

ABSTRACT
Skeletal muscle plays a critical role in physical function and metabolic health. Muscle is a highly adaptable tissue that responds to resistance exercise (RE; loading) by hypertrophying, or during muscle disuse, RE mitigates muscle loss. Resistance exercise training (RET)–induced skeletal muscle hypertrophy is a product of external (e.g., RE programming, diet, some supplements) and internal variables (e.g., mechanotransduction, ribosomes, gene expression, satellite cells activity). RE is undeniably the most potent nonpharmacological external variable to stimulate the activation/suppression of internal variables linked to muscular hypertrophy or countering disuse-induced muscle loss. Here, we posit that despite considerable research on the impact of external variables on RET and hypertrophy, internal variables (i.e., inherent skeletal muscle biology) are dominant in regulating the extent of hypertrophy in response to external stimuli. Thus, identifying the key internal skeletal muscle–derived variables that mediate the translation of external RE variables will be pivotal to determining the most effective strategies for skeletal muscle hypertrophy in healthy persons. Such work will aid in enhancing function in clinical populations, slowing functional decline, and promoting physical mobility. We provide up-to-date, evidence-based perspectives of the mechanisms regulating RET-induced skeletal muscle hypertrophy.
Key Words: SKELETAL MUSCLE, HYPERTROPHY, RESISTANCE EXERCISE, PROTEIN SYNTHESIS, ANABOLIC MECHANISMS
Skeletal muscle plays a critical role in physical function, athletic performance, and metabolic health, and low muscle mass is associated with greater mortality in healthy adults and adults with comorbidities (1). The regulation of skeletal muscle mass is influenced by several variables that can broadly be categorized into external or internal system variables. Resistance exercise training (RET) is the most potent nonpharmacological external means of increasing skeletal muscle mass (2) and is an external system variable. In contrast, internal variables are inherent systemic or, more often, local (within the muscle) biological processes that mechanistically underpin hypertrophy in response to external stimuli like RET (Fig. 1). An important question is to what extent manipulation of external variables influences internal variable responses to affect the outcome—hypertrophy. In our view, identifying the key skeletal muscle molecular targets activated by resistance exercise (RE) that, with repetition, will underpin RET-induced skeletal muscle hypertrophy is critical.
FIGURE 1.
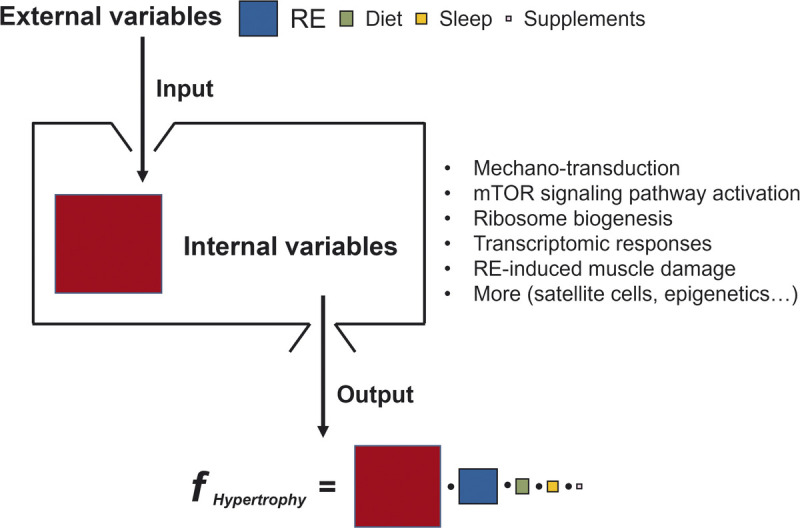
The formulation of external vs internal variables for skeletal muscle hypertrophy. Although external variables (input) are indispensable to activate internal variables for skeletal muscle hypertrophy (output), the response of internal variables stimulated by external variables is the key determinant of skeletal muscle hypertrophy. The size of the color squares reflects the extent of the variables’ contribution to muscle hypertrophy. We are uncertain of the nature of the interaction between the variables and have shown them as multiplicative (•); however, we acknowledge that the function could be additive or more complex (i.e., an unknown function) relationship.
