

Abstract
The EGFR is a prime oncogene that is frequently amplified in GBM. Here, we demonstrate a new tumour suppressive function of EGFR in EGFR-amplified GBMs regulated by EGFR ligands. Constitutive EGFR signaling promotes invasion via activation of a TAB1-TAK1-NF-κB-EMP1 pathway, resulting in large tumours and worse survival in orthotopic models. Ligand-activated EGFR promotes proliferation, and surprisingly, suppresses invasion by upregulating BIN3 which inhibits a DOCK-7 Rho GTPase pathway, resulting in small hyper-proliferating noninvasive tumours and improved survival. TCGA data reveal that in EGFR amplified GBMs, a low level of EGFR ligands confers a worse prognosis, while a high level of EGFR ligands confers an improved prognosis. Thus, increased EGFR ligand shifts the role of EGFR from oncogene to tumour suppressor in EGFR amplified GBMs by suppressing invasion. The tumour suppressive function of EGFR can be activated therapeutically by using tofacitinib, which suppresses invasion by increasing EGFR ligand and upregulating BIN3.
Keywords: Glioblastoma, invasion, proliferation, tumour size, EGFR switch, EGFR, tofacitinib, BIN3, constitutive EGFR signaling, ligand-induced EGFR signaling
Introduction
Glioblastoma (GBM) is a highly invasive cancer. Receptor tyrosine kinases such as EGFR play a particularly important role in GBM invasion and act in part through cytoskeletal reorganization mediated by the Rho GTPase family. The epidermal growth factor receptor (EGFR) is a prime oncogene in GBM. EGFR ligands are known to be variably expressed in GBMs1–3. There is substantial evidence suggesting that the EGFR is an important oncogenic driver in GBM 4, 5. However, multiple clinical trials of EGFR targeting have failed in GBM 6, 7. EGFR activation is thought to play a uniformly oncogenic role in GBM. EGFR gene amplification is found in the classical subtype of GBM8, and is detected in 40–50% of GBMs, resulting in EGFR overexpression. EGFR is also expressed without gene amplification and is detectable in the majority of GBMs 4, 9. Both EGFR wild type (EGFRwt) and constitutively active oncogenic and pro-invasive mutants such as EGFRvIII are expressed 10, 11. EGFRwt is also oncogenic and 12, 13 may be activated by ligand binding or signal constitutively when overexpressed in cancer 2, 13, 14. EGFRwt and EGFRvIII are usually expressed in the same tumour and may activate each other 1, 5, 9, 15, 16. Constitutive signaling is defined here as signaling triggered by EGFR expression in GBM leading to spontaneous dimerization and downstream signaling in the absence of EGFR ligand 14, 17. We have previously reported that that constitutive and ligand-induced EGFR signaling trigger distinct and non-overlapping signaling pathways 2, 17.
We have previously identified Bridging integrator 3 (BIN3) as a protein that is highly induced by ligand-dependent EGFR signaling1. BIN3 is a member of the Bin-Ampiphysin-Rvs (BAR) domain family of proteins that regulate membrane and actin dynamics. BIN3 is ubiquitously expressed and conserved throughout evolution. BAR domain proteins also regulate Rho GTPases that are involved in GBM invasion22, 24, 25. BIN3 maps to chromosome 8p21.3 a tumour suppressor region that is often deleted in non-Hodgkin’s lymphoma and other epithelial cancers 26–28. BIN3 deletion in mice results in increased susceptibility to lymphoma 29.
Seven EGFR ligands are expressed in GBM. EGFR ligands vary in their binding affinity, their kinetics and their function 31, 32. It is currently believed that the presence or level of expression of EGFR ligand(s) in a particular GBM tumour does not have clinical utility, and consequently, is ignored in clinical practice. In this study, we describe a new tumour suppressor function of EGFR in EGFR amplified GBMs that is driven by ligand-dependent EGFR signaling.
Results
Constitutive vs. ligand-induced EGFR signaling elicits distinct biological responses in glioma cells
To understand the biological consequences of constitutive vs. ligand induced EGFR mediated RTK signaling, we examined the effect of EGF on glioma cell invasion using a panel of Mayo PDX lines33 (Fig. 1a, Extended Data Fig. 1a) or primary GBM neurosphere cultures (Fig. 1a). Surprisingly, addition of EGFR ligands suppressed invasion in multiple lines expressing EGFRwt or EGFRvIII plus EGFRwt using a transwell invasion assay (Fig. 1b, Extended Data Fig. 1b–h). siRNA knockdown of EGFR abolished the inhibitory effect of EGF on invasion indicating that the invasion effect is mediated specifically through the EGFR (Fig. 1c–f). Furthermore, siRNA to EGFR without added EGF suppressed invasion demonstrating that the EGFR is driving basal invasion (Fig. 1c–f). The low level of EGFR remaining in the EGFR silenced cells is likely to be EGFRvIII and does not generate ligand-mediated signals (Extended Data Fig. 1i). Additionally, rescue experiments with 3’ UTR targeted siRNA demonstrate that the decreased invasion in EGFR siRNA knockdown cells is abolished following EGFRwt or EGFRvIII overexpression (Fig. 1g–h). Erlotinib, an EGFR TKI, also suppresses invasion (Fig. 1i–j). Erlotinib suppressed invasion transiently and invasion returns to baseline in 48 hours (Fig. 1i–j, Extended Data Fig. 1k–o), likely because of adaptive responses triggered by EGFR inhibition34–38. Overexpression of EGFR increased invasion but if EGF is added, invasion is suppressed (Fig. 1k–l). A scratch wound healing assay and a 3D matrigel invasion assay confirmed that EGF suppresses invasion (Fig. 1t–u, Extended Data Fig. 1z–aa). We also examined signaling events in GBM14 cells, which do not express endogenous EGFR by expressing EGFRwt (Extended Data Fig. 1t–u). Importantly, the invasiveness of GBM overexpressing EGFR cells without ligand is unaffected by cetuximab, an anti-EGFR mAb (Extended Data Fig. 1p–s), although cetuximab pretreatment blocks EGF mediated suppression of invasion (Extended Data Fig. 1v–w). Additionally, expression of EGFRvIII in GBM14 cells also enhances invasion (Extended Data Fig. 1x–y).
Figure 1. Ligand induced EGFR signaling inhibits invasion by upregulation of BIN3.
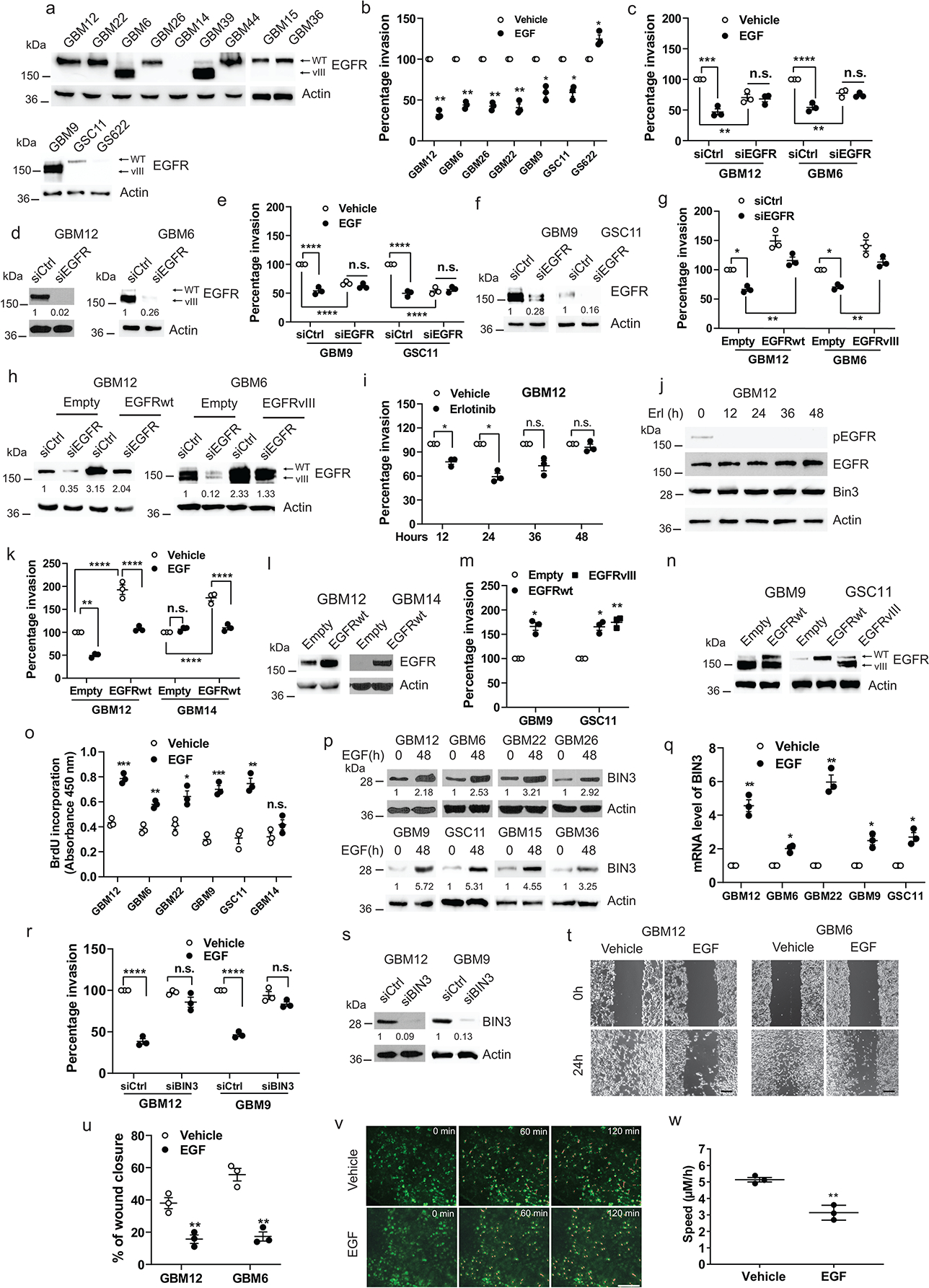
a, EGFR expression in multiple GBM PDXs and neurospheres line. b, Matrigel invasion assay of multiple lines with or without EGF (50 ng/ml). c, Matrigel invasion assay of EGFR siRNA knockdown cells in the presence or absence of EGF. d, Knockdown efficiency of EGFR siRNA was analyzed by Western blot. e-f, Similar experiments in neurospheres. g, Matrigel invasion assay of EGFR siRNA knockdown cells retransfected with EGFRwt or EGFRvIII expression vector. h, Western blot showing knockdown or re-expression efficiency of EGFRwt or EGFRvIII. I-j, Matrigel invasion assay and Western blot of GBM12 with or without erlotinib (1 μM) for indicated times. k-l, Matrigel invasion assay of EGFRwt overexpressing cells in the presence or absence of EGF and Western blot of EGFR expression. m-n, Matrigel invasion assay of EGFRwt or EGFRvIII overexpressing cells and Western blot of EGFR expression. o, BrdU incorporation assay of multiple lines treated with or without EGF (50ng/ml). p-q, Western blot and qPCR analysis of BIN3 in multiple lines treated with EGF (50 ng/ml). r, Matrigel invasion assay of BIN3 siRNA knockdown cells with or without EGF. s, Knockdown efficiency of BIN3 siRNA was analyzed by Western blot. t-u, Representative images and quantitative analysis of scratch assay in cells treated with EGF (50 ng/ml) for 24 hours. Scale bar: 100 μm. v, Representative time-lapse imaging of migrating tumour cells (red lines) through a cranial window. Scale bar: 100 μm. w, Quantification of cell velocity for vehicle and EGF treated group (n=3/group). The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-tailed one-sample Student’s t-test (b, i, m, q), or by two-way analysis of variance (ANOVA) adjusted by Bonferroni’s correction (c, e, g, k, r), or by two-tailed unpaired Student’s t-test (o, u, w). *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Prior studies have reported that addition of EGF results in increased invasiveness of glioma cell lines 39–42. These studies used established GBM cell lines that tend to lose the EGFR amplification 43, 44. Mayo PDX lines and neurospheres tend to retain the EGFR amplification43, 44. We also found that exposing low EGFR expressing cells to EGF leads to increased invasion. However, when the EGFR is overexpressed in these lines, invasion is suppressed by EGF (Fig. 2a–g). Conversely, reducing EGFR levels using different doses of siRNA produces a reversal of the effect of ligand from suppression to promotion of invasion (Fig. 2h–i), suggesting that it is the level of EGFR expression that determines whether the outcome of ligand-dependent EGFR signaling is increased or decreased invasion. EGF has been reported to promote invasion in non-glioma high EGFR expressing cancer cell lines such as A431 cells by a dephosphorylation of FAK45. We found an increase in FAK phosphorylation in response to EGF in GBM lines, while in A431 cells FAK is indeed dephosphorylated (Extended Data Fig. 2a). Thus, there are cell type specific differences in the effects of EGFR signaling on invasion.
Figure 2. EGFR level determines the invasion response to EGFR ligand via BIN3 regulation.
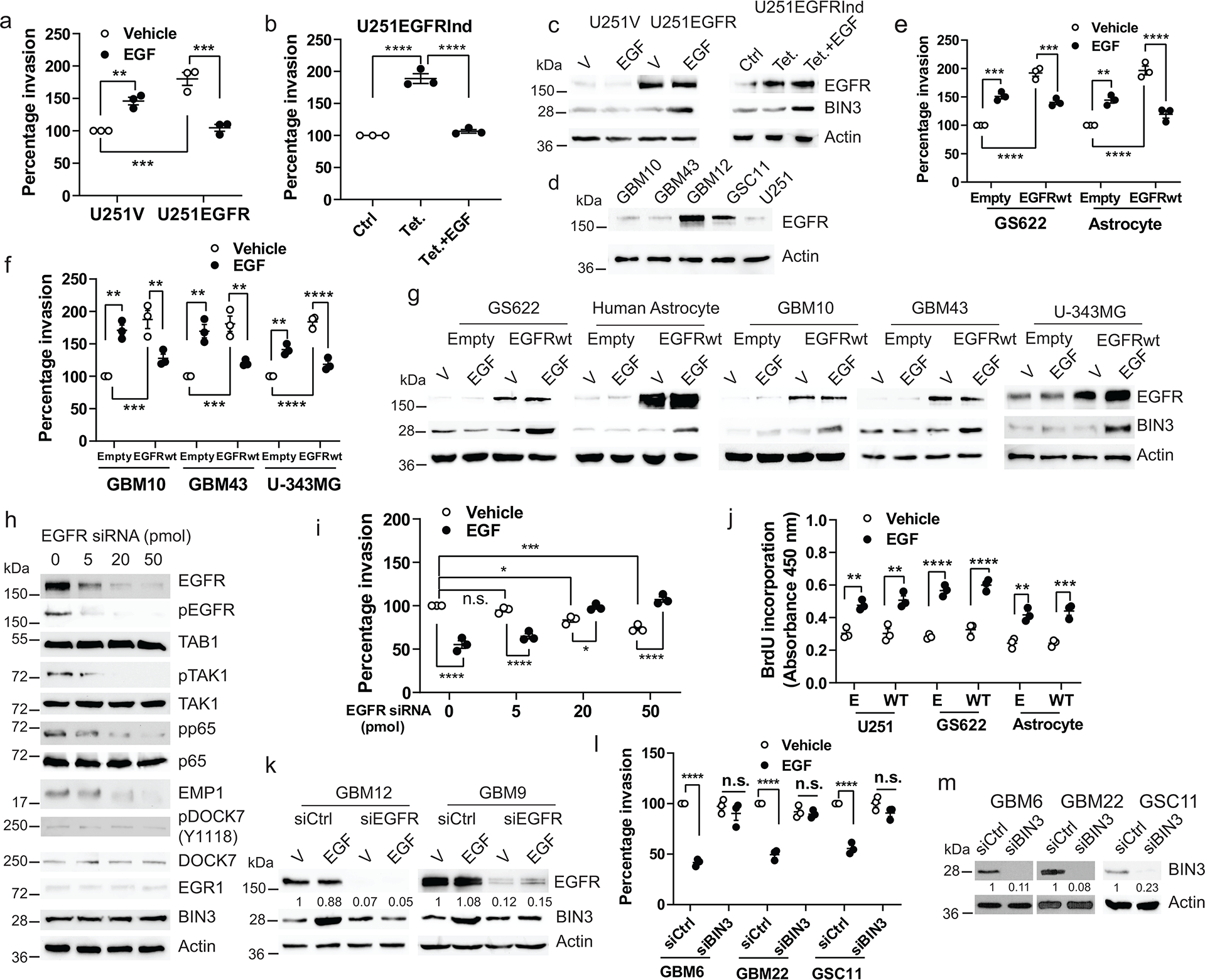
a, Matrigel invasion assay of U251 cells stably transfected with the empty (U251V) or EGFRwt expression vector (U251EGFR) in the presence or absence of EGF (50 ng/ml). b, Matrigel invasion assay of tetracycline (Tet.) induced EGFR overexpressing U251 cells (U251EGFRInd) in the presence or absence of EGF. c, Immunoblot of EGFR and BIN3 expression in cells treated with EGF for 48 hours. d, Immunoblot of EGFR expression in multiple lines. e-f, Matrigel invasion assay of EGFRwt overexpressing cells in the presence or absence of EGF. g, Immunoblot of EGFR and BIN3 expression in EGFRwt overexpressing cells treated with EGF for 48 hours. h, Immunoblot of EGFR, pEGFR and proteins involved in TAB1-BIN3 axis expression in cells transfected with indicated dose of EGFR siRNA. i, Matrigel invasion assay of GBM12 transfected with indicated doses of EGFR siRNA. j, BrdU incorporation assay of cells transiently transfected with empty (E) or EGFRwt (WT) expression vector in the presence or absence of EGF for 48 hours. k, Immunoblot of EGFR and BIN3 expression in EGFR siRNA knockdown cells treated with EGF (50ng/ml). l, Matrigel invasion assay of BIN3 siRNA knockdown cells in the presence or absence of EGF. m, BIN3 siRNA knockdown efficiency was analyzed by Western blot. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-way ANOVA adjusted by Bonferroni’s correction (a, e, f, i, j, l), or by one-way ANOVA adjusted by Bonferroni’s correction (b). * P<0.01, ** P < 0.01, *** P<0.001, **** P<0.0001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Next, we injected GBM12 cells into a live mouse brain and confirmed that EGF suppressed invasion in glioma cells using an in vivo intravital microscopy experiment (Fig. 1v–w). EGF did not induce cell death (Extended Data Fig. 2b). Overexpression of EGFRwt in GBM lines failed to increase proliferation in the absence of ligand (Extended Data Fig. 2c–d). However, EGF induced proliferation in multiple lines expressing high or low EGFR (Fig. 1o, Fig. 2j),
Ligand-activated EGFR mediated suppression of invasion is mediated by upregulation of BIN3
To uncover the downstream signaling mechanisms underlying the effect of EGFR on glioma cell invasion, we examined RNA microarray data from a previous study1. BIN3 was prioritized because it is the most highly upregulated gene when EGF is added (19.2 fold)1, and has a known role in actin organization 46. We confirmed that EGF induced a robust increase in BIN3 by Western blot and qPCR (Fig. 1p–q) that is abolished by siRNA knockdown of the EGFR (Fig. 2k). Importantly, siRNA knockdown of BIN3 blocked the ability of EGF to downregulate GBM invasiveness (Fig. 1r–s, and Fig. 2l–m)., Importantly, in low EGFR expressing lines in which EGF induces invasion, BIN3 is not upregulated (Fig. 2a–g). Since our previous study used an established GBM cell line, we also confirmed the EGF-induced upregulation of BIN3 by RNA-seq in GBM12 cells (Extended Data Fig. 2e). Also, overexpression of BIN3 results in decreased invasion that is not further decreased by EGF (Fig. 3a–b, Extended Data Fig. 3a–b). Overexpression of BIN3 does not affect proliferation or cell death (Extended Data Fig. 2f–h). Next, we identified EGR1 as a highly EGF-induced (13.68 fold) gene from our previous study1. EGR1 sites are present in the BIN3 promoter (Extended Data Fig. 3c). The upregulation of EGR1 and its activation was confirmed by reporter and ChiP assays (Fig. 3c–d, Extended Data Fig. 3d–e). Importantly, siRNA knockdown of EGR1 blocked the ability of EGF to induce BIN3, and to suppress invasion (Fig. 3e–g).
