

Abstract
Radical S-adenosylmethionine (RaS) enzymes have quickly advanced to one of the most abundant and versatile enzyme superfamilies known. Their chemistry is predicated upon reductive homolytic cleavage of a carbon–sulfur bond in cofactor S-adenosylmethionine forming an oxidizing carbon-based radical, which can initiate myriad radical transformations. An emerging role for RaS enzymes is their involvement in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs), a natural product family that has become known as RaS-RiPPs. These metabolites are especially prevalent in human and mammalian microbiomes because the complex chemistry of RaS enzymes gives rise to correspondingly complex natural products with minimal cellular energy and genomic fingerprint, a feature that is advantageous in microbes with small, host-adapted genomes in competitive environments. Herein, we review the discovery and characterization of RaS-RiPPs from the human microbiome with a focus on streptococcal bacteria. We discuss the varied chemical modifications that RaS enzymes introduce onto their peptide substrates and the diverse natural products that they give rise to. The majority of RaS-RiPPs remain to be discovered, providing an intriguing avenue for future investigations at the intersection of metalloenzymology, chemical ecology, and the human microbiome.
Keywords: natural products, RiPPs, radical SAM enzymes, mechanism, microbiome, streptococcus, sequence similarity network
Introduction
Natural products have been indispensable as starting points for drug discovery and sources of inspiration across multiple disciplines.1,2 Aside from structure elucidation, total synthesis, and functional studies, natural product biosynthesis has long emerged as a vibrant and active field.3−6 Especially intriguing are biosynthetic steps that are carried out by metalloenzymes, like those observed in the production of penicillin or vancomycin.7−10 Traditionally, these enzymes were discovered “accidentally” after the natural products were identified through bioactivity-guided screening campaigns. Proceeding in the direction of biological activity → natural product → gene → enzyme is well-established and has provided numerous fascinating enzyme-catalyzed reactions, even if it is indirect with respect to enzyme discovery. Much less traveled is the reverse process, enzyme → gene → natural product → activity. Until recently, genomes and bioinformatic tools were not readily available, and this process has, therefore, only become possible thanks to major advances in DNA sequencing technologies and computational approaches. Even with this information, there are two significant challenges. Finding new metalloenzymes is now easy in a given genome, but for new enzyme families, the corresponding substrates are difficult to intuit based on sequence information alone. Moreover, in the context of natural products, most biosynthetic gene clusters are silent or sparingly expressed, requiring alternative approaches to access the encoded metabolites.11,12
Our natural product biosynthesis investigations have focused on radical S-adenosylmethionine (RaS) metalloenzymes.13−15 In a short period of time, RaS enzymes have advanced to one of the largest and biochemically most versatile enzyme superfamily known.16,17 Underlying this versatile chemistry is a common radical initiation reaction in which cofactor S-adenosylmethionine (SAM), bound via its α-amino and carboxylate groups to a [4Fe–4S]+ cluster, is reductively cleaved to generate, in most cases, a 5′-deoxyadenosyl radical (5′-dA•), which then starts turnover (Figure 1). Seminal early discoveries by Knappe, Barker, and Frey laid the groundwork and led to the characterization of pyruvate formate lyase (PFL) and lysine-2,3-aminomutase (LAM) as enzymes that carried out unusual reactions with the aid of iron and SAM.18−22 In 1984, Knappe and co-workers inferred a catalytic mechanism for the PFL-activating enzyme that looks eerily close to the reaction that we today know RaS enzymes to catalyze, even with an intermediate that approximates the structure of the recently identified intermediate omega.20,23−25 Likewise, the demonstration by Frey and colleagues that LAM uses an Fe–S cluster and SAM to generate the 5′-dA•, the same intermediate formed by adenosylcobalamin-dependent isomerases as previously proposed by Abeles and co-workers, provided a beautiful example of convergent biochemical evolution (Figure 1).21,26−33 These early findings were followed by investigations into a broader set of RaS enzymes, notably biotin synthase,34,35 lipoate synthase,36 anerobic ribonucleotide reductase,37 and spore photoproduct lyase,38 all of which exhibit a similar radical initiation process involving the 5′-dA• (Figure 1). However, it was the visionary analysis by Sofia et al. to which this special issue is dedicated, which linked these enzymes as members of the new RaS enzyme superfamily consisting of 645 members at the time.39 Since then, the superfamily has grown exponentially to nearly one million members, and it provides an exciting frontier of metalloenzymology.
Figure 1.
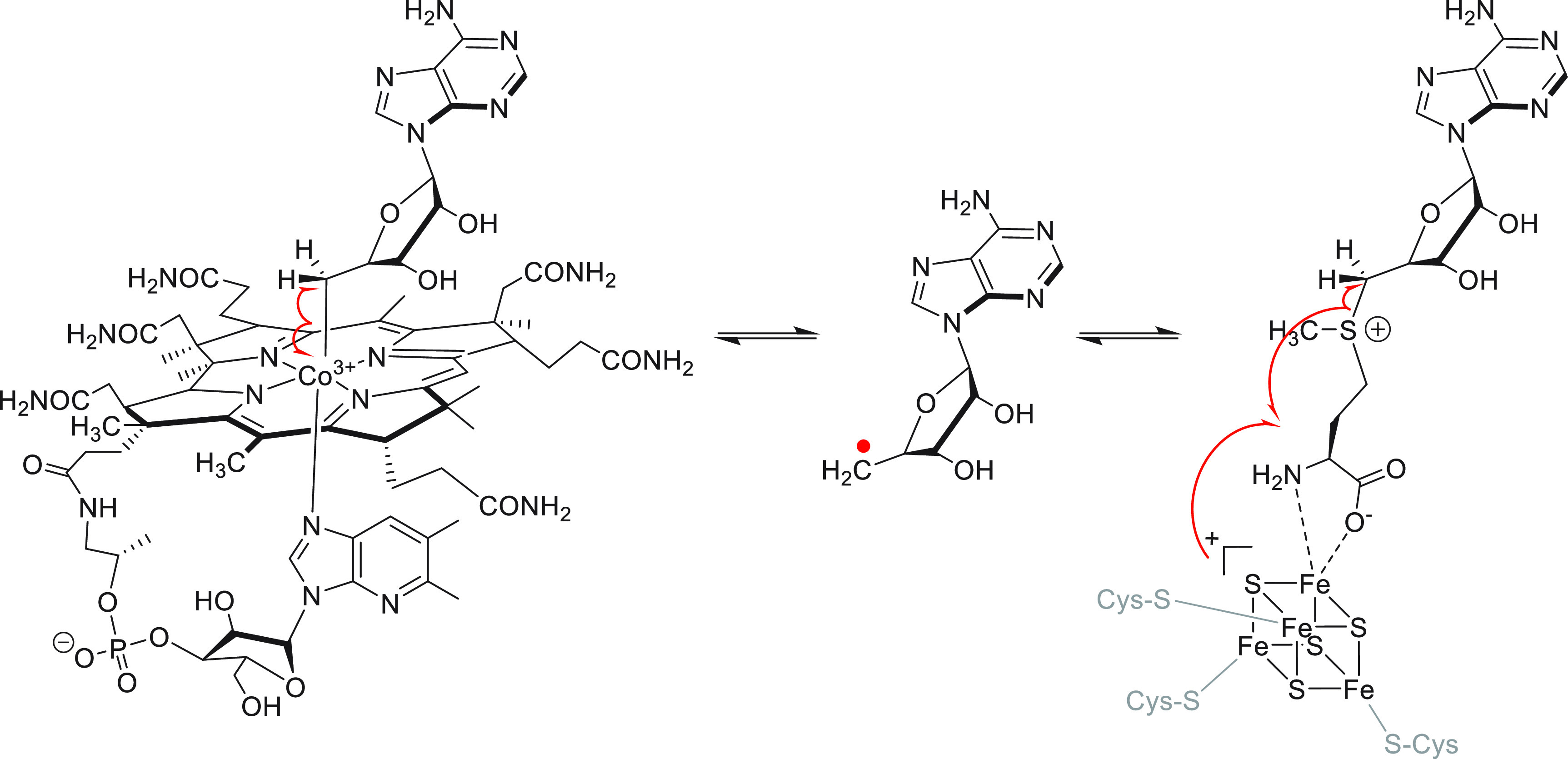
Radical initiation in adenosylcobalamin-dependent enzymes (left) and RaS enzymes (right). Both lead to formation of a 5′-dA• shown in the center, which initiates catalysis. Note that the other two C–S bonds in SAM can be homolyzed as well in a subset of RaS enzymes.
The work of Sofia et al. was a harbinger of the power of bioinformatics. It predicted the involvement of RaS enzymes in diverse physiological pathways from cofactor, DNA precursor, and vitamin biosynthesis to secondary metabolism and catabolic pathways. It also hinted at nature’s expansion of the minimal RaS enzyme scaffold to include additional domains. For example, the combination of vitamin B12 binding sites was described in some superfamily members as well as other N-terminal or C-terminal extensions. What has followed since the report by Sofia et al. are intense investigations and application of numerous kinetic, spectroscopic, and structural studies to elucidate the detailed structure of the metallocofactor, the mechanism of the radical initiation step, detailed mechanisms of the reactions catalyzed in some cases accompanied by high-resolution crystal structures, and a steady stream of some of the most unusual transformations known in biology.13−15,40−48
RaS enzymes participate in diverse pathways in all kingdoms of life, and with only a fraction characterized to date, the reach of this superfamily is all but guaranteed to grow in years to come. Herein, we highlight the intersection of RaS enzymology, the human microbiome, and the biosynthesis of RiPP (ribosomally synthesized and post-translationally modified peptide) natural products,49−55 an area where we expect RaS enzymes to play a prominent role in the future and where we and others have discovered enzymatic reactions in the direction enzyme → gene → natural product → function. We provide a glossary of modifications catalyzed by RaS enzymes onto their respective precursor peptides during RiPP biosynthesis with an emphasis on oral microbiome streptococci, which have provided a rich source of new transformations. The biosynthetic gene clusters for this broader RiPP natural product class, which is now known as RaS-RiPPs, can be detected in nearly all bacterial phyla; we focus on those that are encoded in bacteria associated with human and mammalian microbiomes but note that several novel RaS enzyme-catalyzed reactions—including methylation at unactivated positions,56 α-thioether bond formation,57−60 γ-thioether bond formation,61 epimerization,62−64 tyramine excisions,65 decarboxylative carbon–carbon bond formation,66,67 cyclophane formation,68,69 and others70—have been identified in RiPP biosynthesis outside of mammalian microbiomes (Figure 2). For most RaS-RiPPs discovered, especially those from mammalian microbiota, the detailed functions remain to be elucidated. These natural products offer exciting avenues for further research at the intersection of RiPPs, metalloenzymology, and chemical ecology in the context of human microbiomes.
