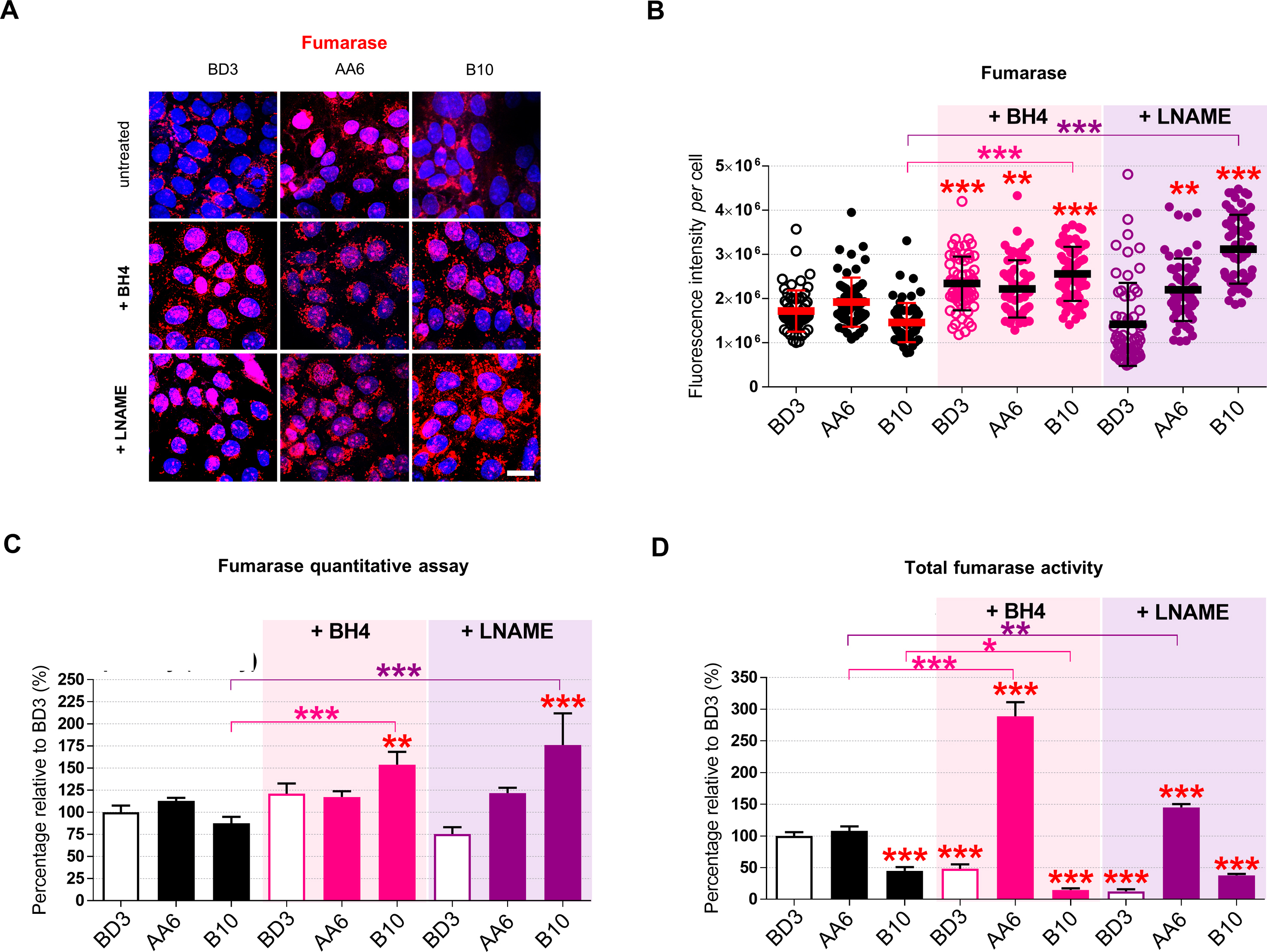
Finally, we assessed whether the fumarate and l-malate pathways are affected by iNOS upon treatment with one or the other molecule. The level of the enzyme fumarase, measured by immunofluorescence (Figs. S6A–B) and sandwich ELISA assay (Fig. S6C), was comparable in untreated cells, and upon BH4 as well as l-NAME treatment it increased in STOX1B cells whereas was essentially not affected in control and STOX1A cells. Despite minor variations in the amount of enzyme, both treatments strongly reduced the fumarase activity in control and STOX1B cells, contrary to STOX1A cells where the opposite effect was observed, and more strikingly upon BH4 treatment (Fig. 4D, Fig. S6D). Thus, BH4 and l-NAME resulted in high levels but poor activity of fumarate in STOX1B cells, raising the possibility that in these cells the enzyme was produced to compensate for lack of activity. Conversely, either treatments strongly increased the fumarase activity in STOX1A cells (despite unchanged amount of the enzyme). The high fumarase activity correlated with increased levels of its product l-malate in STOX1A cells treated with BH4. Conversely, l-NAME treatment decreased the levels of l-malate in these cells. Despite minor differences, control and STOX1 expressing cells, with or without BH4 and l-NAME displayed comparable levels of l-malate (Fig. 4E), indicating that variations in the level of this substrate are not responsible for the amounts of the downstream product (l-malate). In summary, BH4 and l-NAME have globally comparable effect on control and STOX1 overexpressing cells, showing that iNOS modulation affects the fumarate/l-malate pathway in normal as well as preeclamptic conditions. However, only increasing iNOS coupling (treatment with BH4) rescued the levels of l-malate in STOX1A overexpressing cells, whereas l-NAME not only failed to do so but actually reduced the levels of fumarate in these cells (Fig. 4F).
2.5. BH4 restores STOX1-deregulated placental gene expression in vivo
While the mode of action of BH4 in vitro as a NOS coupling agent is well described [14,17], the actual impact of this metabolite in the placenta is not known. We questioned whether in vivo BH4 treatment, provided in drinking water, can restore STOX1-deregulated gene expression in placenta of wild-type (WT) female mice crossed with STOX1A transgenic mice males (heterozygous TgSTOX42), a genetic design leading to a preeclamptic phenotype in the mothers (Fig. 5A) [21,24,27]. Mice placentas at 16.5 days post-coïtus (late gestation) were analyzed at the whole transcriptome level. Total RNAs from pools of three placenta from different mice in four conditions were used (control (Ctrl), Ctrl + BH4, STOX1 (preeclamptic-mimic) mice, and STOX1+BH4) and hybridized to ClariomS mice microarrays.
Fig. 5.

In vivo STOX1 model of preeclampsia and BH4 effect on placenta transcriptomics. (A)in vivo model of preeclampsia and preclinical trial using BH4 supplementation in healthy and preeclamptic pregnant mice. (B) Principal Component Analysis of gene expression in control and STOX1-overexpressing placentas at 16.5 days post-coïtum, with or without BH4 treatment. (C) Enrichment of gene expression of KEGG pathways in STOX1 placentas with or without BH4 treatment. (D) Enrichment of specific pathways related to placental function in this comparison. (E) BH4 rescues the alteration of gene expression induced by STOX1 overexpression in the placenta (threshold chosen: genes with more than two fold changes, and corrected to <30% of the value in control samples).
Out of 22,206 transcripts analyzed, 2476 were modified (ANOVA F.test <0.05). The PCA clustering was able to successfully separate the four conditions (Fig. 5B). Webgestalt ontology analysis was used to characterize the BH4 effects by comparison of the STOX1 and STOX1+BH4 conditions, as shown by comparison of KEGG pathways [60]. Among the highest scored pathways in the STOX1 versus STOX1+BH4 condition, genes in “Oxidative Phosphorylation” and “Glutathione metabolism” were up-regulated, and genes in “Platelet activation” and “Cholesterol metabolism” were down-regulated (Fig. 5C). The GSEA analyses of these pathways are represented in Fig. 5D, showing normalized enrichment scores above 2.
To analyze in more detail the effects of BH4 and STOX1 on various genes, we identified a high proportion of genes that were deregulated by STOX1 and brought back to basal level by BH4 treatment. Overall, 565 genes were modified more than 2-fold between CTLs and STOX1 placentas, and amongst those, 84 (∼15%) were brought back to <30% of their gene expression levels in the control placentas (Fig. 5E). These 84 genes were submitted to network analysis using STRING, and were successfully clustered, revealing several clusters (Interferon-regulated genes, Inflammation regulators, Lipoprotein metabolism) (Fig. S7). In conclusion, these in vivo transcriptomic results prove that BH4 acts by compensating alterations induced by STOX1 at the gene level. Some of these genes are highly relevant in the context of NO physiology, such as Nostrin (NO Synthase Trafficker), a factor that colocalizes with eNOS, and redistributes subcellularly the synthase [61].
In preeclampsia, endothelial cells play a major role since the disease is characterized by a global endothelial dysfunction. Therefore, analyzing the expression data under the angle of endothelial cells is particularly relevant. We submitted our bulk microarray data to the deconvolution algorithm dTangle to characterize the cell types, their proportion and their gene expression profile [62]. To deconvolute these data we feeded dTangle with an available single cell experiment conducted on mice placenta [59]. The algorithm used this reference to identify specific cell markers and the proportion of the different cell types (trophoblasts, endothelial cells, decidual stromal cells, mesencymal fetal cells and blood cells) (Fig. 6A). Using the microarray data, we used deconvolution algorithms from the signature of the major placental gene categories known in mice [63]. Using the 10 prominent genes of five categories (trophoblast, endothelial cells, decidual stromal cells, mesencymal fetal cells and blood cells) on our data, we detected easily the five major gene categories without significant changes in our dataset from the Marsh and Blelloch paper used as a reference [63]. Using these genes we could evaluate the proportion of the different cell types, where endothelial cells represented about 14% of the cells (Fig. 6A). Interestingly (and unexpectedly), BH4 treatment induced a shift in the proportion of trophoblast cells that was increased, while the proportion of decidual stromal cells was decreased. This could contribute to a global improvement of the placental function in BH4 treated mothers, as described in the phenotypic evaluation of the mice described below. Focusing the analysis on the endothelial cells by analyzing the complete set of specific genes characterizing these cells, we could identify specific alterations of their mRNA levels in the endothelial cells under BH4 treatment (Fig. 6B).
Fig. 6.

Cell proportions in the placenta are altered by BH4 treatment, and major endothelial genes are deregulated. (A) Using deconvolution, we were able to dissociate the different major cell types present in the mouse placenta. The proportion of cells was altered by BH4 for trophoblasts and for decidual stromal cells, while the other cell types did not differ quantitatively. (B) In the endothelial cells, several genes were deregulated. The column ‘Specificity’ refers to the endothelial specificity, with *** for genes expressed exclusively in the endothelium, while ** and * refers to genes that are expressed in other tissues, albeit predominantly in the endothelial cells.
