

Abstract
As a consequence of the global rise in the prevalence of adolescent obesity, an unprecedented phenomenon of type 2 diabetes has emerged in pediatrics. At the heart of the development of type 2 diabetes lies a key metabolic derangement: insulin resistance (IR). Despite the wide-spread occurrence of IR affecting an unmeasurable number of youths worldwide, its pathogenesis remains elusive. IR in obese youth is a complex phenomenon that defies explanation by a single pathway. In this review we first describe recent data on the prevalence, severity, and racial/ethnic differences in pediatric obesity. We follow by elucidating the initiating events associated with the onset of IR, and describe a distinct “endophenotype” in obese adolescents characterized by a thin superficial layer of abdominal subcutaneous adipose tissue, increased visceral adipose tissue, marked IR, dyslipidemia, and fatty liver. Further, we provide evidence for the cellular and molecular mechanisms associated with this peculiar endophenotype and its relations to IR in the obese adolescent.
Keywords: Childhood and Adolescent Obesity, Resistance, Adipose Tissue Distribution, Fatty Liver, Adipose Tissue Inflammation, Inflammasome
Childhood obesity with its associated metabolic complications is emerging as a major global health challenge of the 21st century. Despite efforts by government and public health officials, researchers, health care providers, and the media to bring attention to this growing health problem, the number of overweight and obese youth continues to increase unrelentingly. There have been dramatic changes in the nutrition and physical activity habits of children in the US, along with changes in demographics and societal norms, concurrent with the increase in childhood obesity prevalence. Many researchers have placed the origin of the childhood obesity epidemic at the beginning of the 1980s. In fact, between 1976 and 1980 is when the prevalence of obesity started its escalation.1 Recent data from the 2013–2014 National Health and Nutrition Examination Survey, using measured heights and weights, indicate that the prevalence of obesity is 17.0% and extreme obesity 5.8%.2 Obesity rates have decreased in children aged 2 to 5 years since 2003–2004, stabilized in 6- to 11-year-olds since 2007–2008, but steadily increased among adolescents since 1988.3
There are significant racial and ethnic disparities in obesity prevalence among US children and adolescents. The prevalence of childhood obesity among African Americans, Mexican Americans, and Native Americans exceeds that of other ethnic groups. The Centers for Disease Control and Prevention reported that in 2000 the prevalence of obesity was 19% of non-Hispanic black children and 20% of Mexican American children, compared with 11% of non-Hispanic white children. The increase since 1980 is particularly evident among non-Hispanic black and Mexican American adolescents.3 Although the overall prevalence of childhood obesity continued to increase during the first half of this decade (17% in 2004 vs 14% in 2000), the differences by race and ethnicity appear to be diminishing, in part because of rapid increases in obesity in white children: in 2004 the prevalence of childhood obesity was 20% in non-Hispanic blacks, 19% in Mexican Americans, and 16% in non-Hispanic whites, and prevalence was highest in Mexican American boys (22%) and African American girls (24%). Disparities were found in children of other race and ethnicities.2,3
Pubertal maturation is known to impact on obesity development. Girls who mature early have higher body mass index (BMI) and sum skinfolds during their teenage years than girls who mature later,4 and this interaction is strongest in black girls.5 Because black girls undergo pubertal maturation earlier on average than white girls, differences in pubertal maturation stage can account for some racial differences in adolescent obesity.
Severity of Obesity: A Rightward Shift in BMI
Consensus committees have recommended that children and adolescents be considered overweight or obese if the BMI exceeds the 85th or 95th percentiles, on curves generated from the 1963–1965 and 1966–1970 National Health and Nutrition Examination Survey, or exceeds 30 kg/m2 at any age.6–8 The BMI is the accepted screening measure of overweight and obesity for children 2 years of age and older.6 Adults with a BMI between 25 and 30 kg/m2 are considered overweight; those with a BMI ≥30 kg/m2 are considered to be obese. Obesity in adults is subcategorized as class I (BMI ≥30 to 35), class II (BMI ≥35 to 40), and class III (BMI ≥40). Because children grow in height as well as weight, the norms for BMI in children vary with age and sex. More recently, Skinner et al1 used a definition of severe obesity recommended by the American Heart Association as a BMI >120% of the 95th percentile for age and sex or a BMI of ≥35, which represents what is considered class 2 obesity in adults, and class 3 obesity, which is defined as a BMI >140% of the 95th percentile for age and sex or a BMI ≥40, whichever is lower. Notably, this study suggests that determination of the severity of obesity can help identify children and young adults who are at the greatest risk for the negative health effects associated with obesity. Current guidelines for screening do not differentiate among levels of obesity. However, several studies indicate greater risks of insulin resistance (IR), low high-density lipoprotein (HDL) cholesterol level, systolic blood pressure, and glucose metabolism, thus supporting the stratification of risk on the basis of the obesity severity in children and adolescents.1,9,10
The prevalence of severe obesity in female adolescents was approximately 10% in non-Hispanic whites, 20% in non-Hispanic blacks, and 16% in Mexican Americans.1 The rightward shift in the BMI as classified into class 2 and class 3 obesity is particularly notable in adolescents and non-Hispanic black individuals.1 Additionally, overweight and obesity at all levels of severity are increasing significantly among Hispanic girls and non-Hispanic black boys. Future research should determine whether there are specific factors that can be addressed in these high-risk groups. These findings suggest that the causes for the increase in obesity should be sought in part at the population level rather than focusing on individuals. The reasons for the differences in the prevalence of childhood obesity among different ethnic groups are complex, likely involving genetics, physiology, culture, socioeconomic status, environment, and interactions among these variables, as well as others not fully recognized. Although the questions of how might socioeconomic, biological, and cultural factors influence racial or ethnic differences in childhood obesity are of great importance, they are not the focus of this review. Nevertheless, we would recommend reading the following consensus statement written by a 7-member panel of experts in pediatric endocrinology, cardiology, gastroenterology, nutrition, epidemiology, and anthropology, organized by Shaping America’s Health and the Obesity Society to address the evidence base and gaps in knowledge in this area.11
Impact of the Degree of Overall Obesity on Cardiometabolic Risks in Youth
As the prevalence of childhood obesity increases, its health implications are becoming more evident.9–13 Obesity is an important early risk factor for much of adult morbidity and mortality.9,14–16 Childhood obesity frequently persists into adulthood, with up to 80% of obese children reported to become obese adults.14 Many of the metabolic and cardiovascular complications of obesity are already present during childhood and are closely related to the presence of IR and hyperinsulinemia, the most common abnormality of obesity.10,13