At the molecular level, RET-induced skeletal muscle hypertrophy (defined here as an increase in axial cross-sectional area (CSA) of a muscle/muscle fiber) occurs in adult humans because of the accrual of cellular proteins (e.g., myofibrillar, sarcoplasmic, mitochondrial) within preexisting muscle fibers. Surprisingly, we remain largely unaware of the structural adaptations associated with RET-induced skeletal muscle hypertrophy. Nonetheless, an axiom is that hypertrophy requires, among other processes, net muscle protein accretion, which occurs when the rate of muscle protein synthesis (MPS) exceeds that of muscle protein breakdown (MPB)—the algebraic difference between which is commonly referred to as net protein balance. In contrast, extended periods of negative net protein balance (MPB > MPS) manifest as skeletal muscle atrophy, which occurs under a variety of systemic scenarios, such as reduced physical activity and bed rest, or local, including limb immobilization (3). Importantly, many chronic diseases, including cancer, chronic obstructive pulmonary disorder (COPD), cardiovascular disease, sepsis, uremia, and burns, also have muscle-wasting components (4). Therefore, identifying the mechanisms that regulate muscle protein turnover to favor net anabolism is as pertinent a mission clinically as it is for athletes and possibly more so.
The molecular control of MPS and MPB is complex, and many protein signaling cascades dictate net muscle protein balance. Nonetheless, we are still deciphering what signals trigger rises in MPS and thus could potentially contribute to skeletal muscle hypertrophy. The purpose of this review is to provide an up-to-date synopsis of the main molecular mechanisms regulating RET-induced skeletal muscle hypertrophy in humans.
MECHANOTRANSDUCTION
As an external stimulus, RE results in mechanical loading of skeletal muscle; however, the mechanisms by which skeletal muscle ‘senses’ and then initiates responses culminating in hypertrophy are still being unraveled (5). Several protein complexes have been identified as candidate mechanosensors that act as molecular transducers during myofiber contraction. The extracellular matrix is thought to play a critical role in mechanotransducing signals into biochemical signals that ultimately regulate the control of skeletal muscle mass (5).
Costameres connect the extracellular matrix to the sarcolemma of the myofiber, are localized at the Z-disk, and act to transduce force from the sarcomere to the extracellular matrix (5). In the presence of mechanical stimuli, phospholipase Cγ1 colocalizes around focal adhesion kinase (FAK)—a densely localized protein within the costamere—and catalyzes the conversion of phosphatidylinositol 4,5-biphosphate to phosphatidic acid (PA) in HEK293T cells (6,7). The synthesized PA activates the HIPPO pathway effectors Yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) through signaling cascade (7). YAP and TAZ not only control cell growth in Drosophila melanogaster and some mammalian tissues (8,9) but also regulate myoblast proliferation and differentiation (10,11). Furthermore, although no mechanistic link has been elucidated regarding YAP and mechanistic target of rapamycin complex 1 (TORC1) signaling, animal studies demonstrated that elevation of YAP expression is sufficient to augment skeletal muscle mass during inhibition of mTORC1 via rapamyci (12,13). Also, YAP and TAZ may play a role in mechanically induced anabolic signaling through the elevated expression of the genes Slc7a5 and Slc3a2 (14). These genes encode for proteins that are the leucine amino acid transporters, which could sensitize mechanically loaded muscle to leucine stimulated MPS (14). In addition, PA may indirectly modulate skeletal muscle hypertrophy via the HIPPO signaling pathways and activate mTORC1 (15). Researchers demonstrated that RE was sufficient for elevating local PA concentration and inhibiting the production of PA ablated markers of mTORC1 activity after mechanical overload of skeletal muscle (15). In sum, costamere-based protein sensors may be necessary for hypertrophic signaling in the immediate postexercise period.
Titin is a large elastic protein structure that spans half the length of each sarcomere from the Z-disk to the M-band (16) and is a primary contributor to the passive force generated during eccentric contraction (17). Titin contains a stretch-activated kinase domain, and the stretch of the sarcomere during myofibrillar contraction exposes several amino acids in the ATP-binding pocket of titin, thereby activating the protein kinase (18). On the other hand, the role of titin during concentric contraction remains undefined. Because of its mechanosensory properties, titin has been proposed to serve as a mechanosensors and regulator of anabolic stimuli (19,20). However, no existing mechanisms are known connecting titin and mTORC1 signaling. In contrast, titin activation is related to autophagy signaling via muscle ring-finger protein (Murf)1/2-proteasome and therefore may be involved in protein turnover and regulation of muscle mass (21,22).