2.6. BH4 successfully prevents major preeclampsia symptoms in mice in vivo
We then validated whether BH4 treatment in drinking water is beneficial against gestational hypertension, proteinuria, and heart hypertrophy, three major preeclampsia symptoms that are recapitulated in the STOX1 mice model [1,21,24,27]. For the first PE symptom, daily monitoring of the blood pressure (BP) by assessment of systolic arterial pressure throughout gestation [24] showed that preeclamptic pregnant mice developed a gestational hypertension from 5.5 days post-coïtus (early-gestation) compared to WT pregnant mice (Fig. 7A). BH4 treatment succesfully prevented gestational hypertension in preeclamptic pregnant mice and did not change BP in WT pregnant mice compared to the corresponding untreated pregnant mice (Fig. 7A). Analysis of BP by three periods corresponding to early-, mid- and late-gestation showed that BH4 treatment succesfully prevented gestational hypertension in preeclamptic pregnant mice throughout these gestational periods (Fig. S9). In WT pregnant mice BH4 treatment slightly decreased BP during first period (early gestation) whereas slighlty increased BP in the second (mid-gestation) period (Fig. S9).
Fig. 7.

in vivo evidence of BH4 treatment efficiency in preeclampsia in the STOX1 model. (A) Systolic arterial pressure throughout gestation expressed in mm Hg relative to pre-gestation. Black stars compare the mice carrying transgenic animals untreated versus untreated mice carrying WT embryos. Pink stars show a restoration of the blood pressure under BH4 treatment in mice carrying STOX1 overexpressing fetuses. (B) Albumin/creatinine ratio normalized to WT value at E17.5 (late-gestation). (C) Heart weight (mg) to tibia length (mm) ratio. For systolic arterial pressure, data are from n = 9 for WTxWT, n = 7 for WTxWT + BH4, n = 16 for WTxhtTgSTOX42 and n = 12 for WTxhtTgSTOX42 +BH4. For the ACR ratio, data are from n = 7 for WTxWT, n = 5 for WTxWT + BH4, n = 10 for WTxhtTgSTOX42, and n = 9 for WTxhtTgSTOX42 +BH4 mice. For heart weight to tibia length ratio, data are from n = 6 for WTxWT, n = 6 for WTxWT + BH4, n = 4 for WTxhtTgSTOX42, and n = 6 for WTxhtTgSTOX42+BH4 mice; (D) Echographic parameters of the mice comparing STOX1 mice and STOX1 mice treated with BH4. (E) Placental weight in mice carrying transgenic fetuses treated or not by BH4. Mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA, followed by post hoc Dunnett tests using the untreated gestations as a control. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
For the second PE symptom, preeclamptic pregnant mice displayed proteinuria evaluated as the ratio between albumin and creatinine (ACR ratio) in the urine [24] compared to WT pregnant mice. BH4 treatment succesfully prevented proteinuria in preeclamptic pregnant mice without affecting this ratio in WT pregnant mice (Fig. 7B).
The third PE symptom, the cardiac hypertrophy evaluated as the ratio of heart weight over tibia length [27,64] strongly increased in preeclamptic compared to WT pregnant mice (Fig. 7C). BH4 treatment successfully prevented cardiac hypertrophy in preeclamptic mice, and slightly increased the ratio in treated WT pregnant mice (Fig. 7C). Several echographic parameters were measured at 16.5 days of gestation on mice carrying transgenic embryos with or without BH4 treatment (Fig. 7D). Consistently with the actual measures after sacrifice, the heart weight estimated by in vivo echography was reduced to normal level by BH4, and the interventricular septum thickness (a predictor of human coronary disease [65]) was reduced. Several Doppler parameters were corrected by the treatment (increased myocardial performance, increased outflow from the right ventricle and decreased blood velocity for the two sides of the renal arteries).
Furthermore, at term, the placentas were weighed and revealed a decreased placental weight in mice carrying transgenic embryos and this was corrected by BH4 treatment at 18 days post coitus (dpc) (Fig. 7E). There was no reduction in fetal size in this experiment but this is consistent with previous observations showing that in the STOX1 model there is an efficient in utero catch-up growth of the fetus at the end of gestation. The intrauterine growth restriction (IUGR) phenotype detectable clearly at 16.5 dpc, diminishes strongly after 17.5 days [66]. Altogether, this pre-clinical study validated that BH4 treatment throughout gestation successfully prevented the major preeclampsia symptoms.
2.7. BH4 successfully prevents IUGR in the surgical RUPP model of late preeclampsia in rats in vivo
The Reduced Uterine Placental Perfusion (RUPP) model was used to validate the BH4 action on PE symptoms using an independent animal model (Fig. 8A). This surgical procedure is performed at 14 dpc characterizing a clinical phenotype more consistent to late onset preeclampsia (LOPE) rather than to early onset preeclampsia (EOPE) like the STOX1 transgenic mouse. Maternal blood pressure was increased in RUPP animals at 19 days post-coïtum. The LOPE status of the RUPP animals was also illustrated by the fact that the heart weight was not significantly modified, despite reported impaired systolic function in this model by Doppler ultrasound echocardiography [67]. The BH4 did not correct the increased blood pressure in this LOPE model. In the RUPP dams, there was an expected decrease in fetal weight (from 2.34 ± 0.29 g to 2.22 ± 0.25 g). BH4 treatment at 3.5 mg/kg largely compensates this (average weight 2.54 ± 0.3 g), while the high dose (7 mg/kg) appeared deleterious, Fig. 8C. These observations were consistent with placental weight that was partly corrected with BH4 (Fig. 8D), but no differences were found for placental efficiency (Fig. 8E). Also, consistent with these observations was the effect of acetylcholine to assess endothelium-dependent vasorelaxation in isolated uterine artery rings that was brought back to normal in the BH4-treated females (Fig. 8F). No such effect was detectable in the aorta using the same ex-vivo procedure suggesting that the BH4 treatment modulates hemodynamics, at least partly, and specifically in the utero-placental circulation.
Fig. 8.

In vivo evidence of BH4 treatment benefit on IUGR in the RUPP rat model. (A) Structure of the uterine vascularization of the rat uterus. In red are the silver clips that restrict blood flow to 100 μM in the ovarian arteries and to 230 μM in the abdominal aorta. (B, C, D) Mean arterial pressure (MAP), fetal and placental weights analyzed in the RUPP rats. Placental efficiency (D) is calculated by dividing the body weight by the placental weight for every feto-placental unit. (E) Isolated uterine artery function was evaluated using cumulative doses of Acetylcholine (Ach) via wire myography. The area under the curve (AUC from the curve obtained as relaxation against doses of Ach) was considered. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Discussion
There is now a wealth of literature stressing the importance of nitrosative stress in preeclampsia [68,69]. At the center of nitrosative stress are the NOS enzymes, that need coupling with the tetrahydrobiopterin (BH4) cofactor to produce NO, an essential element of vascular health. Therefore insufficient BH4 concentrations were envisaged long ago as causative in PE [70], as mentioned in a recent review [71]. Consistently, specific mutations of eNOS were associated with an increased risk of PE [72]. BH4 production was shown to be negligible in the mature placenta, suggesting that it should originate from other sources, such as the fetus, and will possibly be limiting in pathological pregnancies [73]. A first very elegant preclinical study using BH4 as a treatment in PE was published in 2007 [74]. In this rat model, PE-like symptoms were induced by deoxycortisone acetate (DOCA) injection, and BH4 addition (sepiapterin) was shown to induce relaxation in mesenteric arteries in organ chambers, ex vivo. This descriptive paper nevertheless did not address the fine molecular modification triggered by the treatment, especially in vivo. In the present study, we aimed at a better understanding of BH4 function by cell biology experiments in vitro, and by transcriptome analysis of the placenta in the STOX1 mouse model of EOPE [24], treated or not with BH4. To determine whether any benefits with BH4 are specific for EOPE, we utilized a classical model of LOPE by applying BH4 therapy to the RUPP rat model.
The preeclampsia-associated transcription factor STOX1 is a key regulator of the oxygen-dependent nitroso-redox balance and mitochondrial metabolism [26]. Placental hypoxia associated with oxidative/nitrosative stress is a major feature of preeclampsia [[4], [5], [6]], and STOX1A/STOX1B imbalance has been shown to deregulate nitroso-redox balance and trophoblast function at gene expression level [[4], [5], [6],22]. Here, we show that the preeclamptic condition generated by STOX1A overexpression affects multiple linked gene pathways in trophoblats; in particular it deregulates the HIF1α-driven response in hypoxia, which affects the fumarase-related l-malate pathway, induces iNOS thereby altering NO levels with subsequent nitroso-redox imbalance, metabolic dysregulation, and finally PE-symptoms (Fig. 9). Importantly, we show that under chemical hypoxia only STOX1A overepression reduces the fumarate/fumarase/l-malate pathway, whereas STOX1B overexpression generates the opposite effect, possibly explaining the specificity of STOX1A overexpression in conferring PE-like gene-expression to trophoblastic cell lines [23] and PE symptoms to pregnant mice. Treatement with the NOS coupling agent BH4 (but not the NOS inhibitor l-NAME) increased l-malate in PE trophoblasts, restoring control conditions. Transcriptomic analysis of mice placentas in vivo confirmed that BH4 rescues a high proportion of STOX1-deregulated genes including those involved in glutathione metabolism, oxidative phosphorylation, cholesterol metabolism, inflammation, lipoprotein metabolism, and platelet activation. Finally, with BH4 treatment we successfully corrected gestational hypertension, proteinuria, placental weight and heart hypertrophy in PE pregnant mice as well as Intra-Uterine Growth Restriction and placental dysfunction in rats, demonstrating that BH4 is a potential therapeutic drug for preeclampsia/IUGR.
Fig. 9.

Model for STOX1 effects and BH4 therapeutic opportunity to treat preeclampsia. Increased STOX1 expression induces iNOS accumulation and iNOS uncoupling, NO overproduction and subsequently altered pathways: namely nitroso-redox balance and antioxidant defense, bioenergetic metabolism, the Krebs cycle including citrate synthase activity, fumarase activity and the related l-malate, mitochondrial OXPHOS, in the context of altered stability of the hypoxia effector HIF1A. These alterations are potentially related to preeclampsia symptoms. Supplementation of the NOS cofactor BH4 restores iNOS levels and modulates the other altered patways, opening a new therapeutical opportunity to treat preeclampsia symptoms.
Oxygen-independent alteration of HIF1α stabilization upon STOX1 overexpression indicated that non-canonical regulation of HIF1 α through the fumarase pathway was affected, in agreement with the notion that the activity of this key Krebs cycle enzyme and its related oncometabolites fumarate and l-malate can deregulate the HIF1 α status [[31], [32], [33]]. In support of this model, gestational hypertension is a major PE symptom, and in a rat model of salt-sensitive hypertension due to fumarase insufficiencies, l-malate supplementation attenuated hypertension through the modulation of NOS and NO metabolism [18]. Associated with alteration of fumarase, and the related fumarate and l-malate metabolites, we indeed showed that both STOX1 isoforms strongly affect the NO metabolism, in particular increasing the iNOS content but decreasing the iNOS coupling as could be also in placentas of PE patients [10,11]. These findings strengthen and help clarifying the link between STOX1 overexpression and gestational hypertension in preeclampsia.
The specificity of STOX1A in PE was supported by our findings that STOX1A/STOX1B isoforms oppositely deregulate the stabilization of the hypoxia marker HIF1 α, consistently with the opposite effects of STOX1A and STOX1B overexpression shown on trophoblast fusion [22]. In particular, we observed that STOX1A overexpression decreases l-malate and dysregulates the antioxidant defense with dramatically low levels of catalase in hypoxia-mimentic conditions. Our findings are consistent with the hypothesis that a STOX1-dependent vicious circle of iNOS uncoupling, leading to nitroso-redox imbalance and mitochondrial stress, is a major deregulated pathway in PE. In agreement with this picture, in hypoxia iNOS and NO levels were reduced in STOX1A overexpressing cells, whereas STOX1B overexpressing cells were able to maintain high levels of NO despite reduced amount of the NO synthase (iNOS). Related to iNOS alterations, both STOX1 isoforms strongly affected the nitroso-redox balance including peroxynitrite management, and the antioxidant defence remarkably increasing catalase activity. However, switch of catalase activity from over-activation to control levels in hypoxia mimetic condition, and without relevant changes in the enzyme content, was observed only upon STOX1A overexpression, further linking this factor with altered response upon hypoxia.