To begin assessing the impact of varying degrees of obesity on the prevalence of the cardiometabolic risk factors in children and adolescents, we completed a cross-sectional analysis of the initial metabolic syndrome assessments in our cohort of obese youth.10 As in adults, subjects were classified as having the metabolic syndrome if they met 3 or more of the following criteria for age and gender: (1) BMI > the 97th percentile (BMI z-score above); (2) triglycerides above the 95th percentile; (3) HDL-cholesterol under the fifth percentile; (4) systolic and/or diastolic blood pressure above the 95th percentile; and (5) impaired glucose tolerance (IGT). Fasting glucose levels changed minimally with increasing weight in this cohort. In contrast, the prevalence of IGT greatly increased in the children and adolescents with moderate and severe obesity.10 A similar pattern was also observed for plasma insulin, triglycerides levels, and systolic blood pressure, whereas a significant decrease in HDL-cholesterol was seen with varying degrees of obesity. The degree of obesity and prevalence of the metabolic syndrome were strongly associated, after adjustment for race (P = .009) and race and gender (P = .027). Overall, prevalence of the metabolic syndrome was 38.7% in moderately obese and 49.7% in severely obese subjects. The main findings of the study were that the prevalence of metabolic syndrome appears far more common than previously reported and increases directly with the degree of obesity. Moreover, each element of the syndrome worsens across a spectrum of degrees of obesity, independent of age, gender, and pubertal status. Our results demonstrate a significant adverse effect of worsening obesity on each component of metabolic syndrome, further emphasizing the deleterious impact of increasing weight in this age group. Landmark studies from the Bogalusa Heart Study demonstrated that cardiovascular risk factors present in childhood are predictive of coronary artery disease in adulthood.14,15 Among these risk factors, low-density lipoprotein-cholesterol and BMI measured in childhood were found to predict carotid intima-media thickness in young adults.13–15 There is now substantial evidence that obesity in childhood creates the metabolic platform for adult cardiovascular disease.16 Hyperinsulinemia is commonly associated with obesity; however, it has been difficult to clearly determine which defects comes first. Notably, fasting hyperinsulinemia in prepubertal children in the Pima Indian population has been found to be a risk factor for weight gain.17
Impact of Tissue Lipid Partitioning on Insulin Sensitivity in the Obese Adolescent
Accumulating evidence indicates that the determinant of IR is not the degree of obesity per se but the distribution of fat and, importantly, its accumulation in key insulin-sensitive organs or tissues.18 Interestingly, this phenomenon is not only confined to obese adults, but is also described in obese adolescents with IGT who were found to be more insulin resistant than those with normal glucose tolerance despite similar degree of adiposity.19 Thus, obese adolescents with IGT are characterized by increased intramyocellular lipid content and by increased visceral and decreased subcutaneous fat deposition. Indeed, intramyocellular lipid content and visceral lipid were positively related to the 2-h plasma glucose and inversely related to the glucose disposal and nonoxidative glucose metabolism.19
There is ample evidence indicating that visceral fat accumulation is associated with an impaired insulin action in the obese population.20 Although controversy remains regarding the contribution of visceral and subcutaneous fat to the development of IR,21,22 a previous study by Cruz et al23 showed a direct impact of visceral fat accumulation on insulin sensitivity and secretion, independent of total body adiposity, in obese children with a family history of type 2 diabetes (T2D). Our group subsequently described a new endophenotype that is more relevant to the development of IR in obese adolescents.24 By stratifying a multiethnic cohort of obese adolescents into tertiles based on the proportion of visceral fat in the abdomen (visceral/subcutaneous fat ratio), we observed a significant increase in 2-h glucose and IR (homeostasis model assessment) and decrease in insulin sensitivity (Matsuda index) in obese adolescents with a high proportion of visceral fat and relatively low abdominal subcutaneous fat.24–26 These findings suggest that adolescents at risk for developing alterations in glucose metabolism are not necessarily the most severely obese, but are characterized by an unfavorable lipid partitioning profile.
Despite the demonstrated relationship between intramyocellular lipid content, visceral fat, and metabolic dysfunction, the ectopic fat deposition in the liver is emerging as the most important marker of IR and glucose dysregulation in adults,27,28 as well as in obese pediatric population.26
Role of Fatty Liver in the Obese Youth
Although it remains unclear whether hepatic steatosis is a consequence or a cause of derangements in insulin sensitivity, the presence of steatosis is an important marker of multiorgan IR26–28; moreover, IR is directly related to percent of liver fat. Previously, we reported that rising alanine transaminase levels in obese children and adolescents were associated with deterioration in insulin sensitivity and glucose tolerance.29 Furthermore, abnormal alanine transaminase levels were found in children with T2D.30
To understand the potential role of fatty liver in the onset of T2D in obese youth, we recently assessed whether the severity of hepatic steatosis affects the presence of glucose metabolism dysregulation in a multiethnic cohort of obese adolescents.25 Independent of obesity, the severity of fatty liver was associated with the presence of prediabetes conditions (IGT and impaired fasting glucose/IGT) and predicted prediabetes in obese adolescents. In addition, paralleling the severity of hepatic steatosis, there was a significant decrease in insulin sensitivity and impairment of β-cell function. These findings suggest that intrahepatic fat accumulation is a strong risk factor for T2D, and its early identification is critical to prevent the development of metabolic complications in youth.31
The “Adipose Tissue Expandability” Hypothesis: A Potential Mechanism for IR in Obese Youth
Obesity poses a significant risk factor for the development of cardiovascular disease,32 diabetes,33 and certain cancers,34 thereby shortening life expectancy.35 Nevertheless, many obese individuals do not develop any of these morbidities. One hypothesis explaining this dilemma is that total body fat is not the culprit of adverse health in obesity; rather the relative proportion of lipids in various fat depots is what determines the metabolic risk. Danforth first hypothesized that inadequate subcutaneous adipose tissue (SAT) results in lipid overflow into visceral adipose tissue (VAT), and non-adipose tissues.36 This hypothesis was then elegantly explored by Ravussin and Smith37 in adults. Subsequent studies using the power of mouse models clearly established that adipose tissue (AT) has a defined limit of expansion. Indeed, the “adipose expandability hypothesis”38,39 argues that once AT storage capacity is exceeded then net lipid flux to non-adipose organs will increase, causing lipotoxicity leading to IR and apoptosis.38
Studies from our group performed in obese adolescents indicated that, independent of overall body fat mass, IR is related to a particular abdominal fat distribution and ectopic fat accumulation.40–42 To gain insights in the mechanisms that might be responsible for the inefficient storage of fat in the abdominal SAT, we biopsied the SAT and combined measurements of fat cell size together with transcription of genes regulating lipogenesis/adipogenesis in 2 groups of obese adolescents with similar degrees of obesity but with profound differences in abdominal fat distribution (low VAT/SAT ratio vs high VAT/SAT ratio), As shown in Figure 1, the distribution of abdominal fat is clearly different between the 2 groups: a marked visceral depot and a thin layer of superficial SAT clearly typifies the group with high VAT/SAT ratio. Notably, 3 major differences emerged from the abdominal SAT biopsies: (1) coexistence of large and small adipocytes in the high VAT/SAT group as opposed to a more homogenous cellularity in the low VAT/SAT group; (2) down-regulation of key lipogenic/adipogenic genes; (3) macrophage infiltration and decreased SIRT1 expression.
