

Supplemental Digital Content is Available in the Text.
Key Words: anterior segment dysgenesis, corneal anomalies, glaucoma, megalocornea, corneal decompensation, FOXC1, PITX2
Purpose:
Axenfeld–Rieger syndrome encompasses a group of developmental disorders affecting the anterior chamber structures of the eye, with associated systemic features in some cases. This study aims to compare the difference in anterior segment phenotypes such as those involving the cornea, iris, lens, and anterior chamber angle between cases with disease-causing sequence variations in FOXC1 and PITX2.
Methods:
This cross-sectional study involved 61 individuals, from 32 families with pathogenic FOXC1 or PITX2 variants, who were registered with the Australian and New Zealand Registry of Advanced Glaucoma.
Results:
The median age of the cohort was 39 years at the time of last assessment (range 3–85 years; females, 54%). Thirty-two patients had pathogenic variants in the FOXC1 gene, and 29 patients had pathogenic variants in the PITX2 gene. Corneal abnormalities were more common in individuals with FOXC1 variants (18/36, 50%) than those with PITX2 variants (4/25, 16%; P = 0.007). Iris abnormalities such as hypoplasia (P = 0.008) and pseudopolycoria (P = 0.001) were more common in individuals with PITX2 variants than those with FOXC1 variants. Glaucoma was present in 72% of participants. Corneal decompensation was positively associated with corneal abnormalities (P < 0.001), glaucoma surgery (P = 0.025), and cataract surgery (P = 0.002).
Conclusions:
Corneal abnormalities were more common in individuals with FOXC1 than in those with PITX2 variants and were often associated with early onset glaucoma. These findings highlight that patients with FOXC1 variations require close follow-up and monitoring throughout infancy and into adulthood.
Axenfeld–Rieger syndrome (ARS, MIM 180500) is a developmental disorder that affects the structures of the anterior segment of the eye, including the cornea, iris, anterior chamber angle, and lens, and in some cases has associated systemic features.2 ARS includes the disorders such as Axenfeld anomaly, Axenfeld syndrome, Rieger anomaly, and Rieger syndrome. Because these 4 disorders have overlapping ocular and systemic manifestations and result from variants in the same genes, they are considered to be part of a clinical spectrum. ARS is typically inherited as an autosomal dominant disorder showing phenotypic and genetic heterogeneity, with sequence variants (SVs) or copy number variation in the FOXC1 (MIM 601090) and PITX2 genes (MIM 601542) accounting for 40% to 70% of the cases.3–5 FOXC1 is a member of the fork-head/winged-helix family of transcription factors and is expressed in human tissues, including the eye, brain, and kidney.6 PITX2 is a member of the family of homeobox genes and is expressed in teeth, abdominal organs, brain, heart, and kidneys.7
ARS is associated with both ocular and systemic manifestations.8 Ocular features are the main manifestation of ARS and include posterior embryotoxon, iris stromal hypoplasia, corectopia, pseudopolycoria, and iris processes.9 Posterior embryotoxon is frequently seen in ARS but has been reported to occur as a very mild manifestation in up to 8 to 12% of the normal population.10,11 Glaucoma is the most serious ocular consequence of ARS and develops in up to 50% of the patients.1,12,13 ARS-associated glaucoma is believed to occur because of reduced facility of aqueous outflow caused by maldevelopment of the trabecular meshwork and Schlemm canal.14 In addition to glaucoma, amblyopia and strabismus have also been reported.15 Systemic manifestations of ARS include craniofacial abnormalities (hypertelorism, telecanthus, maxillary hypoplasia, and prognathism),6 dental abnormalities (microdontia, hypodontia, and oligodontia),16,17 cardiac defects (mitral and tricuspid valve disease, atrial septal defects, and tetralogy of Fallot),18–20 skeletal abnormalities (short stature, club feet, and joint abnormalities),21 hearing loss,8 redundant periumbilical skin, umbilical hernia, anal stenosis, and hypospadias.22 Variants in FOXC1 are more commonly associated with isolated ocular abnormalities, but some reports have described associated heart or hearing defects.13,22 Variants in PITX2 have been shown to be associated with ocular, dental, and umbilical abnormalities.22 Corneal abnormalities are commonly reported in Peter anomaly (corneal opacity, iridocorneal synechiae, and an absence of Descemet membrane and corneal endothelium) but are not a common manifestation in ARS. This study aimed to compare detailed anterior segment phenotypes in individuals with ARS caused by FOXC1 and PITX2 variants.