Figure 3. BIN3 inhibits invasiveness of glioma cells through its interaction with DOCK7.
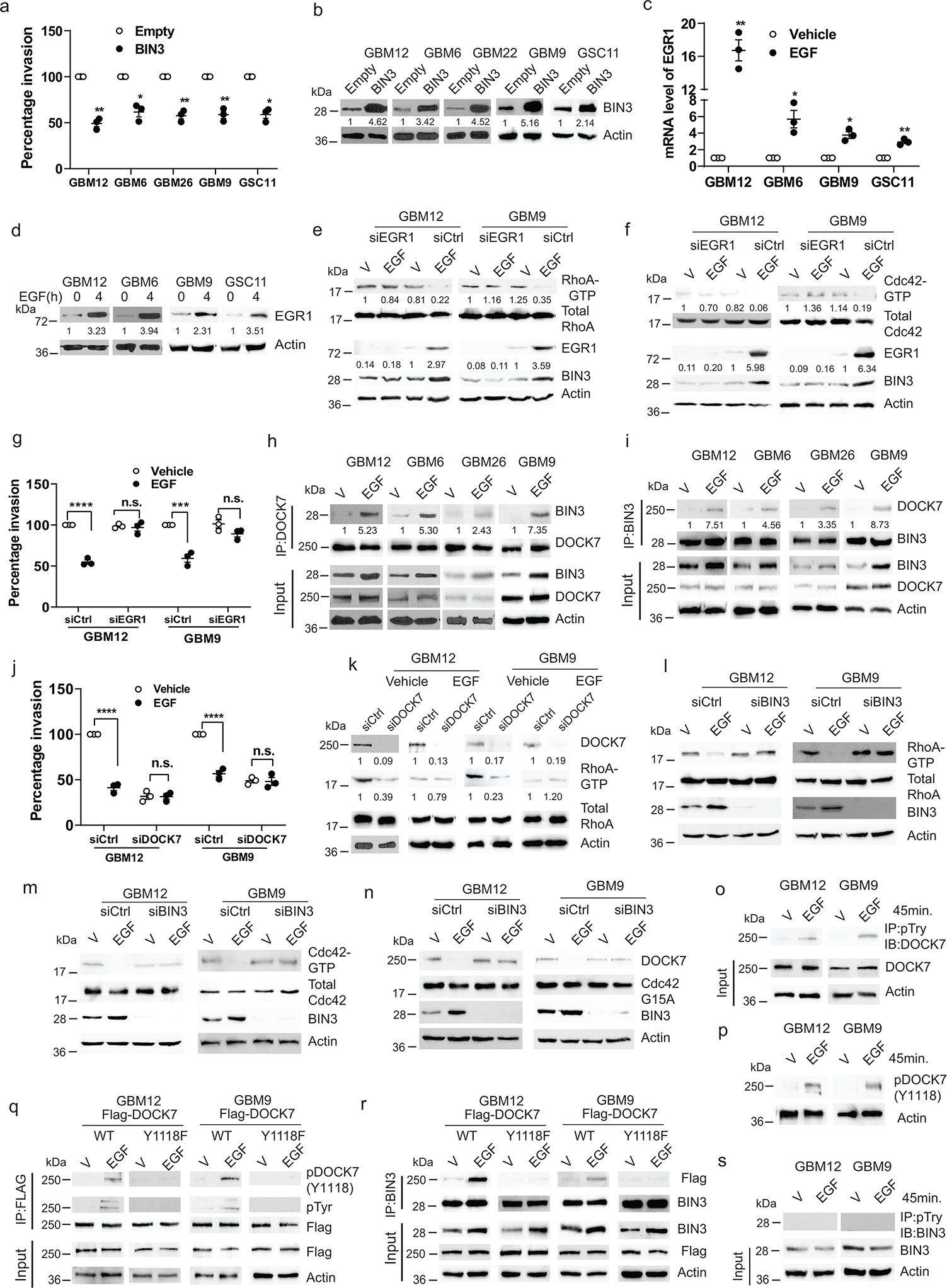
a, Matrigel invasion assay of multiple lines with BIN3 overexpression. b, BIN3 overexpression was analyzed by Western blot. c, EGR1 mRNA levels in multiple lines treated with EGF (50 ng/ml) for 2 hours. d, Immunoblot of EGR1 in cells treated with EGF. e-f, Immunoblot of the indicated proteins in EGR1 siRNA knockdown cells treated with EGF. g, Matrigel invasion assay of EGR1 siRNA knockdown cells in the presence or absence of EGF. h-i, Immunoblot of immunoprecipitated (IP) extracts from multiple lines treated with EGF for 24 hours. j, Matrigel invasion assay of DOCK7 siRNA knockdown GBM12 and GBM9 in the presence or absence EGF (50 ng/ml). k, Immunoblot of the indicated proteins in DOCK7 siRNA knockdown in cells treated with EGF for 24 hours. l-m, Immunoblot of the indicated proteins in BIN3 siRNA knockdown cells treated with EGF (50 ng/ml) for 24 hours. n, Immunoblot of active DOCK7 in EGF treated control or BIN3 siRNA knockdown cells. o, Immunoblot of IP extracts from cells treated with vehicle or EGF. p, Immunoblot of phosphorylated DOCK7(Y1118) in cells treated with vehicle or EGF. q-r, Immunoblot of IP extracts from wild-type or mutant (Y1118F) Flag-DOCK7 overexpressing cells treated with vehicle or EGF. s, Immunoblot of IP extracts from cells treated with vehicle or EGF. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-tailed one-sample Student’s t-test (a, c), or by two-way ANOVA adjusted by Bonferroni’s correction (g, j). *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
BIN3 downregulates invasion via interaction with DOCK7 and inhibition of Rho GTPases
Next, we undertook mass spectrometry to identify proteins associating with BIN3 (Supplementary Table 1). We prioritized an investigation of DOCK7 (dedicator of cytokinesis 7), a member of the DOCK180 family of atypical Rac/Cdc42 guanine nucleotide exchange factors 47, based on a role in neuronal precursor migration48, and in HGF-induced glioblastoma cell invasion via Rac activation49, 50. EGF induced an association between DOCK7 and BIN3 in co-immunoprecipitation experiments (Fig. 3h–i). We hypothesized that DOCK7 constitutively activates Rho GTPases and is inhibited by association with BIN3. We found that siRNA knockdown of RhoA and CDC42 leads to decreased invasiveness (Extended Data Fig. 3f–h). Increased expression of DOCK7 results in increased invasiveness and increased activity of Rho GTPases (Extended Data Fig. 2i–j). Conversely, siRNA knockdown of DOCK7 results in decreased invasiveness and decreased activity of Rho GTPases (Fig. 3j–k, Extended Data Fig. 3i). Addition of EGF did not decrease invasiveness or RhoGTPase activity further in DOCK7 siRNA knockdown cells indicating that DOCK7 is downstream of BIN3 (Fig. 3j–k). siRNA knockdown of DOCK7 has no effect on viability of cells (Extended Data Fig. 3j–k). Next, we found that EGF decreases the activity of both RhoA and Cdc42 (Fig. 3l–m). Rac is not detectable in these cells (Extended Data Fig. 3l). Importantly, siRNA knockdown of BIN3 or EGR1 results in a loss of the EGF-mediated downregulation of Rho GTPase activity (Fig. 3e–f, 3l–m). EGF induces decreased activity of DOCK7 and this is blocked by siRNA knockdown of BIN3 (Fig. 3n). Overexpression of Rho GTPases blocked the EGF induced suppression of invasion (Extended Data Fig. 2k–m). Unlike EGF, HGF increased invasiveness, induced Met phosphorylation, did not induce an association between BIN3 and DOCK7, and increased RhoA activity without affecting BIN3 levels (Extended Data Fig. 3m–p).
Ligand-mediated activation of EGFR results in tyrosine phosphorylation of DOCK7
A previous study has reported tyrosine phosphorylation of DOCK7 on Y1118 in Schwann cells51 We found that EGF induces a tyrosine phosphorylation of DOCK7 on Y1118 in GBM cells (Fig. 3o–p). Mutation of the Y1118 site on DOCK7 result in a loss of tyrosine phosphorylation of DOCK7 in response to EGF and the DOCK7 Y1118F mutant fails to bind to BIN3 in response to EGF (Fig. 3q–r). These data suggest that the tyrosine phosphorylation of DOCK7 and an upregulation of BIN3 by EGFR are both important for the DOCK7 and BIN3 association. We did not detect any tyrosine phosphorylation of BIN3 (Fig. 3s).
Effects of constitutive vs. ligand-activated EGFR signaling in vivo
Next, we examined the impact of EGFR ligand on GBM invasion in an orthotopic mouse model. To induce ligand activation of EGFR in vivo, Mayo PDX GBM12 (EGFRwt) or GBM9 neurospheres (EGFRwt plus EGFRvIII) were stably transfected with TGFα (Fig. 4a, Extended Data Fig. 4a). Since the loss of EGFR amplification in glioma cells in culture has been reported, we confirmed that the EGFR level and EGFR copy number in Mayo PDX lines that have been stably transfected are similar to the parental cells (Extended Data Fig 4b–c). Additionally, we cultured Mayo PDX lines in serum containing medium for up to 2 months and found no reduction in EGFR copy number or expression level or effect on invasion (Extended Data Fig. 4d–f).
Figure 4. TGFα overexpression prolongs survival, reduces invasiveness and increases proliferation in orthotopic glioblastoma mouse model.
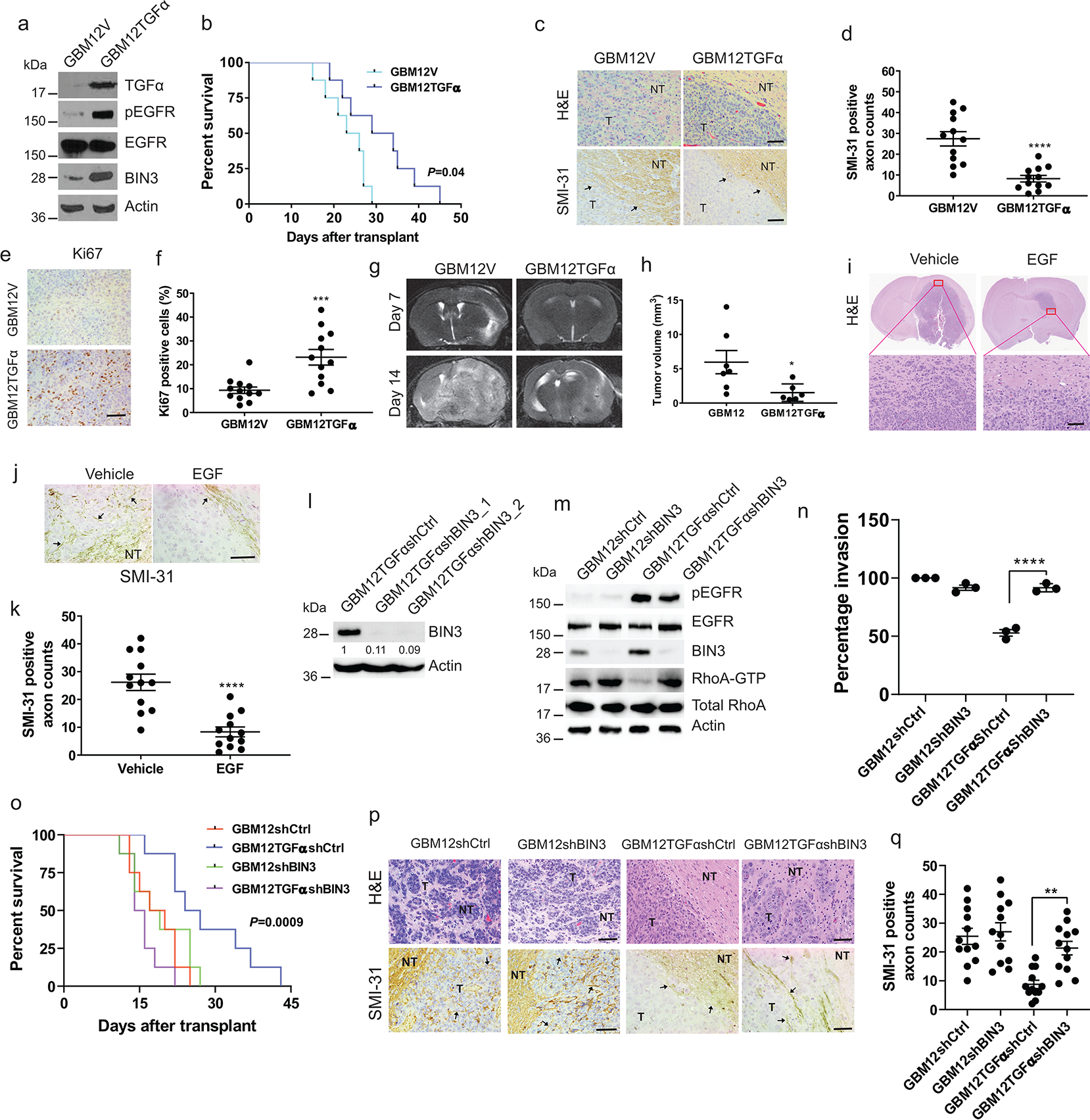
a, Immunoblot of the indicated proteins in GBM12 stably transfected with empty (GBM12V) or TGFα expression vector (GBM12TGFα). b, Kaplan–Meier survival curves of mice with orthotopic xenotransplant model of GBM12TGFα and GBM12V (n=8/group). c, Representative H&E staining and SMI-31 immunostaining in GBM12V and GBM12TGFα tumour tissue sections. T: Tumour; NT: Normal tissue. d, Quantification of SMI-31 counts within tumour region. e-f, Representative images and quantification of Ki67 immunostaining in mouse tumour tissue sections. g, Representative MRI imaging of orthotopic tumour bearing mice obtained at 7 and 14 days after transplantation of GBM12V or GBM12TGFα. h, Tumour volume of two groups after 14 days (GBM12V n=7, GBM12TGFα n=6). i, H&E staining of GBM12 orthotopic tumours from mice intracranially infused with EGF or vehicle (n=4/group). j-k, Representative immunostaining and quantification for SMI-31 in GBM12 orthotopic tumours from mice treated with vehicle or EGF. l, Immunoblot of BIN3 in GBM12TGFα stably transfected with control or BIN3 shRNA. m, Immunblot of the indicated proteins in the indicated cells. n, Matrigel invasion assay of multiple lines. o, Kaplan–Meier survival curves of orthotopic mouse xenotransplant model of the indicated cells (n=8/group). p, Representative H&E staining and SMI-31 immunostaining in tumour tissue sections. q, Quantification of SMI-31 counts. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of Actin. Scale bar: 100 μm. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by log-rank test (b, o), or by two-tailed unpaired Student’s t-test (d, f, h, k), or by two-way ANOVA adjusted by Bonferroni’s correction (n, q). *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
TGFα transfected GBM12 cells or GBM9 neurospheres were injected intracranially in athymic mice, followed by a survival analysis. Increased ligand availability resulted in improved survival (Fig. 4b, Extended Data Fig. 4g) and a sharp decrease in invasiveness as assessed by H&E staining and by staining for disruption of mouse neurofilament (Fig. 4c–d, Extended Data Fig. 4h–i), Proliferation is increased in TGFα expressing tumours (Fig. 4e–f, Extended Data Fig. 4j–k). EGFR levels and copy number were retained in tumours derived from stably transfected cells (Extended Data Fig. 4l–m). Also, serial MRI imaging of tumours reveals much smaller tumours with TGFα expression (Fig. 4g–h). Additionally, exogenous infusion of EGF also resulted in smaller tumours that were noninvasive (Fig. 4i–k). Similar results were found with stable expression of EGF (Extended Data Fig. 4s–y). EGFR ligand did not increase cell death (Extended Data Fig. 4z–cc).
BIN3 is required for ligand-induced EGFR effects on GBM invasion in vivo
Stable overexpression of BIN3 results in decreased invasiveness in transwell invasion assays (Extended Data Fig. 4n–o) and resulted in a better prognosis and non-invasive tumours in an orthotopic mouse model (Extended Data Fig. 4p–r). EGFR copy numbers and levels were maintained in BIN3 overexpressing mouse tumours (Extended Data Fig. 4l–m). Next, BIN3 was stably silenced in GBM12TGFα cells (Fig. 4l). We examined the effects of BIN3 silencing in GBM12TGFa cells on key signals such as phosphorylation of the EGFR, BIN3 and Rho GTPase activity (Fig. 4m). BIN3 silencing in GBM12TGFα overexpressing cells resulted in increased invasiveness (Fig. 4n) and formation of invasive tumours and a worse prognosis (Fig. 4o–q).
Constitutive EGFR signaling drives invasion by upregulating EMP1
Constitutive EGFR signaling does not significantly affect BIN3 levels (Fig. 5a). We previously identified epithelial membrane protein 1 (EMP1) as a gene upregulated by constitutive EGFR signaling1 and confirmed its upregulation by constitutive EGFRwt or EGFRvIII expression but not ligand-activated EGFR (Figure 5a–c). EMP1 activates RhoGTPases and may play a key role in cancer invasiveness 53. siRNA mediated knockdown of EMP1 resulted in a loss of invasiveness resulting from EGFR expression in GBM lines (Fig.5–e). Moreover, constitutive EGFR signaling resulted in RhoA activation and that siRNA knockdown of EMP1 resulted in a loss of EGFR mediated Rho activation (Fig. 4f).
Figure 5. Constitutive EGFR signaling induces TAB1-TAK1-p65-EMP1 pathway.
