

Abstract
The development of pathogenic bacteria resistant to current treatments is a major issue facing the world today. Here, the synthesis and biological activity of fourth generation poly(amidoamine) dendrimers decorated with 1-hexadecyl-azoniabicylo[2.2.2]octane (C16-DABCO), a quaternary ammonium compound known to have antibacterial activity, are described. This highly cationic dendrimer antibiotic was tested against several Gram positive and Gram negative strains of pathogenic bacteria and exhibited activity against both. Higher activity toward the Gram positive strains that were tested was observed. After the antimicrobial activity was assessed, E. coli and B. cereus were subjected to a resistance selection study. This study demonstrated that a multivalent approach to antimicrobial design significantly reduces the likelihood of developing bacterial resistance. Highly cationic dendrimers were also used as pretreatment of a membrane in order to prevent biofilm formation.
Keywords: antibiotic, cationic, dendrimers, multivalency, quaternary ammonium compound
Graphical Abstract
Introduction
Bacteria are dynamic and adaptive. They grow and proliferate in a wide range of pHs and temperatures and in many seemingly inhospitable environments including in vivo.1,2 Over time, the constant exposure of bacteria to antibiotics results in resistance. Antimicrobial resistance is one of the greatest threats to human health worldwide.3,4 Despite an increasing number of drug resistant bacteria being discovered, the pipeline for new antibiotics has slowed.5,6 Essentially, we are losing the evolutionary arms race with pathogenic bacteria. Without appropriate drugs to treat infections, it becomes increasingly likely that an epidemic will occur.7 With bacterial resistance on the rise, new effective antibiotics that can circumvent resistance are becoming highly desirable.8
Bacteria intrinsically have strategies for developing resistance.9,10 One strategy by which bacteria can acquire resistance is by horizontal gene transfer. These genes are usually in the form of a plasmid that can be transferred between one bacterium and another even across genus.10,11 Alternatively, bacteria can produce enzymes that break down certain types of antibiotics (such as β-lactamases breaking down β-lactam scaffold antibiotics).10,12,13 Bacteria are also capable of altering the binding site of certain antibiotic targets, thus reducing the antibiotic’s binding affinity. Methicillin resistant Staphylococcus aureus (MRSA) is an example of bacteria altering a targeted binding site.10 Moreover, bacteria can attenuate metabolic pathways in order to be less susceptible to an antibiotic. Gram negative bacteria, for example, are known to possess efflux pumps that can effectively reduce the accumulation of an antibiotic inside a bacterium.10,12,14
Armed with this knowledge, strategies to circumvent resistance are being developed.9 An antibiotic that has several targets, for example, would decrease the likelihood of resistance since the bacteria would have to make several changes in order to reduce their susceptibility. Alternatively, a strategy that would target the bacterium’s metabolic processes, cell membrane biosynthesis, or another essential target would greatly reduce the likelihood that resistance could be developed. Polymeric and macromolecular systems are currently actively being developed15,16 because of their ability to multivalently17,18 interact with bacterial targets. Dendrimers, macromolecules with branches emanating from the central core,19 are often used as the synthetic multivalent framework for antimicrobials. Several recent reviews of antibacterial dendrimers are available.20,21
Various dendrimer-based antibacterials using this multivalent strategy have been described. Amino acid functionalized dendrimers have been synthesized on a poly(amidoamine) scaffold for drug delivery with low cytotoxicity.22 Ortega et al. have reported the synthesis and activity of amine and ammonium functionalized chloromethylsilane-ended dendrimers effective against Escherichia coli and Staphylococcus aureus.23 Grinstaff et al. prepared amphiphilic anionic dendrimers, with selective activity toward Bacillus subtilis over HUVECs.24 Peptide dendrimers have been reported for drug delivery25 and have been employed against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa.26 Vancomycin conjugated onto a fifth generation poly(amidoamine) showed several orders of magnitude improvement over free vancomycin.27 Furthermore, this multivalent strategy has been employed to tackle difficult to treat viral infections such as anionic dendrimers with activity against HIV type-1 and herpes simplex virus type-2 (HSV-2), which are currently in phase 3 clinical trials.28
Specifically, the dendrimers reported here are fourth generation poly(amidoamine) dendrimers (G(4)-PAMAMs)29 functionalized with quaternary ammonium endgroups (Figure 1). Quaternary ammonium compounds (QACs) represent a well-established type of cationic surfactant known for having antibacterial activity.2 The QAC chosen for the studies reported in this paper is 4-aza-1-hexadecylazoniabicylo-[2.2.2]octane, or C16-DABCO.30 Because the QACs are attached to the dendrimer framework, the effective local concentration of active group that is delivered is much higher than it would be for the same concentration of monomeric QACs in solution. This increase in effective molarity is expected to improve the inhibition of bacterial growth relative to QACs that are not presented multivalently.
Figure 1.

Highly cationic dendrimer antibiotic 1. The red sphere represents the G(4)-PAMAM. The black portion is the linker to the carbohydrate, the mannoside is shown in green, DABCO is shown in red, and the hexadecyl hydrocarbon chain is blue.
In addition to the C16-DABCO endgroups, mannoside endgroups have also been incorporated into the design of the antimicrobial dendrimers since bacteria can be intercepted before they adhere to the host cell surface by multivalent carbohydrates. Small mannose-containing glycodendrimers, for example, inhibit the interaction between E. coli and mannan 10 to 100-fold more effectively than methyl mannoside.31 The C-6 primary hydroxyl group on mannose has the added advantage of providing a convenient point of attachment for the C16-DABCO endgroup. Thus, the C16-DABCO and mannoside functionalized PAMAM dendrimers 1 are designed to deliver a catastrophic dose of positive charge to the bacteria in a highly localized (multivalent) fashion.