MATERIALS AND METHODS
This was a retrospective cross-sectional study. The participants for this study were recruited from the Australian and New Zealand Registry of Advanced Glaucoma (ANZRAG), as previously described.23 ANZRAG is a unique genetic repository that aims to determine the genetic basis of childhood glaucoma, in addition to studying the more prevalent forms of adult onset glaucoma. Individuals with ARS and their family members with FOXC1 and PITX2 pathogenic SVs or copy number variation were included in this study. Individuals with ARS were recruited in the ANZRAG regardless of their glaucoma status. Family members of individuals with ARS were invited to participate in this study, and every effort was made by registry staff and ophthalmologists to characterize a genotype and phenotype for those family members. Participants with ARS-associated variants in genes, other than FOXC1 and PITX2, were excluded. Data collected from the ANZRAG or from medical records directly included age, sex, ethnicity, gene variant, age at diagnosis of glaucoma, ocular manifestations, systemic manifestations, and ocular treatment (laser, glaucoma surgery, corneal graft, and cataract surgery). SVs and copy number variants (CNVs) identified were confirmed in National Association of Testing Authorities–accredited laboratories, and the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines for variant interpretation were used to assess pathogenicity.24 CNVs have been assessed by different methods, including SNP array, multiplex ligation-dependent probe amplification and/or karyotype. Variants and CNVs classified as pathogenic, likely pathogenic, and of uncertain significance (but that have evidence toward pathogenicity) were included (see Supplementary Table 1, Supplemental Digital Content, http://links.lww.com/ICO/B380). In our study, advanced glaucoma was defined as visual field loss of at least 2 of 4 central fixation squares having a pattern standard deviation of 0.5% on a reliable Humphrey 24-2 field or a mean deviation of −15 dB in the worse eye. Nonadvanced glaucoma was defined by glaucomatous visual field defects with corresponding optic disc rim thinning and an enlarged cup-to-disc ratio (CDR) (≥0.7) or CDR asymmetry (≥0.2) between the 2 eyes. Ocular hypertension was defined by an intraocular pressure (IOP) > 21 mm Hg. Megalocornea was defined as corneal diameter >13 mm in the horizontal meridian.25 Corneal abnormality was defined as any corneal lesions present at birth or within 4 years of birth and includes megalocornea, microcornea, corneal opacity, corneal edema, and Haab striae. Corneal decompensation was defined as corneal edema after treatment in a previously normal cornea. We defined early-onset glaucoma as glaucoma developing before the age of 3 years and late-onset glaucoma as glaucoma developing after the age of 3 years. This study was approved by the Southern Adelaide Clinical Human Research Ethics Committee, and all participants provided informed consent. This study adhered to the tenets of the Declaration of Helsinki.
Descriptive statistics of demography and clinical characteristics were performed by using the SPSS software (IBM SPSS Statistics 25, Chicago, IL). The mean and standard deviation were calculated for participants' age at last examination, age at diagnosis of ARS, and age at diagnosis of glaucoma. χ2 and Fisher exact tests were used to determine the association between the categorical variables. Although all statistical analyses were exploratory in nature, a Bonferroni multiple comparison correction was applied where multiple tests were made. A P value of <0.05 was considered as significant.