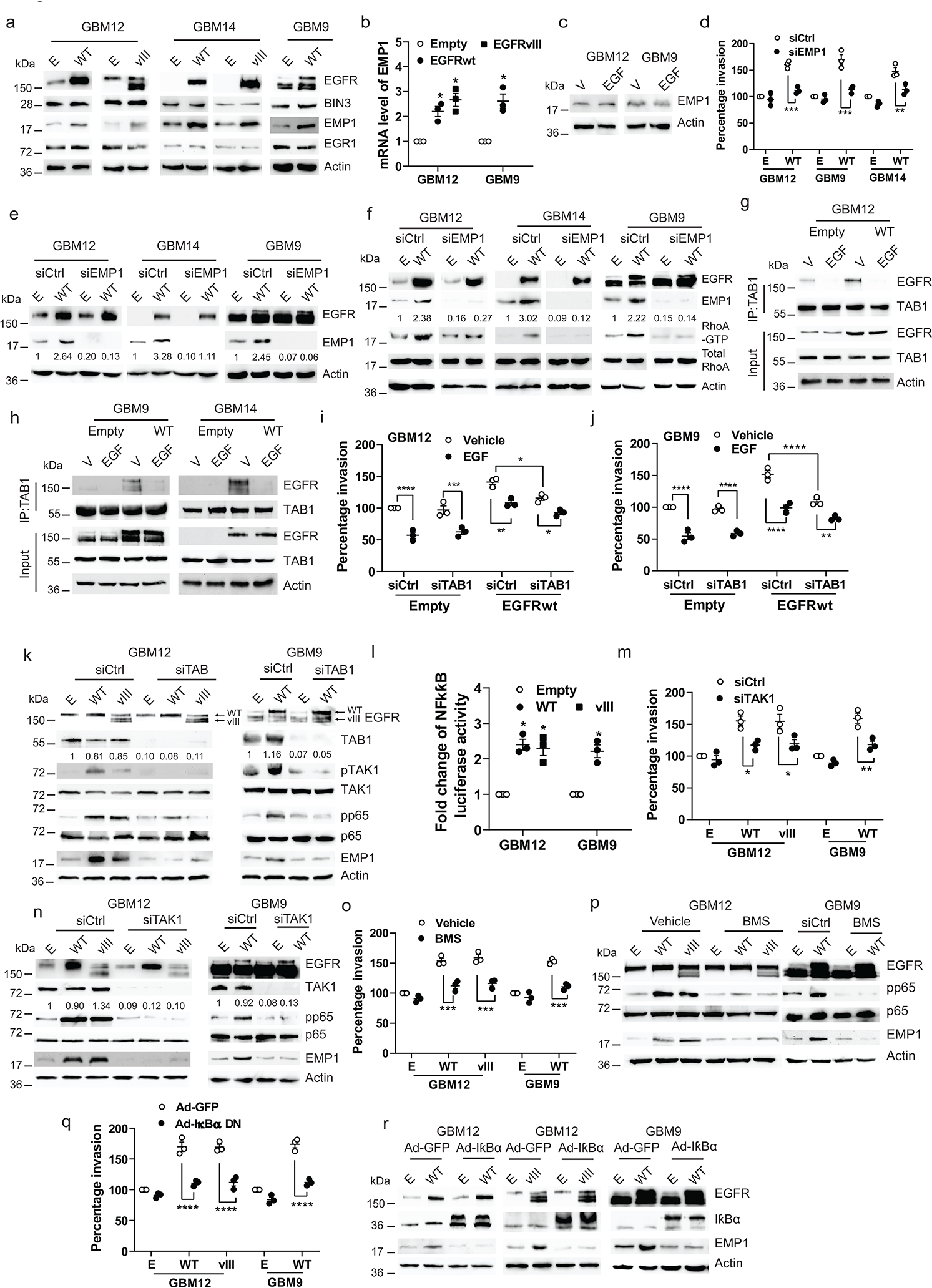
a, Immunoblot of the indicated proteins in cells transiently transfected with empty (E), EGFRwt (WT) or EGFRvIII (vIII) expression vectors. b, mRNA level of EMP1 in cells transiently transfected with empty, EGFRwt or vIII expression vectors. c, Immunoblot of EMP1 protein expression in cells treated with EGF (50 ng/ml) for 24 hours. d, Matrigel invasion of multiple lines transiently transfected with empty or EGFRwt expression vectors, along with control or EMP1 siRNA. e, Efficiency of EMP1 knockdown and EGFRwt overexpression was analyzed by Western blot. f, Immunoblot of the indicated proteins in multiple lines transiently transfected with empty or EGFRwt expression vectors, along with control or EMP1 siRNA. g-h, Immunoblot of immunoprecipitated extracts from EGFRwt overexpressing multiple lines treated with vehicle or EGF for 30 minutes. i-j, Matrigel invasion of EGFRwt or EGFRvIII overexpressing and/or TAB1 siRNA knockdown cells. k. Immunoblot of the indicated proteins in cells as described in i-j. l, NF-κB luciferase reporter activity in EGFRwt or EGFRvIII overexpressing cells. m, Matrigel invasion of EGFRwt or vIII overexpressing and/or TAK1 siRNA knockdown cells. n, Immunoblot of the indicated proteins in cells as described in m. o, Matrigel invasion of EGFRwt or EGFRvIII overexpressing cells treated with NF-κB inhibitor BMS (BMS-345541). p, Immunoblot of the indicated proteins in EGFRwt or vIII overexpressing cells treated with BMS for 48 hours. q, Matrigel invasion of EGFRwt or vIII and/or IκBα dominant negative (DM) overexpressing cells. r, Immunoblot of the indicated proteins in cells as described in q. Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-tailed one-sample Student’s t-test (b, l), or by two-way ANOVA adjusted by Bonferroni’s correction (d, i, j, m, o, q). *P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Next, we undertook mass spectrometry to identify proteins that bind to the EGFR in the absence of EGF (Supplementary Table 2). TAB1 was prioritized because of its known role in NF-κB activation, which it undertakes with its partner TAK154. NF-κB is known to induce GBM invasion55–57. We first confirmed that TAB1 binds to EGFR constitutively and dissociates from it with EGF (Fig. 5g–h). siRNA knockdown of TAB1 results in a loss of constitutive EGFR mediated invasion, activation of TAK1 and phosphorylation of the NF-κB subunit p65 (Fig. 5i–k). We confirmed that EGFR overexpression resulted in increased NF-κB reporter activity (Fig. 5k–l). siRNA knockdown of TAK1 also blocks invasion and p65 phosphorylation (Fig. 5m–n). Finally, chemical inhibition of NF-κB, or a dominant negative IBα mutant, resulted in inhibition of EGFR driven invasion and upregulation of EMP1 (Fig. 5o–r). siRNA knockdown of TAB1 or TAK1 also blocked EGFR-induced upregulation of EMP1 (Fig. 5k, n) indicating that a TAB1-TAK1- NF-κB-EMP1 signaling axis drives ligand-independent EGFR driven invasion.
Effect of EGFR ligands on GBMs expressing both EGFRwt and EGFRvIII
The allelic frequency of EGFRvIII is significantly greater than that of EGFRwt in GBM6 and GBM9 (Extended Data Fig. 5a). Nonetheless, we consistently see a predominantly ligand-dependent signaling in GBM6 and GBM9 when exposed to EGF, with suppression of invasion, upregulation of EGR1, and upregulation of BIN3 (Fig. 1b, Fig. 3d, Fig. 1p). Expression of EGFRvIII or EGFRwtin GBM14 cells resulted in constitutive signaling and increased invasion (Fig. 1k–l, Extended data 5b–c). EGF has no effect on invasion with EGFRvIII expression (Extended data Fig. 1x–y), while EGF suppressed invasion in EGFRwt expressing cells and upregulated BIN3 and EGR1 (Extended data Fig. 5b–c). When we express both EGFRwt and EGFRvIII in GBM14 and express much lower levels of EGFRwt to mimic the conditions in GBM6 and GBM9, we find that addition of EGF results in suppression of invasion, EGR1 and BIN3 upregulation (Extended Data Fig. 5d–e).
Ligand-dependent EGFR signaling mediates proliferation via a Shc driven pathway
Using Mass Spectrometry, we found that Shc association with the EGFR is increased by EGF (Supplementary Table 2). EGF induces association of Shc with the EGFR and tyrosine phosphorylation of Shc (Extended Data Fig. 5f–g). siRNA knockdown of Shc blocks EGF-induced proliferation (Extended Data Fig. 5h). siRNA knockdown of Shc blocked ERK activation in response to EGF (Extended Data Fig. 5i). Also, inhibition of ERK using a chemical inhibitor or siRNA knockdown resulted in a loss of EGF mediated proliferation (Extended Data Fig. 5j–m). Thus, as previously reported, ligand-dependent EGFR signaling induces proliferation through a canonical Shc-ERK pathway.
Phosphorylation of EGFR during constitutive and ligand depend activation:
Next, we examined the tyrosine phosphorylation of specific EGFR residues with or without EGF. We found phosphorylation of the same sites during constitutive and ligand-activated EGFR signaling. However, phosphorylation of all sites was considerably enhanced with ligand-activation in PDX lines (Extended Data Fig. 5n–o, 1t–u). Our data suggest that a low level of EGFR tyrosine phosphorylation seen in constitutively activated EGFR is conducive to activation of an NF-kappa B-EMP1 pathway leading to increased invasion while a high level of EGFR tyrosine phosphorylation detected in ligand-activated EGFR signaling is conducive to activation of an EGR1-BIN3 pathway leading to suppression of invasion. Tofacitinib upregulates BIN3 levels via a ligand-mediated activation of the EGFR and decreases GBM invasion.
Tofacitinib upregulates BIN3 levels via a ligand-mediated activation of the EGFR and decreases GBM invasion
To identify a drug that upregulates BIN3 and specifically inhibits GBM invasion, we examined a panel of drugs that target key components of RTK signaling pathways and found that the Jak1/Jak3 inhibitor tofacitinib strongly upregulated BIN3 levels in multiple lines (Fig. 6a–b) and inhibited invasiveness (Fig. 6c). Previous studies have indicated that inhibition of components of RTK signaling pathways leads to activation of upstream RTKs58. We found that tofacitinib induced a robust activation of the EGFR (Fig. 6d). Furthermore, cetuximab completely inhibits tofacitinib-induced EGFR activation, BIN3 upregulation and suppression of invasion (Fig. 6e–f). Moreover, tofacitinib upregulates the EGFR ligand HB-EGF (Fig. 6g). As expected, tofacitinib blocks phosphorylation of STAT1 and STAT3 (Fig. 6h–i, Extended Data Fig. 5p). siRNA knockdown of STAT3 but not STAT1 mimicked the effect of tofacitinib and resulted in increased HB-EGF secretion and EGFR activation, and suppression of invasion (Fig. 6k–l, Extended Data Fig. 5q–s) that can be blocked by cetuximab, consistent with a recent study reporting that STAT3 inhibition increased the EGFR ligand betacellulin59, although we did not find an upregulation of betacellulin in our model (Extended Data Fig. 5t). STAT3 overexpression blocks the inhibitory effect of tofacitinib on invasion and EGFR activation (Extended Data Fig. 5u–v). Furthermore, tofacitinib induces EGR1 upregulation and activity (Fig. 6m–n). siRNA knockdown of EGR1 results in a loss of tofacitinib-induced BIN3 expression (Fig. 6p). Also, in cells treated with a saturating concentration of EGF, tofacitinib did not induce a further increase in BIN3 level, or increase invasion further (Extended Data Fig. 5w–x). Additionally, tofacitinib induced proliferation in glioma cells (Fig. 5q). Tofacitinib did not induce cell death (Fig. 6r–s). Finally, siRNA knockdown of BIN3 blocks tofacitinib induced suppression of invasion (Fig. 6t–u).
Figure 6. Tofacitinib inhibits invasion of glioma cells by EGR1-mediated BIN3 upregulation.
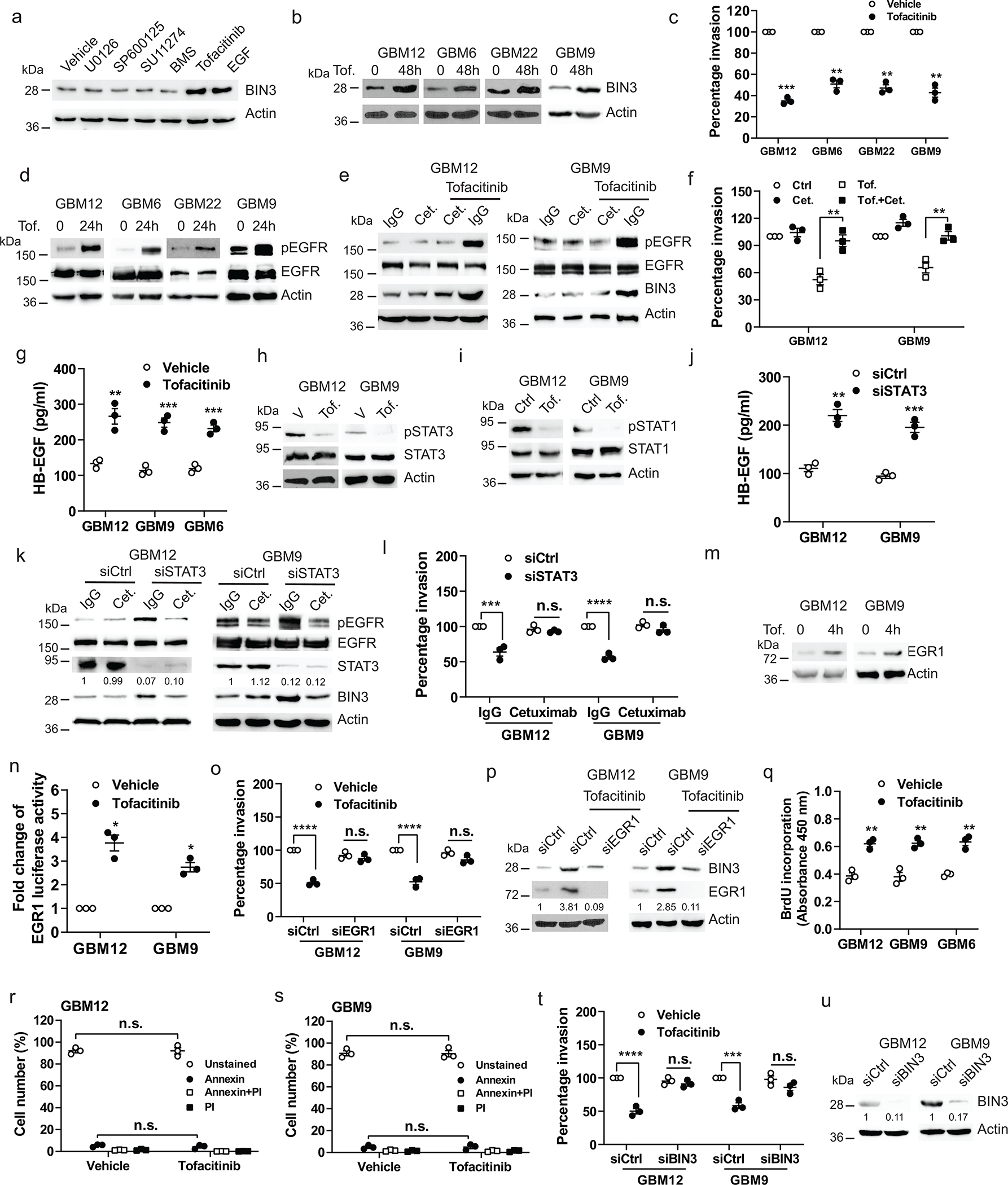
a, Immunoblot of BIN3 expression in GBM12 treated with the indicated drugs for 48 hours. b, Immunoblot of BIN3 expression in multiple lines treated with tofacitinib (1 μM). c, Matrigel invasion of multiple lines in the presence or absence of tofacitinib (1 μM). d, Immunoblot of pEGFR and EGFR in multiple lines treated with tofacitinib (1 μM). e, Immunoblot of the indicated proteins in cells treated with either tofacitinib, cetuximab (cet.), IgG control or their combination for 24 hours. f, Matrigel invasion assay of cells in the presence or absence tofacitinib, cextuximab or their combination. g, ELISA for HB-EGF in the supernatant of tofacitinib treated multiple lines. h-i, Immunoblot of the indicated proteins in cells treated with tofacitinib. j, ELISA for HB-EGF in the supernatant of STAT3 siRNA knockdown cells. k, Immunoblot of the indicated proteins in STAT3 siRNA knockdown cells treated with IgG or cetuximab for 24 hours. l, Matrigel invasion assay of STAT3 siRNA knockdown cells in the presence or absence of IgG or cextuximab. m, Immunoblot of EGR1 expression in cells treated with tofacitinib. n, EGR1 promoter luciferase assay of cells treated with tofacitinib for 2 hours. o, Matrigel invasion assay of EGR1 siRNA knockdown cells in the presence or absence of tofacitinib. p, Immunoblot of BIN3 expression in EGR1 siRNA knockdown cells treated with tofacitinib for 24 hours. q, BrdU incorporation assay of multiple lines treated with tofacitinib. r-s, Annexin V/PI positive staining assay of cells treated with tofacitinib for 24 hours. t, Matrigel invasion assay of BIN3 siRNA knockdown cells in the presence or absence of tofacitinib. u, Knockdown efficiency of BIN3 in cells was analyzed by Western blot. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-tailed one-sample Student’s t-test (c, n), or by two-way ANOVA adjusted by Bonferroni’s correction (f, l, o, r, s, t), or by two-tailed unpaired Student’s t-test (g, j, q). *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, n.s. not significant. Numerical source data, statistics, exact P values and unprocessed blots are available as Source Data.
Next, we compared the effect of tofacitinib on mouse intracranial GBM tumours generated from GBM12TGFα cells or from GBM12V (vector transfected) cells. In GBM12TGFα tumours that already have a high EGFR ligand level, there is no additional benefit from tofacitinib. GBM12V tumours, on the other hand, responded to tofacitinib treatment with a significantly increased survival and decreased invasion (Fig. 7a–c), as did GBM9 neurosphere tumours and GBM6 tumours (Fig. 7f–n). Tofacitinib did not induce significant cell death in the tumours (Fig. 7d–e, i–j, k–m). As expected, tofacitinib increased proliferation in mouse tumours (Extended Data Fig. 5y–aa). Next, we identified two PDX lines that have high endogenous EGFR ligand and high BIN3 levels (Fig. 7n). We found that these lines do not respond to tofacitinib or EGF by upregulation of BIN3 (Fig. 7o). We silenced HB-EGF in GBM39 cells found that tofacitinib or EGF could now upregulate BIN3 in GBM39shHB-EGF cells (Fig. 7p–q). GBM39shHB-EGF cells are more responsive to tofacitinib compared to GBM39shCtrl cells in invasion assays (Fig. 7r). We next examined whether silencing of HB-EGF confers responsiveness to tofacitinib in vivo and found that silencing HB-EGF also renders orthotopic GBM39shHB-EGF tumours more responsive to tofacitinib compared to controls (Fig. 7s–u). These data suggest that tofacitinib is likely to be more effective in EGFR amplified tumours with low levels of EGFR ligand.
Figure 7. Tofacitinib prolongs survival, reduces invasiveness and increases proliferation in an orthotopic glioblastoma mouse model.