In addition to reporting the synthesis and characterization of 1, the minimum inhibitory concentration (MIC) values for 1 were obtained against a series of Gram positive and Gram negative bacteria, and these values are reported here. MIC values for dendrimers were compared to monomeric control compounds. Red blood cell hemolysis and mammalian cell toxicity assays were performed, and results were compared to the MIC values obtained against bacteria. Complete inhibition of the growth of S. aureus biofilm was observed for membranes pre-treated with 1. Finally, in order to determine whether bacteria are more or less likely to develop resistance to multivalent antibacterial compounds such as 1 than to monovalent compounds, MIC values for E. coli and B. cereus that were grown in the maximum tolerable sub-inhibitory concentrations of 1 were determined for up to 50 growth cycles. The MIC values, biofilm inhibition studies, and resistance assays all indicate that 1 is an interesting new multivalent antibacterial compound.
Materials and Methods
Materials.
All standard chemicals and reagents were purchased from Aldrich (Sigma-Aldrich, St. Louis, MO, USA), Alfa Aesar (Johnson Matthey Company, Ward Hill, MA, USA), or Fisher (Fisher Scientific, Hampton, New Hampshire, USA) and were used without further purification. PAMAM dendrimers were purchased from Dendritech (Dendritech, Midland, MI, USA). C16-DABCO monomer was provided by Dr. Robert Engel (R. Engel Laboratory, Queens, NY, USA). Cephalexin was obtained from MP Biomedicals (Santa Ana, California). Both ampicillin and streptomycin were purchased from Fisher (Fisher Scientific, Hampton, New Hampshire, USA). Bacterial cultures and adenocarcinoma cell line (A549) were obtained from the American Type Culture Collection and are as follows: Escherichia coli (ATCC #25922), Bacillus cereus (ATCC #11778), Pseudomonas aeruginosa (ATCC #27853), Staphylococcus aureus (ATCC #29213), Streptococcus oralis (ATCC #35037), and A549 cell line (ATCC #CCL-185). Rabbit blood was purchased from QuadFive (Ryegate, Montana) with EDTA as the anticoagulant.
Methods.
Reactions were monitored via TLC. TLC was performed on silica gel glass plates containing 60 G F −254 and visualization was achieved with a UV light or a cerium ammonium molybdate stain. Column chromatography was performed on Silicycle 230-400 mesh silica gel. 1H spectra were performed on Bruker DRX500 (500 MHz) or Bruker DRX600 (600 MHz). Chemical shifts (δ) were reported in ppm downfield from internal TMS standard. Absorbances were determined on a Molecular Devices Spectramax Plus 384. MALDI spectra were recorded on a Bruker III Biflex with a 337 nm nitrogen laser using freshly recrystallized trans-3-indolacrylic acid as a matrix. Electrophoretic mobility measurements were determined on a Wyatt Technologies Mobiuζ DLS instrument. Critical micelle concentration measurements were taken on a 90 plus Particle Size Analyzer made by Brookhaven Instruments Corporation.
Synthesis of generation 4 PAMAM-based thiourea 1-O-(5-thiourea-3-oxapentyl)-6-(1-hexadecyl-1-azonia-4-azabicyclo-[2.2.2]octane)-a-D-mannopyranoside dendrimer 1.
Mannose functionalized dendrimer (300 mg, 0.01 mmol) was dissolved in DMF (1.5 mL) and cooled to 0 °C. TsCl (115 mg, 0.6 mmol) was added with 2,6-di-tert-butylpyridine (115 mg, 0.134 mL, 0.6 mmol). The reaction was allowed to warm to RT and was stirred for 2.5 h. DMF was removed at reduced pressure leaving a pale yellow fluffy solid. This solid was dissolved in DMPU, 2,6-DTBP (112.8 mg) was added, and C16-DABCO (165.0 mg, 0.488 mmol) was also added. The reaction was allowed to stir for two days. The reaction mixture was dialyzed in water (1000 Mw cutoff), and lyophilized to a white fluffy powder. 1H NMR (500 MHz, DMSO-d6) δ 7.49 (d, J = 7.7 Hz, 3H), 7.12 (d, J = 7.9 Hz, 3H), 4.88 – 4.46 (m, 3H), 3.80 – 3.29 (m, 2H), 3.28 – 3.07 (m, 15H), 3.07 – 2.94 (m, 9H), 2.75 (s, 1H), 2.29 (s, 3H), 1.75 – 1.55 (m, 2H), 1.24 (s, 24H), 0.85 (t, J = 6.7 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 146.14, 146.13, 138.06, 138.06, 128.49, 125.93, 63.81, 51.18, 47.82, 46.97, 44.17, 40.89, 40.57, 40.48, 40.41, 40.32, 40.15, 39.98, 39.82, 39.65, 39.48, 31.72, 29.49, 29.45, 29.37, 29.20, 29.13, 28.92, 26.15, 22.53, 21.59, 21.22, 14.40. MALDI-TOF MS 30,074 g/mol. (NMR data can be found in Figure S3 and MALDI data can be found in Figure S4 of the Supporting Information)
General Procedures for MIC Determinations.