RESULTS
A total of 61 individuals from 32 families with ARS-associated FOXC1 or PITX2 variants were included in this study. The median age of the cohort at last examination was 27 years (range 1–84 years), and 53% were female (Table 1). Most of the participants were of self-reported European ancestry (92%, n = 56). More than half of the participants (59%, n = 36) showed variation in the FOXC1 gene. The common corneal abnormalities seen in this cohort were megalocornea (13%), Haab striae (10%), congenital corneal opacity (13%), and corneal edema (12%) (Fig. 1). The median central corneal thickness (CCT) in individuals with FOXC1 variants was 525 (interquartile range, 509–555 μm), and the median CCT in individuals with PITX2 variants was 541 (interquartile range, 516–602 μm). Interfamilial and intrafamilial variabilities are commonly reported with variants in these genes which support ocular and systemic anomalies being different within family members. The prevalence of megalocornea was significantly higher in individuals with FOXC1 variants (8/36, 22%) than in those with PITX2 variants (0/25, 0%; exact P = 0.010) (Table 2). Corneal abnormalities were seen in 36% of the participants (n = 22) and were significantly more common in individuals with FOXC1 variants (18/36, 50%) than in those with PITX2 variants (4/25, 16%; P = 0.007) (Table 2). The prevalence of Haab striae was 10% (n = 6) and significantly more common in individuals with FOXC1 variants (6/36, 17%) than in those with PITX2 variants (0/25, 0%; exact P = 0.035). More than half of all participants had posterior embryotoxon. The most common iris abnormalities seen in the cohort were iris stromal hypoplasia, iris processes, and corectopia. The prevalence of pseudopolycoria was 16% (n = 10), and this condition was significantly more common in individuals with PITX2 variants (9/25, 36%) than in those with FOXC1 variants (1/36, 3%; P = 0.001). The prevalence of iris stromal hypoplasia was 48% (n = 29) and was significantly more common in individuals with PITX2 variants (17/25, 68%) compared with those FOXC1 variants (12/36, 33%; P = 0.008). The prevalence of cataract was not different between participants with FOXC1 or PITX2 variants (22% vs. 16%, respectively, exact P = 0.548). Cataract reported in the participants were mostly acquired after an intervention, namely, surgery (n = 8). However, the prevalence of cataract was not significantly different between individuals who had surgical intervention and those without surgical interventions (27% vs. 11%, P = 0.134). Twelve individuals had cataract surgery, 7 unilateral and 5 bilateral. However, the prevalence of cataract was 20% and was significantly more common in individuals with corneal abnormalities (9/22, 41%) than in individuals with no corneal abnormalities (3/39, 8%; P = 0.002).
TABLE 1.
Sociodemographic and Clinical Characteristics of the Study Population
| Variable | n = 61 |
| Age at last examination (yr, n (%)) | |
| <50 | 50 (82) |
| Median age, IQR | 27 (13–47) |
| Sex, n (%) | |
| Male | 29 (48) |
| Ethnicity, n (%) | |
| White population | 56 (92) |
| Gene variant, n (%) | |
| FOXC1 | 36 (59) |
| PITX2 | 25 (41) |
| Age at diagnosis of ASD (yr) | |
| Median, IQR | 12 (0–47) |
| Age at diagnosis of glaucoma (yr) | |
| Median, IQR | 13 (1–26) |
ASD, anterior segment dysgenesis; IQR, interquartile range.
FIGURE 1.
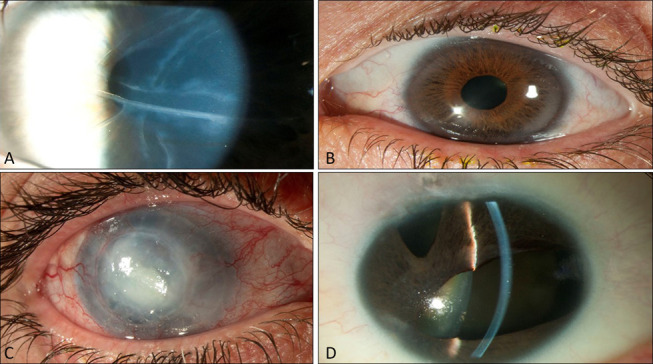
Clinical features of Axenfeld–Rieger syndrome: ocular changes, Haab striae (A), endothelial haze (B), corneal edema (C), corectopia, and polycoria (D).
TABLE 2.
Univariate Analyses for Associated Ocular Features of Axenfeld–Rieger Syndrome (N = 61)
| Variables | Total Prevalence n (%) | FOXC1, n (%) | PITX2, n (%) | P * |
| Corneal abnormality | ||||
| Yes | 22 (36) | 18 (50) | 4 (16) | 0.007 |
| Corneal decompensation | ||||
| Yes | 18 (30) | 10 (28) | 8 (32) | 0.722 |
| Megalocornea | ||||
| Yes | 8 (13) | 8 (22) | 0 (0) | 0.010 |
| Congenital corneal opacity | ||||
| Yes | 8 (13) | 4 (11) | 4 (16) | 0.426 |
| Corneal edema | ||||
| Yes | 7 (12) | 5 (14) | 2 (8) | 0.390 |
| Haab striae | ||||
| Yes | 6 (10) | 6 (17) | 0 (0) | 0.035 |
| Corectopia | ||||
| Yes | 29 (48) | 15 (42) | 14 (56) | 0.270 |
| Pseudopolycoria | ||||
| Yes | 10 (16) | 1 (3) | 9 (36) | 0.001 |
| Ectropion uvea | ||||
| Yes | 5 (8) | 4 (11) | 1 (4) | 0.311 |
| Posterior embryotoxon | ||||
| Yes | 38 (62) | 23 (64) | 15 (60) | 0.758 |
| Iris hypoplasia | ||||
| Yes | 29 (48) | 12 (33) | 17 (68) | 0.008 |
| Peripheral anterior synechiae | ||||
| Yes | 29 (48) | 14 (39) | 15 (60) | 0.104 |
| Iris processes | ||||
| Yes | 16 (26) | 11 (31) | 5 (20) | 0.324 |
| Cataract | ||||
| Yes | 12 (20) | 8 (22) | 4 (16) | 0.548 |
| Glaucoma | ||||
| Yes | 44 (72) | 27 (75) | 17 (68) | 0.549 |
χ2 and Fisher exact tests were used to determine the association between the categorical variables. A P value of <0.05 was considered as significant.