Both STOX1 isoforms strongly reduced citrate synthase (CS) activity, the first enzymatic and pacemaker step of the mitochondrial Krebs cycle, and CS activity was over-activated in hypoxia mimetic conditions, correlated with the activation of mitochondrial respiration. Despite these changes, the global increase of total ATP levels upon STOX1 overexpression appeared essentially due to anaerobic glycolysis. Again, an opposite effect was observed in hypoxia-mimetic conditions upon overexpression of the different STOX1 isoforms, with STOX1A furtherly enhancing total and mitochondrial ATP, whereas STOX1B increased mitochondrial ATP while reducing total ATP levels. These findings indicate that STOX1A overexpression induces a syndrome of preeclampsia of metabolic origin that is exacerbated in hypoxia.
Our findings place iNOS at a central position in PE-related symptoms and reveal iNOS as a major target for PE treatment. However, iNOS coupling (treatment with BH4) rather than iNOS inhibition (treatment with the non-specific NOS inhibitor l-NAME) appears effective in restoring altered control, non-preeclamptic values. In this context, alteration of the l-malate pathway (fumarate/fumarase/l-malate) and the bioenergetic metabolism upon l-NAME helps explaining why this treatment triggers PE symptoms including hypertension in treated rats and promotes, rather than cures, preeclamptic symptoms [8,75]. Moreover, BH4 (but not l-NAME, at least in STOX1B cells) regulated nitroso-redox stress through reduction of peroxynitrite. Further, BH4 increased ATP availability, whereas l-NAME dramatically reduced ATP levels in STOX1A overexpressing cells. Interestingly, a study suggested that NO produced by endothelial NOS (eNOS) is not relevant for the l-NAME effect, suggesting that l-NAME rather acts through iNOS modulation [76]. In our paradigm, however, iNOS inhibition did not restore key altered parameters. Conversely, modulating iNOS (via iNOS coupling) and mitochondrial stress with BH4 restored this multi-step pathway in vitro, also showing that the levels of l-malate and fumarase activity directly depend on NOS coupling and possibly mitochondrial stress.
Nevertheless, inhibition of NOS by l-NAME and increasing NOS coupling with the BH4 cofactor share common effects in vitro, which include a reduction of STOX1 protein accumulation, reduction of iNOS and the central metabolite acetyl-CoA, and stimulation of mitochondrial OXPHOS.
In our first pre-clinical model, we demonstrated that BH4 directly restores STOX1-deregulated gene expression in placenta including glutathione metabolism, oxidative phosphorylation, cholesterol metabolism, inflammation, lipoprotein metabolism and platelet activation, key pathways involved in preeclampsia [7,77,78]. Our results correlate with the metabolomic profiles of women preeclamptic placentas, indicating that glutathione metabolism is one of the key pathways to target in preeclampsia [7]. We show that BH4 is able to target this pathway. To note, we previously performed a transcriptomic analysis to understand the beneficial effects of low-dose aspirin treatment in this mouse model of preeclampsia, and aspirin is now clearly demonstrated to be efficient against preeclampsia [24,79,80]. The aspirin effects were exclusively achieved by alterations of placental gene expression that were disconnected from STOX1 effects, and massively down-regulated genes involved in coagulation, regulated by the HNF1β transcription factor [80]. By contrast, here we show that BH4 directly acts by rescuing STOX1-modulated gene expression. This suggest that BH4 is a more powerful and effective treatment than aspirin, and also that the combination of the two drugs could be in the future the most efficient direction towards a complete protection against preeclampsia symptoms. In the RUPP rats, we showed that the IUGR phenotype is restored by BH4, showing that in this LOPE model, extremely different from the STOX1 mouse model of EOPE, the therapy has beneficial effects as well.
Very recently, Chuaiphichai and coworkers demonstrated that a mouse model deficient for generating BH4, the Gch1 knock-out mice presents clearly with the preeclampsia syndrome features, and induces alterations of the uteroplacental vascular development essential for regulating the blood pressure during pregnancy [81]. These concordant findings strengthen our observations, since different causes of preeclampsia symptoms posit BH4 as an essential molecule in gestation.
In the STOX1 mice, gestational hypertension, proteinuria and heart hypertrophy are all prevented by BH4 therapy in our mouse model that also mimics the long-term cardiovascular complications associated with preeclampsia in women [3,27]. Given the long term cardiovascular effects of PE, and its association with a predisposition to at least strokes, chronic arterial hypertension, heart and renal dysfunction, this novel STOX1A/STOX1B-linked multi-step pathway and the associated BH4 therapy could be of interest also for these complications. Interestingly, STOX1A accumulates in the brain of late-onset Alzheimer's Disease (AD) patients [28] and induces Tau phosphorylation in AD [82]. If the same pathway controlled by STOX1 in preeclampsia is present in Alzheimer's disease, the rescue of STOX1A-dependent altered parameters by BH4 could be beneficial for such disease.
In addition to utilizing the STOX1 model, we strengthened our results implicating BH4 as a beneficial therapy to reduce maternal symptoms of EOPE by including the rat RUPP model that recapitulates LOPE. In this case, we could not induce a maternal amelioration of the blood pressure, while the foetal and placental weight were restored by the BH4 treatment.
4. Conclusions
In summary, STOX1 accumulation in a preeclampsia model impaired iNOS function and the related NO metabolism, which in turn deregulated a multi-step pathway including the nitroso-redox balance with peroxynitrite management, the antioxidant defence, the Krebs cycle including citrate synthase and fumarase activities, the fumarase-related metabolites l-malate and fumarate, and bioenergetic metabolism with acetyl-CoA, mitochondrial OXPHOS, and increased glycolysis to meet the ATP demand, altogether associated with dysregulation of hypoxia HIF1A stabilization. BH4 treatment increases iNOS coupling thereby reverting this STOX1A-dependent multi-step pathway in vitro, rescuing STOX1-deregulated gene expression in the placenta, and treating the major symptoms of preeclampsia in vivo. In an experimental animal model independent of STOX1, the RUPP rat, we showed that the treatment is efficient to correct the IUGR effects of this surgical procedure. Our discoveries suggest that BH4 is a promising therapy to cure adverse maternal-fetal symptoms associated to preeclampsia.
5. Material & methods
5.1. Animals experiments (breeeding, treatment and ethics)
The mouse work (female mice) was performed under the local regulations and ethic committees: Animal Care Committee of the Paris Descartes University (agreement no. 02731.02). Placentas were collected at 16.5 days post coïtum and placed in TriZol prior to RNA extraction [24,66]. BH4 (dose estimated through normal water consumption at 10 mg/kg/day) or pure water was given in the drinking water for two mice per cage by 15 ml frozen and changed twice a day during all the gestational period following the detection of pregnancy by ultrasonography between days E6.5 or E7.5, this dose having been chosen from previously published studies [83]. In more detail, the mice were breed in trios from Friday night to Monday morning with one male mouse (either WT or Tg42 transgenic – heterozygous-) and two WT female mice at 2 months of age, all in the FVB/N genetic background. Overall, they were 9 WT mice without treatment, 7 with BH4, 15 mice carrying transgenic embryos without BH4, and 12 treated with BH4.
To include the RUPP model, studies in rats were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with protocols approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center (agreement no. 1511). Timed-pregnant SAS Sprague-Dawley rats received from Charles River Laboratories (Wilmington, MA, USA) between gestational day 10 and 11 and placed on Envigo 8640 diet. On day 14, the RUPP procedure was performed by first placing rats under isoflurane anesthesia (Butler Schein Animal Health, Dublin, OH, USA). An abdominal incision was made and a silver clip (0.203 mm, internal diameter) placed around the sub-renal abdominal aorta above the uterine arteries along with a clip (0.1 mm, internal diameter) on each branch of the ovarian arteries (Fig. 8). The control group consisted of a Sham surgery on gestational day 14, without clip placement. BH4 was dosed in rats from day 13–18 of gestation. Total number of rats was 13 for Sham 13, 15 for RUPP, 6 for RUPP + BH4 3.5 mg/kg/day and 3 for RUPP + BH4 7 mg/kg/day. BH4 was reconstituted in dionized H2O and frozen as aliquots to administer 2X daily (mornings and evenings) at a final dose of 3.5 or 7 mg/kg body weight given orally by mixing into 0.5 mg peanut butter as vehicle. On gestational day 18, rats were implanted with carotid catheters under isoflurane anesthesia for assessment of conscious blood pressure on day 19 followed by anesthetizing and collecting tissues to weigh fetuses and placentas for calculating pregnancy biometrics. Also, aortas and uterine arteries were isolated and prepared to evaluate endothelium-dependent vasorelaxation in response to increasing concentrations of Acetylcholine. We have previously published specifics regarding these assays for blood pressure and vascular function in pregnant rats [84,85].
5.2. Systolic arterial pressure measurements
Blood pressure was measured by tail-cuff plethysmography using the CODA8 (four channels) device (EMKA Technologies). After five days to one week training, female mice were placed in restrainers on a warming platform (37 °C). The cuff releases progressively the pressure of the cuff and the apparatus records the volume variation of the tail, algorithmically determining the systolic and diastolic pressure, with at least 8 satisfactory measurements per day, taken at the same hour of the day. Systolic and diastolic BP always displayed similar curve profiles in all groups analyzed, therefore only systolic BP is shown.
5.3. Assessment of albumin/creatinine ratio (ACR) in urine
Urine was collected by soft abdominal pressure on the female mice to induce urination, and the samples were stored at −80 °C until use. However, urine samples were difficult to collect from all females at every time point, and therefore results were pooled within each group for each gestational period (early, mid and late). ACR was measured by ELISA using the Albuwell M kit (#1011, Ethos Biosciences) following accurately the specifications of the manufacturer. For ACR ratio, data are from n = 7 for WTxWT, n = 5 for WTxWT + BH4, n = 10 for WTxhtTgSTOX42 and n = 9 for WTxhtTgSTOX42+BH4.
5.4. Heart weight and tibia length measurements
At days 16.5, a part of the mice in each group were sacrificed. The hearts were collected, the blood removed by pressure on an absorbing paper, and weighted. The posterior right tibia was dissected free and measured, to caculate the heart weight to tibia length ratio. Data are from data from n = 6 for WTxWT, n = 6 for WTxWT + BH4, n = 4 for WTxhtTgSTOX42 and n = 6 for WTxhtTgSTOX42+BH4.
5.5. Placental tanscriptome
Total RNAs from pools of three placenta from different mice in the four conditions were used (Ctrl, Ctrl + BH4, STOX1 (preeclamptic-mimic) mice, and STOX1+BH4), and hybridized to ClariomS mice microarrays (#902931, ThermoFisher Scientific), that interrogate more than 22,000 annotated genes. The microarray data was deposited in the EBI database for open accessibility to all the interested researchers. The analyses were carried out using the TAC tool from Affymetrix and the online tool WebGestalt [60] which makes it possible to carry out straightforwardly Gene Set Enrichment Analyses and confront them to Gene Ontology, Molecular Processes or KEGG databases. STRING protein networks analysis was performed using https://string-db.org/cgi/network.pl. Complete transcriptomic data are available under the EBI accession number E-MTAB-9314.