Figure 1.
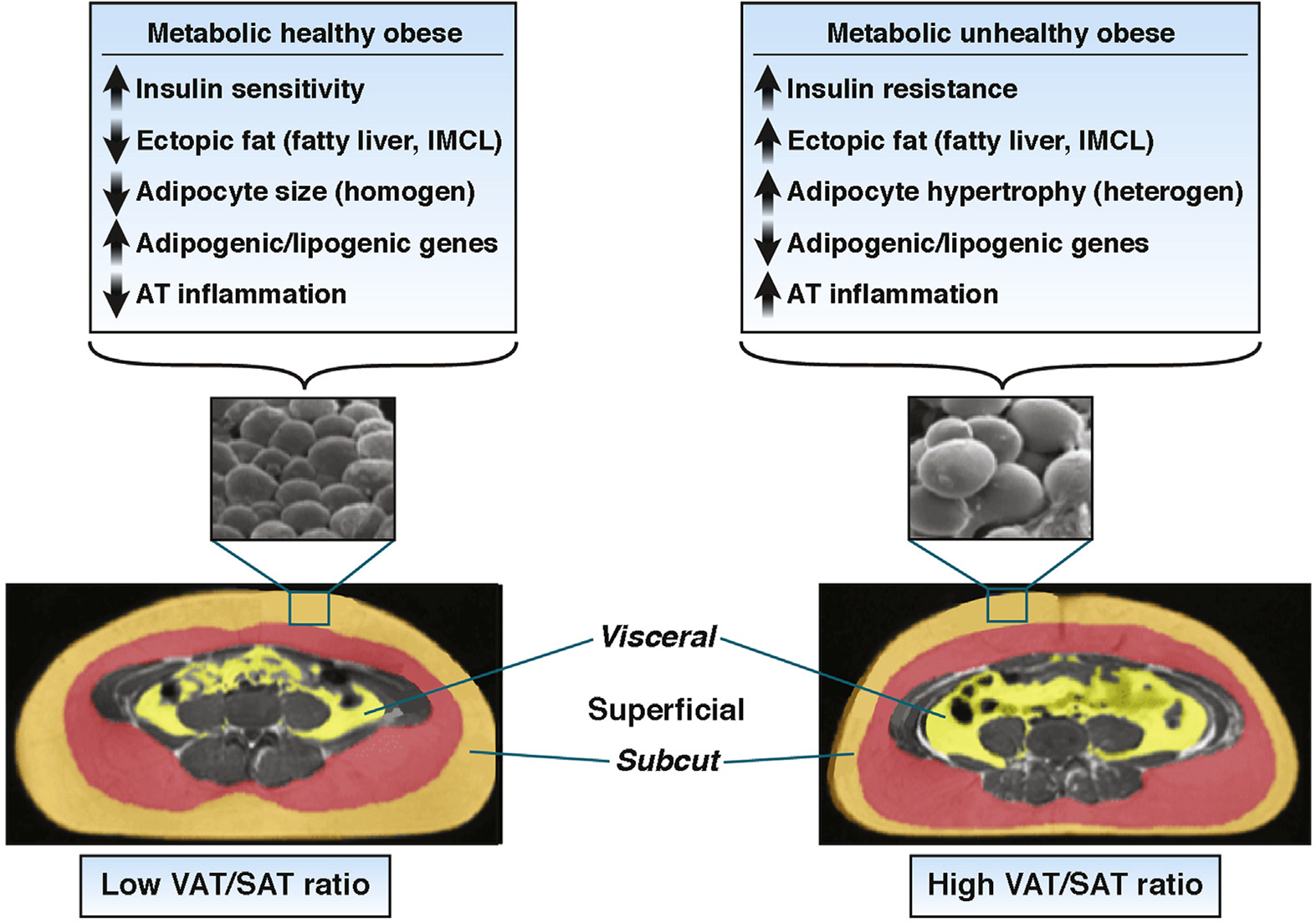
Cellular and molecular differences in the SAT in the obese adolescent with a low VAT/SAT ratio and a high VAT/SAT ratio.
While these studies are instrumental in describing the putative differences characterizing the inadequate storage of fat in the abdominal SAT of obese adolescents, further studies are needed to unravel the underlying mechanisms responsible for the profound remodeling of this depot during this very critical period of development.