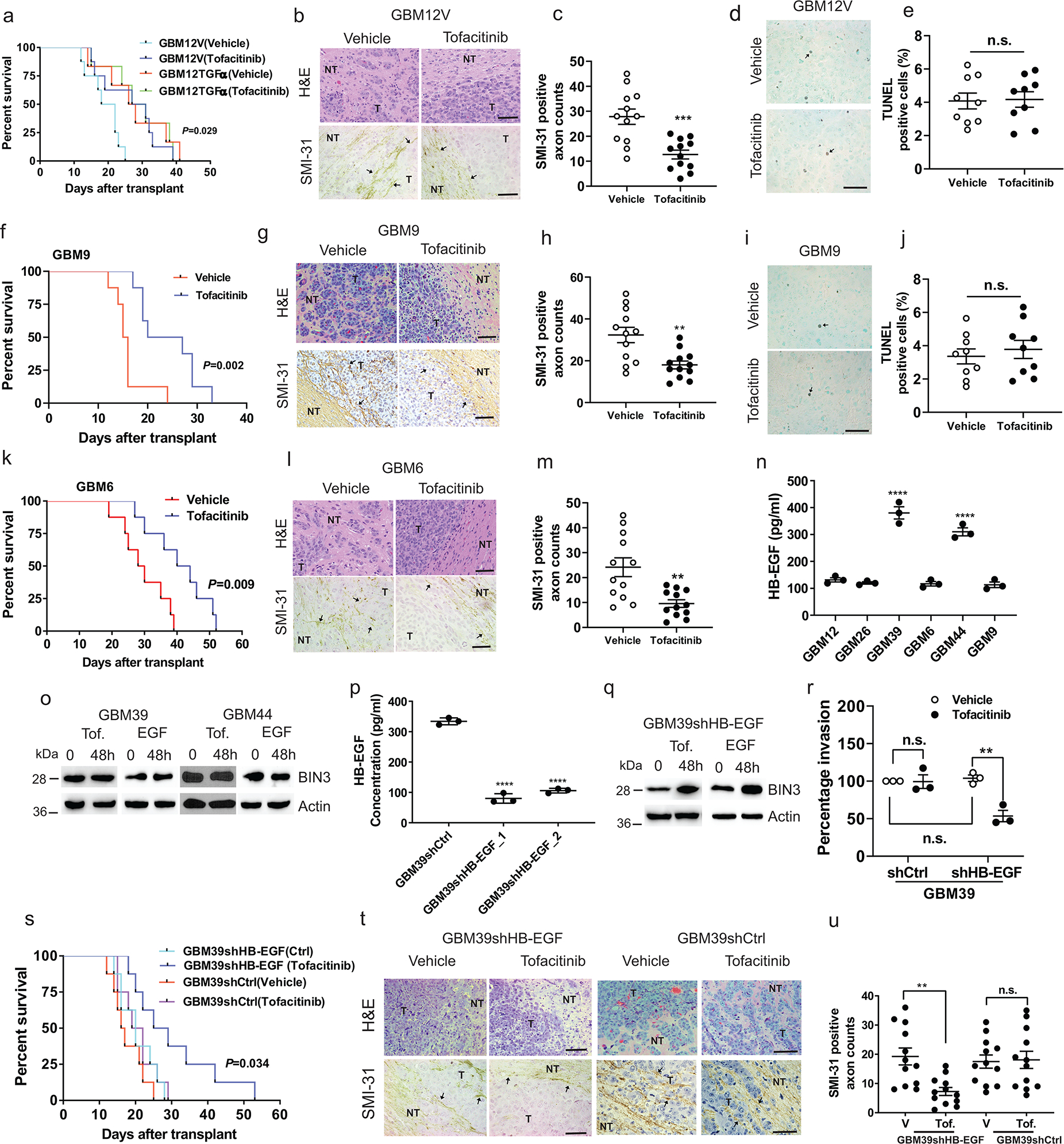
a, Kaplan–Meier survival curves of mice in an orthotopic model of GBM12V and GBM12TGFα treated with vehicle or tofacitinib (50 mg/kg) (n=8 for GBM12TGFα, n=6 for GBM12V). P-value represents vehicle vs. tofacitinib in GBM12TGFα groups. b-c, H&E staining, SMI-31 immunostaining (black arrows) and quantification of SMI-31 counts in GBM12V orthotopic tumours. d-e, TUNEL staining (black arrows) and quantification of TUNEL positive cells in GBM12V orthotropic tumour from vehicle and tofacitinib treated mice. f, Kaplan–Meier survival curves of mice with orthotopic model of GBM9 treated with vehicle or tofacitinib. g-h, H&E staining, SMI-31 immunostaining and quantification of SMI-31 counts in GBM9 orthotopic tumours from vehicle and tofacitinib treated mice. i-j, TUNEL staining and quantification of TUNEL positive cells in a GBM9 orthotopic tumour. k, Kaplan–Meier survival curves of mice with an orthotopic model of GBM6 treated with vehicle or tofacitinib. l-m, H&E staining, SMI-31 immunostaining and quantification of SMI-31 counts in GBM6 orthotopic tumour. n, ELISA for HB-EGF in the supernatants of multiple lines. o, Immunoblot of BIN3 expression in cells treated with tofacitinib or EGF. p, Knockdown efficiency of HB-EGF in GBM39 control shRNA and HB-EGF shRNA clones was confirmed by ELISA. q, Immunoblot of BIN3 expression in GBM39shHB-EGF (GBM39shHB-EGF_1) treated with either tofacitinib or EGF. r, Matrigel invasion assay of the inciated cells in the presence or absence of tofacitinib. s, Kaplan–Meier survival curves of mice with orthotopic models of GBM39shCtrl and GBM39shHB-EGF treated with vehicle or tofacitinib (n=8/group). t-u, H&E staining, SMI-31 immunostaining and quantification of SMI-31 counts in GBM39shHB-EGF and GBM39shCtrl orthotopic tumours from vehicle and tofacitinib treated mice. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. Scale bars: 50 μM. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by log-rank test (a, f, k, s), or by two-tailed unpaired Student’s t-test (c, e, h, j, m, u), or by one-way ANOVA adjusted by Bonferroni’s correction (n, p), or by two-way ANOVA adjusted by Bonferroni’s correction (r). *P <0.05, **P < 0.01, ***P < 0.001, ****P < 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Single cell analysis reveals that ligand-mediated activation of EGFR suppresses invasion
To further investigate the invasive behavior of individual glioma cells we used a nanoplate fabricated with topographic patterns of regular parallel ridges that mimic the ECM environment in brain 60. Exposure of glioma cells to EGF resulted in a significant slowed migration of individual GBM12 and GBM22 cells, but there was a significant heterogeneity among cells (Extended Data Fig. 6a–d, i–l), as reported previously60. The effects of tofacitinib are quite similar to EGF (Extended Data Fig. 6e–g, m–p).
TCGA Analysis demonstrates that EGFR ligands confer an improved prognosis in EGFR amplified GBM
TCGA data indicate that seven EGFR ligands are variably expressed in GBM (Fig. 8a). We found that the combined effects of seven EGFR ligands are oncogenic in EGFR non-amplified GBMs, while, quite unexpectedly, EGFR ligands are tumour suppressive in EGFR amplified GBMs (Fig. 8b–c) while low EGFR ligands, indicative of constitutive EGFR signaling, confer a worse prognosis. EGFR ligands are very strongly oncogenic in non-amplified GBMs (p=0.0009). In EGFR amplified amplified GBMs the role of EGFR ligands is completely reversed with a significant tumour suppressor effect of EGFR ligand (p=0.0355). Importantly, increased phosphorylation of EGFR, suggestive of EGFR activation, correlates with a better prognosis (Fig. 8d). This is likely to primarily reflect ligand mediated activation of EGFRwt or constitutively active EGFR mutants.
Figure 8. TCGA analysis of EGFR and EGFR ligands in GBM.
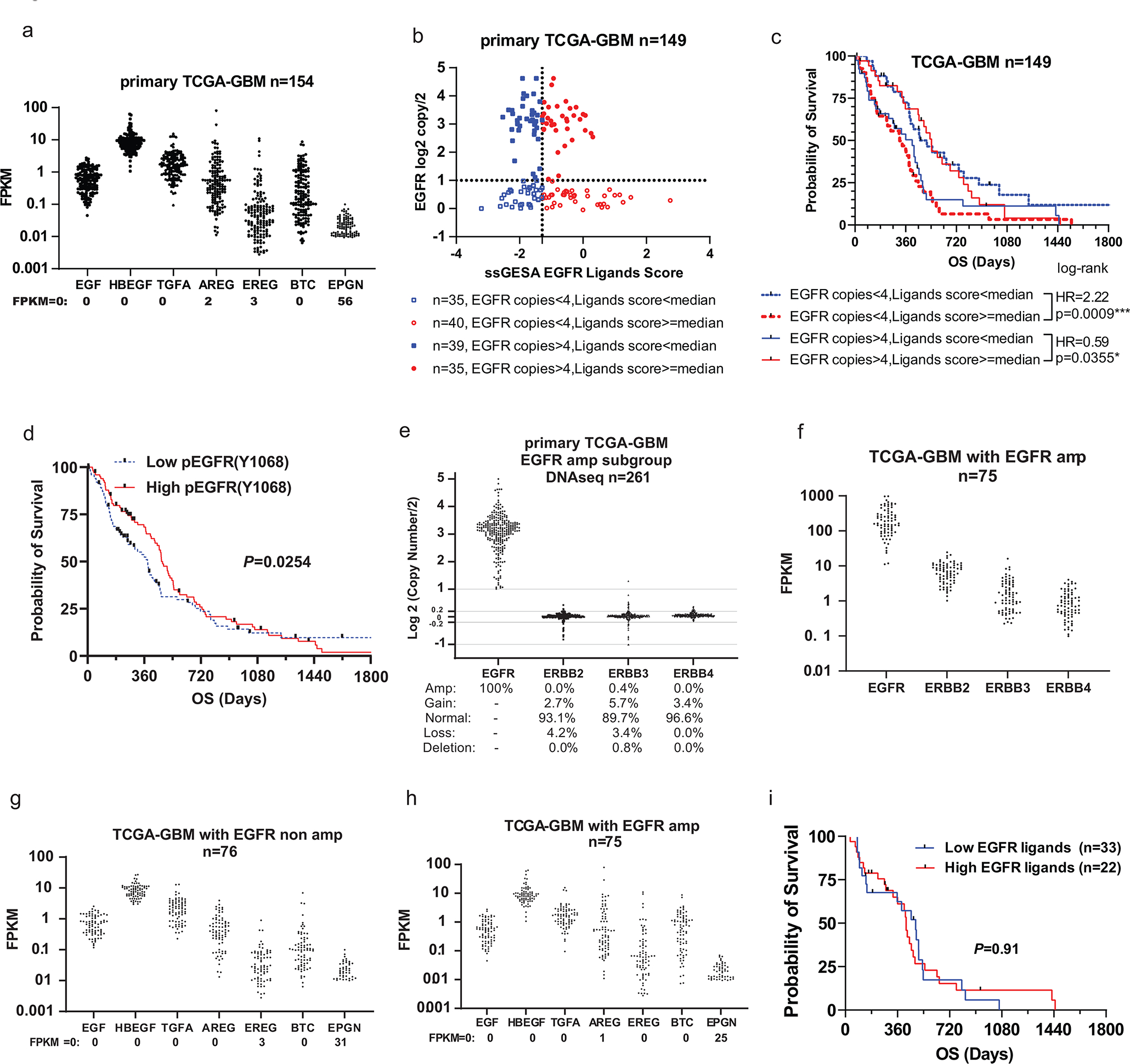
a, The expression distribution of 7 EGFR ligands was represented by Fragments per Kilobase of transcript per Million (FPKM) among 154 primary GBM patients from The Cancer Genome Atlas (TCGA) with RNAseq data. b, Distribution of EGFR ligands signature score and EGFR copy numbers among 149 primary GBM patients from TCGA. EGFR ligands signature score was calculated by Single-sample Gene Set Enrichment Analysis (ssGSEA) of mRNA levels of 7 EGFR ligands. Details are described in the methods. GBM cases with EGFR copy number over 4 were defined as EGFR amplification. c, Kaplan-Meier survival analysis of the four groups of patients as described in b. The Log-rank test was used to identify EGFR ligands’ dual and opposite effects on prognosis between EGFR amp (amplification) and non-amp (non-amplification) GBM patients.* P <0.05, ***: P <0.001. Hazard Ratio (HR): groups with high ligands scores comparing to groups with low scores. d, Overall survival (OS) analysis according to pEGFR expression in GBM patients. The 204 primary TCGA-GBM patients were divided into high-50% and low-50% groups by pEGFR (Y1068) expression level and the effect on survival was examined (N=204). e, EGFR, ERBB2, ERBB3, and ERBB4 mRNA levels in EGFR amplified samples. Primary TCGA-GBM samples with DNAseq and RNAseq data were divided by EGFR copy numbers. f, EGFR, ERBB2, ERBB3 and ERBB4 mRNA levels in EGFR amp samples. g-h, 7 EGFR ligands mRNA levels in EGFR non-amp and amp samples. i, Overall survival analysis according to signature score of 7 EGFR ligands in patients with classical subtype of GBM. Statistical significance was determined by log-rank test (c, i), or by Gehan-Breslow-Wilcoxon test (d). Numerical source data, statistic and unprocessed blots are available as Source Data.
In EGFR amplified GBMs expression of other ErbB receptors is low, suggesting that the EGFR plays the predominant biological role (Fig. 8e–f). Also, EGFR ligand levels are similar in EGFR amplified vs. EGFR non-amplified GBMs(Fig. 8g–h). Also, we did not detect a prognostic value of EGFR ligand in the classical subtype comprising 55 patients (Fig. 8i), likely because 15 out of the 55 patients are not EGFR amplified by our criteria (<4 EGFR copies). Thus it is the EGFR level that is the primary determinant of the prognostic response to EGFR ligands.
Expression of EGFR ligands in GBM
HB-EGF, TGFα and EGF are the most abundant ligands in GBM (Fig. 8a). We examined expression of these three EGFR ligands in 36 resected GBM samples using ELISA (Extended Data Fig. 7a–c). HB-EGF is the most abundant EGFR ligand. Also, HB-EGF is expressed more in the central part of the tumour compared to the leading edge, while expression of the EGFR does not vary significantly (Fig. 7d–g). This pattern of HB-EGF and EGFR expression was also found in the IVY GBM database (Extended Data Fig. 7j–k). Sp1 has a key role in HB-EGF transcription61. We found that Sp1 co-localizes to the same regions and tumour cells as HB-EGF (Extended Data Fig. 7h–i, k–m), suggesting that the regional expression of EGFR ligands reflects availability of transcription factors, Next, we found that Sp1 overexpression increases the expression of HB-EGF while siRNA knockdown of Sp1 results in decreased HB-EGF levels (Extended Data Fig. 7n–q).
BIN3 in GBM
BIN3 does not appear to confer an improved prognosis in EGFR amplified GBMs according to TCGA data (Extended Data Fig. 7r). However, the protein level of BIN3 detected by Western blot in 36 GBMs indicates that increased protein levels of BIN3 confer an improved prognosis (Extended Data Fig. 7s–u). It is known that the correlation between mRNA and protein level can be poor for certain proteins and be dependent on regulatory factors ribosomal density and occupancy etc. 62
Discussion
EGFR is considered a key oncogene in a wide variety of cancers, including GBM. EGFR signaling is considered to be uniformly oncogenic in human cancer. Here, we demonstrate a tumour suppressive function of EGFR in EGFR amplified GBMs promoted by EGFR ligands. Analysis of the TCGA data looking at the combined effect of all seven EGFR ligands, demonstrates that EGFR ligands are tumour suppressive in EGFR amplified GBM. Consistent with this unexpected result, our experimental data demonstrate that ligand-activated EGFR signaling suppresses GBM invasion, and we propose that this suppression of invasion mediates the tumour suppressive effect of ligand-dependent EGFR signaling in EGFR amplified GBMs. Thus, in experimental models, expression of EGFR ligands in GBM cells or infusion of EGF leads to orthotopic tumours that are smaller, less invasive and associated with improved survival. We propose that ligand-dependent EGFR signaling suppresses invasion by upregulation of BIN3.
Previous studies have reported that EGFR stimulates invasion in GBM established cell lines that have lost EGFR amplification. We also found that EGF stimulates invasion in established GBM lines, or low EGFR expressing PDX cells. However, when EGFR is overexpressed in these cells, now EGF suppresses invasion. In most of our PDX models, which usually express significant levels of endogenous EGFR, EGF consistently suppresses invasion. These data indicate that the level of EGFR expression determines whether the outcome of ligand-dependent EGFR signaling is increased or decreased invasion. TCGA analysis show that in non-EGFR amplified GBMs, EGFR ligands are oncogenic while, in a striking reversal, in EGFR amplified GBMs EGFR ligands are tumour suppressive. A key reason is likely to be the differential upregulation of BIN3 in EGFR amplified GBMs. In tumours expressing both EGFRwt and EGFRvIII, adding EGFR ligand results in a phenotype similar to tumours expressing EGFRwt alone.
Although the primary consequence of invasion is tumour recurrence, our experimental findings provide a new insight that invasion may also play a key role in GBM tumour expansion. Thus, increased ligand-dependent EGFR signaling leads to tumours with suppressed invasion that are smaller even though increased proliferation can be detected, suggesting that proliferation alone is not sufficient for tumour expansion and that tumour invasion is required. Ligand-induced EGFR activation leads to upregulation of BIN3 and tyrosine phosphorylation of DOCK7 leading to a physical association of BIN3 and DOCK7. DOCK family members have a RhoGEF domain and function as GEFs for the Rho GTPase family. EGF-induced BIN3-DOCK7 association inhibits the constitutive activity of DOCK7 and downstream Rho GTPases. Thus, ligand induced EGFR activation leads to decreased invasiveness by a BIN3 mediated inhibition of a DOCK7-RhoGTPase pathway.
TCGA data reveal that in EGFR amplified GBMs a low level of EGFR ligands confers a worse prognosis. This is consistent with our experimental findings that demonstrate that constitutive EGFR signaling enhances invasion resulting in larger, more invasive tumours with a worse prognosis in orthotopic models. We have identified that constitutive EGFR signaling triggers a TAB1-TAK1-NF-κB-EMP1 signaling axis resulting in enhanced invasion. There is significant variation in the level of EGFR ligands expressed in GBMs, suggesting that constitutive or ligand-activated signaling may predominate in individual tumours.
Our study suggests that a patient stratification based on the level of EGF receptor and EGFR ligands, may be helpful to predict subsets of patients likely to benefit from targeting the EGFR. Thus, in patients with EGFR amplified GBMs with high EGFR ligands, EGFR inhibition could actually be detrimental, since EGFR ligands and high phosphorylation of the EGFR confer a better prognosis. On the other hand, in EGFR amplified tumours with low ligand, a BBB penetrant drug 63–65. such as tofacitinib, a JAK1/JAK3 that upregulates BIN3 via an upregulation of EGFR ligand and suppresses invasion could be very useful. Tofacitinib could be a unique and effective treatment for GBM that specifically targets invasion. This type of treatment could be used to suppress invasion prior to surgical resection and shrink the main tumour mass. Since invasion is believed to be a key determinant of GBM recurrence, tofacitinib could also be used later in the course of the disease to prevent or treat recurrence. For EGFR non-amplified tumours with high ligand, or possibly for EGFR amplified tumours with low ligand, EGFR inhibition combined with blunting of the accompanying adaptive response could be a useful therapeutic strategy.
Methods
All animal studies were done under Institutional Animal Care and Use Committee-approved protocols.
Cell Culture of Mayo PDXs
As an experimental model, we used GBMs from the widely used Mayo xenograft panel33. In this model, patient derived xenografts (PDXs) are maintained by continuous passage in mice. The Mayo explant cultures were generated from mouse tumours, and generally used without passaging or at early passages for experiments described in this study. Cultures of PDX cells were maintained in DMEM with 10% FBS and 1% Penicillin-Streptomycin (P/S). Cells were grown to 70–80% confluence prior to treatments. GBM9, GSC11 and GS622 neurospheres were cultured in DMEM F12 supplemented with B27 without vitamin A, EGF (20 ng/ml) and FGF (20 ng/ml) as described previously34. Neurospheres were EGF and FGF starved for 72 hours prior to experiments unless otherwise specified. Human astrocyte (SVGp12) and A431 were purchased from ATCC. U251, and U251 constitutively overexpressing EGFR, U251EGFRwt) and U251 cells conditionally overexpressing EGFR in response to tetracycline (U251EGFRind) cells were obtained or generated as described previously2. U343 GBM cell line was a kind gift of Dr. Luiz Penalva (University of Texas Health Science Center, San Antonio, TX).
Plasmids, transfection and generation of stable cell lines
To generate cells stably overexpressing TGFα, EGF or BIN3, cells were transfected with 2 μg of pCMV-TGFα-Flag (Sino Biological, China), pCMV6-EGF-Myc (Origene, MD), pCMV-BIN3-HA (Sino Biological, China) or empty vector using Lipofectamine 2000 according to manufacturer’s instructions. 48 hours after transfection, the cells were selected positivity by hygromycin (200 μg/ml) or neomycin (200 μg/ml). Stable expression colonies were selected and tested for TGFα or BIN3 expression by immunoblotting. Positive colonies for EGF expression were tested by EGF enzyme-linked immunosorbent assay (ELISA) kit. Positive clones were selected for further investigation. Full-length EGR1 promoter-reporter plasmid was a kind gift from Dr. Gail Fraizer (Kent State University, Kent OH). STAT3 expression vector was from ORIGENE (RC215836, Rockville, MD). Sp1-HA expression vector was described previously66. pRK5 Myc-RhoA (15899) and pRK5 Myc-CDC42 (15905) plasmids were from Addgene. The full-length pCAGGS-FLAG-DOCK7 plasmid was a kind gift from Dr. Van Aelst (Cold Spring Harbor Laboratory, NY). DOCK7 Y1118F mutant was constructed by swapping a BbvCI- and PshAI-digested DNA fragment that contains TAT (Y, tyrosine) to TTT (F, phenylalanine) alteration with the corresponding wild-type sequence in pCAGGS-FLAG-DOCK7 that was also digested by the same enzyme pair. The mutation was then confirmed by DNA sequencing.
Western blotting, antibodies and reagents
Whole protein extracts from cells or tumour tissues were analyzed by Western blot as previously described34. Following antibodies were used: EGFR (06–847), TGFα (134A-2B3) and FLAG (F3165) antibodies were from Millipore. DOCK7 (13000-1) antibody was from Proteintech (Rosemont, IL). EGFR (Sepharose Bead Conjugate, 5735), pEGFR (Tyr1068) (2236), pEGFR (Tyr1173) (4407), pEGFR (Tyr845) (6963), ERK (4695), pERK (4370), TAK1 (5206), pTAK1 (4508), EGR1 (4153), STAT1 (9172), Ki-67 (9027), Met (8741), pMet (3077), p65 (8242), pp65 (3033), pShc (2431), Sp1 (9389), pSTAT1 (9167), STAT3 (12640), pSTAT3 (9145), Cdc42 (2466), RhoA (2117), FAK (71433), pFAK (8556), HA-Tag (2367) and Myc-Tag (2276) antibodies were from Cell Signaling Technology (Danvers, MA). EMP-1 antibody (Ab191181) was from Abcam (Cambridge, MA), BIN3 (sc-514396), p65 (sc-109), TAB1 (sc-166138), Shc (sc-967), pTyr(sc-508) and β-Actin (sc-47778) were from Santa Cruz Biotechnology (Dallas, TX). pDOCK7 (Y1118) antibody was obtained from IBL America (Minneapolis, MN). Dilutions of antibodies were listed in Supplementary Table 3.