The broth microdilution method described in ISO/FDIS 20776-1:2006(E)32 was used to determine the minimum inhibitory concentration or MIC. The assay was performed in Nunc brand 96-well plates with Nunclon Delta Surface with an accompanying lid. These plates were chosen because they had the least amount of desiccation after an incubation period (18±2 hours). In addition, the wells closest to the edge were filled with deionized water to prevent the desiccation of the inner wells. Dilutions of each compound and controls were plated in triplicate. Cephalexin was obtained from MP Biomedicals (Santa Ana, California). Both ampicillin and streptomycin were purchased from Fisher Scientific (Hampton, New Hampshire).
General Procedure for Hemolysis Assay.
Rabbit blood was purchased from QuadFive (Ryegate, Montana) in EDTA as the anticoagulant. Red blood cells (RBCs) were separated by centrifugation of 5 ml of received blood for 10 minutes at 900 rpm. Serum fraction was removed and discarded, and RBCs were washed with freshly prepared cold 0.9% saline three times. After the final wash, supernatant was removed and the volume adjusted to 5 ml with cold PBS and furthered diluted 1:10 in PBS as described previously.22 200 μl of RBCs were added to 800 μl of PBS with containing various concentrations of C16-DABCO dendrimer, C16-DABCO monomer, G(4)-PAMAM dendrimer, and mannose functionalized G(4)-PAMAM dendrimer, 1% Triton X-100 as positive control, and PBS alone as negative control. Samples were incubated for 3 hours at 37 °C and mixed by inversion every 30 minutes. After incubation samples were centrifuged for five minutes at 1300 rpm. Absorbance was determined at 540 nm and hemolysis was calculated using equation 1.
| (1) |
General Procedure for Toxicity Assay.
Once a plate of A549 cells became fifty-percent confluent, growth medium was drained from plate and replaced with an EDTA solution. After a ten-minute incubation, cells were removed from the plate and centrifuged until a pellet was formed. EDTA solution was replaced with 2 mL of growth medium. Cells were diluted and stained in order to enumerate cells. 10,000 cells per 100 uL were aliquoted into a 96-well plate in each well. Analytes were added, Promega’s CellTiter 96 AQeous One Solution Cell Proliferation Assay was performed. Using this method, the number of viable cells after exposure to an analyte was determined at A400.
Resistance Selection Studies.
Multistep resistance selection was performed using the broth microdilution method described in ISO/FDIS 20776-1:2006(E).32 The procedure was slightly adapted from Kosowska-Shick et al.33 The multistep resistance selection study is a MIC study with serial daily passages. The bacteria for the next day of the assay came from the MIC plate and the wells chosen were one dilution less than the MIC value. These wells had an optical density that was similar to the growth control wells. Since all of the dilutions were performed in triplicate, all three wells of bacteria (~300 μL) were taken for the next day because the C16-DABCO dendrimer caused noticeable lethargy in growth. The process was repeated daily for a maximum of 50 days or until the MIC had changed appreciably.
General Procedure for MALDI Sample Preparation.
Compound 1 was dissolved in DMF at a concentration of about 6 mg/mL or about 12 pmol/μL. Trans-3-indoleacrylic acid (IAA) was dissolved in DMF at a concentration of 2.25 mg/mL or 12000 pmol/μL. IAA was recrystallized in warm ethanol. Compound 1 was mixed with IAA in concentrations ranging from 1:500 to 1:30000 where 1 μL compound 1 was mixed with 10 μL IAA to obtain a 1:1000. External calibration was performed with proteins such as myoglobin, bovine serum albumen, and trypsinogen in IAA as a matrix to obtain the reported m/z. Standards were dissolved in 80% acetonitrile : 20% water. Spectra were obtained on a Bruker Biflex III with a 337 nm nitrogen laser. Trans-3-indoleacrylic acid, myoglobin, bovine serum albumen, and trypsinogen were all obtained from Sigma-Aldrich.
Results and Discussion
Synthesis of C16-DABCO Dendrimers.
The synthesis of 1 is shown in Scheme 1. Mannose functionalized G(4)-PAMAM dendrimers were prepared using previously described methods.34 The primary hydroxyl group of mannose was tosylated, and SN2 displacement of the tosyl group using chloro 4-aza-1-hexadecyl-azoniabicylo[2.2.2]octane 30 (2) afforded product 1. The degree of mannose functionalization of the substrate dendrimer was determined using MALDI-TOF MS with both the mannose functionalized dendrimers and their precursor peracetylated mannose functionalized dendrimers.34 The average number of C16-DABCO endgroups on 1 was determined using the weight average molecular weight (MW) as determined by MALDI-TOF MS. The ratios of the C16-DABCO and tosyl groups to mannosides were determined using the ratios of methyl groups on both C16-DABCO and tosyl units in the 1H NMR spectra. Chemical transformations were also confirmed by 1H NMR. For dendrimer 1, the MW values indicate that an average of 32 terminal amines of the G(4)-PAMAM dendrimer were functionalized with mannose, and 8 of those 32 received a C16-DABCO. Nine mannosides per dendrimer retained their tosyl group on average. The C16-DABCO was not able to completely displace the tosyl group presumably due to the steric bulk of the dendrimer and the C16-DABCO nucleophile. Zeta potential measurements on the mannose-functionalized dendrimer indicate that the unfunctionalized amines on the mannose-functionalized dendrimer substrate are not surface accessible (Figure S1, Supporting Information). Mannose functionalized PAMAM dendrimers typically have a neutral zeta potential whereas, the addition of C16-DABCO to the dendrimers results in a positive zeta potential indicating these dendrimers are cationic. (Figure S1, Supporting Information)
Scheme 1.