Filamin-C Bag3 is another Z-disk localized protein structure that has been linked with the regulation of muscle size in response to mechanical stimuli (13). Filamin-C is a V-shaped homodimer protein and, in response to mechanical loading, is proposed to interact with Bag3 and, together, regulate two known hypertrophic mechanisms. First, Bag3 increases mechanical-induced activation of YAP through binding-inhibition HIPPO suppressor proteins such as large tumor suppressor kinase 1 (LATS1) and angiomotin-like protein 1 (AMOTL1), augmenting anabolic signaling (12,13,23). Second, Bag3 can increase MPB by signaling chaperone-assisted cell autophagy of damaged Z-disk proteins, a function that may be necessary for adaptation and skeletal muscle hypertrophy (5). Also, Hoffman et al. (24) reported increased phosphorylation of Filamin-C and Bag3 after high-intensity exercise in human muscle; Filimin-C and Bag3 form a mechanosensory complex that may regulate muscle hypertrophy. It appears titin, and Filamin-C Bag3 may play a significant role in regulating muscle mass; however, the mechanisms are far from being understood.
MTOR SIGNALING PATHWAY
The mTOR complex includes a serine/threonine kinase that centers two protein complexes in mammals—mTORC1 and mTOR complex 2 (mTORC2). Both complexes contain the subunits DEP-domain–containing mTOR-interacting protein (DEPTOR) and mammalian lethal with SEC13 protein 8 (mLST8) (25). However, mTORC1 and mTORC2 differ in rapamycin sensitivity, functions, and additional subunits. Broadly, mTORC1 is characterized as a rapamycin-sensitive regulator of cell size with the subunits regulatory-associated protein of mTOR (Raptor) and proline-rich AKT substrate 40 kDa (PRAS40) (25). Upstream stimuli, such as nutrients (i.e., leucine), growth factors (i.e., insuline-like growth factor-1 (IGF-1)) and mechanical stimuli (i.e., RE), are converted to intracellular signals and subsequently detected by mTORC1 subunits, such as Raptor (25). Conversely, mTORC2 is characterized as a rapamycin-insensitive regulator of cytoskeletal structure and cell survival with the subunits: rapamycin-insensitive component of mTOR (Rictor), mitogen-activated protein kinase (MAPK)-interacting protein (mSIN1), and protein associated with Rictor 1 or 2 (PROTOR 1/2) (26).
The mTORC1-induced increases in MPS have been a focal point in the context of skeletal muscle hypertrophy, and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP-1) and p70S6 kinase 1 (S6K1) are two frequently investigated downstream targets. Skeletal muscle hypertrophy has been correlated with basal MPS (27) and RE-induced S6K1 (28) and 4EBP-1 phosphorylation (29). Notably, mTORC1 activation contributes to increases in MPS, but acute short-term (i.e., within hours) RE-induced increases in MPS are not always correlated with chronic RET-induced skeletal muscle hypertrophy (29). Mitchell et al. (29) observed no correlation between untrained men’s acute post-RE MPS rates and muscle hypertrophy after 16 wk of RET. Damas et al. (30) further demonstrated that the MPS after a single bout of RE at baseline were not correlated with the percent change in vastus lateralis CSA (%Δ VL CSA) after 10 wk of RET in young men; however, the MPS after a single bout of RE at weeks 3 and 10 was positively correlated with the %Δ VL CSA after 10 wk of RET. The authors suggested that RE-induced increases in MPS rates largely attenuate muscle damage in the early stages (~3 wk) of RET but, after that, support hypertrophy (30). Overall, mTORC1 seems to play a role in RET-induced skeletal muscle hypertrophy, but the mechanisms underpinning this complex process likely extend beyond merely stimulating mTORC1.