Reagents: Recombinant human EGF (AF-100), HGF (100-39H) and TGFα (100-16A) were obtained from Peprotech (Rocky Hill, NJ). Recombinant human HB-EGF (259-HE) was obtained from R&D systems. Erlotinib (S7786) was purchased from SelleckChem (Houston, TX). Jak inhibitor tofacitinib (1 μM) and ERK inhibitor (U0126, 10 μM) were from Cayman Chemical (Ann Arbor, MI). The JNK inhibitor SP600125 (10 μM), Met inhibitor SU11274 (1 μM) and NF-κB inhibitor BMS-345541 (0.5 μM) were obtained from EMD Millipore (Billerica, MA). Cetuximab was provided by Imclone (New York, NY) and used at a concentration of 100 μg/ml.
Matrigel invasion assay
Invasion of cells was tested with Matrigel Boyden chamber assays (Fisher). 1×105 cells diluted in serum free medium or EGF/FGF free were seeded on Matrigel coated inserts with 10% FBS in the lower chamber as chemo-attractant, EGFR ligands or agents were added to upper chamber. After 24 hours of incubation in a 37 °C tissue culture incubator, invaded cells were stained with the HEMA-3 kit (Fisher) and counted in four to five 20x random fields per well cells and presented as percentage cell invasion. Each invasion assay was done on at least 3 independent occasions.
3D culture and spheroid invasion assay
GBM9 and GSC11 were EGF/FGF starved for 72 hours and embedded in reduced-growth factor medium containing Matrigel (10mg/ml), with medium containing vehicle or EGF (50ng/ml). Spheroids were imaged every 30 minutes over 24 hours with 20x objective on the stage of Andor spinning disk confocal microscope equipped with temperature and CO2 controlling environmental chamber. The invasion distance of cells out of the neurospheres was analyzed using manual track Plugin of ImageJ bundled Java (v 1.8.0_112). Four to five spheroids per treatment from three independent experiments were used to quantify invasion.
Scratch wound assays
Cells were grown to nearly 100% confluence. The cell medium was changed to serum free DMEM containing EGF (50 ng/ml) or vehicle. A scratch wound was then produced on the monolayer using a sterile 200 μl pipette tip. Images of wound were captured 0 and 24 hours after scratch generation.
Bromodeoxyuridine (BrdU) Cell Proliferation Assay, Annexin V/Propidium iodide (PI) assay and Cell viability assay
Cell proliferation was assessed using the BrdU cell kit (Abcam, Cambridge, United Kingdom) according to the manufacturer’s instructions. Cells were serum starved overnight, followed by treatment with or without EGF (50 ng/ml) or tofacitinib (1 μM) for 48 hours before the assay. Annexin V/PI staining and cell viability assay were conducted as previously described45. The representative gating strategy for flow cytometry is shown in Supplementary Fig. 1.
Mass spectrometry, Immunoprecipitation and Chromatin immunoprecipitation
Cells were treated with 50ng/ml EGF for 24 hours, BIN3 (sc-514396) or mouse IgG antibodies were incubated with whole cell lysates overnight at 4°C, and the mixtures were then incubated with 40 μl protein A/G slurry beads (Sigma) for 2 hours at 4°C. Immunoprecipitates were subjected to SDS-PAGE followed by Coomassie stain. Bands were cut from the gel and submitted for Mass Spectrometry Facility. To identify proteins interacting with EGFR, cells were treated with EGF for 30 minutes, cells lysates were immunoprecipitated by EGFR antibody (5735) and subjected to Mass spectrometry analysis.
To identify BIN3 and DOCK7 complexes in cells upon EGF treatment, cell lysates were immunoprecipitated with DOCK7 antibody or BIN3 antibody, and analyzed by Western blot using BIN3 antibody or DOCK7 antibody. EGFR (5735), TAB1 (sc-166138), Shc antibodies (967) were used to detect interaction of EGFR with TAB1 or ShC protein.
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP assay kit (Upstate Biotechnology, Lake Placid, NY, USA). Cells were treated with EGF for 24 hours followed by antibody incubation according to the manufacturer’s protocol. ChIP grade anti-EGR1 (4153) antibody was used to selectively precipitate the corresponding protein-DNA complex. RT-PCR was performed using ViiA 7 Real-Time PCR System (Applied Biosystems) to measure the relative amounts of ChIP DNA and results were quantified relative to inputs. The data are expressed as percentage of input. Followings are the primer sets used: BIN3_Forward: 5’-TTGCAGC-CTGTGTGTCTAAG-3’; BIN3_Reverse: 5’-CTCCAGGAAGTGACGTAAGC-3’.
Analysis of RhoA, CDC42 and DOCK7 activity
The amount of GTP-bound RhoA and Cdc42 were measured using RhoA and Cdc42 pull down assay kit (Cell Signaling) according to the protocol provided by the manufacturer. Briefly, cell lysates were incubated with either glutathione S-transferase (GST)-Rhotekin-RBD or GST-PAK-PBD beads 4 °C for 1 hour to pull down GTP-bound RhoA or Cdc42. Beads were washed four times in washing buffer and re-suspended in lysis buffer. RhoA-GTP and CDC42-GTP were detected by Western blot using RhoA and CDC42 antibodies.
To measure the GEF activity of DOCK7, the cell lysates were incubated with 40 μl CDC42 G15A agarose beads (ab211185, Abcam) at 4°C for 1 hour. The beads were then washed, boiled and the supernatants were used to immunoblot with DOCK7 antibody to determine levels of active DOCK7.
ELISA
Supernatant of 48-hours serum starved cells were collected and concentrated 5–10-fold with Pierce protein concentrator (ThermoFisher). EGFR ligands concentration in supernatant and tumour tissue extracts was determined by ELISA using the corresponding commercial HB-EGF, TGFα, EGF and Betacellulin (BTC) protein detection kits (Thermo Fisher) per the manufacturer’s instructions.
Small interfering RNA (siRNA), shRNA lentiviral particles and Adenovirus
Human BIN3 (sc-77692), HB-EGF (sc-39420), EGFR (sc-29301), DOCK7 (sc-105312), RhoA (sc-29471), Cdc42 (sc-29256), STAT3 (sc-29493), Shc (sc-29480), ERK1(sc-29307), ERK2 (sc-35335), Sp1 (sc-29487), TAB1(sc-61851), TAK1(sc-36606) and scrambled siRNAs (sc-37007) were obtained from Santa Cruz Biotechnology (Dallas, TX). Cells were transfected with the siRNA pool using Lipofectamine2000 (Invitrogen Carlsbad, CA). Experiments were conducted 48 hours after siRNA transfection. The target sequences for two EGFR siRNAs targeting the 3’UTR region were used for rescue experiments. EGFR siRNA-1, GUGGAAUUCAGG-UAGUAAAUAUGAA; EGFRsiRNA-2, GGAAGAUAGUUUUCUCCUUUUACUU. siRNA Knockdown efficiency was confirmed by Western blot.
Human HB-EGF shRNA lentiviral particles (sc-39420-V), DOCK7 shRNA lentiviral activation particles (sc-404461-LAC) and control lentiviral particles (sc-437282) were from Santa Cruz Biotechnology (Dallas, TX). To generate stable HB-EGF shRNA knockdown GBM39, cells were infected with HB-EGF shRNA or control lentiviral particles. Clones were selected in culture medium containing 1 μg/ml puromycin and expanded, knockdown efficiency was determined by HB-EGF ELISA kit. For generation of transient DOCK7 overexpressing GBM12 and GBM6, cells were infected with DOCK7 shRNA or control lentiviral activation particles as described above, except that positive clones were pooled together for further experiments.
Adenovirus-GFP or IĸBα dominant negative (DN) were obtained from Vector Biolabs (Malvern, PA). A Multiplicity of infection (MOI) of 10 was used in the experiments. Cells were exposed to adenovirus, along with transfection of empty, EGFRwt or EGFRvIII vectors.
cDNA Synthesis and real-time PCR
Total RNA from cells was extracted by TRIzol Reagent (Ambion). First-strand cDNA and real- time PCR was performed as described previously45. The expression of each gene was normalized to GAPDH as a reference. The following primers were used. BIN3, 5′-AGATATCCTTGGACTTACTCTC-3′ and 5′-CACCTTTTCCTGATTGAAGG-3′;; GAPDH, 5′-GTGAAGGTCGGAGTCAACGG-3′ and 5′-TGATGACAAGCTTCCCGTTCTC-3′; EGR1, 5’-CTTCAACCCTCAGGCGGACA-3’ and 5’-GGAAAAGCGGCCAGTATAGGT-3’; EMP1, 5’-GTGTTCCAGCTCTTCACCATGG-3’ and 5’-GGAATAGCCGTGGTGATACTGC-3’
Single cell migration assay
For time-lapse analysis of individual cell movement, we used a nano-ridge constructed of transparent polyurethane acrylate (PUA), and fabricated using UV-assisted capillary lithography as previously described 67. Prior to cell seeding, 24 well NanoSurface plate (Curi Bio, Seattle, WA)were coated with laminin (3 μg/cm2, Sigma-Aldrich). Cells were plated at low density (1×104 cells/ml) and incubated at 37°C overnight. For time-lapse imaging, the plate was mounted onto the stage of Andor spinning disk confocal microscope equipped with temperature and CO2 controlling environmental chamber. Six hours after the start of the imaging, EGF (50 ng/ml) or tofacitinib (1 μM) was added to each well without moving the plate. Bright-field images were taken every 30 minutes for 24 hours using a 10x objective. Manual track Plugin of ImageJ bundled Java (v 1.8.0_112) was used to track the movement of cells frame by frame.
Immunohistochemistry, immunofluorescence, TUNEL staining
Tumour tissues were fixed in 10% formalin and embedded in paraffin. Immunohistochemistry analysis was performed using the ABC streptavidin–biotin method with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) as described previously41. The following primary antibodies were used: HB-EGF (sc-365182), Sp1 (9389), SMI-31 (801601), Ki-67 (9027) and EGFR (04–338, Millipore). All antibodies except SMI-31 were incubated overnight at 4°C. SMI-31 was incubated at room temperature for 2 hours. Three to four complete and non-overlapping high magnification (x400) fields were randomly selected for each section.
Human GBM slides stained with antibodies to HB-EGF, Sp1 and EGFR were evaluated under the microscope for signal intensity. The immunostaining score ranges from 0 to 3 based on percentage positive staining (0: 5%, 1:5%–30%, 2: 30%–70%, 3: >70%). Two to three high cellular (tumour central) or low cellular (invasive) regions per tumour section were selected for evaluation.
Immunofluorescence double labeling was performed to demonstrate colocalization of the Sp1 in the nuclei and HB-EGF in the cytoplasm. Human glioblastoma slides were permeabilized by 0.5 % Triton X-100/PBS and blocked in 1% BSA/PBS. The primary antibodies for detecting HB-EGF (1:500) and Sp1 (1:200) were the same used in immunohistochemistry, followed by anti-Rabbit Alexa555-conjugated (4413, Cell Signaling) and anti-Mouse Alexa488-conjugated (ab150113, Abcam) secondary antibodies.
TUNEL staining was performed using the TUNEL assay kit (4810-30-K, R&D Systems, Minneapolis, Minnesota) according to the manufacturer’s instructions.
Luciferase assays
Cells were transfected with EGR1 promoter or empty vector using lipofectamine 2000. Renilla luciferase was co-transfected as an internal control. For NF-κB luciferase assay, NF-κB luciferase reporter and Renilla luciferase were transfected along with EGFRwt or vIII overexpression vectors. A dual-luciferase reporter assay system was used according to manufacturer’s instructions (Promega, Madison WI). Firefly luciferase activity was measured in a luminometer and normalized on the basis of Renilla luciferase activity.
Copy number analysis by quantitative real-time PCR
Genomic DNA was extracted using a standard phenol/chloroform procedure or a QIAamp DNA FFPE Tissue kit (Qiagen, Hilden, Germany). EGFR gene copy number was evaluated using Hs00997424_cn TaqMan assay (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. The TERT locus was used for the internal reference copy number. Human Genomic DNA (Applied Biosystems, Foster City, CA) was used as a normal control.
Allele frequency (AF) analysis by whole exome sequencing (WES)
Genomic DNA extracted from GBM9 was subjected to library preparation using the SureSelect V6 kit (Agilent) following the manufacturer’s instructions. Sequencing was performed on a NextSeq500 with 150 bases of paired-end reads to target 20X of raw depth. BWA-MEM was used to align sequence reads to reference genome GRCh37 (decoy). Post-BAM processing was performed using Picardtools (v2.25.5; https://broadinstitute.github.io/picard/), Samtools v1.122, bedtools v2.30.0 and GATK v3.8. Variants were detected using GATK v3.8, Samtools v1.12, and annotation using WGSA5 Annotation was done using WGSA (whole genome sequencing annotator). The AFs were calculated as follows: AF of EGFRwt=The average coverage for exons 2 to 7/The average coverage for exons 1 and 8 to 28; AF of EGFRvIII=1-AF of EGFRwt.
RNA-seq analysis
GBM12 cells were treated with EGF (50ng/ml) for 90 minutes. RNA was extracted using RNeasy mini kit (Qiagen, Germany). Agilent 2100 Bioanalyzer system and RNA Nano chip kit (5067-1511, Agilent, Santa Clara, CA) was used for RNA Quality measurement. The library for sequencing was generated using Illumina TruSeq® Stranded mRNA Library prep kit (20020594, San Diego CA). The samples were sequenced with 05 flowcell space on Illumina NextSeq 550 sequencer using 85-bp single-end protocol. FPKMs for genes and transcripts were generated by StringTie (v1.3.5), and RSeQC (v3.0.0) was used for generating RNA quality control metrics. Differential gene expression analysis was done with DESeq2 (v1.32.0). A gene was identified as significantly changed if the fold change was greater than 2 (up or down) and the P-value was less than 0.05 in comparison to the vehicle group.
Animal studies
4 to 6 weeks old female nude mice (088) purchased from Charles River Laboratories were used in this study. Cells were injected into the right corpus striatum of the brains of nude mice using a stereotactic frame. For survival experiments, mice were randomly divided into two groups (6–8 mice per group) treated with vehicle or tofacitinib by oral gavage throughout the entire experiment. EGF (33 μg/ml, 0.25 μl/hour) was infused directly into the lateral ventricles using a mini-osmotic pump attached to a cannula for 4 weeks as described previously 68 after which mice were euthanized. To monitor the mice with orthotropic xenografts, MRI was performed at the Mouse MRI Core, Advanced Imaging Research Center, at UTSW.
A two-photon laser intravital microscope combined with cranial window surgery was used to observe cellular movement in vivo.. After the generation of a circular cranial window, 10 μL of 1×104 cells/μl GBM12 stably expressing H2B-GFP were implanted at a depth of 0.5 mm. Following implantation, a 5 mm silicone-based polydimethylsiloxane (PDMS) coverslip was glued to the bone surrounding the cranial window. PDMS (SYLGARD184, Sigma-Aldrich) film was prepared as described by Heo et al.69. Seven days after surgery and 24 hours prior to imaging procedure, 10 ng EGF or vehicle was injected into the mice’s brain through a cranial window. For in vivo imaging, mouse head was stabilized using a customized head frame. Time-lapse z-stack images of the tumour were acquired every 10 minutes for 2 hours using an upright Zeiss LSM780 confocal/multiphoton microscope. Images were imported into Imaris 9.0 to create 3D representations and cell movements were tracked using the Imaris spot detection function.
Data analysis of public databases
The TCGA-GBM clinical data and corresponding RNA-sequencing (RNA-seq) data were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and UCSC Xena browser (https://xena.ucsc.edu). EGFR and pEGFR protein expression data were acquired from the Cancer Proteome Atlas (TCPA, https://tcpaportal.org.). EGFR, HB-EGF and Sp1 mRNA expression in human GBM were acquired from the Ivy Glioblastoma Atlas Project (https://glioblastoma.alleninstitute.org/). Log2 (UQ-FPKM+1) conversion was performed for all RNA-seq data. The GBM subtype information was acquired from GlioVis (https://gliovis.bioinfo.cnio.es). Murat database70 was used as an additional data source to perform gene expression based survival analysis for GBM patients.
Gene Set Enrichment Analysis (ssGSEA)
Overall survival (OS) and RNAseq data is available in 149 out of 154 primary TCGA-GBM patients. The whole transcriptome profiling of upper quartile FPKM (FPKM-UQ) was downloaded from TCGA. Each patient’s 7 EGFR ligands enrichment score was calculated using Single-sample Gene Set Enrichment Analysis (ssGSEA), which represents how the signature (gene-set) of the 7 EGFR ligands is coordinately up- or down-regulated expressed in each case. GBM cases with EGFR copy numbers over 4 were defined as EGFR amplification, 149 patients were divided into EGFR non-amplified with four groups as indicated. The program of ssGSEA was downloaded from https://github.com/broadinstitute/ssGSEA2.0 and run on the platform of RStudio version 1.3.1093.
Statistical analysis
All data were analyzed for significance using GraphPad Prism 8.0 software. Data are presented as means±SEM of three independent experiments. Two-tailed one-sample, two-sample Student’s t-test or Kolmogorov Smimov test were used to compare two data sets. One-way or two-way ANOVA with Bonferroni’s correction was used to compare more than two groups. For the analysis of cell velocity, statistical comparisons were made by Wilcoxon matched pair tests. Kaplan–Meier survival curves were constructed and compared by Log-rank or Gehan’s test. The correlation between mRNA expression and protein expression levels was analyzed by Spearman correlation coefficient. At least 3 independent experiments were performed unless otherwise indicated. Fisher’s exact test was used to determine the association between immunostaining intensity of high and low cellular areas in human tumour sections. P < 0.05 was considered statistically significant. * means that P < 0.05, ** means that P < 0.01, *** means that < 0.001 and **** indicates any P value less than 0.0001.
Data availability
RNA–seq and WES data that support the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) with accession number PRJNA812870 and PRJNA827815. Publicly available WES data were from NCBI SRA with accession number PRJNA543854 (SRX5870263). Previously published microarray data are available in Supplementary Table 1 of Ramnarain et al.1 Mass spectrometry data have been deposited in https://massive.ucsd.edu/ with Dataset Identifier: MSV000089272. The human GBM data were derived from the TCGA Research Network: http://cancergenome.nih.gov/. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Extended Data
Extended Data Fig. 1. Ligand induced EGFR signaling suppresses invasion, while constitutive EGFR signaling induces invasion.