Pearson χ2 test/Fisher exact test.
Glaucoma was present in 72% (44/61) of participants with 56% (34/61) requiring surgical treatment. The median age at diagnosis of glaucoma was 13 years (range, 0–71 years). There was no significant difference in the prevalence of glaucoma between individuals with FOXC1 and those with PITX2 variants (Table 2). However, the median age at diagnosis of glaucoma for FOXC1 was 4 years (range, 0–71 years), whereas the median age at diagnosis of glaucoma for PITX2 was 17 years (range, 3–48 years). Early-onset glaucoma was significantly more common in individuals with corneal abnormalities (11/19, 58%) than in individuals without corneal abnormalities (6/25, 24%; P = 0.022). Early-onset glaucoma was significantly more common in individuals with megalocornea (6/8, 75%) than in individuals with no megalocornea (11/36, 31%; exact P = 0.028). However, the prevalence of Haab striae was not significantly different between individuals with early-onset glaucoma compared with late onset glaucoma (50% vs. 37%, respectively, exact P = 0.426). The most commonly performed glaucoma surgeries were trabeculectomy (51%), glaucoma drainage implants (15%), and goniotomy (3%). Thirty-one individuals had trabeculectomy surgery, 21 unilateral and 9 bilateral. Nine individuals had glaucoma tube implant surgery, 8 unilateral and 1 bilateral. Two individuals had bilateral goniotomy, 7 individuals had both trabeculectomy and tube implant surgeries, and 1 individual had both goniotomy and trabeculectomy.
Corneal decompensation was seen in 30% of the individuals and was more common in eyes with corneal abnormalities (13/22, 59%) than in eyes without corneal abnormalities (5/39, 13%, P < 0.001). The prevalence of corneal decompensation was not different between individuals with FOXC1 and those with PITX2 variants (28% vs. 32%, respectively, P = 0.722). The incidence of corneal decompensation was significantly higher in participants who had undergone glaucoma surgery (14/34, 41%) than in individuals who had not undergone glaucoma surgery (4/27, 15%; P = 0.025). Corneal decompensation was significantly higher in participants who had undergone cataract surgery (8/12, 67%) than in those without cataract surgery (10/49, 20%; P = 0.002). Corneal grafts were more common in patients with corneal abnormalities (5/22, 23%) than in patients without corneal abnormalities (1/39, 3%, exact P = 0.020). The systemic features are summarized in Table 3. The Bonferroni correction was applied for multiple comparison corrections. After adjusting for multiple comparisons (pcorrection = 0.05/25 = 0.002), dental abnormalities, redundant periumbilical skin, umbilical hernia, and pseudopolycoria remained statistically significant with a pcorrection = 0.025.” Each of the variables has a corrected value of 0.025. Comparison of the clinical features between SV and CNV did not show any significance.
TABLE 3.
Univariate Analyses for Associated Systemic Features of Axenfeld–Rieger Syndrome (N = 61)
| Variables | Prevalence, n (%) | FOXC1, n (%) | PITX2, n (%) | P * |
| Dental abnormalities | ||||
| Yes | 28 (46) | 5 (15) | 23 (100) | <0.001 |
| Redundant umbilical skin | ||||
| Yes | 17 (28) | 2 (6) | 15 (88) | <0.001 |
| Umbilical hernia | ||||
| Yes | 10 (16) | 1 (3) | 9 (56) | <0.001 |
| Hearing loss | ||||
| Yes | 14 (23) | 13 (39) | 1 (6) | 0.010 |
| Heart defects | ||||
| Yes | 12 (20) | 10 (30) | 2 (11) | 0.103 |
| Hydrocephalus | ||||
| Yes | 3 (5) | 3 (10) | 0 (0) | 0.262 |
| Learning abnormality | ||||
| Yes | 10 (16) | 8 (24) | 2 (11) | 0.259 |
| Short stature | ||||
| Yes | 6 (10) | 3 (9) | 1 (6) | 0.295 |
| Gastrointestinal abnormalities | ||||
| Yes | 11 (18) | 0 (0) | 8 (42) | 0.005 |
χ2 and Fisher exact tests were used to determine the association between the categorical variables. A P value of <0.05 was considered as significant.