Synthesis of C16-DABCO and mannose functionalized dendrimer 1.
Determination of the Minimum Inhibitory Concentrations.
The antimicrobial activities, in terms of MIC (minimum inhibitory concentration, or the lowest concentration that prevents bacteria from proliferating), of C16-DABCO dendrimer 1 and of the monomeric control compound 2 have been determined and are shown in Table 1. Values in Table 1 are reported on a per active unit (per C16-DABCO basis). In addition, the concentration per molecule/dendrimer is shown in parenthesis following the per C16-DABCO value. MICs for control compounds can be found in Table S1 of the Supporting Information. Both Gram positive (S. oralis, S. aureus, and B. cereus) and Gram negative (P. aeruginosa and E. coli) strains were studied. Of all the strains of bacteria that were tested, Streptococcus oralis had the highest MIC value by at least an order of magnitude compared to the MIC values obtained for the rest of the strains. The MIC for C16-DABCO dendrimer 1 with S. oralis is denoted as greater than 170 μM per active unit (20 μM per dendrimer) because this was the maximum amount of 1 that was readily dissolved, and the MIC value was above the solubility limit. The rest of the Gram positive strains had MIC values with 1 that were two orders of magnitude lower than the MIC value for S. oralis. B. cereus and S. aureus required the lowest concentration of 1 to achieve inhibition at 1.1 μM per active unit (0.13 μM per dendrimer). The MIC values for the two Gram negative strains (P. aeruginosa at 16 μM (2.0 μM per dendrimer) and E. coli at 1.1 μM (0.13 μM per dendrimer)) were one order of magnitude less than the MIC value for S. oralis. C16-DABCO dendrimer 1 appears to be more effective against S. aureus and B. cereus than against P. aeruginosa and E. coli bacteria. S. oralis had the highest MIC tested. The explanation for the high MIC could lie in the composition of the outer surface of the bacteria.
Table 1.
Minimum Inhibitory Concentrations (MICs) for Dendrimer 1 and Monomer 2.
| Gram | Microorganism | MIC C16-DABCO Dendrimer 1 per C16- DABCO (per dendrimer) |
MIC C16-DABCO Monomer 2 |
|---|---|---|---|
| + | Streptococcus oralis | >170 μM (20 μM) | 3000 μM |
| + | Staphylococcus aureus | 1.1 (0.13) | 11 |
| + | Bacillus cereus | 1.1 (0.13) | 17 |
| − | Pseudomonas aeruginosa | 16 (2.0) | 330 |
| − | Escherichia coli | 11 (1.1) | 150 |
When dendrimer 1 and monomer 2 are compared, it becomes clear that multivalency does improve the MIC. In all cases, the MIC value is at least 10-fold higher for the C16-DABCO monomer 2 than for C16-DABCO dendrimer 1 on a per C16-DABCO basis. Both the dendrimer and the monomer are below their critical micelle concentrations (CMC), indicating that they are not acting as aggregated species (see Figure S2 in the Supporting Information). One likely explanation for the multivalent enhancement in activity exhibited by the highly cationic dendrimer antibiotic relative to the monomer is that the polycationic dendrimer displaces native cations associated with surface associated adhesion molecules such as lipopolysaccharides (LPS) for Gram negative bacteria or lipoteichoic acid (LTA) for Gram positive bacteria. This interaction of 1 with LPS or LTA likely alters the standard LPS or LTA cross-linking interactions, destabilizing the outer membrane.35,36 Zeta potential measurements on 1 suggest that the mannose-functionalized dendrimer does not become polycationic until conjugation with C16-DABCO (Figure S1, Supporting Information). Since the polycationic form of an unmodified PAMAM dendrimer generally has more antimicrobial activity than neutral or anionic unmodified PAMAM dendrimers and the data indicates the primary amines are not available for membrane interaction, a positive control using the unmodified PAMAM dendrimer was not performed. However, other groups have performed experiments to determine the antimicrobial activity of an unmodified PAMAM dendrimer.37,38
Hemolysis and Mammalian Cell Toxicity Assays.
Hemolysis of red blood cells (RBCs) serves as a good indication of how compounds interact with cellular membranes, particularly mammalian membranes. RBC hemolysis assays were performed with 1, 2, mannose functionalized G(4)-PAMAM dendrimer, and unfunctionalized G(4)-PAMAM dendrimer (Table 2). Results with dendrimers and 2 were compared to control trials in phosphate buffered saline (PBS) alone (unlysed RBC control) and in PBS with 1% Triton-X100 (near total lysis control). G(4)-PAMAM up to a concentration of 34 μM showed negligible lysis, comparable to the PBS control. Significant lysis occurred only when the concentration of G(4)-PAMAM reached 68 μM. (Figure S5 of the Supporting Information shows a graph of the hemolysis data.)
Table 2.