5.6. Cell culture and treatments
The cell line overexpressing STOX1A (AA6, overexpressing STOX1A) was described by Rigourd et al., 2008 [23], as well as the control (BD3). In the present study we developed another cell line overexpressing STOX1B (B10). For this, we used plasmids encompassing the STOX1B isoform of STOX1, under the control of the CMV promoter. The cells (cultivated in DMEM GlutaMax (GIBCO-Life technologies) + penicillin/streptomycin and 10% heat-decomplemented Fetal Calf Serum, GIBCO-Life technologies), were transfected in the same time with a plasmid encoding a resistance gene to geneticin G-418 in a 1:20 proportion (for a 60 cm2 plate, 10 μg of STOX1 expression plasmid and 0.5 μg of resistance plasmid) to ascertain that the two plasmids entered the cells following transfection with lipofectamin 2000 (GIBCO-Life technologies) in standard conditions. Selection was carried out ∼3 weeks at 500 μg/ml G418 (GIBCO-Life technologies). Resistant clones were isolated and cultivated separately, amplified and maintained in DMEM with Geneticin. The expression of STOX1B was tested by a specific RT-qPCR.
Cells were treated with 0.2 mM cobalt chloride for 24 h to mimic hypoxia-induced HIF1A stabilization, with or without 100 μM BH4 (T4425, Sigma Aldrich) or for 24h with 100 μM l-NAME (N5751, Sigma Aldrich).
5.7. Total RNA extraction and RT-qPCR
Total RNA was isolated from cells using the RNAeasy kit (Qiagen), then reverse-transcribed with SuperScriptIV Reverse transcriptase (Invitrogen). Real-time quantitative PCR was performed using PowerUp Sybr Green PCR Master Mix (ThermoFisher) and the rate of dye incorporation was monitored using the StepOne Plus RealTime PCR system (Applied Biosystems). Three biological replicates were used for each condition. Data were analyzed by StepOne Plus RT PCR software v2.1 and Microsoft Excel. TBP transcript levels were used for normalization of each target (=ΔCT). Real-time PCR CT values were analyzed using the 2-ΔΔCt method to calculate the fold expression. qPCR Primers used to detect STOX1 isoforms mRNA are: STOX1A forward- GCTCTTTGTGCCTTCGACAT, reverse-ATTCTCCACGGACACAGAGT and STOX1B forward-ATGCCAGCTTCCATGACATAT, reverse-TGCCTTCGACATTTTGCAGAA. The primers for STOX1B are expected to amplify a 100bp fragment on STOX1B transcript and not a 2265 bp fragment on STOX1A, which does not occur in RT-qPCR experiments, with elongation time limited to 10 s per cycle.
6. Immunofluorescence, immunoblot, reagents, and antibodies
Cells plated on glass slides were fixed with 2% (wt/vol) paraformaldehyde (PFA) and permeabilized with 0.5% Triton X-100. The glass slides were incubated in blocking buffer [BSA 5% (wt/vol) in PBS] overnight at 4 °C then 1 h at room temperature with the primary antibody, and finally with the secondary antibody and 10 μg/ml Hoechst 33,342 for 1 h at room temperature. STOX1A/B (HPA037845), HIF1A (SAB2101039), MnSOD/SOD2 (polyclonal rabbit-Cy3, S1450), Catalase (SAB4503383), DAF2DA (4,5-Diaminofluorescein Diacetate, D2813), DCF-DA (2′,7′-Dichlorodihydrofluorescein diacetate, D6883), DHE (Dihydroethidium, D7008), DHR123 (Dihydrorhodamine 123, D1054) and Hoechst 33,342 were from Sigma-Aldrich, iNOS/NOS2 (ABN26) from Merck, GAPDH (SC-25778) from Santa Cruz Biotechnology, Fumarase (A21981, Life Technologies), mitoSOX Red (M36008), goat anti-rabbit Alexa Fluor 488-conjugated, goat anti-mouse Alexa Fluor 555-conjugated, goat anti-rabbit HRP-conjugated and goat anti-mouse HRP-conjugated secondary antibodies from Thermo Fisher Scientific.
7. Detection of nitric oxide, the ROS O2− and H2O2, the RNS ONOO− and mitochondrial ROS
Fluorescent molecules were used to measure NO (DAF2DA), ROS anion superoxide O2− (DHE), ROS hydrogen peroxide H2O2 (DCF-DA) and RNS peroxynitrite ONOO− (DHR123) in freshly extracted total proteins. This was done in standard culture conditions, by spectrofluorometry and using microplate reader TECAN Infinite 200. Mitochondrial ROS (anion superoxide O2−) were detected using MitoSOX Red after 1h incubation at 37 °C, and fluorescence intensity was quantified after 3D-confocal microscopy acquisition and reconstruction. Experiments were done in triplicate.
7.1. Fumarate, l-malate and acetyl-coA content measurements
The fumarate content was measured with the Fumarate Assay Kit (Colorimetric assay, MAK060-1 KT, Sigma-Aldrich), the l-malate content with the Malate Assay Kit (Colorimetric assay, MAK067-1 KT, Sigma-Aldrich), and the Acetyl-CoA content with the Acetyl-Coenzyme A Assay Kit (Fluorometric assay, MAK039-1 KT, Sigma-Aldrich) following the manufacturer's instructions. Tests were performed with freshly extracted total proteins. Experiments were done in triplicate.
8. Total ATP measurement and OXPHOS contribution
For ATP levels, cells were treated with 10 μM oligomycin for 1 h (for glycolytic ATP) or untreated (for total ATP levels); then, ATP levels were assessed with the CellTiter-Glo Luminescent assay (Promega), according to supplier instructions.
8.1. Citrate synthase activity, fumarase actvity, SOD activity and catalase activity
Citrate synthase activity was measured with the Citrate Synthase activity Colorimetric Assay kit (K318-100, BioVision). In the same plate, Fumarase levels were tested with sandwich ELISA assay before total and specific fumarase activities with the Fumarase Specific Activity Assay kit (Colorimetric assay, AB110043, ABCAM), Total SOD activity was assessed with the Superoxide Dismutase Activity Colorimetric Assay kit (AB65354, ABCAM) that measures the inhibition activity of xantine oxidase by SOD, and catalase activity with the Catalase Assay kit (AB83464, ABCAM) following the manufacturer's instructions. Tests were performed on freshly extracted total proteins. Experiments were done in triplicate.
8.2. Three-dimensional confocal acquisition, reconstruction, and quantification
For STOX1A/B, iNOS/NOS2, Fumarase, MnSOD/SOD2, Catalase and MitoSOX Red imaging, confocal acquisitions were performed using a spinning-disk Yokogawa CellVoyager CV1000 with a 40X air objective (BioImagerie Photonique UTechS PBI, Institut Pasteur, Paris). Optical slices were taken each 1 μM interval along the z axis covering the whole depth of the cell, at resolution of 1.024/1.024 pixels. Each image was 3D-reconstructed using Maximum Intensity Projection (MIP). Scale bar = 10 μm. Fluorescence quantification was done using a single-imaging frame collection and FIJI ImageJ software (postacquisition analysis). For each condition, 60 cells were analyzed from three independent experiments.
8.3. Protein extraction and immunoblot
Cells were lysed with lysis buffer (50 mM Tris⋅HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM DTT, and protease inhibitor mixture). Lysed cells were not centrifuged, and the whole extract was collected. The protein content was determined with the Bradford reagent (Sigma-Aldrich), and 20 μg of protein were loaded for SDS/PAGE. After blotting, Trans-blot Turbo nitrocellulose membranes (Bio-rad) were probed with primary antibodies, then with HRP-conjugated secondary antibodies. Detection was performed using Clarity Western ECL substrate (Bio-Rad) and ChemiDoc Imaging system (Bio-rad). Protein loading reference was done with GAPDH or with Ponceau. Experiments were done in triplicate, and a representative immunoblot was shown.
8.4. Statistical analysis
The significance of differences between data were determined using one-way ANOVA; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. On cell experiments, Dunnett post-hoc tests were first carried out to compare each group with the untreated BD3 cells, then Student-Newman-Keuls post-hoc tests were performed to identify putative effects between groups the ANOVA yielded a significant p value. For the animal experiments, the non-treated WT females were used as a reference group for post-hoc Dunnett's tests, after ANOVA analysis for each parameter measured.
Author contributions
LC and AD were the major contributors to the experimental data on the cell models; these data were anaysed by LC, AD, MR, and DV. Additional experimental data on the cells were obtained by BC and IG. CC, and AD were at the origin of the first data on oxidative stress metabolites in the cells. AD and DV carried out the experiments on the animal model. DV and FM carried out the transcriptome analysis. SJ from the GENOM’IC platform carried out the transcriptome experiments. IL was in charge of collecting the ultrasound data. The studies in rats were conducted and overseen by FTS, ACP, KCT, ARW, JPG, and DV conceived the experiment. LC, MR and DV drafted the paper.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank the « BioImagerie Photonique UTechS PBI » of Institut Pasteur. This work was supported by The ‘Fondation de France’ through a prize ‘Coeur des Femmes’ attributed to Daniel Vaiman, and through a PRIDE funding from the Département Hospitalo-Universitaire ‘Risques et Grossesse’. Research reported in, and effort toward preparing, this publication was supported by the National Institutes of Health under Award Numbers: P20GM121334 (FTS, JPG, LMA), R00HL130577 (FTS), R01HL148191 (ACP, JPG), R56HL157579 (FTS, JPG), T32HL105324 (JPG), U54GM115428 (JPG). It was also supported by the American Heart Association and Comanche Biopharma in Award Numbers: 19CDA34670055 (LMA) and AWD-001111 (FTS). The content is solely the responsibility of these authors and does not necessarily represent the official views of these funding organizations.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102406.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Hypoxia and STOX1 overexpression affect the the fumarate-dependent L-malate pathway. Quantitative RT-qPCR of (A)STOX1A and (B)STOX1B mRNAs. (C) 3D reconstructions of cells immunostained for STOX1 (green, detection of both STOX1A and STOX1B) counterstained with Hoechst (blue). (D) Quantification of STOX1 (both STOX1A and STOX1B) fluorescence intensity per cell. (E) 3D reconstructions of cells immunostained for fumarase in red. (F) Quantification of fumarase fluorescence intensity per cell. (G) Fumarase quantitative assay measured with sandwich ELISA assay, expressed in percentage relative to the untreated control BD3 cell line. (H) Total fumarase activity (measured with a colorimetric assay) expressed as percentage of the untreated control BD3 cell line. RT-qPCR, quantitative assay (sandwich ELISA for fumarase) and enzyme activity (fumarase), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from 3 independent experiments, scale bar 10μm, mean ± SD. Immunoblot is representative of 3 independent experiments. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars).