Figure 2.

Representative reactions by RaS enzymes catalyzed in RiPP biosynthetic reactions. These transformations have been characterized from bacteria outside of the human microbiome.
RiPP and NRP Natural Products
In the 2001 report, Sofia et al. noted “...many examples from secondary metabolism pathways, such as antibiotic and herbicide biosynthesis, are found, including spectinomycin, subtilosin,...”. Just like the superfamily itself, the number of enzymes involved in secondary metabolism has expanded significantly since then. New approaches are therefore needed to group these enzymes and create an organizing framework with which reactions and mechanisms can be addressed.
With an ability to install complex modifications in a single step, RaS enzymes are ideal tailoring catalysts, especially in RiPP biogenesis. The biosynthetic logic of RiPPs can be contrasted to that of nonribosomal peptide (NRP) natural products. NRPs are synthesized by large, modular assembly line enzymes that build a peptide natural product via the addition of one canonical or noncanonical amino acid at a time.6,71 The minimal unit required to do so is referred to as a module, and it consists of at least three domains, condensation (C) domain, adenylation (A) domain, and peptidyl carrier protein (PCP). Once synthesis of the peptide is complete, it is cleaved from the assembly line by a thioesterase (TE) domain to deliver the mature product. RiPP biogenesis, by contrast, begins with the ribosomal synthesis of a multipartite precursor peptide, consisting of a leader sequence, which is important for enzyme recognition, a core region wherein modifications are installed, and sometimes a follower sequence downstream of the core peptide.49,72,73 After modifications are introduced in the core, typically by a small number of tailoring enzymes, the leader (and follower) is removed to deliver the mature RiPP.
Streptide, a Microbiome RaS-RiPP
The examples of nosiheptide,74 bottromycin,56 subtilosin,75 and polytheonamide76 provided an early glimpse of the unusual chemistry that RaS enzymes can catalyze during RiPP biogenesis. In these cases, much like penicillin and vancomycin, however, the natural products were discovered first followed by the subsequent realization that RaS enzymes played important roles in their biogenesis. Because the substrate for the tailoring enzyme in RiPP biosynthetic pathways is genetically encoded, we reasoned that new RaS enzymes would be easier to characterize in RiPPs compared to other natural product classes when proceeding in the direction enzyme → gene → natural product → function. To avoid working on silent biosynthetic gene clusters (BGCs), we searched the literature for quorum sensing (QS)-regulated RiPP BGCs with one or more RaS enzymes and came across a cluster that we subsequently named str (for streptide) (Figure 3A).77 The cluster codes for a precursor peptide (StrA), a RaS enzyme that had not been characterized (StrB), and a combination peptidase/transporter (StrC); it is controlled by an upstream quorum sensing element, identified and characterized by the Monnet lab,77 suggesting that the mature RiPP is synthesized at high cell densities. Indeed, the product of the str cluster could be detected in culture supernatants; however, the mass obtained by Ibrahim et al. did not match any simple modification, and the structure of mature product remained unknown. Upon isolation from large-scale production cultures and extensive analysis of 1D/2D NMR spectra, streptide was found to contain an unprecedented cross-link between the unactivated β-methylene of a Lys side chain and the C-7 indole of Trp (Figure 3B).78 The α-carbons were found to be S-configured. Absolute configuration of the newly generated chiral center was ultimately determined by de novo total synthesis of the S- and R-diastereomers, with only the latter showing a match with the authentic natural product.79
Figure 3.

Characterization of streptide and its biosynthetic pathway. (A) QS-regulated str BGC. The sequence of the precursor peptide, the internal residues that form the core of streptide, and the two residues to be cross-linked (red) are highlighted. (B) Streptide biosynthetic pathway. See text for details. (C) Proposed mechanism for Lys-Trp cross-link formation. Unmodified amino acids are shown in gray spheres and labeled with one-letter codes. Active site and auxiliary Fe–S clusters are shown in red and blue, respectively.
Biochemical, mechanistic, and structural investigations have provided additional insights into the unusual Lys-Trp cross-linking reaction. StrB is a member of the SPASM-domain RaS enzymes, named after enzymes that are involved in the maturation of subtilosin A, pyrroloquinoline quinone, anaerobic sulfatase, and mycofactocin. These enzymes contain a C-terminal extension capable of binding two “auxiliary” Fe–S clusters.46,80−83 The reaction of StrB was recapitulated in vitro, revealing installation of the Lys-Trp carbon–carbon linkage in a single step with the same regio- and stereochemistry as detected in streptide and in an auxiliary Fe–S cluster-dependent manner.78 The RaS enzymes SuiB and AgaB from orthologous str clusters in Streptococcus suis and Streptococcus agalactiae, respectively, were shown to contain two auxiliary Fe–S clusters and catalyze similar modifications onto precursor peptides that contained the Lys and Trp residue in a K-DGD-W motif, like streptide.84,85 A crystal structure of SuiB visualized the three key domains of the enzyme, the orientation of the two auxiliary clusters relative to the active site Fe–S cluster, and the ligation environment of each, while also providing clues regarding substrate recognition.86 Several mechanisms were considered, and the current working model is shown (Figure 3C). In support of this mechanism is, among other observations, (i) loss of the Lys2 β-2H when side-chain-deuterated Lys is incorporated into the substrate, (ii) formation of 5′-2H-5′-dA with this substrate, (iii) retention of the Lys2 α-1H, and, most importantly, (iv) recent direct observation of the Lys-cross-linked tryptophan radical intermediate by freeze-quench electron paramagnetic resonance (EPR) spectroscopy.87 Together, studies on the str cluster have revealed a new natural product chemotype, a novel reaction for the RaS enzyme superfamily, and an intriguing mechanism involving the Lys-Trp radical intermediate.
RaS-RiPPs Network in Streptococci
How widespread are str-like clusters in streptococcal genomes? To answer this question, Bushin et al. conducted a co-occurrence-based bioinformatic search for all instances of RaS enzymes adjacent to the shp/rgg QS operon, which code for a short hydrophobic peptide (shp) as the autoinducer and the cognate transcriptional regulator (rgg) (see Figure 3A).88 From a total of ∼10,750 RaS enzymes in streptococci, ∼600 were identified that are encoded in RiPP BGCs and controlled by a QS operon. When arranged into a sequence similarity network (SSN), wherein related precursor peptides within the ∼600 BGCs are grouped together via a user-defined similarity threshold using the EFI-EST database,89,90 16 distinct subfamilies of RaS-RiPPs emerged (Figure 4). These have been named based on conserved motifs in the precursor peptide sequence. Some BGCs appear to be species-specific signals like the TQQ cluster that is encoded only in S. suis strains.88 Others are encoded in numerous streptococci, like streptide, suggesting they may form a common interspecies “language”. The network provides a useful organizing theme for genome-guided discovery of new RaS enzymes, proceeding in the direction enzyme → natural product → function.
Figure 4.
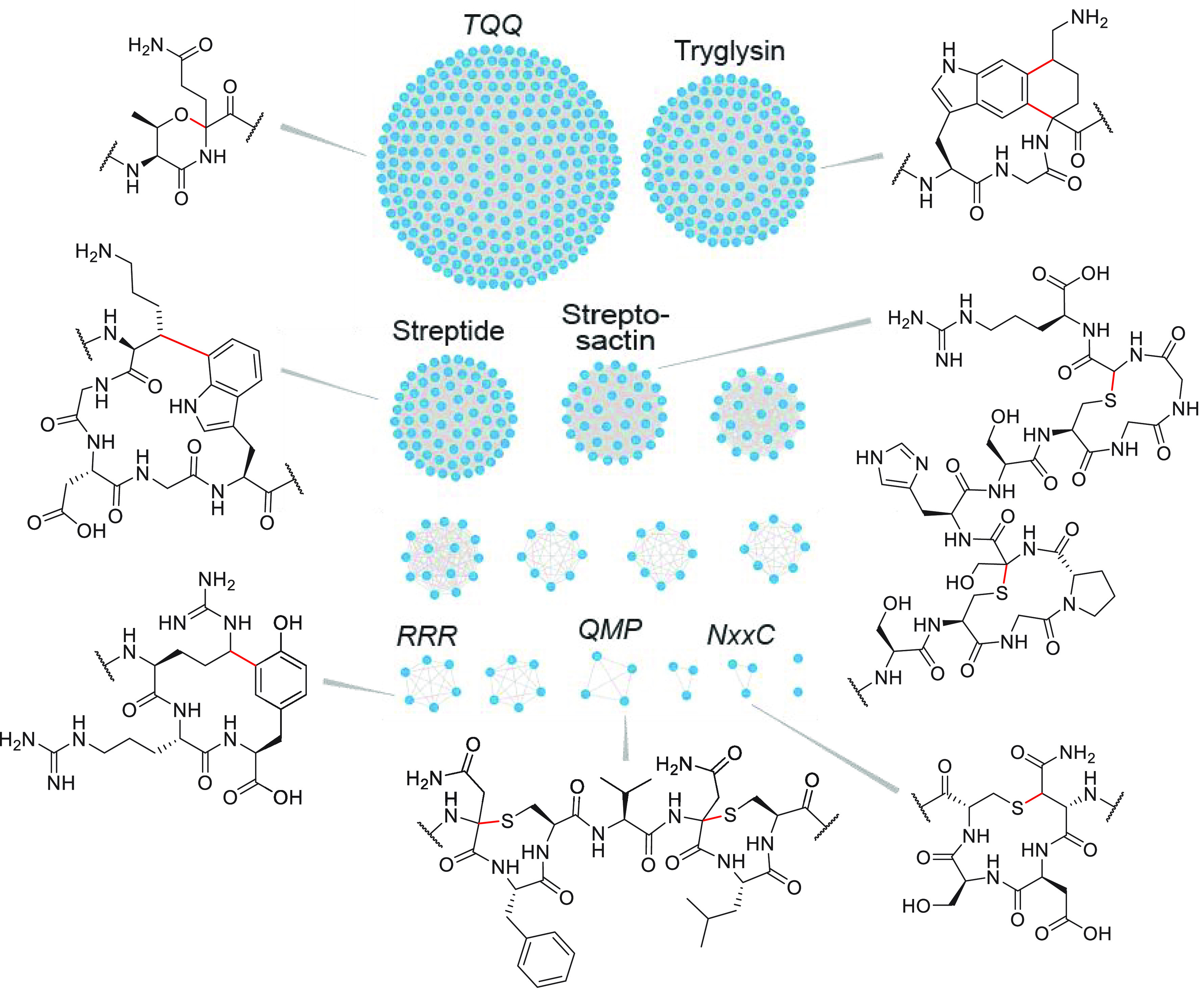
Sequence similarity network of RaS-RiPPs from streptococci associated with mammalian microbiomes.88 The network was generated with an E value of 2, a fraction value of 1, and an alignment cutoff score of 1. Each node represents a unique RaS-RiPP BGC, and the lines connecting them indicate significant sequence similarity in the precursor peptide. Subfamilies for which the mature product has been identified from the original host are labeled with the natural product name; others are labeled with a conserved amino acid motif within the precursor. Enzymatic products are shown for examined subfamilies with the modification introduced by the RaS enzyme indicated in red. These modifications are installed by TqqB (TQQ subfamily), WgkB (tryglysin), StrB, AgaB, and SuiB (streptide), GggB (streptosactin), RrrB (RRR), QmpB (QMP), and NxxcB (NxxC).