Translocation to the lysosome is critical for mTORC1 activation (31), and the intracellular positioning of mTORC1 after anabolic stimuli has been increasingly studied (32). RE induces mTORC1 translocation to the lysosome, and the mTORC1–lysosome complex subsequently translocates to the cell membrane with a proclivity for capillaries (33). mTORC1 relocation to the cell periphery may promote MPS because of increased proximity to upstream activators, translation initiation factors, and microvasculature (i.e., nutrients) (32). Albeit after endurance exercise, work in trained young men suggests that colocalization of mTORC1 with upstream activators could specifically regulate myofibrillar protein synthesis (34), which would certainly contribute to RET adaptations. The potential impact of nutrient provision (33,35,36) and anabolic properties of the lysosome (reviewed elsewhere [37]) remain avenues for future research on spatial regulation of mTORC1 after exercise.
Rapamycin-insensitive, in addition to rapamycin-sensitive components of mTOR signaling, may also contribute to RE-induced increases in MPS and hypertrophy (38), and this notion is supported by evidence from preclinical models. In rats, rapamycin administration ablated RE-induced increases in MPS completely at 6 h post-RE but only partially 18 h post-RE (39); furthermore, rapamycin administration did not completely ablate RET-induced skeletal muscle hypertrophy (40) In a cornerstone article, Drummond et al. (41) observed that rapamycin blunted MPS in humans after acute RE, particularly ~1 h post-RE. Although chronic rapamycin administration is not feasible in humans, these data cumulatively suggest that rapamycin-insensitive processes impact MPS several hours post-RE and chronic RET adaptations. Interestingly, AZD8055, an inhibitor of both mTOR complexes, completely inhibited RE-induced increases of MPS in rats (42), and tripartite motif-containing 28 (TRIM28) has recently been identified as a rapamycin-insensitive regulator of mechanical load-induced hypertrophy (43). Mechanical stimuli activate Ras kinases that subsequently activate extracellular signal-regulated kinases (ERK), which phosphorylate TSC2; thus, MAPK-ERK1/2 signaling may be an additional mechanism by which mTORC1 senses mechanical stimuli (26). ERK1/2 also phosphorylates kinases involved in protein translation (Fig. 2), such as p90 ribosomal protein S6 kinase (p90RSK) and MAP kinase-interacting kinase 1 (MNK1), although these may be mTORC1-dependent processes (26). Both ERK1/2 and mTORC1 may be required to stimulate MPS maximally after RE (41). In sum, the mTOR-related signaling pathways play a major role in skeletal muscle anabolism, and the precise contributions of mTORC1 and mTORC2 continue to be refined.
FIGURE 2.
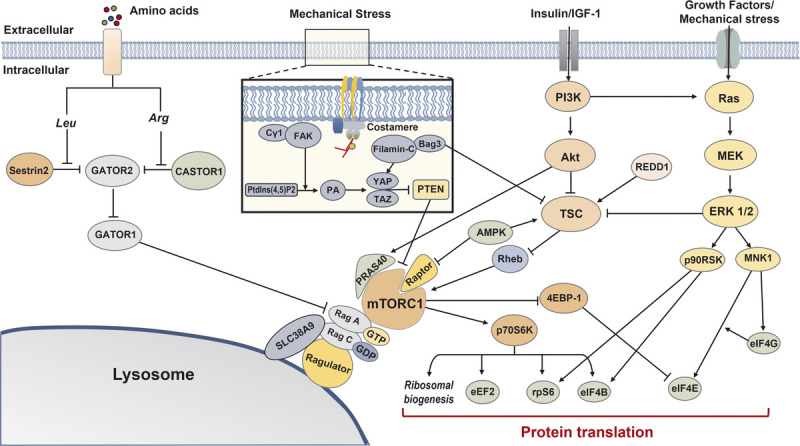
Overview of mTORC1-related protein synthesis pathways. Multiple signaling cascades converge on mTORC1 and contribute to protein translation. Cytosolic leucine and arginine relieve inhibition on GATOR2 by Sestrin2 and CASTOR1, respectively, and subsequently promote mTORC1-induced protein synthesis via GATOR2–GATOR1–Rag signaling. Mechanical stress causes Cγ1 to colocalize around FAK, catalyzing Ptdlns(4,5)P2 conversion to PA, and subsequently activates the HIPPO pathway (YAP and TAZ). Filamin-C and Bag3 interaction also activates the HIPPO pathway. The HIPPO pathway promotes mTORC1 signaling via the suppression of PTEN. The PI3K–Akt–TSC signaling cascade regulates mTORC1 signaling via Rheb, although Akt can directly promote mTORC1 signaling via PRAS40. AMPK and REDD1 inhibit mTORC1 via TSC; AMPK can also inhibit mTORC1 directly via Raptor. mTORC1 promotes protein synthesis by activating several translational and ribosomal components, and Ras–MEK–ERK 1/2 signaling converges on common factors. 