Minimum hemolysis concentrations.
| Compound | Minimum concentration with observed hemolysis |
|---|---|
| Dendrimer 1 | 2.5 μM (0.6 μM per dendrimer) |
| Monomer 2 | 1.2 μM |
| Mannose-functionalized G(4)-PAMAM | Not hemolytic at concentrations studied |
| Unfunctionalized G(4)-PAMAM | 68 μM |
| PBS | Not detected |
| 1% Triton-X100 in PBS | Hemolytic control (near total hemolysis) |
Mannose functionalized G(4)-PAMAM dendrimer displayed significantly lower hemolytic activity when compared to unfunctionalized G(4)-PAMAM dendrimer at similar concentrations most likely due to the buried nature of the remaining primary amines after the addition of mannose (mannose functionalized dendrimers did not induce hemolysis at the concentrations tested). Hemolysis of RBCs was observed at and above 2.5 μM per C16-DABCO unit (0.6 μM per dendrimer) for C16-DABCO functionalized dendrimer 1. The most potently hemolytic compound tested was C16-DABCO monomer control compound 2, and significant hemolysis was observed at 1.2 μM. The concentration required for C16-DABCO dendrimers to lyse RBCs is within the same order of magnitude as the MIC for Gram positive and Gram negative organisms tested, indicating broad rather than selective activity for 1. Since hemolysis was not observed for mannose functionalized dendrimers prior to the addition of C16-DABCO, the activity of the dendrimer was attributed to the C16-DABCO endgroups.
Additional studies to determine toxicity of C16-DABCO dendrimers and C16-DABCO monomers were performed using MTS and A549 human lung carcinoma cells (ATCC# CCL-185). Observable toxicity of C16-DABCO dendrimer 1 toward A549 cancer cells was seen at 8.8 μM per active unit (1.1 μM per dendrimer). This value is similar to the MIC values obtained against the Gram negative bacteria tested and is almost an order of magnitude larger than the MIC values for 1 with E. coli and P. aeruginosa (Table 1). On per C16-DABCO basis, the C16-DABCO dendrimer 1 was more than an order of magnitude more potent than monomer 2 (8.8 μM and 190 μM for 1 and 2, respectively).
Taken together, the results from the hemolysis and the toxicity assays indicate that 1 is active against mammalian cells, presumably their membranes. In terms of toxicity, addition of C16-DABCO to the dendrimer scaffold generally increases the toxicity with the exception being red blood cells. The multivalent presentation of C16-DABCO appears to increase membrane activity. Changing alkyl chain length on the DABCO subunit or altering the ratios of unfunctionalized mannose and C16-DABCO residues could increase the selectivity for bacteria over human cells. Alternatively, compounds such as 2 may be more ideally suited for broader decontamination protocols in which activity against human cells and microorganisms is desired.
Biofilm Disruption and Formation Studies.
Experiments involving biofilm disruption and inhibition of formation were carried out to determine if the C16-DABCO dendrimers had activity against biofilms. To determine whether 1 disrupts biofilm stability after it has been established, the established biofilms were treated during day 1 and day 3 with C16-DABCO dendrimer 1, C16-DABCO monomer 2, or mannose functionalized G(4)-PAMAM dendrimer. No inhibition or knock-back of biofilms in several species (Gram positive and negative) was observed, as determined by log-kill platings (data shown in Figure S7 of the Supporting Information). The most likely explanation for the dendrimers’ lack of activity in biofilm disruption assays is that the dendrimers are too large to penetrate into the biofilm and are restricted to the biofilm surface. This result is as expected, as relatively few compounds are lethal to established biofilms.39,40,35,41
Experiments aimed at inhibiting biofilm growth at the membrane surface where biofilms are inoculated and attach exhibited much more dramatic results. Membranes pre-treated with a 1 mg/mL solution of C16-DABCO dendrimer 1 (274 μM per active unit, 33.3 μM per dendrimer), a 1 mg/mL solution of C16-DABCO, a 1 mg/mL solution of mannose functionalized G(4)-PAMAM dendrimer, a saline control (negative) or a 10% bleach control (positive) were inoculated with S. aureus and allowed to grow for three growth cycles, after which membranes were disrupted and diluted in DE neutralizing broth or PBS for log plating. Figure 2 shows a representative trial for triplicate experiments revealing the results of dilution log platings of day three S. aureus biofilms from membranes pre-treated with 10% bleach (left column), 274 μM per C16-DABCO (33.3 μM per dendrimer) 1 (middle column) and saline (right column). No visible biofilms were present on membranes pre-treated with either 10% bleach or C16-DABCO dendrimer 1 at day three, and no colonies grew from dilution log platings representing those membranes (Figure 2 left and middle columns). In contrast, robust biofilms were observed on membranes pre-treated with saline, and colony growth was evident from dilution log platings of those membranes, as expected (Figure 2 right column). Pre-treatment of membranes with C16-DABCO dendrimer 1 (1 mg/mL solution, i.e. 137 μM per C16-DABCO or 16.6 μM per dendrimer) or with mannose functionalized G(4)-PAMAM dendrimers lacking the C16-DABCO endgroups (also 1mg/mL) did not inhibit biofilm formation, nor did pre-treatment with a 1 mg/mL (i.e. 2962 μM) solution of 2.
Figure 2.

Dilution plating of S. aureus day three biofilms. Membranes were pre-treated with 10% bleach (column 1), 1mg/mL C16-DABCO dendrimer 1 (column 2) or saline (column 3) and allowed to dry before inoculation with S. aureus.
As shown in Figure 2, the pre-treatment of the biofilm inoculation surface with an appropriate dose of 1 causes complete inhibition of the growth of S. aureus biofilms, and these results may have important implications for development of new materials that inhibit biofilm growth for medical and other commercial applications.40,42
Multistep Resistance Selections Studies.