A Role of the Inflammasome in the Low Storage Capacity of the Abdominal SAT in Obese Adolescents
Recognition of IR catastrophic health sequelae has engendered intense interest in its pathophysiology. Despite the daunting complexity of the field, chronic low-level inflammation in particular, innate immunity, has emerged as a primary determinant of obesity-related pathology including the full spectrum of diabetic disease.43–47 However, these studies have been performed in AT obtained mainly from rodents and adult individuals. Little is known about AT tissue inflammation and particularly whether it is affected by the type of fat depot in obese youth, possibly because of the difficulties in obtaining AT samples. To date, there has been only one study examining immune cells in the SAT of children48 and one study examining collagen deposition.49 Adolescence is a critical period for the development of adiposity and IR, perhaps because of the accelerated growth of this tissue.50 Indeed, the plasticity of the AT during this developmental stage is at its highest potential with maximum hypertrophy and hyperplasia occurring. This is in contrast to the fact that the number of adipocytes stays constant in adulthood in lean and obese individuals, even after marked weight loss.51 Indeed, many adipocyte biologists believe that the total number of fat cells present in most individuals is set during adolescence and that changes in fat mass generally reflect increased lipid storage in a fixed number of adipocytes.52,53
The innate immune cell sensor leucine-rich–containing family, pyrin domain containing 3 (NLRP3) inflammasome controls the activation of caspase-1, and the release of proinflammatory cytokines interleukin (IL) 1β and IL18.54–59 The NLRP3 inflammasome is implicated in adipose tissue inflammation and the pathogenesis of IR.54,55 We recently tested the hypothesis that adipose tissue inflammation and NLRP3 inflammasome are linked to the down-regulation of SAT adipogenesis/lipogenesis in obese adolescents with altered abdominal fat partitioning.60 We performed abdominal SAT biopsies on 58 obese adolescents and grouped them by magnetic resonance imaging-derived visceral fat to VAT plus SAT (VAT/VAT+SAT) ratio (cutoff, 0.11). Adolescents with a high VAT/VAT+SAT ratio showed higher SAT macrophage infiltration and higher expression of the NLRP3 inflammasome-related genes (ie, TLR4, NLRP3, IL1B, and CASP1) (Figure 2). The increase in inflammation markers was paralleled by a decrease in genes related to insulin sensitivity (ADIPOQ, GLUT4, PPARG2, and SIRT1) and lipogenesis (SREBP1c, ACC, LPL, and FASN) (Figure 2). Furthermore, SAT ceramide concentrations correlated with the expression of CASP1 and IL1B. Infiltration of macrophages and up-regulation of the NLRP3 inflammasome, together with the associated high ceramide content in the plasma and SAT of obese adolescents with a high VAT/VAT+SAT, may contribute to the limited expansion of the subcutaneous abdominal adipose depot and the development of IR.60
Figure 2.
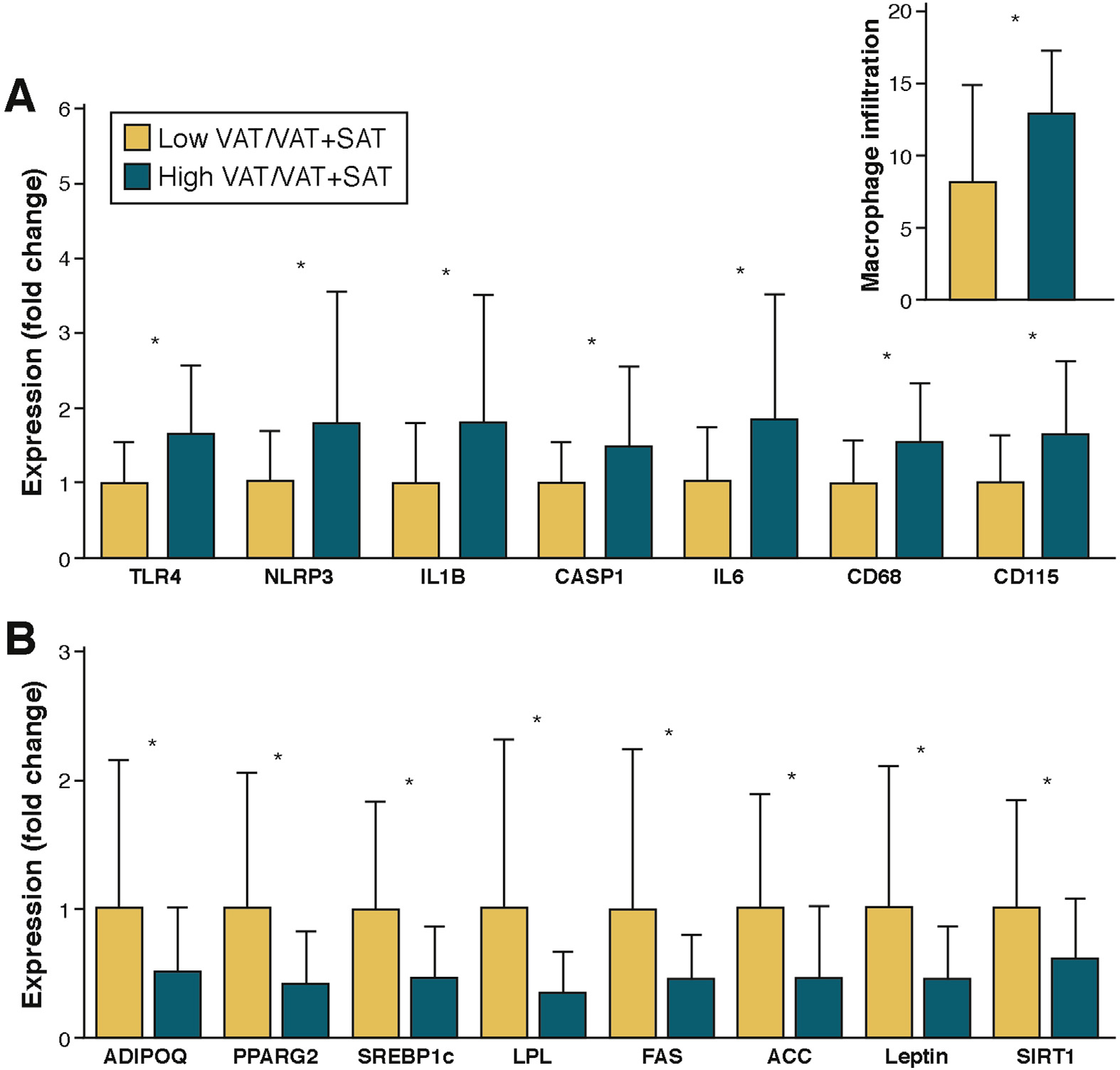
(A) Increased inflammatory gene expression and macrophage infiltration in abdominal SAT of the high VAT/VAT+SAT group. (B) Decreased lipogenic gene expression in abdominal SAT of the high VAT/VAT+SAT group. Subcutaneous expression of specific genes was normalized to the expression of 18S ribosomal RNA and based on the expression of a human control adipose tissue (2ΔΔCt). Expression values of the low VAT/VAT+SAT group (white bars) were set to 1, and values for the high VAT/VAT+SAT group (black bars) are expressed as fold changes compared with 1 (mean ± SD, n = 58). Macrophage infiltration (inset in A) was assessed by immunohistochemistry. *Indicates that the t test between the two groups was significant at the <.05 level.