The str, aga, and sui BGCs, which encode RaS enzymes that introduce Lys-Trp linkages, colocated to the streptide subfamily in the network, suggesting distinct chemical reactions may be catalyzed by RaS enzymes in each of the remaining 15 subfamilies. This has turned out to be the case after examination of six additional subfamilies thus far. The RaS enzyme in the tryglysin cluster (WgkB) carries out a complex modification, in which Trp and Lys residues, arranged in a WGK motif, are connected with two carbon–carbon bonds between the indole C-5 and C-6 and the Lys α-C and δ-C, respectively, giving rise to a unique tetrahydrobenzindole moiety (Figure 4).88 Single cross-links are not observed, suggesting both modifications are introduced in one turnover or that the enzyme has enhanced affinity and/or specificity for the singly cross-linked product(s).
Assessment of NxxcB, the tailoring RaS enzyme in the NxxC subfamily, revealed the first β-thioether linkage introduced by a RaS enzyme (Figure 4).91 Shortly thereafter, additional examples of β-thioether and novel γ-thioether connections were identified, introduced by the RaS enzymes PapB and CteB/BeiB, respectively.61,92,93 α-Thioether linkages, known as sactionine bridges, were already known and were first identified in subtilosin, where they are introduced by the sactisynthase AlbA.57 β-Thioethers are a hallmark of lanthipeptides, but in this compound family, they are introduced heterolytically via ATP-dependent formation of a dehydroalanine or dehydrobutyrine, followed by conjugate addition by Cys onto the acceptor.72,73 Because Ser and Thr are dehydrated to form the Michael acceptor, lanthipeptide β-thioethers occur at Ser/Thr acceptor residues. As NxxcB employs a radical mechanism, the acceptor can in theory be any amino acid with a β-carbon. In the native NxxcB substrate, the β-thioether acceptor is Asn, though the enzyme also tolerates Ala, Gln, and Asp at this position.91 A working model has emerged for the mechanism of NxxcB based on preliminary studies. Much like the pathway proposed for sactisynthases,57,60 NxxcB activates the Cys-thiol via chelation to an auxiliary Fe–S cluster, which can be observed by altered UV–vis absorption properties upon incubation of NxxcB with its substrate NxxcA.91 Following radical initiation, 5′-dA• is proposed to abstract the β-H of Asn, which then reacts with the Fe–S-activated Cys-thiol to generate the C–S bond concomitant with reduction of the auxiliary Fe–S cluster. In contrast to the observed flexibility at the acceptor residue, NxxcB does not accept Ser or Thr in place of Cys. The basis for this strict requirement is not yet known; similar results have been observed with sactisynthases.60
In addition to the tryglysin and NxxC subfamilies, the first ether modification was observed with TqqB, the RaS enzyme in the largest TQQ subfamily in the streptococcal RaS-RiPPs network.94 TqqB links adjacent Thr-Gln residues within the TQQ sequence via an aliphatic ether connection, thereby introducing a backbone morpholine modification into the peptide (Figure 4). Here again, the enzyme exhibited some degree of promiscuity at the acceptor residue with Ala, Asn, and N-Me-Gln yielding turnover similar to that of Gln in wild-type TqqA. However, TqqB did not react with Cys and only marginally with Ser when these were substituted for Thr. Initial studies with TqqB suggest a mechanism akin to that of the sactisynthases and NxxcB, with an auxiliary Fe–S cluster activating the Thr side chain alcohol for C–O bond formation.
The RaS enzyme (RrrB) in the RRR subfamily modifies a 41mer precursor peptide, the synthesis of which proved difficult.95 Therefore, a heterologous approach was employed in which rrrA and rrrB were coexpressed in Escherichia coli, the former with an N-terminal hexa-His maltose binding protein purification tag and a protease cleavage sequence between the tag and the precursor. Upon expression, the modified peptide was purified using the tag, proteolyzed, and analyzed by NMR spectroscopy, revealing formation of an Arg-Tyr cross-link at the C-terminus of the peptide forming a 16-membered ring macrocycle.95 The key cross-link occurs between the γ-C of Arg and the ortho-position (relative to phenolic-OH) of a tyrosine–phenol (Figure 4). RrrB was shown to accept significantly shorter RrrA substrates that are truncated at the N-terminus, making biochemical and mechanistic studies possible in the future.
Finally, the RaS enzymes from the GGG (streptosactin) and QMP subfamilies, termed GggB and QmpB, respectively, have been characterized as well and shown to introduce two sactionine bridges onto the corresponding precursor peptides GggA and QmpA (Figure 4).96,97 However, as opposed to Type 1 sactipeptides, which incorporate nested macrocycles, wherein the most upstream (N-terminal) Cys residue reacts with the most downstream (C-terminal) acceptor, GggB and QmpB introduce a distinct “bicycle” topology of unnested macrocycles where the Cys donors and acceptors alternate along the core peptide. These are referred to as type 2 sactipeptides.
Additional RaS enzymes from the RaS-RiPPs network are currently being investigated. The studies thus far have expanded the chemical repertoire of RaS enzymes and demonstrate the advantages of this enzyme-first discovery approach.
Novel RaS-RiPPs from Streptococci
The RaS-RiPP network is not just a source of new enzymatic chemistry. Each BGC codes for a natural product, and, other than streptide, the mature RiPPs from two other clusters have been reported. Knowledge of the reaction carried out by WgkB enabled identification of the mature product in culture supernatants of Streptococcus ferus. Termed tryglysin A, the product consists of the unusual tetrahydrobenzindole modification between the Trp and Lys side chains on a 7mer peptide backbone (Figure 5).98 Tryglysin B is the suspected product of the wgk BGC from S. mutans. Bioactivity assays with tryglysin A have revealed surprisingly potent and specific antimicrobial activity. Tryglysin A inhibits the growth of Streptococcus pneumonia, the causative agent of bacterial pneumonia, with a minimal inhibitory concentration of <100 nM. It is similar in potency to the clinically used antibiotic ciprofloxacin. However, whereas ciprofloxacin is broad spectrum and kills commensal and pathogenic bacteria alike, tryglysin does not affect another 15 streptococci tested nor other commensal bacteria. It does exhibit strong and bacteriostatic growth inhibition against the producing strain, an observation that remains to be explained.
Figure 5.
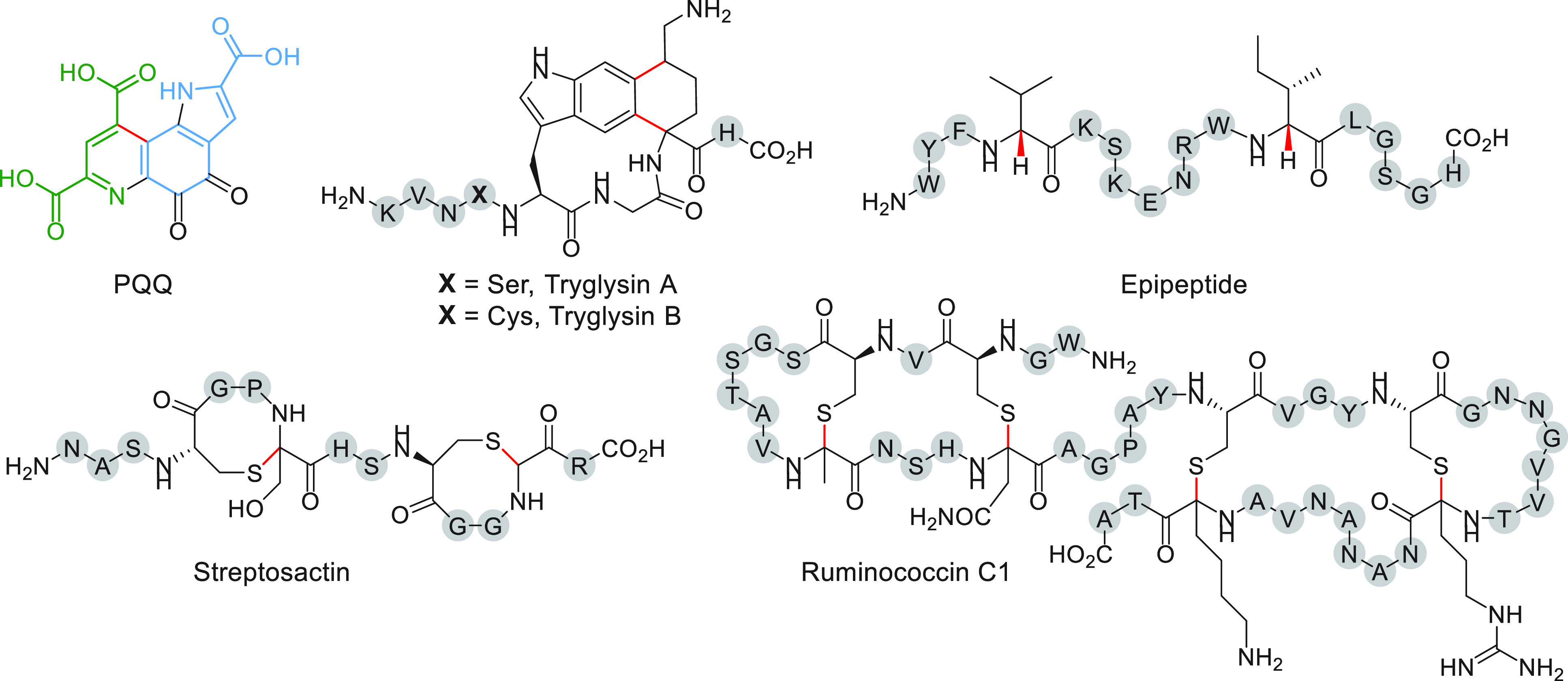
Structures of mature RaS-RiPPs isolated from original hosts that reside in human or mammalian microbiomes. Pyrroloquinoline quinone (PQQ) has been identified from K. pneumoniae, tryglysin A from S. ferus, streptosactin from S. thermophilus along with streptide (see Figure 3), and ruminococcin C1 from Ruminococcus gnavus. Tryglysin B is the predicted product from S. mutans. Epipeptide has been isolated from the soil-dwelling Bacillus subtilis; its BGC can be observed in the human microbiome. Bonds installed by RaS enzymes are shown in red. PQQ has been color-coded to emphasize its amino acid origins from Glu (green) and Tyr (blue).