Outie is defined as an umbilical tip protruding past the periumbilical skin. Umbilical hernia is a defect in the abdominal wall caused by failure of umbilical ring closure which causes the abdominal contents to protrude out. Redundant umbilical skin is excessive skin surrounding the umbilicus.
Pearson χ2/Fisher exact test.
DISCUSSION
This study characterizes the detailed corneal phenotypes of individuals with ARS-associated FOXC1 and PITX2 variants. Within the spectrum of anterior segment dysgenesis, corneal abnormalities such as congenital corneal opacities, microcornea, and iridocorneal adhesions are common in Peter anomaly but are uncommon in ARS.26 Posterior embryotoxon is the only peripheral corneal change that has been frequently reported in ARS; the absence of megalocornea and congenital corneal opacity are often useful criteria to distinguish ARS from other anterior segment disorders.6 Others have previously reported that ARS is not associated with corneal abnormalities,6 whereas Fuchs dystrophy/cornea guttatae can coexist in individuals with ARS-associated PITX2 variants.27 In this study, we further extended the phenotypic characterization to assess differences in corneal findings and management in FOXC1 and PITX2 carriers. We reported that more than one-third of individuals had corneal abnormalities, with megalocornea the most common. Moreover, we found that corneal abnormalities were more common in individuals with FOXC1 variants than in those with PITX2 variants, with only 16% of individuals with PITX2 variants having corneal abnormalities compared with 50% of those with FOXC1 variants. We identified that iris abnormalities, including iris stromal hypoplasia and pseudopolycoria, were more common in people with PITX2 than FOXC1 variants. Corectopia has been reported to be common in PITX2 variants,28 but our study is the first to show that iris hypoplasia and pseudopolycoria were common in individuals with PITX2 variants. The absence of pseudopolycoria in individuals with FOXC1 variants is consistent with previous smaller case series.29,30
Early-onset glaucoma in individuals with FOXC1 variants is consistent with previous studies.28,31 FOXC1 has been recently identified as a primary open-angle glaucoma susceptibility locus and an essential regulator of lymphangiogenesis,32 and this could explain the increased prevalence of glaucoma and lower age at diagnosis in the FOXC1 carriers. FOXC1 and PITX2 are developmental genes that are critical for normal corneal development,33 and variants involving these genes can result in reduced CCT.34 In our study, the median CCT in individuals with FOXC1 and PITX2 was slightly below that of normal population. Studies have shown that corneal abnormalities such as megalocornea, keratoconus, pellucid marginal degeneration, Fuchs endothelial dystrophy, and posterior polymorphous dystrophy can be associated with glaucoma.35–38 The mechanism of glaucoma development in these corneal conditions is multifactorial and includes oxidative stress, abnormal scleral biomechanical properties, and maldevelopment of angle structures. In ARS, glaucoma development is mostly attributed to the maldevelopment of the angle structures.14 Glaucoma can also cause corneal changes such as megalocornea, Haab striae, corneal edema, and reduced endothelial cell count. In our study, most of the individuals with megalocornea had an early-onset glaucoma suggesting that the increased corneal diameter may be attributed to the increased IOP. Endothelial cell loss has been reported in primary open-angle glaucoma, primary angle-closure glaucoma, and secondary glaucoma and is attributed to increased IOP, antiglaucoma medications, and incisional glaucoma surgery.35 In addition, in our study, corneal decompensation was seen commonly in participants with glaucoma and in those who had undergone glaucoma and cataract surgeries.