4EBP-1, eukaryotic translation initiation factor 4E-binding protein 1; Akt, protein kinase B; AMPK, AMP-activated protein kinase; Arg, arginine; eEF2, eukaryotic elongation factor 2; eIF4B, eukaryotic translation initiation factor 4B; eIF4E, eukaryotic translation initiation factor 4E; eIF4G, eukaryotic translation initiation factor 4G; ERK 1/2, extracellular signal-related kinase 1/2; FAK, focal adhesion kinase; IGF-1, insulin-like growth factor 1; Leu, leucine; MEK, mitogen-activated protein kinase; MNK1, MAP kinase-interacting kinase 1; mTORC1, mechanistic target of rapamycin complex 1; p70S6K, p70 S6 kinase 1; p90RSK, p90 ribosomal protein S6 kinase; PI3K, phosphoinositide 3-kinase; PRAS40, proline-rich AKT substrate 40 kDa; PtdIns(4,5)P2, phosphatidylinositol 4,5-biphosphate; PTEN, phosphatase and tensin homolog; Raptor, regulatory-associated protein of mTOR; REDD1, regulated in development and DNA damage responses 1; Rheb, Ras homolog enriched in brain; rpS6, ribosomal Protein S6; TAZ, transcriptional coactivator with PDZ-binding motif; TSC, tuberous sclerosis complex; YAP, Yes-associated protein 1.
RIBOSOMAL BIOGENESIS
A ribosome is a protein- and RNA-containing (ribosomal RNA–rRNA) molecular machine that plays an indispensable role in protein translation. A translationally competent ribosome (80S) contains two subunits (one large (60S) and one small (40S)), formed by the intricate association of over 80 ribosomal-associated proteins and 4 rRNAs (44). Ribosome biogenesis consumes a large proportion of cellular energy and is the only molecular process requiring coordinated activation of all three RNA polymerases (45). The RE-induced increase in MPS occurs via two mechanisms (46). Increased translational efficiency is an increased rate of messenger RNA (mRNA) translation with a fixed pool of ribosomes, whereas increased translational capacity occurs when increased numbers of ribosomes are available to translate mRNA. Importantly, mTOR-dependent and mTOR-independent mechanisms regulate translational efficiency and capacity (39).
Ribosomal biogenesis has emerged as a regulatory of RET-induced skeletal muscle hypertrophy (47). Given that rRNA makes up ~85% of total RNA (48), any change in total RNA concentration reflects changes in ribosomal biogenesis. Work in preclinical synergist ablation (SA) models of skeletal muscle overload demonstrate that increased total RNA concentration is associated with skeletal muscle hypertrophy (47). Acknowledging that SA models are pertinent for identifying potential cellular and molecular mechanisms regulating rapid skeletal muscle hypertrophy, the extent of hypertrophy with SA is extreme and in the range of 40%–70% hypertrophy within days (49) to weeks (47). Such rates of hypertrophy do not reflect magnitudes of muscle growth observed in human models of RET. Work from animal models (39) suggests that both translational efficiency and capacity are important in sustaining increases in MPS. However, recent work from Kotani et al. (50) showed no increase in MPS, although three bouts of RE was sufficient to increase ribosomal content.
In RE-trained humans, one bout of RE resulted in no measurable increase in total RNA content, despite elevated rates of MPS (46). Indeed, markers of ribosomal gene expression and transcription plateau after ~2 wk of RET (51) and, in some instances, return to baseline after 12 wk of RET (52). It raises the possibility that the rise in ribosomal content in response to unaccustomed exercise is initially rapid and is nonspecific to the exercise stimulus. Nonetheless, as RET progresses, ribosomal content declines, as does non–stimulus-specific mRNA content, the protein synthetic response becomes more efficient and specific to the exercise stimulus, and translational efficiency is elevated (Fig. 3) (53). Brook et al. (54) recently observed integrated (i.e., days to weeks) RNA synthesis, which would predominantly reflect ribosomal biogenesis, was increased above basal rates over the 0- to 6-wk period with RET, whereas MPS was not significantly increased above basal level during this period; however, this observation does not necessarily mean that ribosomal biogenesis is not relevant for RET-induced skeletal muscle hypertrophy (55). In sum, protein synthetic responses and transcriptional programs rapidly adapt to the RET stimulus, and further increasing translational capacity would not be required and would likely decline. The pertinent question is, do underlying changes in translational capacity with RET limit skeletal muscle hypertrophy?