The product of the ggg gene cluster has been identified as well from culture supernatants of Streptococcus thermophilus. Termed streptosactin, it consists of a 14mer peptide with a pair of 4-residue sactionine macrocycles (Figure 5).96 It is produced at picomolar titers, and its presence was therefore confirmed using a standard generated heterologously in E. coli. When screened against a panel of bacteria, including over a dozen streptococci, streptosactin exhibited growth-inhibitory activity only toward the producing host and its closest relatives. The biosynthesis of streptosactin is linked to the expression of early competence genes. In Streptococcus pneumoniae, fratricidal agents are produced in this growth phase, a process by which competent cells kill noncompetent sibling cells possibly as a means of increasing genetic diversity.99,100 The timing of production of streptosactin, its potent self-killing activity, and other phenotypes observed have led to the proposal that streptosactin may act as a fratricidal agent in S. thermophilus.
RaS-RiPPs from the Human Microbiome
The preceding paragraphs have focused on the reactions catalyzed by RaS enzymes involved in RiPP biosynthesis in mammalian microbiome streptococci. RaS-RiPPs, however, go far beyond streptococci. As the corresponding BGCs typically consist of a small number of genes and have a minimal genomic footprint, they are over-represented in bacteria with small, host-adapted genomes, including Ruminococcus and Enterococcus to name some.101 Indeed, several other RiPPs have been found from members of mammalian microbiomes. The sactipeptide ruminococcin C1 is the first sactipeptide identified from the human microbiome (Figure 5).102 It was purified from the cecal contents of rats that were monoassociated with the human gut symbiont Ruminococcus gnavus E1. Biochemical studies showed that it contains four sactionine bridges forming a double hairpin structure, a novel topology for sactipeptides. NMR studies in conjunction with CYANA-based calculations suggested that ruminococcin C1 contains S-configured α-carbons at each of the four cross-linked sites.103 Two SPASM-domain containing RaS enzymes with a total of three [4Fe–4S] clusters, MC1 and MC2, were shown to processively install the four α-thioether bonds to form the double hairpin.104,105 Interestingly, production of active ruminococcin C1 requires an additional proteolytic cleavage by host-derived trypsin, making this metabolite a symbiotic product of R. gnavus and human host cells (Figure 5).102,106 Ruminococcin C1 shows potent antibiotic activity against Staphylococcus aureus, Enterococcus faecalis, and notably Clostridium difficile, Clostridium perfringens, and Clostridium botulinum with MIC values in the 0.4–12.5 μM range.102 No toxicity was observed against eukaryotic cells, suggesting ruminococcin C1 may serve as an appealing candidate for drug development.
Another RaS-RiPP produced by members of human microbiota is pyrroloquinoline quinone (PQQ) (Figure 5). It is a redox-active cofactor for bacterial methanol, methylamine, or glucose dehydrogenases and is found in many Gram-negative bacteria, including the opportunistic pathogen Klebsiella pneumoniae.107−109 The dehydrogenases use PQQ to oxidize these substrates forming reduced PQQH2 in the process. The electrons then enter the electron transport chain, allowing the host to derive energy from these substrates. Though devoid of peptide bonds, PQQ is indeed a RiPP synthesized according to RiPP biosynthetic logic. Early isotope labeling studies suggested it was derived from Glu and Tyr.110−113 Then, in an early demonstration of heterologous expression of an entire BGC, the pqq operon from Acinetobacter cacoaceticus and K. pneumoniae was expressed in E. coli, thereby identifying all genes required for synthesis of PQQ.114,115 Although the required genes were identified, PQQ biosynthesis remained largely unknown until recent reports by the Klinman lab showed that PqqE, in a strict RiPP recognition element (RRE)-dependent fashion via protein PqqD,116 installs a C–C linkage between the Glu γ-carbon and the ortho-position of the tyrosine side chain within the precursor peptide, PqqA.117 Structural and spectroscopic studies showed unusual auxiliary cluster ligation in the Methylobacterium extorquens PqqE with AuxI and AuxII consisting of a [2Fe–2S] cluster ligated by 4 Cys residues and a [4Fe–4S] cluster ligated by 3 Cys and 1 Asp residues, respectively.118 The unusual ligation environment has been proposed to modulate the redox potential of these Fe–S clusters, making them more oxygen-tolerant. Reactions of several other pqq tailoring enzymes have been demonstrated, as well,119−123 leading to further insights into the biosynthetic pathway of this unusual cofactor.
Lastly, epipeptides form a class of RaS-RiPPs that were identified in Bacillus subtilis 168 (Figure 5).62 While their production has not yet been demonstrated from human microbiota, we highlight them here as epipeptide BGCs are abundant in human microbiome firmicutes, notably Staphylococcus, Corynebacterium, Streptococcus, and Enterococcus.62 In the B. subtilis epipeptide, the RaS enzyme YydG epimerizes two residues forming d-Val and d-Ile in the mature 17mer product. Epimerization occurs via abstraction of the α-H followed by H atom donation by a key Cys residue that is proposed to form a transient cysteinyl radical. This radical is then proposed to be re-reduced by an external reductant mediated by an auxiliary Fe–S cluster. Epipetide permeabilizes the membrane leading to dissipation of the proton motive force and activation of the cell envelope stress response via the LiaRS two-component system.124 It is thought to act as either a cannibalism-related antimicrobial peptide, like sporulation delaying protein (SDP) and the sporulation killing factor (SKF),125,126 or a fratricidal agent. Interestingly, like streptosactin and tryglysin, epipeptide primarily targets the producing cell. The significance of such toxins in the human microbiome remains to be determined.
Uncharacterized RaS-RiPPs in the Human Microbiome
Although only a small number of RaS-RiPPs have been identified from human microbiota, simple genome gazing reveals many potential ones. We highlight three such BGCs (Figure 6). The first occurs in Parabacteroidetes and contains an upstream two-component regulatory system, a 61mer precursor peptide, a RaS enzyme, a TonB-dependent receptor, a peptidase-domain-containing ABC transporter, and two nucleotidyltransferase genes. Another is observed in Clostridia, notably in Clostridium perfringens, and consists of a simple RaS-RiPP architecture with a 55mer precursor peptide, a RaS enzyme, and a transporter. Finally, a third uncharacterized cluster can be seen in Clostridium sporogenes with a similarly simple architecture.
Figure 6.

Select uncharacterized RaS-RiPP BGCs in the human microbiome. Shown are gene clusters from Parabacteroides distasonis (top), Clostridium perfringens (middle), and Clostridium sporogenes (bottom). Genes are color-coded and labeled. The precursor peptide sequence is shown in each case. Note that BGCs homologous to these are not found in streptococci; they are, therefore, not observed in the SSN in Figure 4.
How common are RaS-RiPP BGCs in human microbiomes and what might the entire universe of RaS-RiPPs look like? Several SSNs have been generated for RaS-RiPP subclasses, such as the sactipeptides using RODEO,61 epipeptides,62 QS-regulated streptococcal RaS-RiPPs,88 and for other RaS enzyme subclasses, such as the Gly• enzymes.127,128 However, a map for the entire genomic landscape of RaS-RiPPs has not yet been developed because of lack of unifying genomic features, difficulty in identifying precursor peptides with current methods, and the computational resources necessary for working with the very large RaS enzyme superfamily. For these reasons, it has been much easier to identify members of already known families rather than entirely new ones. However, an all-encompassing network would be highly valuable and provide an organizing framework for future research into microbiome RaS-RiPPs while at the same time facilitating natural product discovery in the direction enzyme → gene → natural product → function.
Conclusions
It is well-established that bacteria communicate with a chemical language consisting of small molecules.129 The molecules represent “words”, and through their effect on neighboring organisms, they convey “meaning”. In the competitive context of animal microbiomes, the ability to generate complex molecules for communication and competition with minimal genomic footprint and cellular energy is highly advantageous. For these reasons, RiPPs are over-represented in the human microbiome, where most strains have diminished genomes and compete with hundreds of other species for nutrients.101,130,131 Among this class of natural products, the RaS-RiPPs are especially intriguing for the unusual chemistry that the RaS enzymes catalyze leading to structurally novel metabolites. The report by Sofia et al., insights into the biosynthesis of RiPP natural products, the genome sequencing revolution, and advances in our understanding of the complexity of microbiomes have all generated an exciting forefront of research at the confluence of RaS enzymology and RiPP natural products in the context of human and animal microbiomes. We anticipate that many more RaS-RiPPs will be discovered from human microbiota, unveiling yet new chemistry catalyzed by RaS enzymes and, importantly, providing insights into the functions of these RiPPs and their roles in human health and disease.
Acknowledgments
The authors thank the Eli Lilly–Edward C. Taylor Fellowship in Chemistry (to K.A.C.), the National Science Foundation GRFP Award (to L.B.B.), and the National Science Foundation CAREER Award (to M.R.S.) for financial support.
The authors declare no competing financial interest.