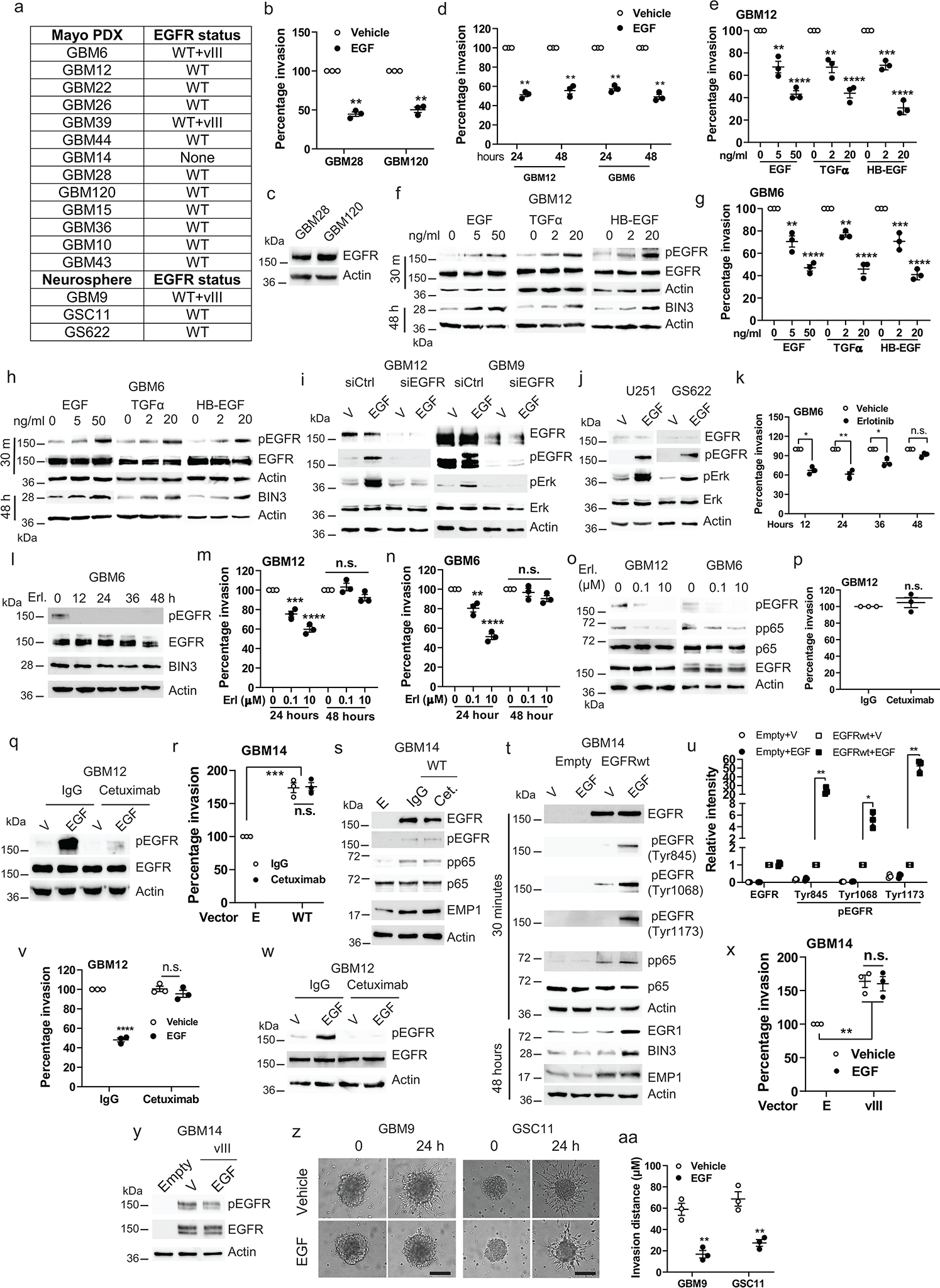
a, PDXs and neurospheres used in the study. b, Matrigel invasion assay of cells in the presence or absence of EGF(50 ng/ml). c, Immunoblot of EGFR expression. d, Matrigel invasion assay of cellstreated with EGFfor the indicated times. e, Matrigel invasion assay of GBM12 with various EGFR ligands. f, Immunoblot of the indicated proteins in GBM12 treated with EGFR ligands. g-h, Similar experiments in GBM6. i-j, Immunoblot of pEGFR and pERK in EGFR siRNA knockdown cellsand in U251 and GS622 cells treated with vehicle or EGF for 5 minutes. k, Matrigel invasion assay of GBM6 treated with erlotinib (1 μM) for the indicated times. l, Efficacy of erlotinib was analyzed by Western blot. m-n, Matrigel invasion assay of cells treated with the indicated concentrations of erlotinib. o, Immunoblot demonstrating efficacy of erlotinib (Erl). p, Matrigel invasion assay of GBM12 with IgG or cetuximab. q, Immunoblot of pEGFR and EGFR expression in GBM12 treated with EGF, IgG or Cetuximab (5 min.). r, Matrigel invasion assay of EGFRwt overexpressing GBM14 with IgG or cetuximab. s, Immunoblot of the indicated proteins in EGFR overexpressing GBM14 treated withEGF, IgG or cetuximab for 48 hours. t, Immunoblot of the indicated proteins in EGFR overexpressing GBM14 treated with or without EGF. u, Quantification of Western blot band intensity with actin as the reference protein. v, Matrigel invasion assay of GBM12 with the indicated drugs. w, Immunoblot of EGFR and pEGFR in cetuximab pretreated GBM12 with the indicated drugs. x, Matrigel invasion assay of EGFRvIII overexpressing GBM14 in the presence or absence of EGF. y, Immunoblot of EGFR and pEGFR in EGFRvIII overexpressing GBM14 treated with vehicle or EGF for 5 minutes. z, Representative static images showing progression of invasion in cellswith EGF. aa, Quantification of invasion distance of neurospheres. Western blot images are representative of three independent biological replicates. Actin served as the loading control. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined bytwo-tailed one-sample Student’s t-test (b, d, k, p, u), or by one-way ANOVA adjusted by Bonferroni’s correction (e, g, m, n, r, v, x), or by two-tailed unpaired Student’s t-test (aa). *P<0.05, **P< 0.01, ***P< 0.001, ****P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Extended Data Fig. 2. Suppression of invasion induced by EGF is mediated by an EGFR-BIN3-DOCK7 pathway.
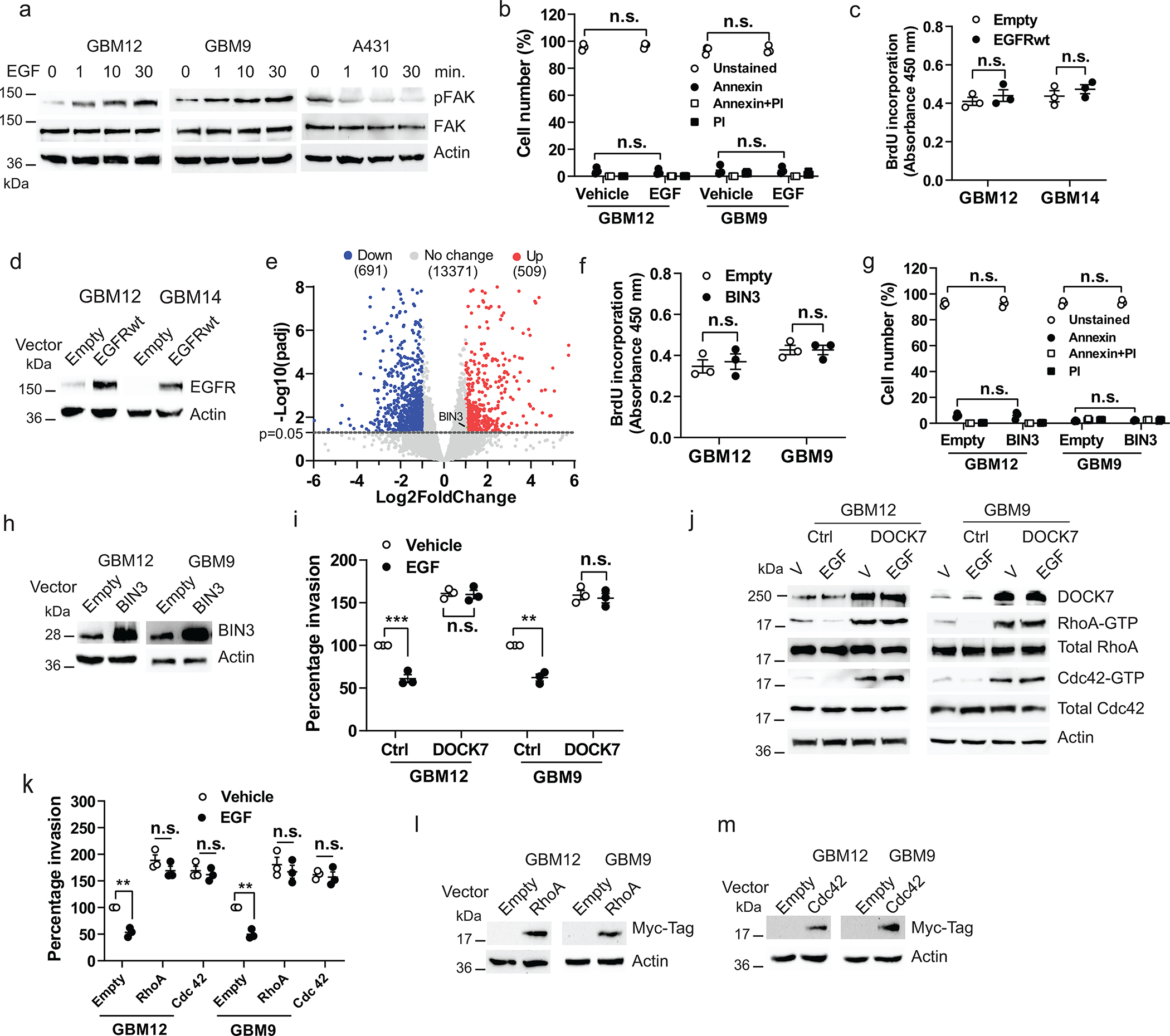
a, Immunoblot of pFAK and FAK expression in cells treated with EGF (50 ng/ml) for the indicated times. b, Annexin V/PI positive staining assay of cells treated with EGF. c, BrdU incorporation assay of cells transiently transfected with empty or EGFRwt expression vectors. d, Overexpression of EGFRwt in cells was analyzed by Western blot. e, Volcano plot of differentially expressed genes as assessed by RNA-seq in vehicle or EGF treated GBM12. Significantly upregulated BIN3 is highlighted in green (Fold change=2.1, p=0.03). f, BrdU incorporation assay ofcells transiently transfected with empty or BIN3 vectors. g, Annexin V/PI positive staining assay of cells transiently transfected with empty or BIN3 expression vectors. h, BIN3 overexpression in cells was analyzed by Western blot. i, Matrigel invasion assay of DOCK7 overexpressing cells. Cells were infected with control or DOCK7 lentiviral activation particles, and 72 hours after infection invasion assay was performed in the presence or absence of EGF. j, Immunoblot of the indicated proteins in DOCK7 overexpressing cells treated with EGF for 24 hours. k, Matrigel invasion assay of cells transiently transfected with empty, Myc-RhoA or Myc-Cdc42 expression vectors in the presence or absenceof EGF. l-m, Overexpression of Myc-RhoA and Myc-Cdc42 was analzyed by Western blot. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined bytwo-way ANOVA adjusted by Bonferroni’s correction (b, g, i, k), or by two-tailed unpaired Student’s t-test (c, f). *P<0.05, **P< 0.01, ***P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Extended Data Fig. 3. Regulation and biological effects of BIN3.
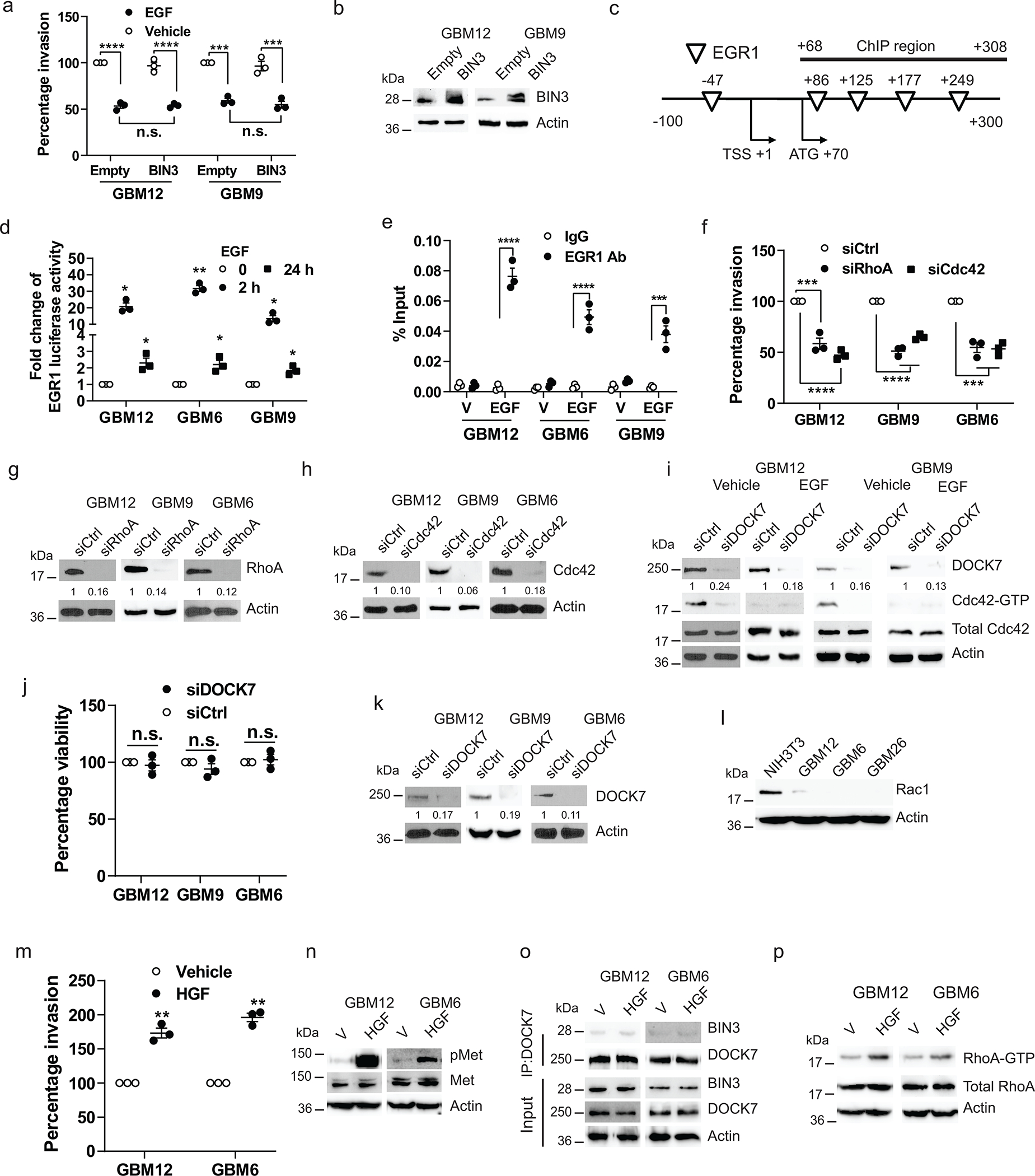
a, Matrigel invasion of BIN3 overexpressing cells in the presence or absenceof EGF (50 ng/ml). b, BIN3 overexpression was analyzed by Western blot. c, Schematic diagram of the putative EGR binding sites in BIN3 promoter region together with the corresponding ChIP-qPCR amplicons. d, EGR1 luciferase reporter activity in multiple linestreated with EGF. e, Percentage input done by ChIP-qPCR to assess the EGR-1 occupancy of BIN3 gene in multiple lines treated with vehicle (V) or EGF for 24 hours. IgG was used as negative control. f, Matrigel invasion assay of Cdc42 or RhoA siRNA knockdown in multiple lines. g-h, Knockdown efficiency of RhoA and Cdc42 siRNA was analyzed by Western blot. i, Immunoblot of Cdc42-GTP and total Cdc42 expression in DOCK7 siRNA knockdown cells. j, Cell viability assay of control and DOCK7 siRNA knockdown multiple lines. k, Knockdown efficiency of DOCK7 was analyzed by Western blot. l, Immunoblot of Rac1 expression in multiple lines. m, Matrigel invasion assay of cells in the presence or absence of HGF (20ng/ml). n, Immunoblot of pMet and Met expression in cells treated with HGF for 1 hour. o, Immunoblot of immunoprecipitated extracts from cellstreated with HGF for 24 hours. p, Immunoblot of RhoA-GTP expression in cells treated with HGF (20 ng/ml) for 24 hours. The Western blot images are representative of three independent biological replicates. The numbers below the blots indicate the relative band intensity of protein against that of actin. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-way ANOVA adjusted by Bonferroni’s correction (a, e), or by two-tailed one- sample Student’s t-test (d, j), or one-way ANOVA adjusted by Bonferroni’s correction (f). *P<0.05, **P< 0.01, ***P< 0.001, ****P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Extended Data Fig. 4. EGFR ligand overexpression prolongs survival, reduces invasiveness and increases proliferation in orthotopic glioblastoma mouse model.
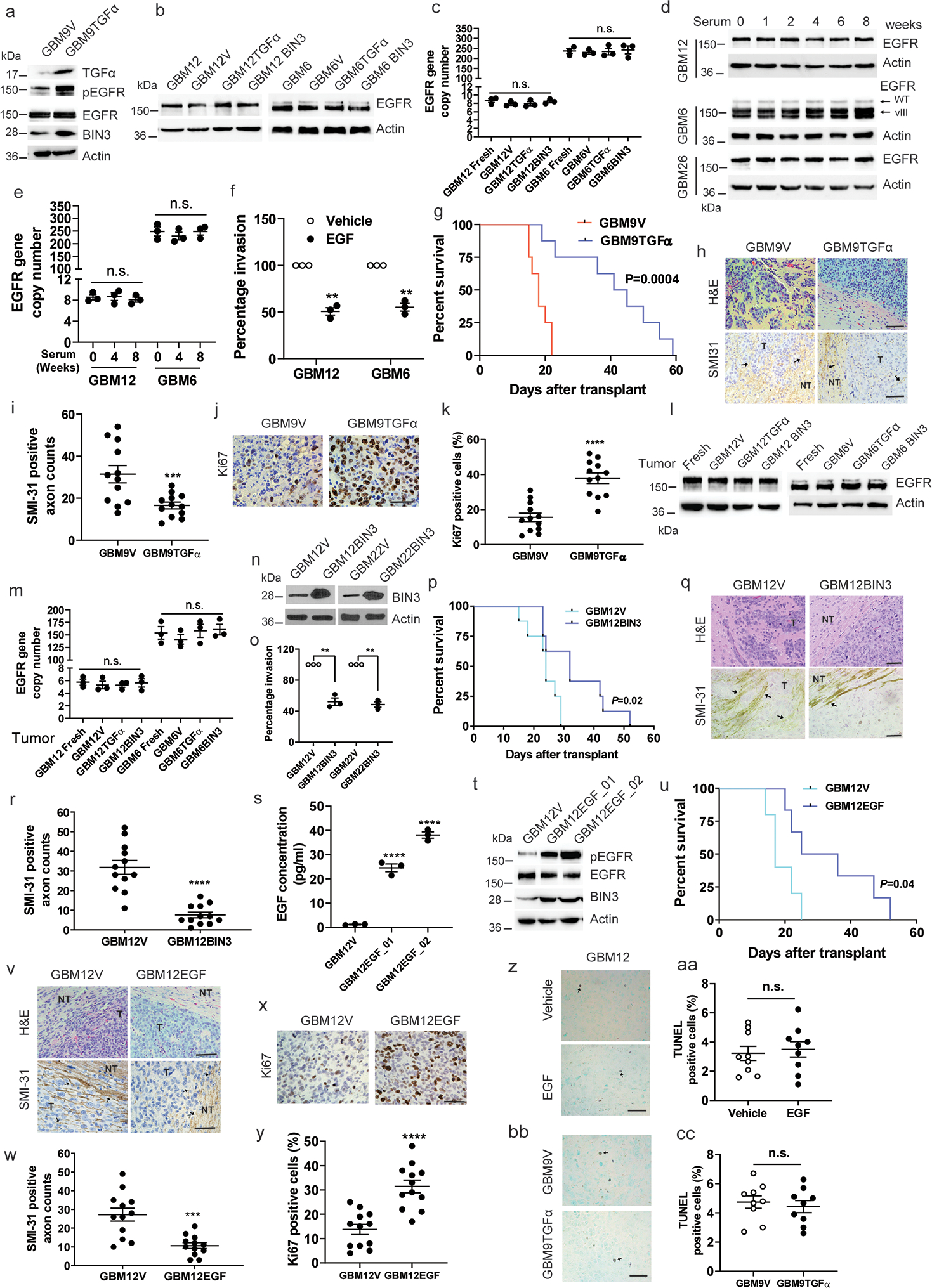
a, Immunoblot of the indicated proteins in GBM9 stably transfected with empty (GBM9V) or TGFα expression vector (GBM9TGFα). b, Immunoblot of EGFR expression in cells stably transfected with empty, TGFα, or BIN3 expression vectors. c, EGFR copy numbers in cells described in b. d, Immunoblot of EGFR expression in multiple lines cultured in 10% serum for the indicated times. e, EGFR copy numbers in cellsdescribed in d. f, Matrigel invasion assay of cells cultured in 10% serum for 8 weeks in the presence or absence of EGF. g, Kaplan–Meier survival curves of mice with GBM9V and GBM9TGFα tumours (n=8/group). h-i, H&E staining, SMI-31 immunostaining (black arrow)and quantification of SMI-31 countsin mouse tumours. j-k, Ki67 immunostaining and quantification of Ki67 positive cellsin mouse tumours. l, Immunoblot of EGFR expression in various tumours.. m, EGFR copy numbers in mouse tumours compared to fresh explants cultures. n, Immunoblot of BIN3 expression in cells stably overexpressing BIN3 or empty vector. o, Matrigel invasion assay of multiple lines. p, Kaplan–Meier survival curves of mice with GBM12V and GBM12BIN3 tumours (n=8/group). q-r, H&E staining, SMI-31immunostaining and quantification of SMI-31 counts in mouse tumours. s, ELISA for EGF in GBM12 stably transfected with EGF overexpressing or empty vector. t, Immunoblot of the indicated proteins in EGF-overexpressing GBM12 clones. u, Kaplan–Meier survival curves of mice with GBM12V and GBM12EGF (GBM12EGF_02) tumours (n=6/group). v, H&E staining, SMI-31 immunostaining and quantification of SMI-31 countsin mouse tumours. x-y, Ki67 immunostaining and quantification of Ki67 postive cells in mouse tumours. z-aa, Representative TUNEL staining (black arrows) and quantification of TUNEL positive cells in GBM12 orthotopic tumours from vehicle orEGF treated mice. bb-cc, Representative TUNEL staining and quantification of TUNEL positivecells in mouse tumours. Scale bars: 50 μM. Western blot images are representative of three independent biological replicates. Actin served as the loading control. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by one-way ANOVA adjusted by Bonferroni’s correction (c, e, m, s), or by two-tailed one-sample Student’s t-test (f, o), or two-tailed unpaired Student’s t-test (i, k, r, w, y, aa, cc). *P<0.05, **P< 0.01, ***P< 0.001, ****P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Extended Data Fig. 5. Biological effects of EGFR and STAT activation in GBM cells.