The SAT has been proposed to act as a “sink,” being able to accommodate excess energy as triglycerides in the adipocyte and thus prevent the flow of lipid to other places.38,39 Of note, it represents the largest home for storage of energy in children and adolescents (90% or more of total fat mass), whereas the visceral fat is quite small, representing about 5% of total fat mass.61,62 At puberty, both the total fat mass and its subcutaneous distribution changes, particularly in girls. Driven by the rise in sex steroids, differences among the superficial fat depots emerge at puberty. For example, the gluteofemoral SAT region increases in girls, whereas the VAT depot increases more in boys with very little enlargement of the gluteofemoral depot.61,62 Given the growth of the AT and its remodeling in this development stage, adolescence is considered one of the critical periods for obesity and its associated IR. Our group has been studying the puberty-induced changes in abdominal fat patterning in relation to the development of IR in adolescents for the past 15 years. Most of our studies have focused mainly on measuring the distribution of intra-abdominal and subcutaneous fat and we have seldom measured the gluteofemoral region. Currently undergoing studies in our group are measuring and determining if the storage capacity of gluteofemoral AT plays a role in determining the level of central adiposity in obese adolescents during puberty. We have shown that in some obese adolescents this protective mechanism of the abdominal SAT may not be functioning, which may be responsible for the ectopic fat storage and IR.40,41 The limited storage capacity was shown by the down-regulation of a series of lipogenic/adipogenic genes.40 Although these studies indicated some defects in the transcription of genes regulating lipogenesis and adipogenesis, direct proof of their functional defects is still lacking. Assessing the in vivo dynamic flux of lipid synthesis and adipocyte proliferation, coupled with robust measures of insulin sensitivity and imaging, are needed for a thorough understanding of this very complex endophenotype in obese adolescents.
Inflammation of the SAT: A Potential Mediator of Hepatic IR in the Obese Adolescent
Hepatic steatosis and IR are among the most prevalent metabolic disorders and are tightly associated with obesity and T2D, even early in adolescence.25,26,31 However, the underlying mechanisms linking obesity to hepatic lipid accumulation and IR are incompletely understood. Using a dog model, Rebrin et al first demonstrated the concept that insulin, by regulating adipocyte lipolysis, controls liver glucose production.63 Thus, the adipocyte emerged as a potential mediator between insulin and liver glucose output. Recent studies by Perry et al64 tested the hypothesis that insulin’s ability to suppress lipolysis in white adipose tissue (WAT) may be critical for the suppression of hepatic glucose production (HGP) by reducing fatty acid flux to the liver, resulting in decreased hepatic acetyl coenzyme A (CoA) concentrations and decreased pyruvate carboxylase activity resulting in decreased conversion of pyruvate to glucose. To examine this hypothesis, Perry et al64 developed a comprehensive metabolomics flux approach to simultaneously measure whole-body rates of lipolysis, glucose turnover, and intrahepatic fluxes, as well as hepatic acetyl CoA content, in rapidly freeze-clamped livers. Using this approach, they demonstrated that hepatic acetyl CoA content is a key regulator of HGP and that an insulin-mediated reduction of hepatic acetyl CoA concentrations, through inhibition of WAT lipolysis, is the major mechanism by which insulin suppresses HGP in vivo. These are the first studies that demonstrated insulin regulation of pyruvate carboxylase activity through modulation of hepatic acetyl CoA concentrations, further demonstrating that this mechanism is responsible for insulin suppression of HGP in vivo. Additionally, in that same study, they next examined whether increased hepatic acetyl CoA concentrations might be responsible for increased rates of HGP in IR and might link inflammation and macrophage-induced lipolysis in WAT with fasting and postprandial hyperglycemia through a similar mechanism. Consistent with this hypothesis, they showed that high-fat–fed rats manifested a 2-fold increase in plasma and WAT IL6 concentrations, which caused hyperglycemia by increasing lipolysis, hepatic acetyl CoA, and HGP both before and during a hyperinsulinemic-euglycemic clamp.
To determine whether the findings described by Perry et al in rats would translate to humans,64 our group measured rates of HGP and lipolysis in obese insulin-resistant adolescents during the hyperinsulinemic-euglycemic clamp. As shown in Figure 3, compared with weight-matched obese insulin-sensitive control adolescents, insulin-resistant obese adolescents displayed fasting hyperglycemia and hyperinsulinemia that were associated with increased rates of HGP, impaired insulin-mediated suppression of lipolysis, and impaired insulin-mediated suppression of HGP. Impaired insulin suppression of lipolysis was associated with an increased diameter of large adipocytes and increased macrophage infiltration into the subcutaneous white adipose tissue (Figure 3A–I). Consistent with the findings in insulin-resistant high-fat–fed rodents, insulin-resistant obese adolescents displayed increased plasma IL6 concentrations, and an approximately 50% increase in IL6 concentrations in WAT, which were more than 20 times higher than plasma IL6 concentrations (Figures 3J and K). Similar to the HFD rats, the increased rates of lipolysis and HGP in obese insulin-resistant adolescents were associated with increased CGI-58 protein expression in WAT (Figure 3L). Altogether, these results translate to humans by demonstrating that WAT inflammation is associated with increased IL6 concentrations in WAT, increased rates of lipolysis, increased rates of HGP, and hepatic IR assessed by a hyperinsulinemic-euglycemic clamp in obese insulin-resistant adolescents (Figure 4). These results are consistent with previous studies that have observed that increased plasma IL6 concentrations are strong predictors of IR in obese adolescents. These results have important clinical implications for the pathogenesis of T2D and identify macrophage-induced WAT lipolysis and hepatic acetyl CoA as novel potential therapeutic targets for T2D.
Figure 3.