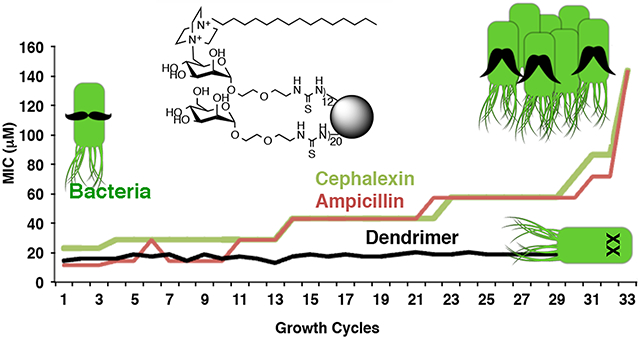
Because development of resistance is such a widespread problem for antimicrobial agents,3,8,11 studies were performed to determine the degree to which bacteria are able to develop resistance to dendrimer 1. Following the protocol put forward by the International Organization for Standardization,32 bacteria were grown in the highest tolerated sub-inhibitory concentration of 1 or 2, and MIC values were determined after each growth cycle. The results for the multistep resistance selection studies are shown in Figure 3, where MIC values are shown as a function of growth cycle. Figure 3a shows the effect that repeatedly growing the E. coli in the highest tolerated sub-inhibitory concentration of 2 has on the MIC values against the E. coli for 2, for cephalexin, and for ampicillin. Figure 3a reveals that the MIC value of the monomeric C16-DABCO control compound 2 increased significantly more over the seventeen growth cycles than the MIC values increase for cephalexin and ampicillin for E. coli grown in the presence of 2.
Figure 3.

All graphs represent bacteria grown in sub inhibitory concentration of antibiotics and show the MIC changes over time. (A) E. coli grown in sub inhibitory concentration of C16-DABCO monomer 2. (B) E. coli grown in sub inhibitory concentration of C16-DABCO dendrimer 1. (C) B. cereus grown in sub inhibitory concentrations of C16-DABCO dendrimers 1.
In Figures 3b and 3c, respectively, results are shown for E. coli and B. cereus that were repeatedly grown in the presence of the highest tolerated sub-inhibitory concentration of 1. As shown in Figure 3b, the MIC value for 1 only increased from 1.1 μM per C16-DABCO unit (0.13 μM per dendrimer) to 1.6 μM per C16-DABCO unit (0.20 μM per dendrimer) over a 33 day period, while the MIC values for cephalexin and ampicillin increased much more: 22 μM to 137 μM for cephalexin and 11 μM to 134 μM for ampicillin. As shown in Figure 3c, the MIC value for 1 increased from 0.11 μM per C16-DABCO unit (0.013 μM per dendrimer) to 0.60 μM per C16-DABCO (0.073 μM per dendrimer) over a 50 day period, while the MIC values for cephalexin and ampicillin increased much more: 41 μM to 164 μM for cephalexin and 27 μM to 162 μM for ampicillin.
The graphs in Figures 3b and 3c reveal a pattern in which the MIC values do not change for several growth cycles, and then an increase in MIC value is observed. These increases in the MIC values most likely occur because of alterations undergone by the bacteria in order to decrease their susceptibility to the multivalent dendrimer antibiotic. When compared to the ampicillin and cephalexin controls, a much smaller increase in MIC value was obtained for 1 with both E. coli and B. cereus. The MIC values for 1 remained significantly lower for the duration of the assay than the values for cephalexin and ampicillin.
The difference between the results with multivalent 1 when compared to monomeric 2 and to the cephalexin and ampicillin controls suggests that multivalency is a viable concept for the development of antibacterial compounds to which bacteria are less likely to develop resistance.
Conclusion
The synthesis and characterization of a mannose functionalized G(4)-PAMAM dendrimer bearing quaternary ammonium endgroups comprised of DABCO with a hexadecyl hydrocarbon chain are reported. This dendrimer (compound 1) presumably utilizes both the mannoside and the quaternary ammonium endgroups to interact detrimentally with the bacterial cell wall. The antibacterial properties of the dendrimer were assessed using both Gram positive and Gram negative strains of bacteria. Minimum inhibitory concentration values for 1 against S. oralis, S. aureus, B. cereus, P. aeruginosa, and E. coli were obtained and were compared to the MIC values of monomeric C16-DABCO control compound 2. On a per active unit basis, dendrimer 1 is in all cases at least 10 times more potent as an antibiotic than the monomeric control compound 2. Of the bacterial strains tested, dendrimer 1 is most active against Gram positive bacteria S. aureus and B. cereus, which both have MIC values in the low micromolar range. Toxicity studies with A549 lung cancer cells and hemolysis assays with red blood cells indicate compound 1 is active against mammalian cells as well. When presented multivalently, C16-DABCO is more toxic to bacteria and mammalian cells. Although biofilm disruption was not observed using 1, complete inhibition of the growth of S. aureus biofilm was achieved for membranes that were pre-treated with 1. Finally, using a series of resistance assays, bacteria were shown to be less likely to successfully develop resistance to multivalent antibacterial compound 1 than to the monovalent comparison compound 2 or to other small molecule antibiotics. MIC values for E. coli and B. cereus that were grown in the maximum tolerable sub-inhibitory concentrations of 1 were determined for up to 50 growth cycles. Overall, the results reported here for C16-DABCO functionalized dendrimer 1 indicate that multivalent displays of antimicrobial agents afford macromolecules with increased activity against bacteria both in solution and in biofilm relative to the monomeric endgroups (active group) alone and that multivalency can be used to thwart the development of bacterial resistance.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the National Institute of General Medical Science for support (GM62444). W. M. D. acknowledges the Montana Board of Research and Commercialization Technology for stipend support.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS website.