References
- Clardy J.; Walsh C. Lessons from Natural Molecules. Nature 2004, 432, 829–837. 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Nivina A.; Yuet K. P.; Hsu J.; Khosla C. Evolution and Diversity of Assembly-Line Polyketide Synthases. Chem. Rev. 2019, 119, 12524–12547. 10.1021/acs.chemrev.9b00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F.; Marahiel M. A. Macrocyclization Strategies in Polyketide and Nonribosomal Peptide Biosynthesis. Nat. Prod. Rep. 2007, 24, 735–749. 10.1039/b613652b. [DOI] [PubMed] [Google Scholar]
- Herbst D. A.; Townsend C. A.; Maier T. The Architectures of Iterative Type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. 10.1039/C8NP00039E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach M. A.; Walsh C. T. Assembly-Line Enzymology for Polyketide and Nonribosomal Peptide Antibiotics: Logic, Machinery, and Mechanisms. Chem. Rev. 2006, 106, 3468–3496. 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Schofield C. J.; Baldwin J. E.; Byford M. F.; Clifton I.; Hajdu J.; Hensgens C.; Roach P. Proteins of the Penicillin Biosynthesis Pathway. Curr. Opin. Struct. Biol. 1997, 7, 857–864. 10.1016/S0959-440X(97)80158-3. [DOI] [PubMed] [Google Scholar]
- Hubbard B. K.; Walsh C. T. Vancomycin Assembly: Nature’s Way. Angew. Chem., Int. Ed. 2003, 42, 730–765. 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- Roach P. L.; Clifton I. J.; Hensgens C. M. H.; Shibata N.; Schofield C. J.; Hajdu J.; Baldwin J. E. Structure of IsopenicillinN Synthase Complexed with Substrate and the Mechanism Ofpenicillin Formation. Nature 1997, 387, 827–830. 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- Forneris C. C.; Seyedsayamdost M. R. In Vitro Reconstitution of OxyC Activity Enables Total Chemoenzymatic Syntheses of Vancomycin Aglycone Variants. Angew. Chem., Int. Ed. Engl. 2018, 57, 8048–8052. 10.1002/anie.201802856. [DOI] [PubMed] [Google Scholar]
- Rutledge P. J.; Challis G. L. Discovery of Microbial Natural Products by Activation of Silent Biosynthetic Gene Clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- Covington B. C.; Xu F.; Seyedsayamdost M. R. A Natural Product Chemist’s Guide to Unlocking Silent Biosynthetic Gene Clusters. Annu. Rev. Biochem. 2021, 90, 763–788. 10.1146/annurev-biochem-081420-102432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Radical S-Adenosylmethionine Enzymes. Chem. Rev. 2014, 114, 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf B. J.; McCarthy E. L.; Booker S. J. Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem. 2016, 85, 485–514. 10.1146/annurev-biochem-060713-035504. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Booker S. J. Radical Mechanisms of S-Adenosylmethionine-Dependent Enzymes. Adv. Protein Chem. 2001, 58, 1–45. 10.1016/S0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- Akiva E.; Brown S.; Almonacid D. E.; Barber A. E.; Custer A. F.; Hicks M. A.; Huang C. C.; Lauck F.; Mashiyama S. T.; Meng E. C.; Mischel D.; Morris J. H.; Ojha S.; Schnoes A. M.; Stryke D.; Yunes J. M.; Ferrin T. E.; Holliday G. L.; Babbitt P. C. The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42, D521–D530. 10.1093/nar/gkt1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday G. L.; Akiva E.; Meng E. C.; Brown S. D.; Calhoun S.; Pieper U.; Sali A.; Booker S. J.; Babbitt P. C. Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a ″Plug and Play″ Domain. Methods Enzymol. 2018, 606, 1–71. 10.1016/bs.mie.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe J.; Bohnert E.; Brümmer W. S-Adenosyl-l-Methionine, a Component of the Clastic Dissimilation of Pyruvate in Escherichia Coli. Biochim. Biophys. Acta BBA - Gen. Subj. 1965, 107, 603–605. 10.1016/0304-4165(65)90205-9. [DOI] [PubMed] [Google Scholar]
- Knappe J.; Schacht J.; Möckel W.; Höpner Th.; Vetter H. Jr.; Edenharder R. Pyruvate Formate-Lyase Reaction in Escherichia Coli. Eur. J. Biochem. 1969, 11, 316–327. 10.1111/j.1432-1033.1969.tb00775.x. [DOI] [PubMed] [Google Scholar]
- Knappe J.; Neugebauer F. A.; Blaschkowski H. P.; Gänzler M. Post-Translational Activation Introduces a Free Radical into Pyruvate Formate-Lyase. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 1332–1335. 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M.; Frey P. A. The Role of S-Adenosylmethionine in the Lysine 2,3-Aminomutase Reaction. J. Biol. Chem. 1987, 262, 14859–14862. 10.1016/S0021-9258(18)48103-3. [DOI] [PubMed] [Google Scholar]
- Chirpich T. P.; Zappia V.; Costilow R. N.; Barker H. A. Lysine 2,3-Aminomutase: Purification and Properties of a Pyridoxal Phosphate and S-adenosylmethionine-activated Enzyme. J. Biol. Chem. 1970, 245, 1778–1789. 10.1016/S0021-9258(19)77160-9. [DOI] [PubMed] [Google Scholar]
- Horitani M.; Shisler K.; Broderick W. E.; Hutcheson R. U.; Duschene K. S.; Marts A. R.; Hoffman B. M.; Broderick J. B. Radical SAM Catalysis via an Organometallic Intermediate with an Fe-[5′-C]-Deoxyadenosyl Bond. Science 2016, 352, 822–825. 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick W. E.; Hoffman B. M.; Broderick J. B. Mechanism of Radical Initiation in the Radical S-Adenosyl-l-Methionine Superfamily. Acc. Chem. Res. 2018, 51, 2611–2619. 10.1021/acs.accounts.8b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byer A. S.; Yang H.; McDaniel E. C.; Kathiresan V.; Impano S.; Pagnier A.; Watts H.; Denler C.; Vagstad A. L.; Piel J.; Duschene K. S.; Shepard E. M.; Shields T. P.; Scott L. G.; Lilla E. A.; Yokoyama K.; Broderick W. E.; Hoffman B. M.; Broderick J. B. Paradigm Shift for Radical S-Adenosyl-l-Methionine Reactions: The Organometallic Intermediate Ω Is Central to Catalysis. J. Am. Chem. Soc. 2018, 140, 8634–8638. 10.1021/jacs.8b04061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Magnusson O. Th. S-Adenosylmethionine: A Wolf in Sheep’s Clothing, or a Rich Man’s Adenosylcobalamin?. Chem. Rev. 2003, 103, 2129–2148. 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- Frey P. A. Travels with Carbon-Centered Radicals. 5′-Deoxyadenosine and 5′-Deoxyadenosine-5′-Yl in Radical Enzymology. Acc. Chem. Res. 2014, 47, 540–549. 10.1021/ar400194k. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Abeles R. H. The Role of the B12 Coenzyme in the Conversion of 1,2-Propanediol to Propionaldehyde. J. Biol. Chem. 1966, 241, 2732–2733. 10.1016/S0021-9258(18)96600-7. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Essenberg M. K.; Abeles R. H. Studies on the Mechanism of Hydrogen Transfer in the Cobamide Coenzyme-Dependent Dioldehydrase Reaction. J. Biol. Chem. 1967, 242, 5369–5377. 10.1016/S0021-9258(18)99437-8. [DOI] [PubMed] [Google Scholar]
- Finlay T. H.; Valinsky J.; Mildvan A. S.; Abeles R. H. Electron Spin Resonance Studies with Dioldehydrase: Evidence for Radical Intermediates in Reactions Catalyzed by Coenzyme B12. J. Biol. Chem. 1973, 248, 1285–1290. 10.1016/S0021-9258(19)44295-6. [DOI] [PubMed] [Google Scholar]
- Baraniak J.; Moss M. L.; Frey P. A. Lysine 2,3-Aminomutase: Support for a Mechanism of Hydrogen Transfer Involving S-Adenosylmethionine. J. Biol. Chem. 1989, 264, 1357–1360. 10.1016/S0021-9258(18)94194-3. [DOI] [PubMed] [Google Scholar]
- Moss M. L.; Frey P. A. Activation of Lysine 2,3-Aminomutase by S-Adenosylmethionine. J. Biol. Chem. 1990, 265, 18112–18115. 10.1016/S0021-9258(17)44724-7. [DOI] [PubMed] [Google Scholar]
- Wu W.; Booker S.; Lieder K. W.; Bandarian V.; Reed G. H.; Frey P. A. Lysine 2,3-Aminomutase and Trans-4,5-Dehydrolysine: Characterization of an Allylic Analogue of a Substrate-Based Radical in the Catalytic Mechanism. Biochemistry 2000, 39, 9561–9570. 10.1021/bi000658p. [DOI] [PubMed] [Google Scholar]
- Duin E. C.; Lafferty M. E.; Crouse B. R.; Allen R. M.; Sanyal I.; Flint D. H.; Johnson M. K. [2Fe-2S] to [4Fe-4S] Cluster Conversion in Escherichia Coli Biotin Synthase. Biochemistry 1997, 36, 11811–11820. 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- Ollagnier-De Choudens S.; Sanakis Y.; Hewitson K. S.; Roach P.; Baldwin J. E.; Münck E.; Fontecave M. Iron-Sulfur Center of Biotin Synthase and Lipoate Synthase. Biochemistry 2000, 39, 4165–4173. 10.1021/bi992090u. [DOI] [PubMed] [Google Scholar]
- Miller J. R.; Busby R. W.; Jordan S. W.; Cheek J.; Henshaw T. F.; Ashley G. W.; Broderick J. B.; Cronan J. E.; Marletta M. A. Escherichia Coli LipA Is a Lipoyl Synthase: In Vitro Biosynthesis of Lipoylated Pyruvate Dehydrogenase Complex from Octanoyl-Acyl Carrier Protein. Biochemistry 2000, 39, 15166–15178. 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- Ollagnier S.; Meier C.; Mulliez E.; Gaillard J.; Schuenemann V.; Trautwein A.; Mattioli T.; Lutz M.; Fontecave M. Assembly of 2Fe-2S and 4Fe-4S Clusters in the Anaerobic Ribonucleotide Reductase from Escherichia Coli. J. Am. Chem. Soc. 1999, 121, 6344–6350. 10.1021/ja990073m. [DOI] [Google Scholar]