{kind=link}
NO metabolism and antioxidant defense upon STOX1 overexpression and hypoxia.(A) 3D reconstructions of cells immunostained for iNOS (green), counterstained with Hoechst (blue) (quantification in Fig. 2A). (B) 3D reconstructions of cells immunostained for mitochondrial MnSOD/SOD2 (red), counterstained with Hoechst (blue). (C) Quantification of mitochondrial MnSOD/SOD2 fluorescence intensity per cell. (D) ROS hydrogen peroxide (H2O2) detected with the DCFDA probe expressed as percentage relative to the untreated control BD3 cell line. (E) 3D reconstructions of cells immunostained for catalase (green), counterstained with Hoechst (blue). (F) Quantification of catalase fluorescence intensity per cell. Dosage (H2O2), n= 6 for untreated BD3/AA6/B10 in H2O2 assays n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from 3 independent experiments, scale bar 10μm, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars).
{kind=link}
The BH4 metabolite counteracts STOX1 effects through iNOS regulation and rescue of L-malate levels. Quantitative RT-qPCR of (A) STOX1A and (B) STOX1B. (C) 3D reconstructions of cells immunostained for STOX1 (green, STOX1A/B), counterstained with Hoechst (blue). (D) Quantification of STOX1 fluorescence intensity per cell. (E) 3D reconstructions of cells immunostained for iNOS (green), counterstained with Hoechst (blue). (F) ROS hydrogen peroxide (H2O2) expressed as percent of the untreated control BD3 cell line. (G) 3D reconstructions of cells stained for mitochondrial ROS with MitoSOX (red), counterstained with Hoechst (blue). Data of untreated cells in panel A, B, D are from the corresponding panels in Fig. S1. RT-qPCR and dosage (H2O2), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (pink or purple stars).
{kind=link}
The BH4 metabolite counteracts STOX1 alteration of the antioxidant defense.(A) 3D reconstructions of cells immunostained for SOD2/MnSOD (red), counterstained with Hoechst (blue). (B) Quantification of SOD2/MnSOD fluorescence intensity per cell. (C) Total SOD activity (measured as inhibition of the activity of SOD, percentage). (D) 3D reconstructions of cells immunostained for catalase (green), counterstained with Hoechst (blue). (E) Quantification of catalase fluorescence intensity per cell. (F) Catalase activity expressed in nmol/min/ml. Data of untreated cells are from the corresponding panels in Fig. S2C (SOD2), Fig. 2E (SOD activity), Fig. S2F (catalase levels), Fig. 2F (catalase activity). Enzyme activities (SOD and catalase), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (black stars) or the corresponding untreated condition (pink or purple stars).
{kind=link}
The BH4 metabolite and L-NAME increase the mitochondrial ATP fraction in STOX1A overexpressing cells.(A) Citrate synthase activity expressed in mU/mg of protein. (B) Acetyl-CoA expressed in nmol/g of protein, blue histograms are for non-treated cells in chemical hypoxia. (C) Percentage of the OXPHOS fraction of ATP. Data of untreated cells are from the corresponding panels in Fig. 3D (citrate synthase activity), Fig. 3E (Acetly-CoA), Fig. 3F (ATP levels), Fig. 3F (OXPHOS fraction). (D) Total ATP in the various cells under BH4 and LNAME treatments with estimation of the part attributable to the respiratory chain (OXPHOS). For the dosages (ATP and acetyl-CoA), and the enzyme activity (citrate synthase), mean ± SD of n = 3 independent experiments are represented,. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA, and post hoc Dunnett test using the untreated control as reference (black stars, on D, red stars on A, B, and C) or the corresponding untreated condition (pink or purple stars).
{kind=link}
The BH4 metabolite and L-NAME increase fumarase activity in STOX1A overexpressing cells.(A) 3D reconstructions of cells immunostained for fumarase (red), counterstained with Hoechst (blue) and (B) quantification of fumarase fluorescence intensity per cell. (C) Fumarase (measured by sandwich ELISA assay) expressed as percentage of the untreated control BD3 cell line. (D) Percentage of fumarase activity relative to the untreated control BD3 cell line. Data of untreated are from Fig. S1F (fumarase), Fig. S1G (fumarase quantity), Fig. S1H (fumarase activity). Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. Quantity assay by sandwich ELISA (fumarase), and enzyme (fumarase) activity, n = 3 independent experiments, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001; based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (pink or purple stars).
{kind=link}
Network enrichment of genes modified by STOX1 and corrected by BH4 treatment in mice placenta. The genes of figure 5E were significantly associated in a network (PPI = 3.7 10-8). The enrichment in molecular functions revealed enrichment of the following terms: Hormone activity, Cholesterol transporter activity, and several Gene Ontology terms connected with lipoprotein physiology. In terms of Biological processes, meaningfully enriched terms were Protein oxidation, Response to stress, and Homeostatic processes.
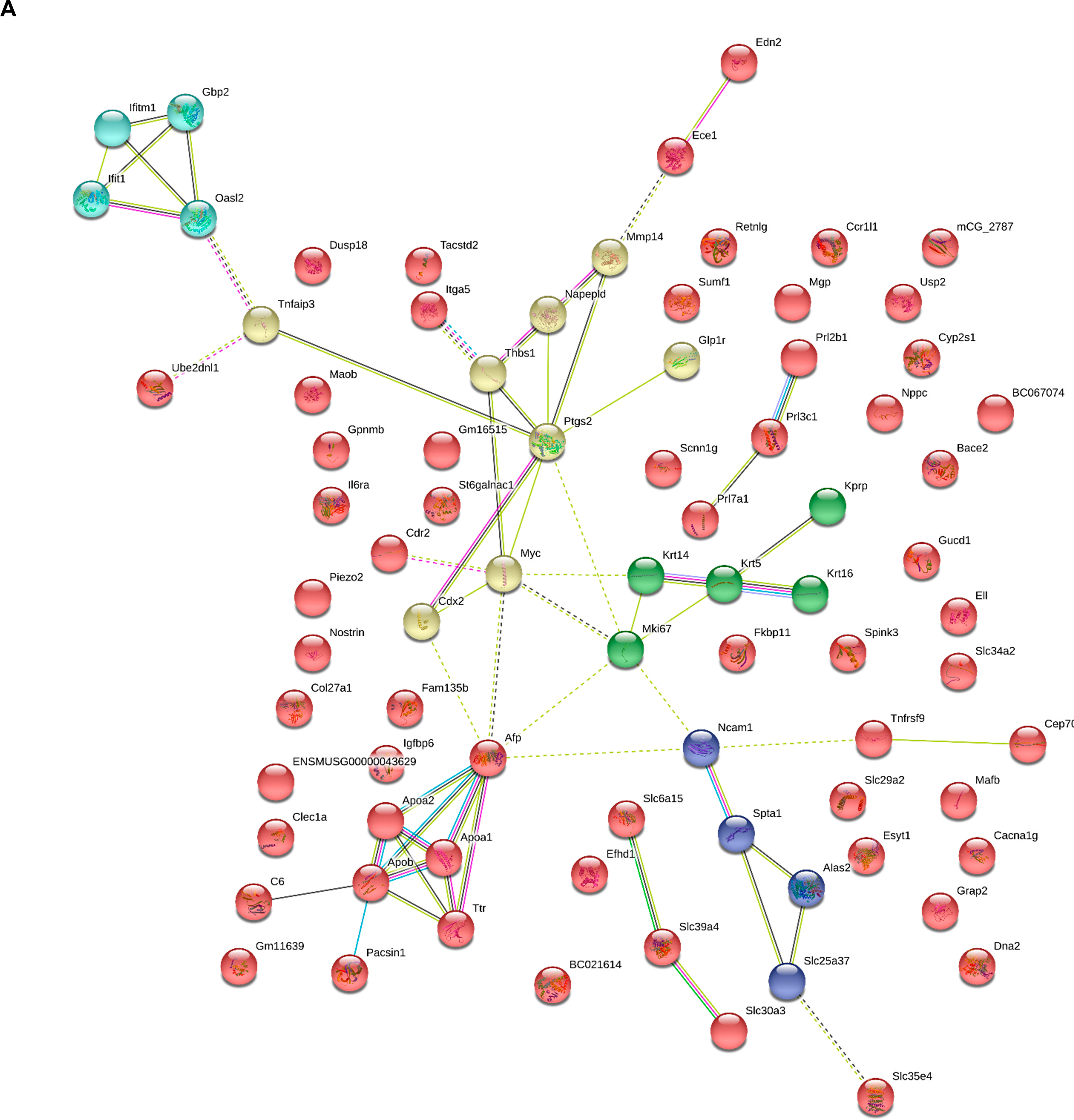{kind=link}
Modulations of blood pressure in early, mid and late gestation in mice. Systolic arterial pressure in periods E1.5-5.5, E6.5-12.5 and E13.5-18.5 during gestation expressed in mm Hg relative to pre-gestation. Data from n= 9 for WTxWT, n=7 for WTxWT +BH4, n=16 for WTxhtTgSTOX42, and n=12 for WTxhtTgSTOX42 +BH4. Mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (black stars) or the corresponding untreated condition (pink stars).
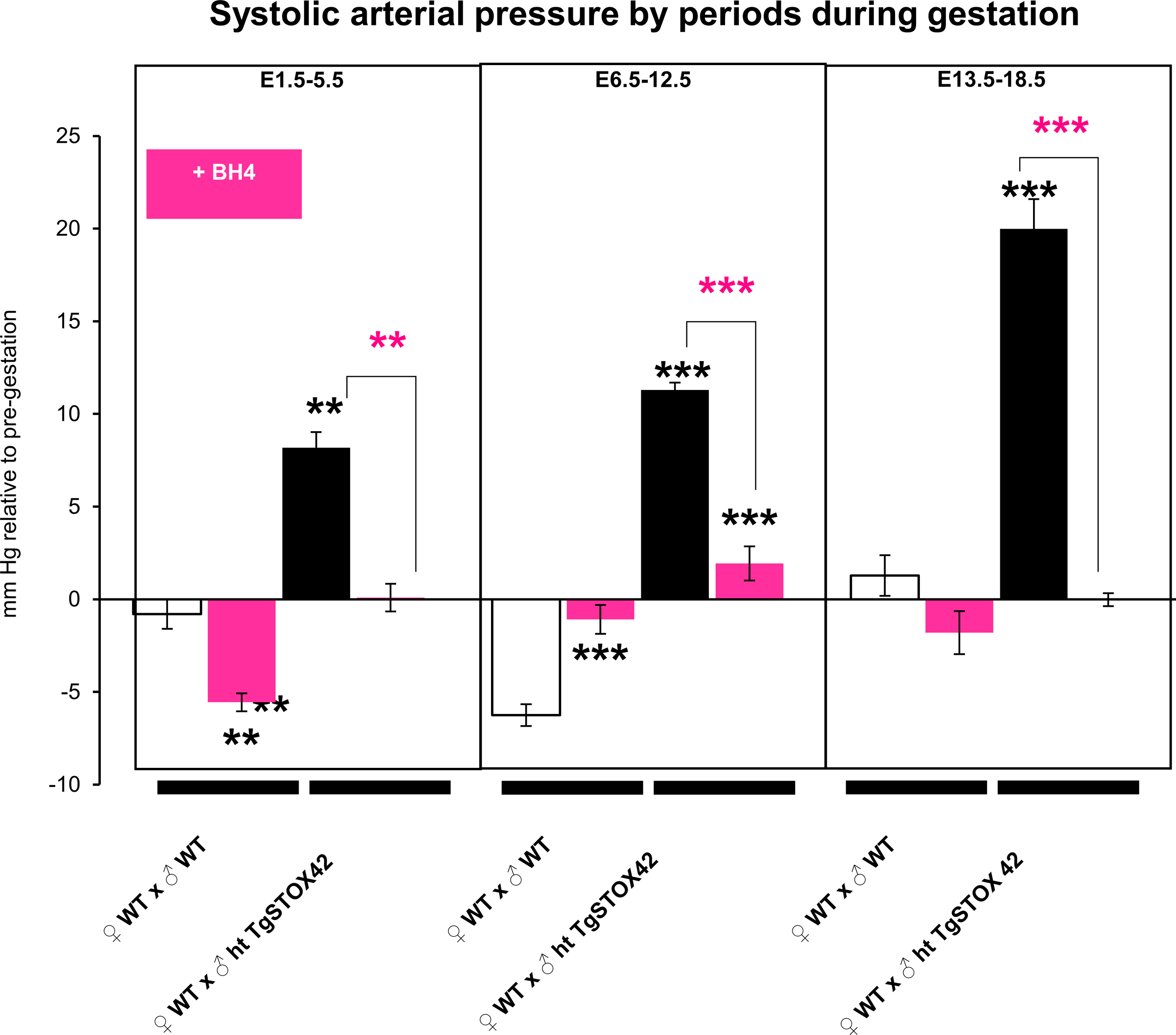{kind=link}
Full membrane immunoblots.