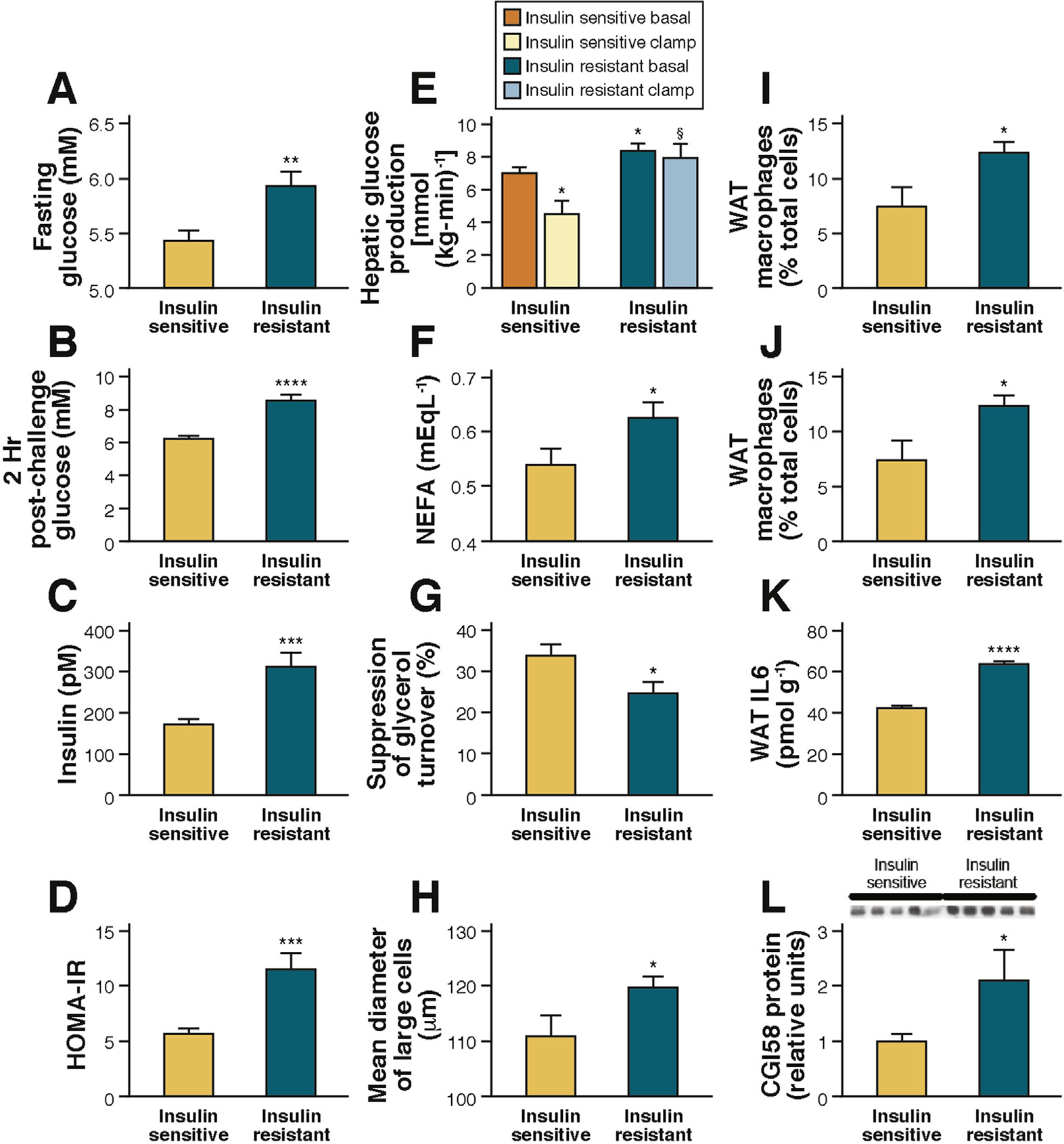
(A–L) Insulin-resistant obese adolescents have increased lipolysis and impaired suppression of HGP associated with increased WAT IL6 concentrations.
Figure 4.
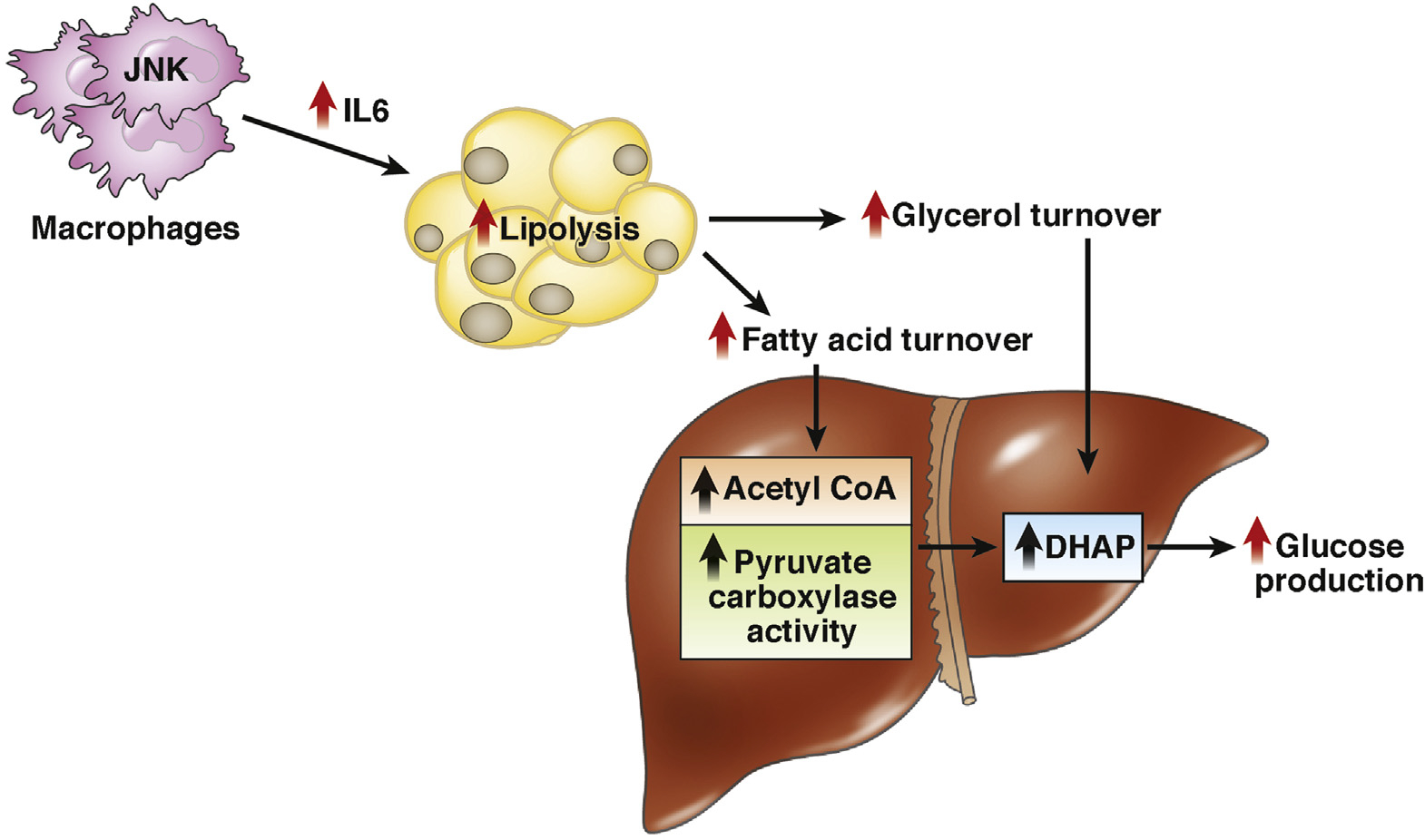
Insulin-resistant obese adolescents. A model to explain the role of SAT Inflammation, activating lipolysis in AT, thereby increasing acetyl-CoA and pyruvate carboxylase activity in the liver, which ultimately enhances HGP in the obese insulin-resistant adolescent.