ARS has a significant impact on vision because of disruption of the visual axis and can be associated with glaucoma, cataracts, and corneal decompensation. Our findings show that corneal abnormalities were more common in individuals with FOXC1 variants than in those with PITX2 variants, and individuals with corneal abnormalities were more likely to require corneal transplantation for corneal decompensation. Moreover, ARS-associated FOXC1 variant carriers with glaucoma were diagnosed at a younger age and were more likely to require glaucoma procedures and corneal grafts than PITX2 carriers. Therefore, individuals with FOXC1 variants require close follow-up and monitoring throughout infancy and into adulthood. Raised IOP, glaucoma surgery, cataract surgery, and corneal grafting are all associated with endothelial cell loss.39–41 Therefore, individuals with FOXC1 variants should have endothelial cell density (ECD) assessment by using specular microscopy.
ARS is generally diagnosed by ophthalmologists, and the diagnosis is primarily clinical. Phenotypic and genotypic heterogeneity and overlap of clinical presentations can make diagnosing ARS very challenging, especially in young children who may require an examination under anesthesia to assess angle abnormalities. ARS can present with subtle iris and corneal features and sometimes can be misdiagnosed as primary congenital glaucoma42 or primary open-angle glaucoma31 when ARS ocular features are subtle. Although some would argue that the presence of posterior embryotoxon is essential to make a diagnosis of ARS, our study showed that posterior embryotoxon was not always identified with FOXC1 and PITX2 developmental abnormalities. As a result, genotype–phenotype correlations and the presence of systemic features can assist clinicians in making a correct diagnosis and disease classification. In this study, systemic features were present in more than half of the patients with FOXC1 variants, but in all individuals with PITX2 variants, and this has been reported on an overlapping set of ANZRAG patients in the past.31,43 It has been previously reported that while the systemic features are variably expressed between both genes, the ocular features are believed to be similar between both genes. Tumer et al, Strungaru et al, and D'Haene et al previously reported phenotypic differences between the FOXC1 and PITX2 variants, both associated with ARS.6,22,28 Patients with ARS have a 50% to 60% lifetime risk of developing glaucoma, which can occur anytime during infancy, childhood, early adulthood, or middle age.44 Therefore, patients with ARS should be examined annually for the changes in IOP and the optic nerve head. Patients with ARS with corneal abnormalities and those who have undergone surgical interventions are at risk of developing corneal decompensation. Cataract in ARS could either be developmental or acquired after intervention such as glaucoma drainage surgery. Therefore, these patients should be followed regularly with repeated corneal ECD, corneal morphology, and assessment for cataract.
There were some limitations in this study. Because it was a retrospective cross-sectional study, collection of data relied on information provided by ophthalmologists to the ANZRAG or on their record in medical notes when available, and some of the data such as age at diagnosis of ARS and age at diagnosis of glaucoma were missing. However, variables with a large component of missing data were not included in the final analysis. All the participants were drawn from the ANZRAG, and although the registry includes participants with ARS irrespective of their glaucoma status, the recruitment may have introduced bias toward glaucoma. Furthermore, this study may have been underpowered to detect some potentially significant findings because of the small sample size, multiple features examined, and the stringency of the Bonferroni multiple measures correction. This is expected in a rare disease such as ARS. However, all statistical tests were exploratory in nature, and further studies with a larger sample size should be performed to support our findings. Although there are 3 types of ARS, our study was limited to types 1 and 3 because no gene associated with type 2 ARS has been identified. However, variations in the FOXC1 and PITX2 account for 40 to 70% of the cases. Another limitation was that our study did not report the corneal ECD between the FOXC1 and PITX2 variants because our attempt to collect this information has been complicated by the small sample size, age of participants, availability of equipment, and the integrity of the cornea which has prevented clear imaging. However, future research should aim to investigate the corneal ECD in these individuals. To conclude, this study compared the anterior segment phenotypes of individuals with ARS-associated pathogenic FOXC1 and PITX2 gene variants. Our findings show that 36% of affected individuals had corneal abnormalities and that FOXC1 variants were significantly more associated with corneal abnormalities compared with PITX2 variants. The prevalence of cataracts was significantly higher in individuals with corneal abnormalities. Corneal decompensation was positively associated with corneal abnormalities, glaucoma surgeries, and cataract surgeries. These findings have important implications for assisting clinicians in managing patients with ARS.
ACKNOWLEDGMENTS
The authors thank Angela Chappell and Carly Emerson for the ophthalmic photographs.
Footnotes
Supported by the Australian National Health and Medical Research Council (NHMRC) Centres of Research Excellence Grant 1116360 (2017–2021) and the Flinders Foundation Health Seed Research Grant. Jamie E Craig was an NHMRC Practitioner Fellow (APP1154824), David A Mackey is an NHMRC Practitioner Fellow (APP1154518), and Emmanuelle Souzeau has an Early Career Fellowship from the Hospital Research Foundation. We are grateful to the research participants and the referring practitioners.
The authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.corneajrnl.com).
Contributor Information
Lachlan S. W. Knight, Email: lachlan.wheelhouseknight@flinders.edu.au.
Deepa Taranath, Email: deepa.taranath@gmail.com.
David A. Mackey, Email: david.mackey@uwa.edu.au.
Jonathan B. Ruddle, Email: jruddle@unimelb.edu.au.
Mark Y. Chiang, Email: ym_chiang@hotmail.com.
Owen M. Siggs, Email: owen.siggs@flinders.edu.au.
Emmanuelle Souzeau, Email: emmanuelle.souzeau@flinders.edu.au.
Jamie E. Craig, Email: jamie.craig@flinders.edu.au.
REFERENCES
- 1.Sowden JC. Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond). 2007;21:1310–1318. [DOI] [PubMed] [Google Scholar]
- 2.Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reis LM, Tyler RC, Volkmann Kloss BA, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20:1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisschuh N, De Baere E, Wissinger B, et al. Clinical utility gene card for: Axenfeld-Rieger syndrome. Eur J Hum Genet. 2011:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisschuh N, Dressler P, Schuettauf F, et al. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2006;47:3846–3852. [DOI] [PubMed] [Google Scholar]
- 6.Tumer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009;17:1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semina EV, Reiter R, Leysens NJ, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham ET, Jr, Eliott D, Miller NR, et al. Familial Axenfeld-Rieger anomaly, atrial septal defect, and sensorineural hearing loss: a possible new genetic syndrome. Arch Ophthalmol. 1998;116:78–82. [DOI] [PubMed] [Google Scholar]
- 9.Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130:107–115. [DOI] [PubMed] [Google Scholar]
- 10.Waring GO, III, Rodrigues MM, Laibson PR. Anterior chamber cleavage syndrome. A stepladder classification. Surv Ophthalmol. 1975;20:3–27. [DOI] [PubMed] [Google Scholar]
- 11.Burian HM, Rice MH, Allen L. External visibility of the region of Schlemm's canal; report on a family with developmental anomalies of cornea, iris, and chamber angle. AMA Arch Ophthalmol. 1957;57:651–658. [PubMed] [Google Scholar]
- 12.Idrees F, Vaideanu D, Fraser SG, et al. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006;51:213–231. [DOI] [PubMed] [Google Scholar]
- 13.Souzeau E, Siggs OM, Zhou T, et al. Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. Eur J Hum Genet. 2017;25:839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson DR. The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans Am Ophthalmol Soc. 1981;79:458–485. [PMC free article] [PubMed] [Google Scholar]
- 15.Chang TC, Summers CG, Schimmenti LA, et al. Axenfeld-Rieger syndrome: new perspectives. Br J Ophthalmol. 2012;96:318–322. [DOI] [PubMed] [Google Scholar]
- 16.Jena AK, Kharbanda OP. Axenfeld-Rieger syndrome: report on dental and craniofacial findings. J Clin Pediatr Dent. 2005;30:83–88. [DOI] [PubMed] [Google Scholar]
- 17.O'Dwyer EM, Jones DC. Dental anomalies in Axenfeld-Rieger syndrome. Int J Paediatr Dent. 2005;15:459–463. [DOI] [PubMed] [Google Scholar]
- 18.Gürbüz-Köz O, Atalay T, Köz C, et al. Axenfeld-Rieger syndrome associated with truncus arteriosus: a case report. Turk J Pediatr. 2007;49:444–447. [PubMed] [Google Scholar]
- 19.Antevil J, Umakanthan R, Leacche M, et al. Idiopathic mitral valve disease in a patient presenting with Axenfeld-Rieger syndrome. J Heart Valve Dis. 2009;18:349–351. [PubMed] [Google Scholar]
- 20.Bekir NA, Güngör K. Atrial septal defect with interatrial aneurysm and Axenfeld-Rieger syndrome. Acta Ophthalmol Scand. 2000;78:101–103. [DOI] [PubMed] [Google Scholar]
- 21.Kannu P, Oei P, Slater HR, et al. Epiphyseal dysplasia and other skeletal anomalies in a patient with the 6p25 microdeletion syndrome. Am J Med Genet A. 2006;140:1955–1959. [DOI] [PubMed] [Google Scholar]
- 22.D'Haene B, Meire F, Claerhout I, et al. Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest Ophthalmol Vis Sci. 2011;52:324–333. [DOI] [PubMed] [Google Scholar]