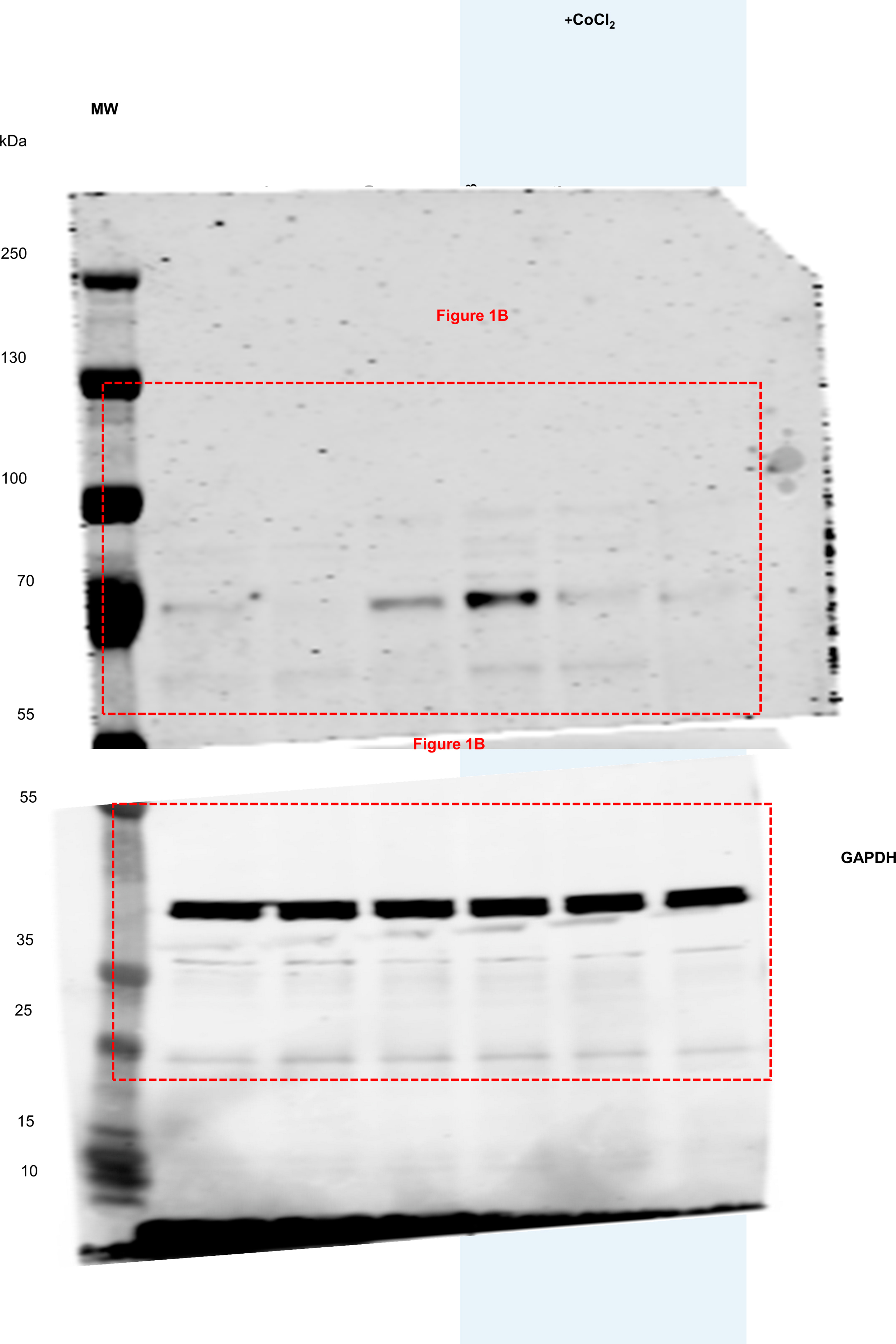{kind=link}
References
- 1.Phipps E.A., Thadhani R., Benzing T., Karumanchi S.A. Pre-eclampsia: pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019;15:275–289. doi: 10.1038/s41581-019-0119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen C.W., Jaffe I.Z., Karumanchi S.A. Pre-eclampsia and cardiovascular disease. Cardiovasc. Res. 2014;101:579–586. doi: 10.1093/cvr/cvu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basit S., Wohlfahrt J., Boyd H.A. Pre-eclampsia and risk of dementia later in life: nationwide cohort study. BMJ. 2018;363:k4109. doi: 10.1136/bmj.k4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta S., Agarwal A., Sharma R.K. The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet. Gynecol. Surv. 2005;60:807–816. doi: 10.1097/01.ogx.0000193879.79268.59. [DOI] [PubMed] [Google Scholar]
- 5.Myatt L., Cui X. Oxidative stress in the placenta. Histochem. Cell Biol. 2004;122:369–382. doi: 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy C., Kenny L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016;6 doi: 10.1038/srep32683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawasaki K., Kondoh E., Chigusa Y., Kawamura Y., Mogami H., Takeda S., Horie A., Baba T., Matsumura N., Mandai M., et al. Metabolomic profiles of placenta in preeclampsia. Hypertension. 2019;73:671–679. doi: 10.1161/HYPERTENSIONAHA.118.12389. [DOI] [PubMed] [Google Scholar]
- 8.Yallampalli C., Garfield R.E. Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am. J. Obstet. Gynecol. 1993;169:1316–1320. doi: 10.1016/0002-9378(93)90299-x. [DOI] [PubMed] [Google Scholar]
- 9.Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amaral L.M., Pinheiro L.C., Guimaraes D.A., Palei A.C., Sertorio J.T., Portella R.L., Tanus-Santos J.E. Antihypertensive effects of inducible nitric oxide synthase inhibition in experimental pre-eclampsia. J. Cell Mol. Med. 2013;17:1300–1307. doi: 10.1111/jcmm.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du L., He F., Kuang L., Tang W., Li Y., Chen D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J. Hum. Hypertens. 2017;31:49–55. doi: 10.1038/jhh.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palei A.C., Granger J.P., Spradley F.T. Placental ischemia says "NO" to proper NOS-mediated control of vascular tone and blood pressure in preeclampsia. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222011261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J., Druhan L.J., Zweier J.L. Reactive oxygen and nitrogen species regulate inducible nitric oxide synthase function shifting the balance of nitric oxide and superoxide production. Arch. Biochem. Biophys. 2010;494:130–137. doi: 10.1016/j.abb.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey J., Shaw A., Fischer R., Ryan B.J., Kessler B.M., McCullagh J., Wade-Martins R., Channon K.M., Crabtree M.J. A novel role for endothelial tetrahydrobiopterin in mitochondrial redox balance. Free Radic. Biol. Med. 2017;104:214–225. doi: 10.1016/j.freeradbiomed.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinho S., Adao R., Leite-Moreira A.F., Bras-Silva C. Persistent pulmonary hypertension of the newborn: pathophysiological mechanisms and novel therapeutic approaches. Front. Pediatr. 2020;8:342. doi: 10.3389/fped.2020.00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanet H., Capuron L., Castanon N., Calon F., Vancassel S. Tetrahydrobioterin (BH4) pathway: from metabolism to neuropsychiatry. Curr. Neuropharmacol. 2021;19:591–609. doi: 10.2174/1570159X18666200729103529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kukor Z., Valent S., Toth M. Regulation of nitric oxide synthase activity by tetrahydrobiopterin in human placentae from normal and pre-eclamptic pregnancies. Placenta. 2000;21:763–772. doi: 10.1053/plac.2000.0584. [DOI] [PubMed] [Google Scholar]
- 18.Hou E., Sun N., Zhang F., Zhao C., Usa K., Liang M., Tian Z. Malate and aspartate increase L-arginine and nitric oxide and attenuate hypertension. Cell Rep. 2017;19:1631–1639. doi: 10.1016/j.celrep.2017.04.071. [DOI] [PubMed] [Google Scholar]
- 19.Camarena Pulido E.E., Garcia Benavides L., Panduro Baron J.G., Pascoe Gonzalez S., Madrigal Saray A.J., Garcia Padilla F.E., Totsuka Sutto S.E. Efficacy of L-arginine for preventing preeclampsia in high-risk pregnancies: a double-blind, randomized, clinical trial. Hypertens. Pregnancy. 2016;35:217–225. doi: 10.3109/10641955.2015.1137586. [DOI] [PubMed] [Google Scholar]
- 20.van Dijk M., Mulders J., Poutsma A., Konst A.A., Lachmeijer A.M., Dekker G.A., Blankenstein M.A., Oudejans C.B. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat. Genet. 2005;37:514–519. doi: 10.1038/ng1541. [DOI] [PubMed] [Google Scholar]
- 21.Vaiman D., Miralles F. Targeting STOX1 in the therapy of preeclampsia. Expert Opin. Ther. Targets. 2016;20:1433–1443. doi: 10.1080/14728222.2016.1253682. [DOI] [PubMed] [Google Scholar]
- 22.Ducat A., Couderc B., Bouter A., Biquard L., Aouache R., Passet B., Doridot L., Cohen M.B., Ribaux P., Apicella C., et al. Molecular mechanisms of trophoblast dysfunction mediated by imbalance between STOX1 isoforms. iScience. 2020;23 doi: 10.1016/j.isci.2020.101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rigourd V., Chauvet C., Chelbi S.T., Rebourcet R., Mondon F., Letourneur F., Mignot T.M., Barbaux S., Vaiman D. STOX1 overexpression in choriocarcinoma cells mimics transcriptional alterations observed in preeclamptic placentas. PLoS One. 2008;3 doi: 10.1371/journal.pone.0003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doridot L., Passet B., Mehats C., Rigourd V., Barbaux S., Ducat A., Mondon F., Vilotte M., Castille J., Breuiller-Fouche M., et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension. 2013;61:662–668. doi: 10.1161/HYPERTENSIONAHA.111.202994. [DOI] [PubMed] [Google Scholar]
- 25.Ducat A., Doridot L., Calicchio R., Mehats C., Vilotte J.L., Castille J., Barbaux S., Couderc B., Jacques S., Letourneur F., et al. Endothelial cell dysfunction and cardiac hypertrophy in the STOX1 model of preeclampsia. Sci. Rep. 2016;6 doi: 10.1038/srep19196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doridot L., Chatre L., Ducat A., Vilotte J.L., Lombes A., Mehats C., Barbaux S., Calicchio R., Ricchetti M., Vaiman D. Nitroso-redox balance and mitochondrial homeostasis are regulated by STOX1, a pre-eclampsia-associated gene. Antioxidants Redox Signal. 2014;21:819–834. doi: 10.1089/ars.2013.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miralles F., Collinot H., Boumerdassi Y., Ducat A., Duche A., Renault G., Marchiol C., Lagoutte I., Bertholle C., Andrieu M., et al. Long-term cardiovascular disorders in the STOX1 mouse model of preeclampsia. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-48427-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Dijk M., van Bezu J., Poutsma A., Veerhuis R., Rozemuller A.J., Scheper W., Blankenstein M.A., Oudejans C.B. The pre-eclampsia gene STOX1 controls a conserved pathway in placenta and brain upregulated in late-onset Alzheimer's disease. J. Alzheimers Dis. 2010;19:673–679. doi: 10.3233/JAD-2010-1265. [DOI] [PubMed] [Google Scholar]
- 29.Tripathi V.K., Subramaniyan S.A., Hwang I. Molecular and cellular response of Co-cultured cells toward cobalt chloride (CoCl2)-induced hypoxia. ACS Omega. 2019;4:20882–20893. doi: 10.1021/acsomega.9b01474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srinivasan S., Dunn J.F. Stabilization of hypoxia-inducible factor-1alpha in buffer containing cobalt chloride for Western blot analysis. Anal. Biochem. 2011;416:120–122. doi: 10.1016/j.ab.2011.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai Z.J., Gao J., Ma X.B., Yan K., Liu X.X., Kang H.F., Ji Z.Z., Guan H.T., Wang X.J. Up-regulation of hypoxia inducible factor-1alpha by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J. Exp. Clin. Cancer Res. 2012;31:28. doi: 10.1186/1756-9966-31-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iommarini L., Porcelli A.M., Gasparre G., Kurelac I. Non-canonical mechanisms regulating hypoxia-inducible factor 1 alpha in cancer. Front. Oncol. 2017;7:286. doi: 10.3389/fonc.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaacs J.S., Jung Y.J., Mole D.R., Lee S., Torres-Cabala C., Chung Y.L., Merino M., Trepel J., Zbar B., Toro J., et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 34.Pollard P.J., Briere J.J., Alam N.A., Barwell J., Barclay E., Wortham N.C., Hunt T., Mitchell M., Olpin S., Moat S.J., et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 35.Sudarshan S., Linehan W.M., Neckers L. HIF and fumarate hydratase in renal cancer. Br. J. Cancer. 2007;96:403–407. doi: 10.1038/sj.bjc.6603547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bardella C., Olivero M., Lorenzato A., Geuna M., Adam J., O'Flaherty L., Rustin P., Tomlinson I., Pollard P.J., Di Renzo M.F. Cells lacking the fumarase tumor suppressor are protected from apoptosis through a hypoxia-inducible factor-independent, AMPK-dependent mechanism. Mol. Cell Biol. 2012;32:3081–3094. doi: 10.1128/MCB.06160-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng X., Chen M., Li X., Yang P., Zhao X., Ouyang Y., Yang Z., Liang M., Hou E., Tian Z. Insufficient fumarase contributes to hypertension by an imbalance of redox metabolism in Dahl salt-sensitive rats. Hypertens. Res. 2019;42:1672–1682. doi: 10.1038/s41440-019-0290-y. [DOI] [PubMed] [Google Scholar]