The Future of Pediatric Obesity Research
The key to successfully preventing and treating childhood and adolescent obesity ultimately lies in developing and funding a targeted research agenda. Research should focus on the mechanisms that regulate body fat distribution during adolescence, as well as gender and ethnicity differences in body composition and fat distribution. Other key research areas include the differing susceptibility to weight gain during childhood and adolescence; the underlying changes in physical activity at puberty; more clinical studies on the efficacy of specific prevention and treatment programs; and the effort to move from efficacy to broad effectiveness.
Until recently, childhood obesity has been considered a clinical problem for specialist pediatricians. Now, however, the problem must be approached in a more global manner. The public health community must consider the urgent need to institute preventive programs. Given the reluctance of policymakers to institute changes, particularly those that are unpopular or expensive, it is important to establish objective evidence of the beneficial impact of any preventive or treatment programs. To stop the epidemic of childhood obesity, acting on all levels—medical, social, political, and educational—is fundamental. A broad range of action would include conducting nutrition education campaigns, regulating the marketing of junk food to children, eliminating energy-dense foods and sodas from schools, and promoting physical activity.
Acknowledgments
The authors thank all of the volunteers, as well as Karin Allen and Bridget Pierpont from Yale University for their skillful help in this study.
Funding
This study was supported by National Institutes of Health (NIH) National Institute of Child Health and Human Development grants R01-HD-40787, R01-HD-28016, and K24-HD-01464 to S.C.; Clinical and Translational Science Award grant UL1-RR-0249139 from the National Center for Research Resources, a component of the NIH; grant R01-EB006494 (Bioimage Suite); and Distinguished Clinical Scientist Award from the American Diabetes Association (S.C.), as well as grants DK-49230 and DK-085638 (G.I.S.), grants DK-090556 and AG045712 (V.D.D.), the Diabetes Research Center grant P30-DK-045735.
Abbreviations used in this paper:
- AT
adipose
- BMI
body mass index
- CoA
coenzyme A
- HDL
high-density lipoprotein
- HGP
hepatic glucose production
- IGT
impaired glucose tolerance
- IL
interleukin
- IR
insulin resistance
- SAT
subcutaneous adipose tissue
- T2D
type 2 diabetes
- VAT
visceral adipose tissue
- WAT
white adipose tissue
Footnotes
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Skinner A, Skelton JA. Prevalence and trends in obesity and severe obesity among children in the United States, 1999–2012. JAMA Pediatr 2014;168:561–566. [DOI] [PubMed] [Google Scholar]
- 2.Ogden CL, Carroll MD, Lawman HG, et al. Trends in obesity prevalence among children and adolescents in the United States, 1988–1994 through 2013–2014. JAMA 2016;315:2292–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogden CL, Flegal KM, Carroll MD, et al. Prevalence and trends in overweight among U.S. children and adolescents, 1999–2000. JAMA 2002;288:1728–1732. [DOI] [PubMed] [Google Scholar]
- 4.Biro FM, McMahon RP, Striegel-Moore R, et al. Impact of timing of pubertal maturation on growth in black and white female adolescents: The National Heart, Lung, and Blood Institute Growth and Health Study. J Pediatr 2001;138:636–643. [DOI] [PubMed] [Google Scholar]
- 5.Bacha F, Saad R, Gungor N, et al. Obesity, regional fat distribution, and syndrome X in obese black versus white adolescents: race differential in diabetogenic and atherogenic risk factors. J Clin Endocrinol Metab 2003;88:2534–2540. [DOI] [PubMed] [Google Scholar]
- 6.Barlow SE; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120(suppl 4):S164–S192. [DOI] [PubMed] [Google Scholar]
- 7.Freedman DS, Mei Z, Srinivasan SR, et al. Cardiovascular risk factors and excess adiposity among overweight children and adolescents: the Bogalusa Heart Study. J Pediatr 2007;150:12–17.e2. [DOI] [PubMed] [Google Scholar]
- 8.Whitlock EP, Williams SB, Gold R, et al. Screening and interventions for childhood overweight: a summary of evidence for the US Preventive Services Task Force. Pediatrics 2005;116:e125–e144. [DOI] [PubMed] [Google Scholar]
- 9.Skinner AC, Perrin EM, Moss LA, et al. Cardiometabolic risks and severity of obesity in children and young adults. N Engl J Med 2015;373:1307–1317. [DOI] [PubMed] [Google Scholar]
- 10.Weiss R, Dziura J, Burgert TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 2004;350:2362–2374. [DOI] [PubMed] [Google Scholar]
- 11.Caprio S, Daniels SR, Drewnowski A, et al. Influence of race, ethnicity, and culture on childhood obesity: implications for prevention and treatment: a consensus statement of Shaping America’s Health and the Obesity Society. Diabetes Care 2008;31:2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha R, Fisch G, Teague B, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med 2002;346:802–810. [DOI] [PubMed] [Google Scholar]
- 13.Steinberger J, Moran A, Hong CP, et al. Adiposity in childhood predicts obesity and insulin resistance in young adulthood. J Pediatr 2001;138:469–473. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasan SR, Bao W, Wattigney WA, et al. Adolescent overweight is associated with adult overweight and related multiple cardiovascular risk factors: the Bogalusa Heart Study. Metabolism 1996;45:235–240. [DOI] [PubMed] [Google Scholar]
- 15.Freedman DS, Khan LK, Dietz WH, et al. Relationship of childhood obesity to coronary heart disease risk factors in adulthood: the Bogalusa Heart Study. Pediatrics 2001;108:712–718. [DOI] [PubMed] [Google Scholar]
- 16.Litwin SE. Childhood obesity and adult cardiovascular disease. J AM Coll Cardio 2014;64:1588–1590. [DOI] [PubMed] [Google Scholar]
- 17.Odeleye OE, de Courten M, Pettit D, et al. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes 1997;46:1341–1345. [DOI] [PubMed] [Google Scholar]
- 18.Shulman G Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 2014;371:2237–2238. [DOI] [PubMed] [Google Scholar]
- 19.Weiss R, Dufour S, Taksali SE, et al. Pre-type 2 diabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance and altered myocellular and abdominal fat partitioning. Lancet 2003;362:951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Despres JP. Is visceral obesity the cause of the metabolic syndrome? Ann Med 2006;38:52–63. [DOI] [PubMed] [Google Scholar]