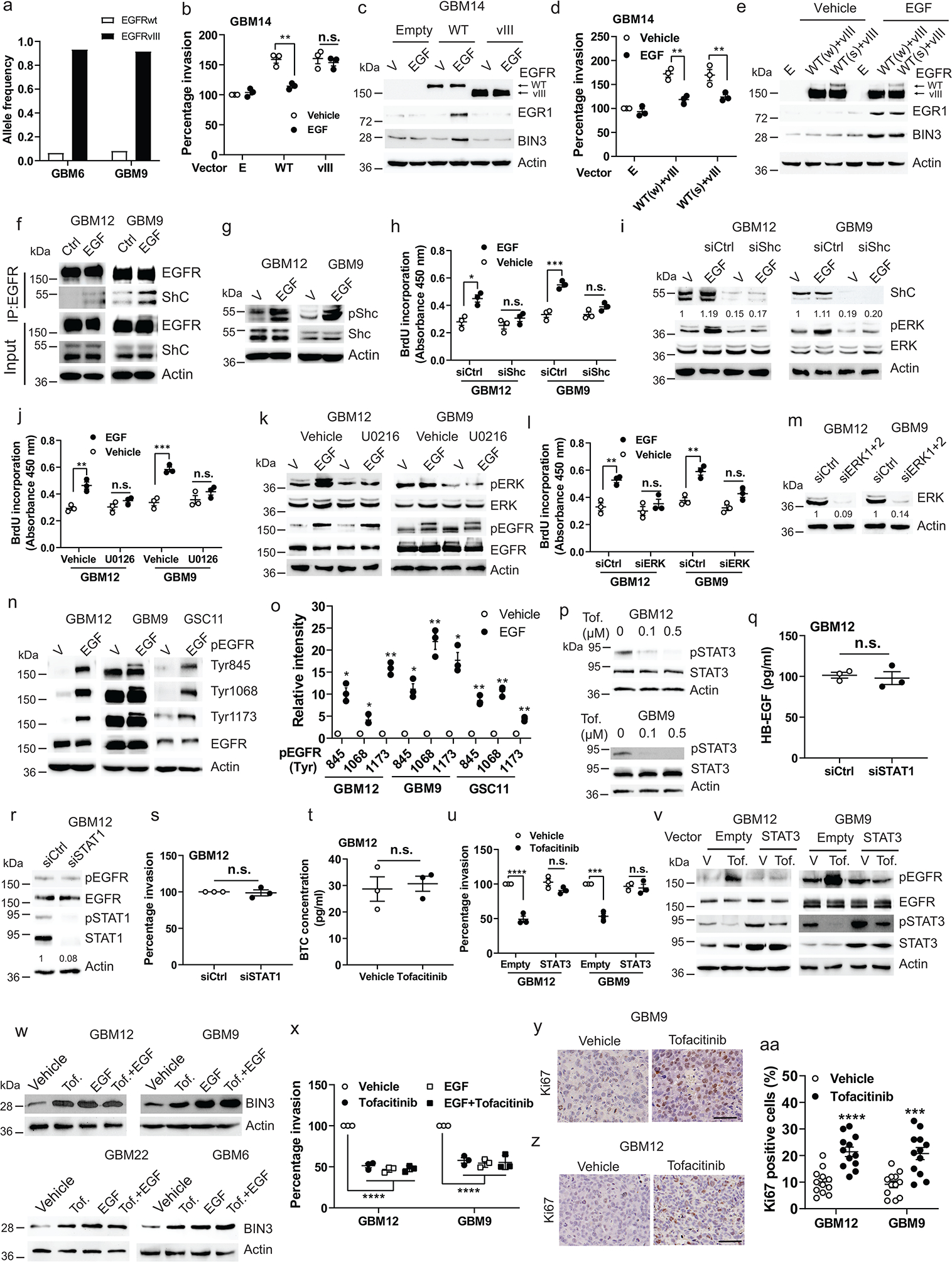
a, The allele frequency of EGFRwt and EGFRvIII in GBM6 and GBM9. b, Matrigel invasion assay of EGFRwt or EGFRvIII overexpressing GBM14 with or without EGF (50 ng/ml). c, Immunoblot of the indicated proteins in EGFRwt or vIII overexpressing GBM14 cells treated with EGF (24 h). d, Matrigel invasion assay of EGFRwt and vIII overexpressing GBM14 with or without of EGF.WT(w): weak expression of WT; WT(s): strong expression of WT. e, Immunoblot of the indicated proteins in EGFRwt and vIII overexpressing GBM14 treated with EGF (24h). f, Immunoblot of immunoprecipitated extracts from cells treated with EGF (30 min.). g, Immunoblot of the indicated proteins in cells treated with EGF (30 min.)h, BrdU incorporation assay of Shc siRNA knockdown cellstreated with EGF. i, Immunoblot of the indicated proteins in Shc siRNA knockdown cells treated with EGF (30 min.). j, BrdU incorporation assay of cells treated with EGF, U0126 or a combination. k, Immunoblot of the indicated proteins in cells treated with EGF or U0126 or a combination (30 min.). l, BrdU incorporation assay of ERK siRNA knockdown cells treated with EGF. m, Knockdown efficiency of ERK was analyzed by Western blot. n, Immunoblot of different Tyr resides of pEGFR in multiple lines treated with EGF (30 min.). o, Quantification of Western blot band intensity with actin as reference. p, Immunoblot of the indicated proteins in cells treated with tofacitinib (Tof.). q, ELISA for HB-EGF in the supernatant of STAT1 siRNA knockdown GBM12. r, Immunoblot of pEGFR and pSTAT1 in STAT1 siRNA knockdown GBM12. s, Matrigel invasion assay of control or STAT1 siRNA knockdown GBM12. t, ELISA for BTC in the supernatants of GBM12 treated with tofacitinib (72 h). u, Matrigel invasion assay of STAT3 overexpressing GBM12 in response to tofacitinib. v, Immunoblot of the indicated proteins in STAT3 overexpressingcells treated with tofacitinib. w, Immunoblot of BIN3 in multiple lines treated with EGF, tofacitinib or both (48 h). x, Matrigel invasion assay with EGF or tofacitinib or both. y-aa, Ki67 immunostaining and quantification of KI67 positive cells in GBM12 and GBM9 tumours from tofacitinib treated mice. Scale bars: 50 μM. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Data are represented as mean ± SEM from three independent experiments. Statistical significance was determined by two-way ANOVA adjusted by Bonferroni’s correction (b, d, h, j, l, u), or by two-tailed one-sample Student’s t-test (o, s), or by two-tailed unpaired Student’s t-test (q, aa), or by one-way ANOVA adjusted by Bonferroni’s correction (x). *P<0.05, **P< 0.01, ***P< 0.001, ****P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Extended Data Fig. 6. EGF and tofacitinib suppress migration in single cell analysis.
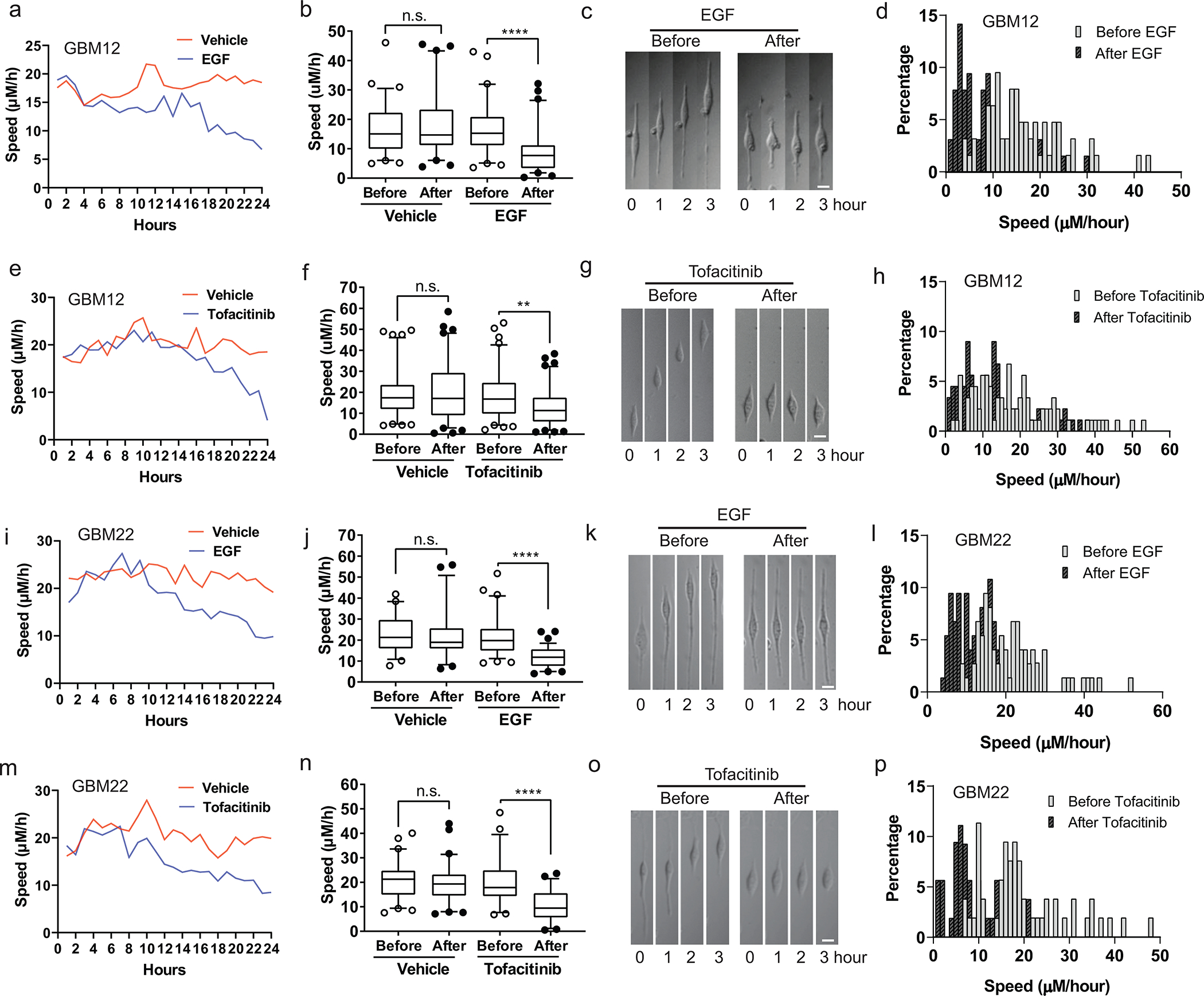
a, Time-lapse migration speed in GBM12 before and after addition of vehicle (n=67) or EGF (50 ng/ml)(n=63). b, Quantification of migration velocity of GBM12 before and after addition of vehicle or EGF. Cell migration velocity was calculated over a period of 6 hours. c, Representative static time-lapse images of the same cell migration before and after addition of EGF. d, Percentage of cell migration speed before and after addition of EGF. e, Time-lapse migration speed in GBM12 before and after addition of vehicle (n=81) or tofacitinib (1 μM) (n=89). f, Quantification of migration velocity of GBM12 before and after addition of vehicle or tofacitinib. g, Representative static time-lapse images of the same cell migration before and after addition of tofacitinib. h, Percentage of cell migration speed before and after addition of tofacitinib. i, Time-lapse migration speed in GBM22 before and after addition of vehicle (n=57) or EGF (n=74). j, Quantification of migration velocity of GBM22 before and after addition of vehicle or EGF. k, Representative static time-lapse images of the same cell migration before and after addition of EGF. l, Percentage of cell migration speed before and after addition of EGF. m, Time-lapse migration speed in GBM22 before and after addition of vehicle (n=61) or tofacitinib (n=53). n, Quantification of migration velocity of GBM22 before and after addition of vehicle or tofacitinib. o, Representative static time-lapse images of the same cell migration before and after addition of tofacitinib. p, Percentage of cell migration speed before and after addition of tofacitinib. Scale bars: 10 μM (c, g, k and o). Whiskers of the boxplot mark the 5th and 95th percentiles, the box marks the 25th to the 75th percentiles with the median (b, f, j, m). **P < 0.001, **** P < 0.0001, n.s. not significant, P values were determined using two-tailed Wilcoxon matched pair tests(b, f, j, n). The results are representative of two independently repeated experiments. Numerical source data, statistic and exact P values are available as Source Data.
Extended Data Fig. 7. EGFR ligands, EGFR, BIN3 and Sp1 expression in human glioblastoma.
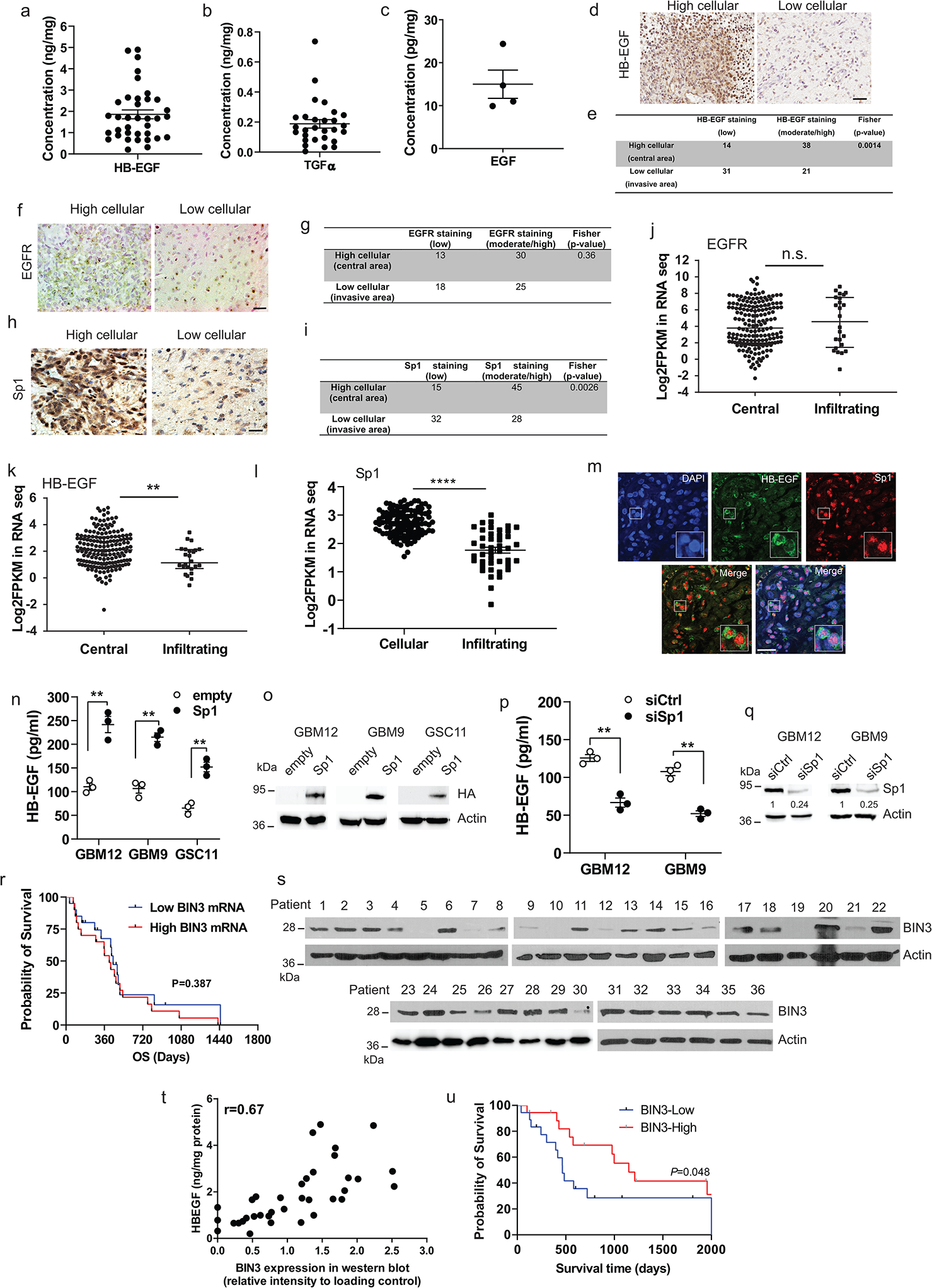
a-c, ELISA for HB-EGF, TGFα and EGF in human glioblastoma lysates. d, Representative immunohistochemical staining of HB-EGF in human glioblastoma. e, Summary of HB-EGF staining in high and low cellular areas across the tissue sections from 20 samples. Score 0 and 1 are defined as low, 2 and 3 are defined as moderate/high. Staining intensity in the two areas were compared using Fisher’s exact test. f, Representative EGFR immunostaining in high cellular (central) and low cellular (infiltrating) areas of human glioblastoma. g, Summary of EGFR staining of the tissue sections from 20 samples. h, Representative Sp1 immunostaining in high cellular and low cellular areas of human glioblastoma. i, Summary of Sp1 staining of the tissue sections from 20 samples. j-l, Ivy Atlas distribution of EGFR, HB-EGF and Sp1 mRNA expression in central and infiltrating areas of human GBM. m, Representative images of fluorescent double staining of HB-EGF (green) and Sp1 (red) in human GBM tissue sections. n, ELISA for HB-EGF in multiple lines transiently transfected with empty or HA-Sp1 vectors. o, HA-Sp1 overexpression was analyzed by Western blot. p, ELISA for HB-EGF in Sp1 siRNA knockdown cells. q, Sp1 siRNA knockdown was analyzed by Western blotting. r, Overall survival (OS) analysis according to BIN3 mRNA levels in classical GBM patients with amplified EGFR. s, Immunoblot of BIN3 expression in human glioblastoma extracts. t, Scatter plot of HB-EGF and BIN3 expression in human glioblastoma lysates (n=36). u, Kaplan-Meier curves of survival rates for high and low levels of BIN3 assessed by Western blot. The Western blot images are representative of three independent biological replicates. Actin served as the loading control. The numbers below the blots indicate the relative band intensity of protein against that of actin. Scale bars: 50 μM. Data are represented as mean ± SEM from three independent experiments. Significance was determined by Kolmogorov Smimov test (j, k, l), or by two-tailed unpaired Student’s t-test (n, p), or by log-rank test (r, u). **P< 0.01, ****P< 0.001, n.s. not significant. Numerical source data, statistic, exact P values and unprocessed blots are available as Source Data.