- 38.Mateo J., Garcia-Lecea M., Cadenas S., Hernandez C., Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003;376:537–544. doi: 10.1042/BJ20031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strijdom H., Muller C., Lochner A. Direct intracellular nitric oxide detection in isolated adult cardiomyocytes: flow cytometric analysis using the fluorescent probe, diaminofluorescein. J. Mol. Cell. Cardiol. 2004;37:897–902. doi: 10.1016/j.yjmcc.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X., Kim W.S., Hatcher N., Potgieter K., Moroz L.L., Gillette R., Sweedler J.V. Interfering with nitric oxide measurements. 4,5-diaminofluorescein reacts with dehydroascorbic acid and ascorbic acid. J. Biol. Chem. 2002;277:48472–48478. doi: 10.1074/jbc.M209130200. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes D.C., Wosniak J., Jr., Pescatore L.A., Bertoline M.A., Liberman M., Laurindo F.R., Santos C.X. Analysis of DHE-derived oxidation products by HPLC in the assessment of superoxide production and NADPH oxidase activity in vascular systems. Am. J. Physiol. Cell Physiol. 2007;292:C413–C422. doi: 10.1152/ajpcell.00188.2006. [DOI] [PubMed] [Google Scholar]
- 42.Chatre L., Biard D.S., Sarasin A., Ricchetti M. Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E2910–E2919. doi: 10.1073/pnas.1422264112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markiewicz R., Litowczenko J., Gapinski J., Wozniak A., Jurga S., Patkowski A. Nanomolar nitric oxide concentrations in living cells measured by means of fluorescence correlation spectroscopy. Molecules. 2022;27 doi: 10.3390/molecules27031010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulandavelu S., Dulce R.A., Murray C.I., Bellio M.A., Fritsch J., Kanashiro-Takeuchi R., Arora H., Paulino E., Soetkamp D., Balkan W., et al. S-nitrosoglutathione reductase deficiency causes aberrant placental S-nitrosylation and preeclampsia. J. Am. Heart Assoc. 2022;11 doi: 10.1161/JAHA.121.024008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gheddouchi S., Mokhtari-Soulimane N., Merzouk H., Bekhti F., Soulimane F., Guermouche B., Meziane Tani A., Narce M. Low SOD activity is associated with overproduction of peroxynitrite and nitric oxide in patients with acute coronary syndrome. Nitric Oxide. 2015;49:40–46. doi: 10.1016/j.niox.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 46.Miller A.F. Superoxide dismutases: ancient enzymes and new insights. FEBS Lett. 2012;586:585–595. doi: 10.1016/j.febslet.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Branicky R., Noe A., Hekimi S. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018;217:1915–1928. doi: 10.1083/jcb.201708007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cortese-Krott M.M., Koning A., Kuhnle G.G.C., Nagy P., Bianco C.L., Pasch A., Wink D.A., Fukuto J.M., Jackson A.A., van Goor H., et al. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxidants Redox Signal. 2017;27:684–712. doi: 10.1089/ars.2017.7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glorieux C., Calderon P.B. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017;398:1095–1108. doi: 10.1515/hsz-2017-0131. [DOI] [PubMed] [Google Scholar]
- 50.Gebicka L., Didik J. Catalytic scavenging of peroxynitrite by catalase. J. Inorg. Biochem. 2009;103:1375–1379. doi: 10.1016/j.jinorgbio.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 51.Bauer G. Increasing the endogenous NO level causes catalase inactivation and reactivation of intercellular apoptosis signaling specifically in tumor cells. Redox Biol. 2015;6:353–371. doi: 10.1016/j.redox.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arena G., Cisse M.Y., Pyrdziak S., Chatre L., Riscal R., Fuentes M., Arnold J.J., Kastner M., Gayte L., Bertrand-Gaday C., et al. Mitochondrial MDM2 regulates respiratory complex I activity independently of p53. Mol. Cell. 2018;69:594–609 e598. doi: 10.1016/j.molcel.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larsen S., Nielsen J., Hansen C.N., Nielsen L.B., Wibrand F., Stride N., Schroder H.D., Boushel R., Helge J.W., Dela F., et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pietrocola F., Galluzzi L., Bravo-San Pedro J.M., Madeo F., Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metabol. 2015;21:805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 55.Williams N.C., O'Neill L.A.J. A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front. Immunol. 2018;9:141. doi: 10.3389/fimmu.2018.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown G.C., Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 2007;75:283–290. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 58.Litvinova L., Atochin D.N., Fattakhov N., Vasilenko M., Zatolokin P., Kirienkova E. Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 2015;6:20. doi: 10.3389/fphys.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T., Zhang M., Mukosera G.T., Borchardt D., Li Q., Tipple T.E., Ishtiaq Ahmed A.S., Power G.G., Blood A.B. L-NAME releases nitric oxide and potentiates subsequent nitroglycerin-mediated vasodilation. Redox Biol. 2019;26 doi: 10.1016/j.redox.2019.101238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zimmermann K., Opitz N., Dedio J., Renne C., Muller-Esterl W., Oess S. NOSTRIN: a protein modulating nitric oxide release and subcellular distribution of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 2002;99:17167–17172. doi: 10.1073/pnas.252345399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunt G.J., Freytag S., Bahlo M., Gagnon-Bartsch J.A. dtangle: accurate and robust cell type deconvolution. Bioinformatics. 2019;35:2093–2099. doi: 10.1093/bioinformatics/bty926. [DOI] [PubMed] [Google Scholar]
- 63.Marsh B., Blelloch R. Single nuclei RNA-seq of mouse placental labyrinth development. Elife. 2020;9 doi: 10.7554/eLife.60266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin F.C., Spurgeon H.A., Rakusan K., Weisfeldt M.L., Lakatta E.G. Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am. J. Physiol. 1982;243:H941–H947. doi: 10.1152/ajpheart.1982.243.6.H941. [DOI] [PubMed] [Google Scholar]
- 65.Huang B.T., Peng Y., Liu W., Zhang C., Huang F.Y., Wang P.J., Zuo Z.L., Liao Y.B., Chai H., Huang K.S., et al. Increased interventricular septum wall thickness predicts all-cause death in patients with coronary artery disease. Intern. Med. J. 2015;45:275–283. doi: 10.1111/imj.12667. [DOI] [PubMed] [Google Scholar]
- 66.Collinot H., Marchiol C., Lagoutte I., Lager F., Siauve N., Autret G., Balvay D., Renault G., Salomon L.J., Vaiman D. Preeclampsia induced by STOX1 overexpression in mice induces intrauterine growth restriction, abnormal ultrasonography and BOLD MRI signatures. J. Hypertens. 2018;36:1399–1406. doi: 10.1097/HJH.0000000000001695. [DOI] [PubMed] [Google Scholar]
- 67.Bakrania B.A., Hall M.E., Shahul S., Granger J.P. The Reduced Uterine Perfusion Pressure (RUPP) rat model of preeclampsia exhibits impaired systolic function and global longitudinal strain during pregnancy. Pregn. Hyper. 2019;18:169–172. doi: 10.1016/j.preghy.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taysi S., Tascan A.S., Ugur M.G., Demir M. Radicals, oxidative/nitrosative stress and preeclampsia. Mini Rev. Med. Chem. 2019;19:178–193. doi: 10.2174/1389557518666181015151350. [DOI] [PubMed] [Google Scholar]
- 69.Aouache R., Biquard L., Vaiman D., Miralles F. Oxidative stress in preeclampsia and placental diseases. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toth M. [Role of tetrahydrobiopterin in the regulation of activity of human placental nitric oxide synthase in normal and pre-eclamptic pregnancies] Orv. Hetil. 2002;143:391–398. [PubMed] [Google Scholar]
- 71.Guerby P., Tasta O., Swiader A., Pont F., Bujold E., Parant O., Vayssiere C., Salvayre R., Negre-Salvayre A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021;40 doi: 10.1016/j.redox.2021.101861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kukor Z., Valent S. [Nitric oxide and preeclampsia] Orv. Hetil. 2010;151:2125–2135. doi: 10.1556/OH.2010.29012. [DOI] [PubMed] [Google Scholar]
- 73.Iwanaga N., Yamamasu S., Tachibana D., Nishio J., Nakai Y., Shintaku H., Ishiko O. Activity of synthetic enzymes of tetrahydrobiopterin in the human placenta. Int. J. Mol. Med. 2004;13:117–120. [PubMed] [Google Scholar]
- 74.Mitchell B.M., Cook L.G., Danchuk S., Puschett J.B. Uncoupled endothelial nitric oxide synthase and oxidative stress in a rat model of pregnancy-induced hypertension. Am. J. Hypertens. 2007;20:1297–1304. doi: 10.1016/j.amjhyper.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 75.Paulis L., Zicha J., Kunes J., Hojna S., Behuliak M., Celec P., Kojsova S., Pechanova O., Simko F. Regression of L-NAME-induced hypertension: the role of nitric oxide and endothelium-derived constricting factor. Hypertens. Res. 2008;31:793–803. doi: 10.1291/hypres.31.793. [DOI] [PubMed] [Google Scholar]