- 21.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000;106:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry RJ, Samuel VT, Petersen KF, et al. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014;510:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruz ML, Bergman RN, Goran MI. Unique effect of visceral fat on insulin sensitivity in obese Hispanic children with a family history of type 2 diabetes. Diabetes Care 2002;25:1631–1636. [DOI] [PubMed] [Google Scholar]
- 24.Taksali S, Caprio S, Dziura J, et al. High visceral and low abdominal subcutaneous fat stores in the obese adolescent: a determinant of an adverse metabolic syndrome. Diabetes 2008;57:367–371. [DOI] [PubMed] [Google Scholar]
- 25.Cali AMG, De Oliveira A, Kim H, et al. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology 2009;49:1896–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D’Adamo E, Cali AM, Weiss R, et al. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 2010;33:1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fabbrini E, Magkos F, Mohammed BS, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 2009;106:15430–15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010;51:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgert TS, Taksali SE, Dziura J, et al. Alanine aminotransferase levels and fatty liver in childhood obesity: associations with insulin resistance, adiponectin, and visceral fat. J Clin Endocrinol Metab 2006;91:4287–4294. [DOI] [PubMed] [Google Scholar]
- 30.Nadeau KJ, Klingensmith G, Zeitler P. Type 2 diabetes in children is frequently associated with elevated alanine aminotransferase. J Pediatr Gastroenterol Nutr 2005;41:94–98. [DOI] [PubMed] [Google Scholar]
- 31.Newton KP, Hou J, Crimmins N, et al. Prevalence of prediabetes and type 2 diabetes in children with nonalcoholic fatty liver disease. JAMA Pediatr 2016;170:e161971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eckel RH. Obesity and heart disease: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation 1997;96:3248–3250. [DOI] [PubMed] [Google Scholar]
- 33.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006;444:840–846. [DOI] [PubMed] [Google Scholar]
- 34.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 2004;4:579–591. [DOI] [PubMed] [Google Scholar]
- 35.Peeters A, Barendregt JJ, Willekens F, et al. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Ann Intern Med 2003;138:24–32. [DOI] [PubMed] [Google Scholar]
- 36.Danforth E Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet 2000;26:13. [DOI] [PubMed] [Google Scholar]
- 37.Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci 2002;967:363–378. [DOI] [PubMed] [Google Scholar]
- 38.Gray SL, Vidal-Puig AJ. Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr Rev 2007;65:S7–S12. [DOI] [PubMed] [Google Scholar]
- 39.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome–an allostatic perspective. Biochim Biophys Acta 2010;1801:338–349. [DOI] [PubMed] [Google Scholar]
- 40.Kursawe R, Eszlinger M, Narayan D, et al. Cellularity and adipogenic profile of the abdominal subcutaneous adipose tissue from obese adolescents: association with insulin resistance and hepatic steatosis. Diabetes 2010;59:2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kursawe R, Caprio S, Giannini C, et al. Decreased transcription of ChREBP-alpha/beta isoforms in abdominal subcutaneous adipose tissue of obese adolescents with prediabetes or early type 2 diabetes: associations with insulin resistance and hyperglycemia. Diabetes 2013;62:837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillum MP, Kotas ME, Erion DM, et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011;60:3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444:860–867. [DOI] [PubMed] [Google Scholar]
- 44.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011;121:2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab 2008;4:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246. [DOI] [PubMed] [Google Scholar]
- 47.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sbarbati A, Osculati F, Silvagni D, et al. Obesity and inflammation: evidence for an elementary lesion. Pediatrics 2006;117:220–223. [DOI] [PubMed] [Google Scholar]
- 49.Tam CS, Tordjman J, Divoux A, et al. Adipose tissue remodeling in children: the link between collagen deposition and age-related adipocyte growth. J Clin Endocrinol Metab 2012;97:1320–1327. [DOI] [PubMed] [Google Scholar]
- 50.Dietz WH. Critical periods in childhood for the development of obesity. Am J Clin Nutr 1994;59:955–959. [DOI] [PubMed] [Google Scholar]
- 51.Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature 2008;453:783–787. [DOI] [PubMed] [Google Scholar]
- 52.Bjorntorp P Effects of age, sex, and clinical conditions on adipose tissue cellularity in man. Metabolism 1974;23:1091–1102. [DOI] [PubMed] [Google Scholar]
- 53.Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab 1976;5:299–311. [DOI] [PubMed] [Google Scholar]
- 54.Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A 2011;108:15324–15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stienstra R, Joosten LA, Koenen T, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 2010;12:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youm YH, Adijiang A, Vandanmagsar B, et al. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011;152:4039–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011;12:408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 2010;327:296–300. [DOI] [PubMed] [Google Scholar]
- 60.Kursawe R, Dixit VD, Scherer PE, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes 2016;65:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang TT, Johnson MS, Figueroa-Colon R, et al. Growth of visceral fat, subcutaneous abdominal fat, and total body fat in children. Obes Res 2001;9:283–289. [DOI] [PubMed] [Google Scholar]
- 62.Bennett B, Larson-Meyer DE, Ravussin E, et al. Impaired insulin sensitivity and elevated ectopic fat in healthy obese vs. nonobese prepubertal children. Obesity (Silver Spring) 2012;20:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rebrin K, Steil GM, Mittelman SD, et al. Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest 1996;98:741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perry RJ, Camporez JP, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015;160:745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]