- Cheek J.; Broderick J. B. Direct H Atom Abstraction from Spore Photoproduct C-6 Initiates DNA Repair in the Reaction Catalyzed by Spore Photoproduct Lyase: Evidence for a Reversibly Generated Adenosyl Radical Intermediate. J. Am. Chem. Soc. 2002, 124, 2860–2861. 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Radical SAM, a Novel Protein Superfamily Linking Unresolved Steps in Familiar Biosynthetic Pathways with Radical Mechanisms: Functional Characterization Using New Analysis and Information Visualization Methods. Nucleic Acids Res. 2001, 29, 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Woldring R. P.; Román-Meléndez G. D.; McClain A. M.; Alzua B. R.; Marsh E. N. G. Recent Advances in Radical SAM Enzymology: New Structures and Mechanisms. ACS Chem. Biol. 2014, 9, 1929–1938. 10.1021/cb5004674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S.; Fedoseyenko D.; Mahanta N.; Manion H.; Naseem S.; Dairi T.; Begley T. P. Novel Enzymology in Futalosine-Dependent Menaquinone Biosynthesis. Curr. Opin. Chem. Biol. 2018, 47, 134–141. 10.1016/j.cbpa.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Mehta A. P.; Abdelwahed S. H.; Mahanta N.; Fedoseyenko D.; Philmus B.; Cooper L. E.; Liu Y.; Jhulki I.; Ealick S. E.; Begley T. P. Radical S-Adenosylmethionine (SAM) Enzymes in Cofactor Biosynthesis: A Treasure Trove of Complex Organic Radical Rearrangement Reactions. J. Biol. Chem. 2015, 290, 3980–3986. 10.1074/jbc.R114.623793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley T. P.; Xi J.; Kinsland C.; Taylor S.; McLafferty F. The Enzymology of Sulfur Activation during Thiamin and Biotin Biosynthesis. Curr. Opin. Chem. Biol. 1999, 3, 623–629. 10.1016/S1367-5931(99)00018-6. [DOI] [PubMed] [Google Scholar]
- Yokoyama K.; Lilla E. A. C–C Bond Forming Radical SAM Enzymes Involved in the Construction of Carbon Skeletons of Cofactors and Natural Products. Nat. Prod. Rep. 2018, 35, 660–694. 10.1039/C8NP00006A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott T. A.; Piel J. The Hidden Enzymology of Bacterial Natural Product Biosynthesis. Nat. Rev. Chem. 2019, 3, 404. 10.1038/s41570-019-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell T. A. J.; Goldman P. J.; Drennan C. L. SPASM and Twitch Domains in S-Adenosylmethionine (SAM) Radical Enzymes. J. Biol. Chem. 2015, 290, 3964–3971. 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat S. S.; Williams H. J.; Raushel F. M. Intermediates in the Transformation of Phosphonates to Phosphate by Bacteria. Nature 2011, 480, 570–573. 10.1038/nature10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhu X.; Torelli A. T.; Lee M.; Dzikovski B.; Koralewski R. M.; Wang E.; Freed J.; Krebs C.; Ealick S. E.; Lin H. Diphthamide Biosynthesis Requires an Fe-S Enzyme-Generated Organic Radical. Nature 2010, 465, 891. 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K.-D.; Fischbach M. A.; Garavelli J. S.; Goransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Muller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H.-G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Sussmuth R. D.; Tagg J. R.; Tang G.-L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalban-Lopez M.; Scott T. A.; Ramesh S.; Rahman I. R.; van Heel A. J.; Viel J. H.; Bandarian V.; Dittmann E.; Genilloud O.; Goto Y.; Grande Burgos M. J.; Hill C.; Kim S.; Koehnke J.; Latham J. A.; Link A. J.; Martinez B.; Nair S. K.; Nicolet Y.; Rebuffat S.; Sahl H.-G.; Sareen D.; Schmidt E. W.; Schmitt L.; Severinov K.; Sussmuth R. D.; Truman A. W.; Wang H.; Weng J.-K.; van Wezel G. P.; Zhang Q.; Zhong J.; Piel J.; Mitchell D. A.; Kuipers O. P.; van der Donk W. A. New Developments in RiPP Discovery, Enzymology and Engineering. Nat. Prod. Rep. 2021, 38, 130–239. 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J. A.; Donia M. S.; Schmidt E. W. Ribosomal Peptide Natural Products: Bridging the Ribosomal and Nonribosomal Worlds. Nat. Prod. Rep. 2009, 26, 537–559. 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham J. A.; Barr I.; Klinman J. P. At the Confluence of Ribosomally Synthesized Peptide Modification and Radical S-Adenosylmethionine (SAM) Enzymology. J. Biol. Chem. 2017, 292, 16397–16405. 10.1074/jbc.R117.797399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N.; Hudson G. A.; Mitchell D. A. Radical S-Adenosylmethionine Enzymes Involved in RiPP Biosynthesis. Biochemistry 2017, 56, 5229–5244. 10.1021/acs.biochem.7b00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A.; Balty C.; Berteau O. Radical SAM Enzymes in the Biosynthesis of Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs). Front. Chem. 2017, 5, 87. 10.3389/fchem.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A.; Berteau O. Radical SAM Enzymes and Ribosomally-Synthesized and Post-translationally Modified Peptides: A Growing Importance in the Microbiomes. Front. Chem. 2021, 9, 474. 10.3389/fchem.2021.678068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L.; Rachid S.; Stadler M.; Wenzel S. C.; Müller R. Synthetic Biotechnology to Study and Engineer Ribosomal Bottromycin Biosynthesis. Chem. Biol. 2012, 19, 1278–1287. 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Flühe L.; Knappe T. A.; Gattner M. J.; Schäfer A.; Burghaus O.; Linne U.; Marahiel M. A. The Radical SAM Enzyme AlbA Catalyzes Thioether Bond Formation in Subtilosin A. Nat. Chem. Biol. 2012, 8, 350–357. 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- Flühe L.; Burghaus O.; Wieckowski B. M.; Giessen T. W.; Linne U.; Marahiel M. A. Two [4Fe-4S] Clusters Containing Radical SAM Enzyme SkfB Catalyze Thioether Bond Formation during the Maturation of the Sporulation Killing Factor. J. Am. Chem. Soc. 2013, 135, 959–962. 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- Kincannon W. M.; Bruender N. A.; Bandarian V. A Radical Clock Probe Uncouples H Atom Abstraction from Thioether Cross-Link Formation by the Radical S-Adenosyl-l-Methionine Enzyme SkfB. Biochemistry 2018, 57, 4816–4823. 10.1021/acs.biochem.8b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flühe L.; Marahiel M. A. Radical S-Adenosylmethionine Enzyme Catalyzed Thioether Bond Formation in Sactipeptide Biosynthesis. Curr. Opin. Chem. Biol. 2013, 17, 605–612. 10.1016/j.cbpa.2013.06.031. [DOI] [PubMed] [Google Scholar]
- Hudson G. A.; Burkhart B. J.; DiCaprio A. J.; Schwalen C. J.; Kille B.; Pogorelov T. V.; Mitchell D. A. Bioinformatic Mapping of Radical S-Adenosylmethionine-Dependent Ribosomally Synthesized and Post-Translationally Modified Peptides Identifies New Cα, Cβ, and Cγ-Linked Thioether-Containing Peptides. J. Am. Chem. Soc. 2019, 141, 8228–8238. 10.1021/jacs.9b01519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A.; Guillot A.; Ruffié P.; Leprince J.; Berteau O. Post-Translational Modification of Ribosomally Synthesized Peptides by a Radical SAM Epimerase in Bacillus subtilis. Nat. Chem. 2017, 9, 698–707. 10.1038/nchem.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagstad A. L.; Kuranaga T.; Püntener S.; Pattabiraman V. R.; Bode J. W.; Piel J. Introduction of D-Amino Acids in Minimalistic Peptide Substrates by an S-Adenosyl-l-Methionine Radical Epimerase. Angew. Chem., Int. Ed. Engl. 2019, 58, 2246–2250. 10.1002/anie.201809508. [DOI] [PubMed] [Google Scholar]
- Parent A.; Benjdia A.; Guillot A.; Kubiak X.; Balty C.; Lefranc B.; Leprince J.; Berteau O. Mechanistic Investigations of PoyD, a Radical S-Adenosyl-l-Methionine Enzyme Catalyzing Iterative and Directional Epimerizations in Polytheonamide A Biosynthesis. J. Am. Chem. Soc. 2018, 140, 2469–2477. 10.1021/jacs.7b08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka B. I.; Lakis E.; Verest M.; Helf M. J.; Scalvenzi T.; Vagstad A. L.; Sims J.; Sunagawa S.; Gugger M.; Piel J. Natural Noncanonical Protein Splicing Yields Products with Diverse β-Amino Acid Residues. Science 2018, 359, 779–782. 10.1126/science.aao0157. [DOI] [PubMed] [Google Scholar]
- Khaliullin B.; Ayikpoe R.; Tuttle M.; Latham J. A. Mechanistic Elucidation of the Mycofactocin-Biosynthetic Radical S-Adenosylmethionine Protein, MftC. J. Biol. Chem. 2017, 292, 13022–13033. 10.1074/jbc.M117.795682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruender N. A.; Bandarian V. The Radical S-Adenosyl-l-Methionine Enzyme MftC Catalyzes an Oxidative Decarboxylation of the C-Terminus of the MftA Peptide. Biochemistry 2016, 55, 2813–2816. 10.1021/acs.biochem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. Q. N.; Tooh Y. W.; Sugiyama R.; Nguyen T. P. D.; Purushothaman M.; Leow L. C.; Hanif K.; Yong R. H. S.; Agatha I.; Winnerdy F. R.; Gugger M.; Phan A. T.; Morinaka B. I. Post-Translational Formation of Strained Cyclophanes in Bacteria. Nat. Chem. 2020, 12, 1042–1053. 10.1038/s41557-020-0519-z. [DOI] [PubMed] [Google Scholar]