- 23.Souzeau E, Goldberg I, Healey PR, et al. Australian and New Zealand registry of advanced glaucoma: methodology and recruitment. Clin Exp Ophthalmol. 2012;40:569–575. [DOI] [PubMed] [Google Scholar]
- 24.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29:385–394. [DOI] [PubMed] [Google Scholar]
- 26.Bhandari R, Ferri S, Whittaker B, et al. Peters anomaly: review of the literature. Cornea. 2011;30:939–944. [DOI] [PubMed] [Google Scholar]
- 27.Kniestedt C, Taralczak M, Thiel MA, et al. A novel PITX2 mutation and a polymorphism in a 5-generation family with Axenfeld-Rieger anomaly and coexisting Fuchs' endothelial dystrophy. Ophthalmology. 2006;113:1791:e1791–1798. [DOI] [PubMed] [Google Scholar]
- 28.Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007;48:228–237. [DOI] [PubMed] [Google Scholar]
- 29.Komatireddy S, Chakrabarti S, Mandal AK, et al. Mutation spectrum of FOXC1 and clinical genetic heterogeneity of Axenfeld-Rieger anomaly in India. Mol Vis. 2003;9:43–48. [PubMed] [Google Scholar]
- 30.Honkanen RA, Nishimura DY, Swiderski RE, et al. A family with Axenfeld-Rieger syndrome and Peters Anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol. 2003;135:368–375. [DOI] [PubMed] [Google Scholar]
- 31.Souzeau E, Siggs OM, Zhou T, et al. Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. Eur J Hum Genet. 2017;25:1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fatima A, Wang Y, Uchida Y, et al. Foxc1 and Foxc2 deletion causes abnormal lymphangiogenesis and correlates with ERK hyperactivation. J Clin Invest. 2016;126:2437–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinoza HM, Cox CJ, Semina EV, et al. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Mol Genet. 2002;11:743–753. [DOI] [PubMed] [Google Scholar]
- 34.Asai-Coakwell M, Backhouse C, Casey RJ, et al. Reduced human and murine corneal thickness in an Axenfeld-Rieger syndrome subtype. Invest Ophthalmol Vis Sci. 2006;47:4905–4909. [DOI] [PubMed] [Google Scholar]
- 35.Cohen EJ. Keratoconus and normal-tension glaucoma: a study of the possible association with abnormal biomechanical properties as measured by corneal hysteresis (An AOS Thesis). Trans Am Ophthalmol Soc. 2009;107:282–299. [PMC free article] [PubMed] [Google Scholar]
- 36.Désir J, Sznajer Y, Depasse F, et al. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet. 2010;18:761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagarsheth M, Singh A, Schmotzer B, et al. Relationship between Fuchs endothelial corneal dystrophy severity and glaucoma and/or ocular hypertension. Arch Ophthalmol. 2012;130:1384–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourgeois J, Shields MB, Thresher R. Open-angle glaucoma associated with posterior polymorphous dystrophy. A clinicopathologic study. Ophthalmology. 1984;91:420–423. [DOI] [PubMed] [Google Scholar]
- 39.Wirbelauer C, Wollensak G, Pham DT. Influence of cataract surgery on corneal endothelial cell density estimation. Cornea. 2005;24:135–140. [DOI] [PubMed] [Google Scholar]
- 40.Bourne RR, Minassian DC, Dart JK, et al. Effect of cataract surgery on the corneal endothelium: modern phacoemulsification compared with extracapsular cataract surgery. Ophthalmology. 2004;111:679–685. [DOI] [PubMed] [Google Scholar]
- 41.Arnavielle S, Lafontaine PO, Bidot S, et al. Corneal endothelial cell changes after trabeculectomy and deep sclerectomy. J Glaucoma. 2007;16:324–328. [DOI] [PubMed] [Google Scholar]
- 42.Siggs OM, Souzeau E, Pasutto F, et al. Prevalence of FOXC1 variants in individuals with a suspected diagnosis of primary congenital glaucoma. JAMA Ophthalmol. 2019;137:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Souzeau E, Siggs OM, Pasutto F, et al. Gene-specific facial dysmorphism in Axenfeld-Rieger syndrome caused by FOXC1 and PITX2 variants. Am J Med Genet A. 2021;185:434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmological Soc. 1983;81:736–784. [PMC free article] [PubMed] [Google Scholar]