Experimental procedures for determining critical micelle concentration and zeta potential, and plots of electrophoretic mobility, CMC, 1H NMR, 13C NMR, and MALDI-TOF MS spectra of compound 1, hemolysis data, and MIC values. ChemDraw figure showing the structure of G(4)-PAMAM.
REFERENCES
- 1.National Institute of Health (NIH), Human Microbiome Project Defines Normal Bacterial Makeup of the Body. June 13, 2012. https://www.nih.gov/news-events/news-releases/nih-human-microbiome-project-defines-normal-bacterial-makeup-body (accessed June 21, 2016).
- 2.McDonnell G; Russell AD Antiseptics and Disinfectants: Activity, Action, and Resistance. Clin. Microbiol. Rev 1999, 12 (1), 147–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization (WHO), Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: France, 2014. http://www.who.int/drugresistance/documents/surveillancereport/en/ (accessed June 21, 2016) [Google Scholar]
- 4.Magiorakos AP; Srinivasan A; Carey RB; Carmeli Y; Falagas ME; Giske CG; Harbarth S; Hindler JF; Kahlmeter G; Olsson-Liljequist B; Paterson DL; Rice LB; Stelling J; Struelens MJ; Vatopoulos A; Weber JT; Monnet DL Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: an International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infection 2012, 18 (3), 268–281. [DOI] [PubMed] [Google Scholar]
- 5.Boucher HW; Talbot GH; Bradley JS; Edwards JE Jr.; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. J. Clin. Infect. Dis 2009, 48 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert DN; Guidos RJ; Boucher HW; Talbot GH; Spellberg B; Edwards JE Jr.; Scheld WM; Bradley JS; Bartlett JG The 10 x '20 Initiative: Pursuing a Global Commitment to Develop 10 New Antibacterial Drugs by 2020. Clin. Infect. Dis 2010, 50 (8), 1081–1083. [DOI] [PubMed] [Google Scholar]
- 7.Appelbaum PC 2012 and Beyond: Potential for the Start of a Second Pre-Antibiotic Era? J. Antimicrob. Chemother 2012, 67 (9), 2062–2068. [DOI] [PubMed] [Google Scholar]
- 8.MacDougall C; Polk RE Antimicrobial Stewardship Programs in Health Care Systems. Clin. Microbiol. Rev 2005, 18 (4), 638–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engler AC; Wiradharma N; Ong ZY; Coady DJ; Hedrick JL; Yang Y-Y Emerging Trends in Macromolecular Antimicrobials to Fight Multi-Drug-Resistant Infections. Nano Today 2012, 7 (3), 201–222. [Google Scholar]
- 10.Neu HC The Crisis in Antibiotic-Resistance. Science 1992, 257 (5073), 1064–1073. [DOI] [PubMed] [Google Scholar]
- 11.Davies J; Davies D Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev 2010, 74 (3), 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh C Molecular Mechanisms that Confer Antibacterial Drug Resistance. Nature 2000, 406 (6797), 775–781. [DOI] [PubMed] [Google Scholar]
- 13.Drawz SM; Bonomo RA Three Decades of beta-Lactamase Inhibitors. Clin. Microbiol. Rev 2010, 23 (1), 160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole K Efflux-Mediated Antimicrobial Resistance. J. Antimicrob. Chemother 2005, 56 (1), 20–51. [DOI] [PubMed] [Google Scholar]
- 15.Ganewatta MS; Tang C Controlling Macromolecular Structures Towards Effective Antimicrobial Polymers. Polymer 2015, 63, A1–A29. [Google Scholar]
- 16.Munoz-Bonilla A; Fernandez-Garcia M The Roadmap of Antimicrobial Polymeric Materials in Macromolecular Nanotechnology. Eur. Polym. J 2015, 65, 46–62. [Google Scholar]
- 17.Mammen M; Choi SK; Whitesides GM Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem. Int. Ed 1998, 37 (20), 2755–2794. [DOI] [PubMed] [Google Scholar]
- 18.Page MI; Jencks WP Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and Chelate Effect. Proc. Nat. Acad. Sci 1971, 68 (8), 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newkome G; Moorefield C; Vogtle F Dendrimers and Dendrons: Concepts, Syntheses, Applications. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2001; p 623. [Google Scholar]
- 20.Mintzer MA; Dane EL; O'Toole GA; Grinstaff MW Exploiting Dendrimer Multivalency To Combat Emerging and Re-Emerging Infectious Diseases. Mol. Pharm 2012, 9 (3), 342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kannan RM; Nance E; Kannan S; Tomalia DA Emerging Concepts in Dendrimer-Based Nanomedicine: from Design Principles to Clinical Applications. J. Int. Med 2014, 276 (6), 579–617. [DOI] [PubMed] [Google Scholar]
- 22.Navath RS; Menjoge AR; Wang B; Romero R; Kannan S; Kannan RM Amino Acid-Functionalized Dendrimers with Heterobifunctional Chemoselective Peripheral Groups for Drug Delivery Applications. Biomacromolecules 2010, 11 (6), 1544–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortega P; Copa-Patino JL; Munoz-Fernandez MA; Soliveri J; Gomez R; de la Mata FJ Amine and Ammonium Functionalization of Chloromethylsilane-Ended Dendrimers. Antimicrobial Activity Studies J. Org. Biomol. Chem 2008, 6 (18), 3264–3269. [DOI] [PubMed] [Google Scholar]