- Imai Y.; Meyer K. J.; Iinishi A.; Favre-Godal Q.; Green R.; Manuse S.; Caboni M.; Mori M.; Niles S.; Ghiglieri M.; Honrao C.; Ma X.; Guo J. J.; Makriyannis A.; Linares-Otoya L.; Böhringer N.; Wuisan Z. G.; Kaur H.; Wu R.; Mateus A.; Typas A.; Savitski M. M.; Espinoza J. L.; O’Rourke A.; Nelson K. E.; Hiller S.; Noinaj N.; Schäberle T. F.; D’Onofrio A.; Lewis K. A New Antibiotic Selectively Kills Gram-Negative Pathogens. Nature 2019, 576, 459–464. 10.1038/s41586-019-1791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. K.; Jochimsen A. S.; Lefave S. J.; Young A. P.; Kincannon W. M.; Roberts A. G.; Kieber-Emmons M. T.; Bandarian V. New Role for Radical SAM Enzymes in the Biosynthesis of Thio(Seleno)Oxazole RiPP Natural Products. Biochemistry 2021, 60, 3347–3361. 10.1021/acs.biochem.1c00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahiel M. A. A Structural Model for Multimodular NRPS Assembly Lines. Nat. Prod. Rep. 2016, 33, 136–140. 10.1039/C5NP00082C. [DOI] [PubMed] [Google Scholar]
- Knerr P. J.; van der Donk W. A. Discovery, Biosynthesis, and Engineering of Lantipeptides. Annu. Rev. Biochem. 2012, 81, 479–505. 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- Repka L. M.; Chekan J. R.; Nair S. K.; van der Donk W. A. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem. Rev. 2017, 117, 5457–5520. 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Zhou S.; Liu W. Opportunities and Challenges from Current Investigations into the Biosynthetic Logic of Nosiheptide-Represented Thiopeptide Antibiotics. Curr. Opin. Chem. Biol. 2013, 17, 626–634. 10.1016/j.cbpa.2013.06.021. [DOI] [PubMed] [Google Scholar]
- Kawulka K.; Sprules T.; McKay R. T.; Mercier P.; Diaper C. M.; Zuber P.; Vederas J. C. Structure of Subtilosin A, an Antimicrobial Peptide from Bacillus Subtilis with Unusual Posttranslational Modifications Linking Cysteine Sulfurs to α-Carbons of Phenylalanine and Threonine. J. Am. Chem. Soc. 2003, 125, 4726–4727. 10.1021/ja029654t. [DOI] [PubMed] [Google Scholar]
- Freeman M. F.; Gurgui C.; Helf M. J.; Morinaka B. I.; Uria A. R.; Oldham N. J.; Sahl H.-G.; Matsunaga S.; Piel J. Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides. Science 2012, 338, 387–390. 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.; Guillot A.; Wessner F.; Algaron F.; Besset C.; Courtin P.; Gardan R.; Monnet V. Control of the Transcription of a Short Gene Encoding a Cyclic Peptide in Streptococcus Thermophilus: A New Quorum-Sensing System?. J. Bacteriol. 2007, 189, 8844–8854. 10.1128/JB.01057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramma K. R.; Bushin L. B.; Seyedsayamdost M. R. Structure and Biosynthesis of a Macrocyclic Peptide Containing an Unprecedented Lysine-to-Tryptophan Crosslink. Nat. Chem. 2015, 7, 431–437. 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isley N. A.; Endo Y.; Wu Z.-C.; Covington B. C.; Bushin L. B.; Seyedsayamdost M. R.; Boger D. L. Total Synthesis and Stereochemical Assignment of Streptide. J. Am. Chem. Soc. 2019, 141, 17361–17369. 10.1021/jacs.9b09067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K. Biological Systems Discovery in Silico: Radical S-Adenosylmethionine Protein Families and Their Target Peptides for Posttranslational Modification. J. Bacteriol. 2011, 193, 2745–2755. 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H. Bioinformatic Evidence for a Widely Distributed, Ribosomally Produced Electron Carrier Precursor, Its Maturation Proteins, and Its Nicotinoprotein Redox Partners. BMC Genomics 2011, 12, 21. 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. L.; Lee K.-H.; St. Clair J.; Krebs C.; Booker S. J. In Vitro Characterization of AtsB, a Radical SAM Formylglycine-Generating Enzyme That Contains Three [4Fe-4S] Clusters. Biochemistry 2008, 47, 7523–7538. 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman P. J.; Grove T. L.; Sites L. A.; McLaughlin M. I.; Booker S. J.; Drennan C. L. X-Ray Structure of an AdoMet Radical Activase Reveals an Anaerobic Solution for Formylglycine Posttranslational Modification. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8519–8524. 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramma K. R.; Seyedsayamdost M. R. Lysine-Tryptophan-Crosslinked Peptides Produced by Radical SAM Enzymes in Pathogenic Streptococci. ACS Chem. Biol. 2017, 12, 922–927. 10.1021/acschembio.6b01069. [DOI] [PubMed] [Google Scholar]
- Schramma K. R.; Forneris C. C.; Caruso A.; Seyedsayamdost M. R. Mechanistic Investigations of Lysine–Tryptophan Cross-Link Formation Catalyzed by Streptococcal Radical S-Adenosylmethionine Enzymes. Biochemistry 2018, 57, 461–468. 10.1021/acs.biochem.7b01147. [DOI] [PubMed] [Google Scholar]
- Davis K. M.; Schramma K. R.; Hansen W. A.; Bacik J. P.; Khare S. D.; Seyedsayamdost M. R.; Ando N. Structures of the Peptide-Modifying Radical SAM Enzyme SuiB Elucidate the Basis of Substrate Recognition. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 10420–10425. 10.1073/pnas.1703663114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balo A. R.; Caruso A.; Tao L.; Tantillo D. J.; Seyedsayamdost M. R.; Britt R. D. Trapping a Cross-Linked Lysine–Tryptophan Radical in the Catalytic Cycle of the Radical SAM Enzyme SuiB. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2101571118. 10.1073/pnas.2101571118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushin L. B.; Clark K. A.; Pelczer I.; Seyedsayamdost M. R. Charting an Unexplored Streptococcal Biosynthetic Landscape Reveals a Unique Peptide Cyclization Motif. J. Am. Chem. Soc. 2018, 140, 17674–17684. 10.1021/jacs.8b10266. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A. Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence–Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. 10.1021/acs.biochem.7b00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A.; Bouvier J. T.; Davidson D. B.; Imker H. J.; Sadkhin B.; Slater D. R.; Whalen K. L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A Web Tool for Generating Protein Sequence Similarity Networks. Biochim. Biophys. Acta 2015, 1854, 1019–1037. 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso A.; Bushin L. B.; Clark K. A.; Martinie R. J.; Seyedsayamdost M. R. Radical Approach to Enzymatic β-Thioether Bond Formation. J. Am. Chem. Soc. 2019, 141, 990–997. 10.1021/jacs.8b11060. [DOI] [PubMed] [Google Scholar]
- Grove T. L.; Himes P. M.; Hwang S.; Yumerefendi H.; Bonanno J. B.; Kuhlman B.; Almo S. C.; Bowers A. A. Structural Insights into Thioether Bond Formation in the Biosynthesis of Sactipeptides. J. Am. Chem. Soc. 2017, 139, 11734–11744. 10.1021/jacs.7b01283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Yang Y.; Ji X.; Zhao R.; Li G.; Gu Y.; Shi A.; Jiang W.; Zhang Q. The SCIFF-Derived Ranthipeptides Participate in Quorum Sensing in Solventogenic Clostridia. Biotechnol. J. 2020, 15, 2000136. 10.1002/biot.202000136. [DOI] [PubMed] [Google Scholar]
- Clark K. A.; Bushin L. B.; Seyedsayamdost M. R. Aliphatic Ether Bond Formation Expands the Scope of Radical SAM Enzymes in Natural Product Biosynthesis. J. Am. Chem. Soc. 2019, 141, 10610–10615. 10.1021/jacs.9b05151. [DOI] [PubMed] [Google Scholar]
- Caruso A.; Martinie R. J.; Bushin L. B.; Seyedsayamdost M. R. Macrocyclization via an Arginine-Tyrosine Crosslink Broadens the Reaction Scope of Radical S-Adenosylmethionine Enzymes. J. Am. Chem. Soc. 2019, 141, 16610–16614. 10.1021/jacs.9b09210. [DOI] [PubMed] [Google Scholar]
- Bushin L. B.; Covington B. C.; Rued B. E.; Federle M. J.; Seyedsayamdost M. R. Discovery and Biosynthesis of Streptosactin, a Sactipeptide with an Alternative Topology Encoded by Commensal Bacteria in the Human Microbiome. J. Am. Chem. Soc. 2020, 142, 16265–16275. 10.1021/jacs.0c05546. [DOI] [PubMed] [Google Scholar]
- Caruso A.; Seyedsayamdost M. R. Radical SAM Enzyme QmpB Installs Two 9-Membered Ring Sactionine Macrocycles during Biogenesis of a Ribosomal Peptide Natural Product. J. Org. Chem. 2021, 86, 11284–11289. 10.1021/acs.joc.1c01507. [DOI] [PubMed] [Google Scholar]
- Rued B. E.; Covington B. C.; Bushin L. B.; Szewczyk G.; Laczkovich I.; Seyedsayamdost M. R.; Federle M. J. Quorum Sensing in Streptococcus Mutans Regulates Production of Tryglysin, a Novel RaS-RiPP Antimicrobial Compound. mBio 2021, 12, e02688-20. 10.1128/mBio.02688-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverys J.-P.; Håvarstein L. S. Cannibalism and Fratricide: Mechanisms and Raisons d’être. Nat. Rev. Microbiol. 2007, 5, 219–229. 10.1038/nrmicro1613. [DOI] [PubMed] [Google Scholar]
- Claverys J.-P.; Martin B.; Håvarstein L. S. Competence-Induced Fratricide in Streptococci. Mol. Microbiol. 2007, 64, 1423–1433. 10.1111/j.1365-2958.2007.05757.x. [DOI] [PubMed] [Google Scholar]