- 76.Mattson D.L., Meister C.J. Sodium sensitivity of arterial blood pressure in L-NAME hypertensive but not eNOS knockout mice. Am. J. Hypertens. 2006;19:327–329. doi: 10.1016/j.amjhyper.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 77.Lee S.M., Moon J.Y., Lim B.Y., Kim S.M., Park C.W., Kim B.J., Jun J.K., Norwitz E.R., Choi M.H., Park J.S. Increased biosynthesis and accumulation of cholesterol in maternal plasma, but not amniotic fluid in pre-eclampsia. Sci. Rep. 2019;9:1550. doi: 10.1038/s41598-018-37757-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kazmi R.S., Cooper A.J., Lwaleed B.A. Platelet function in pre-eclampsia. Semin. Thromb. Hemost. 2011;37:131–136. doi: 10.1055/s-0030-1270339. [DOI] [PubMed] [Google Scholar]
- 79.Rolnik D.L., Wright D., Poon L.C., O'Gorman N., Syngelaki A., de Paco Matallana C., Akolekar R., Cicero S., Janga D., Singh M., et al. Aspirin versus placebo in pregnancies at high risk for preterm preeclampsia. N. Engl. J. Med. 2017;377:613–622. doi: 10.1056/NEJMoa1704559. [DOI] [PubMed] [Google Scholar]
- 80.Ducat A., Vargas A., Doridot L., Bagattin A., Lerner J., Vilotte J.L., Buffat C., Pontoglio M., Miralles F., Vaiman D. Low-dose aspirin protective effects are correlated with deregulation of HNF factor expression in the preeclamptic placentas from mice and humans. Cell Death Dis. 2019;5:94. doi: 10.1038/s41420-019-0170-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chuaiphichai S., Yu G.Z., Tan C.M.J., Whiteman C., Douglas G., Dickinson Y., Drydale E.N., Appari M., Zhang W., Crabtree M.J., et al. Endothelial GTPCH (GTP cyclohydrolase 1) and tetrahydrobiopterin regulate gestational blood pressure, uteroplacental remodeling, and fetal growth. Hypertension. 2021;78:1871–1884. doi: 10.1161/HYPERTENSIONAHA.120.17646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Abel D., Michel O., Veerhuis R., Jacobs M., van Dijk M., Oudejans C.B. Direct downregulation of CNTNAP2 by STOX1A is associated with Alzheimer's disease. J. Alzheimers Dis. 2012;31:793–800. doi: 10.3233/JAD-2012-120472. [DOI] [PubMed] [Google Scholar]
- 83.Hattori Y., Hattori S., Wang X., Satoh H., Nakanishi N., Kasai K. Oral administration of tetrahydrobiopterin slows the progression of atherosclerosis in apolipoprotein E-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:865–870. doi: 10.1161/01.ATV.0000258946.55438.0e. [DOI] [PubMed] [Google Scholar]
- 84.Palei A.C., Martin H.L., Wilson B.A., Anderson C.D., Granger J.P., Spradley F.T. Impact of hyperleptinemia during placental ischemia-induced hypertension in pregnant rats. Am. J. Physiol. Heart Circ. Physiol. 2021;320:H1949–H1958. doi: 10.1152/ajpheart.00724.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang W., Li Q., Duncan J.W., Bakrania B.A., Bradshaw J.L., Granger J.P., Rana S., Spradley F.T. Luteolin-induced vasorelaxation in uterine arteries from normal pregnant rats. Pregn. Hyper. 2021;23:11–17. doi: 10.1016/j.preghy.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hypoxia and STOX1 overexpression affect the the fumarate-dependent L-malate pathway. Quantitative RT-qPCR of (A)STOX1A and (B)STOX1B mRNAs. (C) 3D reconstructions of cells immunostained for STOX1 (green, detection of both STOX1A and STOX1B) counterstained with Hoechst (blue). (D) Quantification of STOX1 (both STOX1A and STOX1B) fluorescence intensity per cell. (E) 3D reconstructions of cells immunostained for fumarase in red. (F) Quantification of fumarase fluorescence intensity per cell. (G) Fumarase quantitative assay measured with sandwich ELISA assay, expressed in percentage relative to the untreated control BD3 cell line. (H) Total fumarase activity (measured with a colorimetric assay) expressed as percentage of the untreated control BD3 cell line. RT-qPCR, quantitative assay (sandwich ELISA for fumarase) and enzyme activity (fumarase), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from 3 independent experiments, scale bar 10μm, mean ± SD. Immunoblot is representative of 3 independent experiments. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars).
NO metabolism and antioxidant defense upon STOX1 overexpression and hypoxia.(A) 3D reconstructions of cells immunostained for iNOS (green), counterstained with Hoechst (blue) (quantification in Fig. 2A). (B) 3D reconstructions of cells immunostained for mitochondrial MnSOD/SOD2 (red), counterstained with Hoechst (blue). (C) Quantification of mitochondrial MnSOD/SOD2 fluorescence intensity per cell. (D) ROS hydrogen peroxide (H2O2) detected with the DCFDA probe expressed as percentage relative to the untreated control BD3 cell line. (E) 3D reconstructions of cells immunostained for catalase (green), counterstained with Hoechst (blue). (F) Quantification of catalase fluorescence intensity per cell. Dosage (H2O2), n= 6 for untreated BD3/AA6/B10 in H2O2 assays n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from 3 independent experiments, scale bar 10μm, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (blue stars).
The BH4 metabolite counteracts STOX1 effects through iNOS regulation and rescue of L-malate levels. Quantitative RT-qPCR of (A) STOX1A and (B) STOX1B. (C) 3D reconstructions of cells immunostained for STOX1 (green, STOX1A/B), counterstained with Hoechst (blue). (D) Quantification of STOX1 fluorescence intensity per cell. (E) 3D reconstructions of cells immunostained for iNOS (green), counterstained with Hoechst (blue). (F) ROS hydrogen peroxide (H2O2) expressed as percent of the untreated control BD3 cell line. (G) 3D reconstructions of cells stained for mitochondrial ROS with MitoSOX (red), counterstained with Hoechst (blue). Data of untreated cells in panel A, B, D are from the corresponding panels in Fig. S1. RT-qPCR and dosage (H2O2), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (pink or purple stars).
The BH4 metabolite counteracts STOX1 alteration of the antioxidant defense.(A) 3D reconstructions of cells immunostained for SOD2/MnSOD (red), counterstained with Hoechst (blue). (B) Quantification of SOD2/MnSOD fluorescence intensity per cell. (C) Total SOD activity (measured as inhibition of the activity of SOD, percentage). (D) 3D reconstructions of cells immunostained for catalase (green), counterstained with Hoechst (blue). (E) Quantification of catalase fluorescence intensity per cell. (F) Catalase activity expressed in nmol/min/ml. Data of untreated cells are from the corresponding panels in Fig. S2C (SOD2), Fig. 2E (SOD activity), Fig. S2F (catalase levels), Fig. 2F (catalase activity). Enzyme activities (SOD and catalase), n = 3 independent experiments, mean ± SD. Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (black stars) or the corresponding untreated condition (pink or purple stars).
The BH4 metabolite and L-NAME increase the mitochondrial ATP fraction in STOX1A overexpressing cells.(A) Citrate synthase activity expressed in mU/mg of protein. (B) Acetyl-CoA expressed in nmol/g of protein, blue histograms are for non-treated cells in chemical hypoxia. (C) Percentage of the OXPHOS fraction of ATP. Data of untreated cells are from the corresponding panels in Fig. 3D (citrate synthase activity), Fig. 3E (Acetly-CoA), Fig. 3F (ATP levels), Fig. 3F (OXPHOS fraction). (D) Total ATP in the various cells under BH4 and LNAME treatments with estimation of the part attributable to the respiratory chain (OXPHOS). For the dosages (ATP and acetyl-CoA), and the enzyme activity (citrate synthase), mean ± SD of n = 3 independent experiments are represented,. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA, and post hoc Dunnett test using the untreated control as reference (black stars, on D, red stars on A, B, and C) or the corresponding untreated condition (pink or purple stars).
The BH4 metabolite and L-NAME increase fumarase activity in STOX1A overexpressing cells.(A) 3D reconstructions of cells immunostained for fumarase (red), counterstained with Hoechst (blue) and (B) quantification of fumarase fluorescence intensity per cell. (C) Fumarase (measured by sandwich ELISA assay) expressed as percentage of the untreated control BD3 cell line. (D) Percentage of fumarase activity relative to the untreated control BD3 cell line. Data of untreated are from Fig. S1F (fumarase), Fig. S1G (fumarase quantity), Fig. S1H (fumarase activity). Immunofluorescence, n = 60 cells from three independent experiments, mean ± SD. Scale bar 10μm. Quantity assay by sandwich ELISA (fumarase), and enzyme (fumarase) activity, n = 3 independent experiments, mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001; based on one-way ANOVA versus the untreated control (red stars) or the corresponding untreated condition (pink or purple stars).
Network enrichment of genes modified by STOX1 and corrected by BH4 treatment in mice placenta. The genes of figure 5E were significantly associated in a network (PPI = 3.7 10-8). The enrichment in molecular functions revealed enrichment of the following terms: Hormone activity, Cholesterol transporter activity, and several Gene Ontology terms connected with lipoprotein physiology. In terms of Biological processes, meaningfully enriched terms were Protein oxidation, Response to stress, and Homeostatic processes.
Modulations of blood pressure in early, mid and late gestation in mice. Systolic arterial pressure in periods E1.5-5.5, E6.5-12.5 and E13.5-18.5 during gestation expressed in mm Hg relative to pre-gestation. Data from n= 9 for WTxWT, n=7 for WTxWT +BH4, n=16 for WTxhtTgSTOX42, and n=12 for WTxhtTgSTOX42 +BH4. Mean ± SD. *p ≤ 0.05 **p ≤ 0.01 ***p ≤ 0.001 based on one-way ANOVA versus the untreated control (black stars) or the corresponding untreated condition (pink stars).
Full membrane immunoblots.