- 24.Meyers SR; Juhn FS; Griset AP; Luman NR; Grinstaff MW, Anionic Amphiphilic Dendrimers as Antibacterial Agents. J. Am. Chem. Soc 2008, 130 (44), 14444–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadler K; Tam J Peptide dendrimers: Applications and Synthesis. Mol. Biotechnol 2002, 90, 195–229. [DOI] [PubMed] [Google Scholar]
- 26.Pires J; Siriwardena T; Stach M; Tinguely R; Kasraian S; Luzarro F; Leib SL; Darbre T; Reymond JL; Endimiani A In Vitro activity of the Novel Antimicrobial Peptide Dendrimer G3KL Against Multidrug-Resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2015, 59:7915–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi SK; Myc A; Silpe JE; Sumit M; Wong PT; McCarthy K; Desai AM; Thomas TP; Kotlyar A; Holl MMB; Orr BG; Baker JR Jr. Dendrimer-Based Multivalent Vancomycin Nanoplatform for Targeting the Drug-Resistant Bacterial Surface. ACS Nano 2013, 7 (1), 214–228. [DOI] [PubMed] [Google Scholar]
- 28.Telwatte S; Moore K; Johnson A; Tyssen D; Sterjovski J; Aldunate M; Gorry PR; Ramsland PA; Lewis GR; Paull JRA; Sonza S; Tachedjian G Virucidal Activity of the Dendrimer Microbicide SPL7013 against HIV-1. Antiviral Res. 2011, 90 (3), 195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esfand R; Tomalia DA Poly(amidoamine) (PAMAM) Dendrimers: From Biomimicry to Drug Delivery and Biomedical Applications. Drug Discov Today 2001, 6 (8), 427–436. [DOI] [PubMed] [Google Scholar]
- 30.Abel T; Cohen JI; Engel R; Filshtinskaya M; Melkonian A; Melkonian K Preparation and Investigation of Antibacterial Carbohydrate-Based Surfaces. Carbohydr. Res 2002, 337 (24), 2495–2499. [DOI] [PubMed] [Google Scholar]
- 31.Heidecke CD; Lindhorst TK, Iterative Synthesis of Spacered Glycodendrons as Oligomannoside Mimetics and Evaluation of their Antiadhesive Properties. Chem. Eur. J 2007, 13 (32), 9056–9067. [DOI] [PubMed] [Google Scholar]
- 32.ISO/FDIS; ISO 20776-1:2006; Clinical Laboratory Testing and in Vitro Diagnostic Test Systems -- Susceptibility Testing of Infectious Agents and Evaluation of Performance of Antimicrobial Susceptibility Test Devices -- Part 1: Reference Method for Testing the in Vitro Activity of Antimicrobial Agents Against Rapidly Growing Aerobic Bacteria Involved in Infectious Diseases. International Organization of Standardization, Geneva, Switzerland. http://www.iso.org/iso/catalogue_detail.htm?csnumber=41630 (accessed August 29, 2016) [Google Scholar]
- 33.Kosowska-Shick K; Clark C; Pankuch GA; McGhee P; Dewasse B; Beachel L; Appelbaum PC Activity of Telavancin Against Staphylococci and Enterococci Determined by MIC and Resistance Selection Studies. Antimicrob. Agents Chemother 2009, 53 (10), 4217–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woller EK; Walter ED; Morgan JR; Singel DJ; Cloninger MJ Altering the Strength of Lectin Binding Interactions and Controlling the Amount of Lectin Clustering Using Mannose/Hydroxyl-Functionalized Dendrimers. J. Am. Chem. Soc 2003, 125 (29), 8820–8826. [DOI] [PubMed] [Google Scholar]
- 35.Hancock REW Alterations in Outer-Membrane Permeability. Annu. Rev. Microbiol 1984, 38, 237–264. [DOI] [PubMed] [Google Scholar]
- 36.Percy MG; Grundling A; Lipoteichoic Acid Synthesis and Function in Gram-Positive Bacteria. Annu. Rev. Microbiol 2014, 68, 81–100. [DOI] [PubMed] [Google Scholar]
- 37.Wang B; Navath RS; Manjoge AR; Balakrishnan B; Bellair R; Dai H; Romero R; Kannan S; Kannan R Inhibition of Bacterial Growth and Intramniotic Infection in a Guinea Pig Model of Chorioamnionitis Using PAMAM Dendrimers. Int. J. Pharm 2010, 395, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zarena AS; Gopal S Dendrimers a New Dimension in Targeting Biofilms. Mini. Rev. Med. Chem 2013, 13, 1448–1461. [DOI] [PubMed] [Google Scholar]
- 39.Mah TFC; O'Toole GA Mechanisms of Biofilm Resistance to Antimicrobial Agents. Trends Microbiol. 2001, 9 (1), 34–39. [DOI] [PubMed] [Google Scholar]
- 40.Stewart PS; Franklin MJ Physiological Heterogeneity in Biofilms. Nature Rev. Microbiol 2008, 6 (3), 199–210. [DOI] [PubMed] [Google Scholar]
- 41.Stewart PS; Costerton JW Antibiotic Resistance of Bacteria in Biofilms. Lancet 2001, 358 (9276), 135–138. [DOI] [PubMed] [Google Scholar]
- 42.Mah TF; Pitts B; Pellock B; Walker GC; Stewart PS; O'Toole GA A Genetic Basis for Pseudomonas Aeruginosa Biofilm Antibiotic Resistance. Nature 2003, 426 (6964), 306–310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.