- Donia M. S.; Cimermancic P.; Schulze C. J.; Wieland Brown L. C.; Martin J.; Mitreva M.; Clardy J.; Linington R. G.; Fischbach M. A. A Systematic Analysis of Biosynthetic Gene Clusters in the Human Microbiome Reveals a Common Family of Antibiotics. Cell 2014, 158, 1402–1414. 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiumento S.; Roblin C.; Kieffer-Jaquinod S.; Tachon S.; Leprètre C.; Basset C.; Aditiyarini D.; Olleik H.; Nicoletti C.; Bornet O.; Iranzo O.; Maresca M.; Hardré R.; Fons M.; Giardina T.; Devillard E.; Guerlesquin F.; Couté Y.; Atta M.; Perrier J.; Lafond M.; Duarte V. Ruminococcin C, a Promising Antibiotic Produced by a Human Gut Symbiont. Sci. Adv. 2019, 5, eaaw9969. 10.1126/sciadv.aaw9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roblin C.; Chiumento S.; Bornet O.; Nouailler M.; Muller C. S.; Jeannot K.; Basset C.; Kieffer-Jaquinod S.; Coute Y.; Torelli S.; Le Pape L.; Schunemann V.; Olleik H.; De La Villeon B.; Sockeel P.; Di Pasquale E.; Nicoletti C.; Vidal N.; Poljak L.; Iranzo O.; Giardina T.; Fons M.; Devillard E.; Polard P.; Maresca M.; Perrier J.; Atta M.; Guerlesquin F.; Lafond M.; Duarte V. The Unusual Structure of Ruminococcin C1 Antimicrobial Peptide Confers Clinical Properties. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 19168–19177. 10.1073/pnas.2004045117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balty C.; Guillot A.; Fradale L.; Brewee C.; Boulay M.; Kubiak X.; Benjdia A.; Berteau O. Ruminococcin C, an Anti-Clostridial Sactipeptide Produced by a Prominent Member of the Human Microbiota Ruminococcus Gnavus. J. Biol. Chem. 2019, 294, 14512–14525. 10.1074/jbc.RA119.009416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balty C.; Guillot A.; Fradale L.; Brewee C.; Lefranc B.; Herrero C.; Sandström C.; Leprince J.; Berteau O.; Benjdia A. Biosynthesis of the Sactipeptide Ruminococcin C by the Human Microbiome: Mechanistic Insights into Thioether Bond Formation by Radical SAM Enzymes. J. Biol. Chem. 2020, 295, 16665–16677. 10.1074/jbc.RA120.015371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crost E. H.; Ajandouz E. H.; Villard C.; Geraert P. A.; Puigserver A.; Fons M. Ruminococcin C, a New Anti-Clostridium Perfringens Bacteriocin Produced in the Gut by the Commensal Bacterium Ruminococcus Gnavus E1. Biochimie 2011, 93, 1487–1494. 10.1016/j.biochi.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Salisbury S. A.; Forrest H. S.; Cruse W. B.; Kennard O. A Novel Coenzyme from Bacterial Primary Alcohol Dehydrogenases. Nature 1979, 280, 843–844. 10.1038/280843a0. [DOI] [PubMed] [Google Scholar]
- Duine J. A.; Jzn J. F.; van Zeeland J. K. Glucose Dehydrogenase from Acinetobacter Calcoaceticus: A “Quinoprotein.. FEBS Lett. 1979, 108, 443–446. 10.1016/0014-5793(79)80584-0. [DOI] [PubMed] [Google Scholar]
- Duine J. A. The PQQ Story. J. Biosci. Bioeng. 1999, 88, 231–236. 10.1016/S1389-1723(00)80002-X. [DOI] [PubMed] [Google Scholar]
- van Kleef M. A. G.; Duine J. A. L-Tyrosine Is the Precursor of PQQ Biosynthesis in Hyphomicrobium X. FEBS Lett. 1988, 237, 91–97. 10.1016/0014-5793(88)80178-9. [DOI] [PubMed] [Google Scholar]
- Houck D. R.; Hanners J. L.; Unkefer C. J. Biosynthesis of Pyrroloquinoline Quinone. 1. Identification of Biosynthetic Precursors Using Carbon-13 Labeling and NMR Spectroscopy. J. Am. Chem. Soc. 1988, 110, 6920–6921. 10.1021/ja00228a070. [DOI] [Google Scholar]
- Houck D. R.; Hanners J. L.; Unkefer C. J. Biosynthesis of Pyrroloquinoline Quinone. 2. Biosynthetic Assembly from Glutamate and Tyrosine. J. Am. Chem. Soc. 1991, 113, 3162–3166. 10.1021/ja00008a053. [DOI] [Google Scholar]
- Houck D. R.; Hanners J. L.; Unkefer C. J.; van Kleef M. A. G.; Duine J. A. PQQ: Biosynthetic Studies InMethylobacterium AM1 AndHyphomicrobium X Using Specific13C Labeling and NMR. Antonie Van Leeuwenhoek 1989, 56, 93–101. 10.1007/BF00822589. [DOI] [PubMed] [Google Scholar]
- Goosen N; Horsman H P; Huinen R G; van de Putte P Acinetobacter calcoaceticus Genes Involved in Biosynthesis of the Coenzyme Pyrrolo-Quinoline-Quinone: Nucleotide Sequence and Expression in Escherichia coli K-12. J. Bacteriol. 1989, 171, 447–455. 10.1128/jb.171.1.447-455.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J. J. M.; Sellink E.; Loenen W. A. M.; Riegman N. H.; van Kleef M.; Postma P. W. Cloning of Klebsiella Pneumoniae Pqq Genes and PQQ Biosynthesis in Escherichia coli. FEMS Microbiol. Lett. 1990, 71, 337–343. 10.1111/j.1574-6968.1990.tb03847.x. [DOI] [PubMed] [Google Scholar]
- Kloosterman A. M.; Shelton K. E.; van Wezel G. P.; Medema M. H.; Mitchell D. A. RRE-Finder: A Genome-Mining Tool for Class-Independent RiPP Discovery. mSystems 2020, 5, e00267-20. 10.1128/mSystems.00267-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham J. A.; Iavarone A. T.; Barr I.; Juthani P. V.; Klinman J. P. PqqD Is a Novel Peptide Chaperone That Forms a Ternary Complex with the Radical S-Adenosylmethionine Protein PqqE in the Pyrroloquinoline Quinone Biosynthetic Pathway. J. Biol. Chem. 2015, 290, 12908–12918. 10.1074/jbc.M115.646521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Walker L. M.; Tao L.; Iavarone A. T.; Wei X.; Britt R. D.; Elliott S. J.; Klinman J. P. Structural Properties and Catalytic Implications of the SPASM Domain Iron–Sulfur Clusters in Methylorubrum Extorquens PqqE. J. Am. Chem. Soc. 2020, 142, 12620–12634. 10.1021/jacs.0c02044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Klinman J. P. Biogenesis of the Peptide-Derived Redox Cofactor Pyrroloquinoline Quinone. Curr. Opin. Chem. Biol. 2020, 59, 93–103. 10.1016/j.cbpa.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson O. T.; Toyama H.; Saeki M.; Schwarzenbacher R.; Klinman J. P. The Structure of a Biosynthetic Intermediate of Pyrroloquinoline Quinone (PQQ) and Elucidation of the Final Step of PQQ Biosynthesis. J. Am. Chem. Soc. 2004, 126, 5342–5343. 10.1021/ja0493852. [DOI] [PubMed] [Google Scholar]
- Koehn E. M.; Latham J. A.; Armand T.; Evans R. L.; Tu X.; Wilmot C. M.; Iavarone A. T.; Klinman J. P. Discovery of Hydroxylase Activity for PqqB Provides a Missing Link in the Pyrroloquinoline Quinone Biosynthetic Pathway. J. Am. Chem. Soc. 2019, 141, 4398–4405. 10.1021/jacs.8b13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson O. T.; Toyama H.; Saeki M.; Rojas A.; Reed J. C.; Liddington R. C.; Klinman J. P.; Schwarzenbacher R. Quinone Biogenesis: Structure and Mechanism of PqqC, the Final Catalyst in the Production of Pyrroloquinoline Quinone. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7913–7918. 10.1073/pnas.0402640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnot F.; Iavarone A. T.; Klinman J. P. Multistep, Eight-Electron Oxidation Catalyzed by the Cofactorless Oxidase, PqqC: Identification of Chemical Intermediates and Their Dependence on Molecular Oxygen. Biochemistry 2013, 52, 4667–4675. 10.1021/bi4003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp P. F.; Benjdia A.; Strahl H.; Berteau O.; Mascher T. The Epipeptide YydF Intrinsically Triggers the Cell Envelope Stress Response of Bacillus subtilis and Causes Severe Membrane Perturbations. Front. Microbiol. 2020, 11, 151. 10.3389/fmicb.2020.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Pastor J. E.; Hobbs E. C.; Losick R. Cannibalism by Sporulating Bacteria. Science 2003, 301, 510–513. 10.1126/science.1086462. [DOI] [PubMed] [Google Scholar]
- Ellermeier C. D.; Hobbs E. C.; Gonzalez-Pastor J. E.; Losick R. A Three-Protein Signaling Pathway Governing Immunity to a Bacterial Cannibalism Toxin. Cell 2006, 124, 549–559. 10.1016/j.cell.2005.11.041. [DOI] [PubMed] [Google Scholar]
- Levin B. J.; Huang Y. Y.; Peck S. C.; Wei Y.; Martinez-del Campo A.; Marks J. A.; Franzosa E. A.; Huttenhower C.; Balskus E. P. A Prominent Glycyl Radical Enzyme in Human Gut Microbiomes Metabolizes trans-4-Hydroxy-L-Proline. Science 2017, 355, eaai8386. 10.1126/science.aai8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N.; Gerlt J. A. Discovery of New Enzymatic Functions and Metabolic Pathways Using Genomic Enzymology Web Tools. Curr. Opin. Biotechnol. 2021, 69, 77–90. 10.1016/j.copbio.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler B. L.; Losick R. Bacterially Speaking. Cell 2006, 125, 237–246. 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Wilson M. R.; Zha L.; Balskus E. P. Natural Product Discovery from the Human Microbiome. J. Biol. Chem. 2017, 292, 8546–8552. 10.1074/jbc.R116.762906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleti G.; Baker J. L.; Tang X.; Alvarez R.; Dinis M.; Tran N. C.; Melnik A. V.; Zhong C.; Ernst M.; Dorrestein P. C.; Edlund A. Identification of the Bacterial Biosynthetic Gene Clusters of the Oral Microbiome Illuminates the Unexplored Social Language of Bacteria During Health and Disease. mBio 2019, 10, e00321-19. 10.1128/mBio.00321-19. [DOI] [PMC free article] [PubMed] [Google Scholar]