Supplementary Material
Acknowledgements
This work was supported in part by funding from the Department of Veteran’s Affairs to AH (2I01BX002559-08) and from the National Institutes of Health (1R01CA244212-01A1 and 1R01NS119225-01A1) to AH. DEG is supported by a National Cancer Institute (NCI) Midcareer Investigator Award in Patient-Oriented Research, K24CA201543-01. SB is supported by grants from the National Institutes of Health (R01CA258381 and R01CA246807) and the National Aeronautics and Space Administration (80NSSC20K0732). CMC’s research is supported by NIH grant 1RO1CA251698 and CPRIT grant RP190077. DZ was supported by NIH grant R01 CA194578, SKM is supported by awards from the Cancer Prevention and Research Institute of Texas (RR190034) and the NCI (K22CA237752). Research reported in this publication was supported in part by the Harold C. Simmons Comprehensive Cancer Center’s Biomarker Research Core, which are supported by NCI Cancer Center Support Grant 1P30 CA142543-03. JNS was supported by the MIT/Mayo Physical Sciences Center for Drug Distribution and Efficacy in Brain Tumours (U54CA210180). We acknowledge NIH shared instrumentation grant 1S10OD023552-01 that funded the MRI equipment. We thank Dr. Shi-Yuan Cheng (Northwestern University) for a kind gift of GSC11 cells. We thank Dr. Deok-Ho Kim (Johns Hopkins University) for advice on the use of patterned nanosurfaces.
Footnotes
Conflict of Interest disclosure: The Department of Veteran’s Affairs has filed a patent on the use of tofacitinib in glioblastoma, with Amyn Habib as the inventor. The other authors declare no competing interests.
References
- [1].Ramnarain DB, Park S, Lee DY, Hatanpaa KJ, Scoggin SO, Otu H, Libermann TA, Raisanen JM, Ashfaq R, Wong ET, Wu J, Elliott R, and Habib AA (2006) Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells, Cancer Res 66, 867–874. [DOI] [PubMed] [Google Scholar]
- [2].Chakraborty S, Li L, Puliyappadamba VT, Guo G, Hatanpaa KJ, Mickey B, Souza RF, Vo P, Herz J, Chen MR, Boothman DA, Pandita TK, Wang DH, Sen GC, and Habib AA (2014) Constitutive and ligand-induced EGFR signalling triggers distinct and mutually exclusive downstream signalling networks, Nat Commun 5, 5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mishima K, Higashiyama S, Asai A, Yamaoka K, Nagashima Y, Taniguchi N, Kitanaka C, Kirino T, and Kuchino Y (1998) Heparin-binding epidermal growth factor-like growth factor stimulates mitogenic signaling and is highly expressed in human malignant gliomas, Acta Neuropathol (Berl) 96, 322–328. [DOI] [PubMed] [Google Scholar]
- [4].Hatanpaa KJ, Burma S, Zhao D, Habib AA (2010) Epidermal growth factor receptor (EGFR) in glioma: Signal transduction, neuropathology, imaging and radioresistance Neoplasia 12, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].An Z, Aksoy O, Zheng T, Fan QW, and Weiss WA (2018) Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies, Oncogene 37, 1561–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karpel-Massler G, Schmidt U, Unterberg A, and Halatsch ME (2009) Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: where do we stand?, Mol Cancer Res 7, 1000–1012. [DOI] [PubMed] [Google Scholar]
- [7].Reardon DA, Wen PY, and Mellinghoff IK (2014) Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges, Neuro Oncol 16 Suppl 8, viii7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, and Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1, Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, Chen J, Lau J, Knobbe-Thomsen C, Weller M, Jura N, Reifenberger G, Shokat KM, and Weiss WA (2013) EGFR phosphorylates tumour-derived EGFRvIII driving STAT3/5 and progression in glioblastoma, Cancer Cell 24, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Roos A, Dhruv HD, Peng S, Inge LJ, Tuncali S, Pineda M, Millard N, Mayo Z, Eschbacher JM, Loftus JC, Winkles JA, and Tran NL (2018) EGFRvIII-Stat5 Signaling Enhances Glioblastoma Cell Migration and Survival, Mol Cancer Res 16, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jahani-Asl A, Yin H, Soleimani VD, Haque T, Luchman HA, Chang NC, Sincennes MC, Puram SV, Scott AM, Lorimer IA, Perkins TJ, Ligon KL, Weiss S, Rudnicki MA, and Bonni A (2016) Control of glioblastoma tumourigenesis by feed-forward cytokine signaling, Nat Neurosci 19, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Talasila KM, Soentgerath A, Euskirchen P, Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO, Sakariassen PO, Eskilsson E, Stieber D, Keunen O, Brekka N, Moen I, Nigro JM, Vintermyr OK, Lund-Johansen M, Niclou S, Mork SJ, Enger PO, Bjerkvig R, and Miletic H (2013) EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis, Acta neuropathologica 125, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, Woolfenden S, Boskovitz A, Raval A, Bronson RT, Pfannl R, Whittaker CA, Housman DE, and Charest A (2011) Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation, Cancer Res 71, 7198–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, Pelton JG, Shan Y, Shaw DE, Wemmer DE, Groves JT, and Kuriyan J (2013) Conformational coupling across the plasma membrane in activation of the EGF receptor, Cell 152, 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Biernat W, Huang H, Yokoo H, Kleihues P, and Ohgaki H (2004) Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas, Brain pathology 14, 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Inda MD, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, Tan P, Depinho RA, Cavenee W, and Furnari F (2010) Tumour heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma, Genes Dev 24, 1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao D, and Habib AA (2015) Ligand-Independent EGFR Signaling, Cancer Res 75, 3436–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huang PH, Xu AM, and White FM (2009) Oncogenic EGFR signaling networks in glioma, Sci Signal 2, re6. [DOI] [PubMed] [Google Scholar]
- [19].Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK, Pan E, Aliwarga E, Chong ST, Kok CYL, Endaya BB, Habib AA, Horibe T, Ng WH, Ho IAW, Hui KM, Kordula T, and Lam PYP (2017) Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme, Nat Commun 8, 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Armento A, Ehlers J, Schotterl S, and Naumann U (2017) Molecular Mechanisms of Glioma Cell Motility, In Glioblastoma (De Vleeschouwer S, Ed.), Brisbane (AU). [PubMed] [Google Scholar]
- [21].Vollmann-Zwerenz A, Leidgens V, Feliciello G, Klein CA, and Hau P (2020) Tumour Cell Invasion in Glioblastoma, Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, and Tran NL (2013) Implications of Rho GTPase Signaling in Glioma Cell Invasion and Tumour Progression, Front Oncol 3, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, Yiin JJ, Lu S, Keezer S, Fenton T, Furnari FB, Hamilton RL, Vuori K, Sarkaria JN, Nagane M, Nishikawa R, Cavenee WK, and Cheng SY (2011) Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRalpha-stimulated glioma tumourigenesis in mice and humans, J Clin Invest 121, 4670–4684. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [24].de Kreuk BJ, and Hordijk PL (2012) Control of Rho GTPase function by BAR-domains, Small GTPases 3, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simionescu-Bankston A, Leoni G, Wang Y, Pham PP, Ramalingam A, DuHadaway JB, Faundez V, Nusrat A, Prendergast GC, and Pavlath GK (2013) The N-BAR domain protein, Bin3, regulates Rac1- and Cdc42-dependent processes in myogenesis, Dev Biol 382, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Birnbaum D, Adelaide J, Popovici C, Charafe-Jauffret E, Mozziconacci MJ, and Chaffanet M (2003) Chromosome arm 8p and cancer: a fragile hypothesis, The lancet oncology 4, 639–642. [DOI] [PubMed] [Google Scholar]
- [27].Chang BL, Liu W, Sun J, Dimitrov L, Li T, Turner AR, Zheng SL, Isaacs WB, and Xu J (2007) Integration of somatic deletion analysis of prostate cancers and germline linkage analysis of prostate cancer families reveals two small consensus regions for prostate cancer genes at 8p, Cancer Res 67, 4098–4103. [DOI] [PubMed] [Google Scholar]
- [28].Ye H, Pungpravat N, Huang BL, Muzio LL, Mariggio MA, Chen Z, Wong DT, and Zhou X (2007) Genomic assessments of the frequent loss of heterozygosity region on 8p21.3-p22 in head and neck squamous cell carcinoma, Cancer Genet Cytogenet 176, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ramalingam A, Duhadaway JB, Sutanto-Ward E, Wang Y, Dinchuk J, Huang M, Donover PS, Boulden J, McNally LM, Soler AP, Muller AJ, Duncan MK, and Prendergast GC (2008) Bin3 deletion causes cataracts and increased susceptibility to lymphoma during aging, Cancer Res 68, 1683–1690. [DOI] [PubMed] [Google Scholar]
- [30].Prendergast GC, Muller AJ, Ramalingam A, and Chang MY (2009) BAR the door: cancer suppression by amphiphysin-like genes, Biochim Biophys Acta 1795, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Singh B, Carpenter G, and Coffey RJ (2016) EGF receptor ligands: recent advances, F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu C, Leche CA 2nd, Kiyatkin A, Yu Z, Stayrook SE, Ferguson KM, and Lemmon MA (2022) Glioblastoma mutations alter EGFR dimer structure to prevent ligand bias, Nature 602, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carlson BL, Pokorny JL, Schroeder MA, and Sarkaria JN (2011) Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery, Curr Protoc Pharmacol Chapter 14, Unit 14 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guo G, Gong K, Ali S, Ali N, Shallwani S, Hatanpaa KJ, Pan E, Mickey B, Burma S, Wang DH, Kesari S, Sarkaria JN, Zhao D, and Habib AA (2017) A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma, Nat Neurosci 20, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Guo G, Gong K, Puliyappadamba VT, Panchani N, Pan E, Mukherjee B, Damanwalla Z, Bharia S, Hatanpaa KJ, Gerber DE, Mickey BE, Patel TR, Sarkaria JN, Zhao D, Burma S, and Habib AA (2019) Efficacy of EGFR plus TNF inhibition in a preclinical model of temozolomide-resistant glioblastoma, Neuro Oncol 21, 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun C, and Bernards R (2014) Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies, Trends in biochemical sciences 39, 465–474. [DOI] [PubMed] [Google Scholar]
- [37].Gong K, Guo G, Panchani N, Bender ME, Gerber DE, Minna JD, Fattah F, Gao B, Peyton M, Kernstine K, Mukherjee B, Burma S, Chiang C-M, Zhang S, Amod Sathe A, Xing C, Dao KH, Zhao D, Akbay EA, and Habib AA (2020) EGFR inhibition triggers an adaptive response by co-opting antiviral signaling pathways in lung cancer, Nature Cancer 1, 394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gong K, Guo G, Beckley NA, Yang X, Zhang Y, Gerber DE, Minna JD, Burma S, Zhao D, Akbay EA, and Habib AA (2021) Comprehensive targeting of resistance to inhibition of RTK signaling pathways by using glucocorticoids, Nat Commun 12, 7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Misek SA, Chen J, Schroeder L, Rattanasinchai C, Sample A, Sarkaria JN, and Gallo KA (2017) EGFR Signals through a DOCK180-MLK3 Axis to Drive Glioblastoma Cell Invasion, Mol Cancer Res 15, 1085–1095. [DOI] [PubMed] [Google Scholar]
- [40].Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, Zhou Q, Majumder S, Bi E, Liu DX, Huang S, and Lu Z (2014) PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells, Nat Commun 5, 5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lund-Johansen M, Bjerkvig R, Humphrey PA, Bigner SH, Bigner DD, and Laerum OD (1990) Effect of epidermal growth factor on glioma cell growth, migration, and invasion in vitro, Cancer Res 50, 6039–6044. [PubMed] [Google Scholar]
- [42].Westermark B, Magnusson A, and Heldin CH (1982) Effect of epidermal growth factor on membrane motility and cell locomotion in cultures of human clonal glioma cells, J Neurosci Res 8, 491–507. [DOI] [PubMed] [Google Scholar]
- [43].Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, and Fine HA (2006) Tumour stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumours than do serum-cultured cell lines, Cancer Cell 9, 391–403. [DOI] [PubMed] [Google Scholar]
- [44].Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, and James CD (2005) Patient tumour EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme, Neuro Oncol 7, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lu Z, Jiang G, Blume-Jensen P, and Hunter T (2001) Epidermal growth factor-induced tumour cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase, Mol Cell Biol 21, 4016–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Coll PM, Rincon SA, Izquierdo RA, and Perez P (2007) Hob3p, the fission yeast ortholog of human BIN3, localizes Cdc42p to the division site and regulates cytokinesis, EMBO J 26, 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gadea G, and Blangy A (2014) Dock-family exchange factors in cell migration and disease, Eur J Cell Biol 93, 466–477. [DOI] [PubMed] [Google Scholar]
- [48].Nakamuta S, Yang YT, Wang CL, Gallo NB, Yu JR, Tai Y, and Van Aelst L (2017) Dual role for DOCK7 in tangential migration of interneuron precursors in the postnatal forebrain, J Cell Biol 216, 4313–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Murray DW, Didier S, Chan A, Paulino V, Van Aelst L, Ruggieri R, Tran NL, Byrne AT, and Symons M (2014) Guanine nucleotide exchange factor Dock7 mediates HGF-induced glioblastoma cell invasion via Rac activation, Br J Cancer 110, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yamamoto K, Murata H, Putranto EW, Kataoka K, Motoyama A, Hibino T, Inoue Y, Sakaguchi M, and Huh NH (2013) DOCK7 is a critical regulator of the RAGE-Cdc42 signaling axis that induces formation of dendritic pseudopodia in human cancer cells, Oncology reports 29, 1073–1079. [DOI] [PubMed] [Google Scholar]
- [51].Yamauchi J, Miyamoto Y, Chan JR, and Tanoue A (2008) ErbB2 directly activates the exchange factor Dock7 to promote Schwann cell migration, J Cell Biol 181, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Witusik-Perkowska M, Rieske P, Hulas-Bigoszewska K, Zakrzewska M, Stawski R, Kulczycka-Wojdala D, Bienkowski M, Stoczynska-Fidelus E, Gresner SM, Piaskowski S, Jaskolski DJ, Papierz W, Zakrzewski K, Kolasa M, Ironside JW, and Liberski PP (2011) Glioblastoma-derived spheroid cultures as an experimental model for analysis of EGFR anomalies, J Neurooncol 102, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ahmat Amin MKB, Shimizu A, and Ogita H (2019) The Pivotal Roles of the Epithelial Membrane Protein Family in Cancer Invasiveness and Metastasis, Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hirata Y, Takahashi M, Morishita T, Noguchi T, and Matsuzawa A (2017) Post-Translational Modifications of the TAK1-TAB Complex, Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Raychaudhuri B, Han Y, Lu T, and Vogelbaum MA (2007) Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype, J Neurooncol 85, 39–47. [DOI] [PubMed] [Google Scholar]
- [56].Westhoff MA, Zhou S, Nonnenmacher L, Karpel-Massler G, Jennewein C, Schneider M, Halatsch ME, Carragher NO, Baumann B, Krause A, Simmet T, Bachem MG, Wirtz CR, and Debatin KM (2013) Inhibition of NF-kappaB signaling ablates the invasive phenotype of glioblastoma, Mol Cancer Res 11, 1611–1623. [DOI] [PubMed] [Google Scholar]
- [57].Puliyappadamba VT, Hatanpaa KJ, Chakraborty S, and Habib AA (2014) The role of NF-kappaB in the pathogenesis of glioma, Mol Cell Oncol 1, e963478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, and Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR, Nature 483, 100–103. [DOI] [PubMed] [Google Scholar]
- [59].Fan Q, An Z, Wong RA, Luo X, Lu ED, Baldwin A, Mayekar MK, Haderk F, Shokat KM, Bivona TG, and Weiss WA (2020) Betacellulin drives therapy resistance in glioblastoma, Neuro Oncol 22, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Smith CL, Kilic O, Schiapparelli P, Guerrero-Cazares H, Kim DH, Sedora-Roman NI, Gupta S, O’Donnell T, Chaichana KL, Rodriguez FJ, Abbadi S, Park J, Quinones-Hinojosa A, and Levchenko A (2016) Migration Phenotype of Brain-Cancer Cells Predicts Patient Outcomes, Cell Rep 15, 2616–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Miyata K, Yotsumoto F, Nam SO, Odawara T, Manabe S, Ishikawa T, Itamochi H, Kigawa J, Takada S, Asahara H, Kuroki M, and Miyamoto S (2014) Contribution of transcription factor, SP1, to the promotion of HB-EGF expression in defense mechanism against the treatment of irinotecan in ovarian clear cell carcinoma, Cancer Med 3, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Maier T, Guell M, and Serrano L (2009) Correlation of mRNA and protein in complex biological samples, FEBS Lett 583, 3966–3973. [DOI] [PubMed] [Google Scholar]
- [63].Konoeda F, Shichita T, Yoshida H, Sugiyama Y, Muto G, Hasegawa E, Morita R, Suzuki N, and Yoshimura A (2010) Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model, Biochem Biophys Res Commun 402, 500–506. [DOI] [PubMed] [Google Scholar]
- [64].Fukuyama T, Tschernig T, Qi Y, Volmer DA, and Baumer W (2015) Aggression behaviour induced by oral administration of the Janus-kinase inhibitor tofacitinib, but not oclacitinib, under stressful conditions, Eur J Pharmacol 764, 278–282. [DOI] [PubMed] [Google Scholar]
- [65].Paik J, and Deeks ED (2019) Tofacitinib: A Review in Psoriatic Arthritis, Drugs 79, 655–663. [DOI] [PubMed] [Google Scholar]
- [66].Ramnarain DB, Paulmurugan R, Park S, Mickey BE, Asaithamby A, Saha D, Kelliher MA, Mukhopadhyay P, Banani F, Madden CJ, Wright PS, Chakravarty S, and Habib AA (2008) RIP1 links inflammatory and growth factor signaling pathways by regulating expression of the EGFR, Cell Death Differ 15, 344–353. [DOI] [PubMed] [Google Scholar]
- [67].Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, Kone L, Farber H, Lee DY, An SS, Levchenko A, and Quinones-Hinojosa A (2012) Regulation of brain tumour dispersal by NKCC1 through a novel role in focal adhesion regulation, PLoS Biol 10, e1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, and DePinho RA (2002) Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis, Cancer Cell 1, 269–277. [DOI] [PubMed] [Google Scholar]
- [69].Heo C, Park H, Kim YT, Baeg E, Kim YH, Kim SG, and Suh M (2016) A soft, transparent, freely accessible cranial window for chronic imaging and electrophysiology, Sci Rep 6, 27818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, Hainfellner JA, Heppner FL, Dietrich PY, Zimmer Y, Cairncross JG, Janzer RC, Domany E, Delorenzi M, Stupp R, and Hegi ME (2008) Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma, J Clin Oncol 26, 3015–3024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA–seq and WES data that support the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) with accession number PRJNA812870 and PRJNA827815. Publicly available WES data were from NCBI SRA with accession number PRJNA543854 (SRX5870263). Previously published microarray data are available in Supplementary Table 1 of Ramnarain et al.1 Mass spectrometry data have been deposited in https://massive.ucsd.edu/ with Dataset Identifier: MSV000089272. The human GBM data were derived from the TCGA Research Network: http://cancergenome.nih.gov/. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.