FIGURE 3.
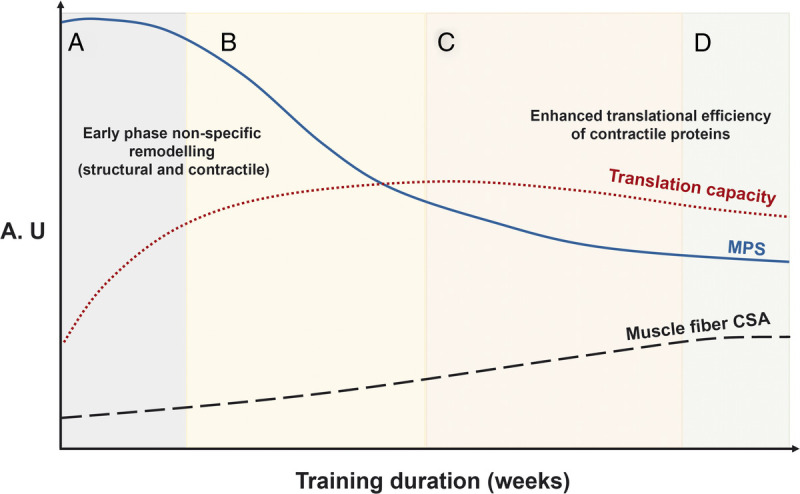
A proposed framework of changes in MPS, translational capacity, and muscle fiber CSA in response to RET. The overarching concept is that initial increases in MPS are a biological response to support remodeling of damaged muscle protein and eventually muscle hypertrophy. A, The early-stage increases in MPS are sustained partly by a concomitant elevated translational capacity to support the remodeling of damaged structural and contractile elements of the muscle proteome. B, After the attenuation of exercise-induced muscle damage, there is a reduction in the contribution of MPS to the remodeling of proteins related to the structural and architectural apparatus toward contractile muscle proteins. C, After a relatively short period of time, the rates of MPS are subsequently regulated by the adaptive increase in translational efficiency. D, The result is a detectable increase in skeletal muscle size and mass. All these responses are designed to support an expansion of the muscle protein pool, that is, fiber CSA. The schematic and legend are adapted (with permission) from McGlory et al. (53). A.U., arbitrary units.
RET may lead to a heterogenous hypertrophic response across individuals. Phillips et al. (56) clustered individuals who had completed 20 wk of RET into quartiles of RET-induced changes in lean body mass and demonstrated that individuals with the greatest hypertrophy had a downregulation of genes encoding ribosomal proteins. In addition, several studies demonstrated similar increases in RNA content between individuals who show no change, or a profound increase in skeletal muscle hypertrophy, after RET (57,58). Furthermore, the genes encoding rRNAs (45S and 5S) are tandemly repeated, meaning individuals have numerous copies of rRNA genes; interestingly, there seems to be significant individual heterogeneity in rDNA copy number (59). Figueiredo et al. (60) demonstrated that rDNA copy number was positively correlated with 45S pre-rRNA expression 24 h after a bout of RE. Furthermore, after a bout of RE, hypomethylation was observed at rDNA enhancer(s) sites and binding domains for the transcription factor MYC (60), which is implicated in RNA polymerase I activity and ribosome biogenesis. A limitation with the studies mentioned previously is that the lack of a control group makes it difficult to discern how much change is due to the intervention (i.e., RET), and how much change is simply due to random error (61,62). Nonetheless, although the work is suggestive that differential responders to RET exist and that ribosomal biogenesis may be an important determinant for explaining differential responders to RET, the current data are, in our view, inconclusive, and future work is required.
GENE EXPRESSION
With the advent of “omics” technologies providing a global and unbiased perspective on understanding molecular transducers of skeletal muscle adaptations, we know that exercise results in changes in the abundance of more than 2000 gene transcripts (of a possible 45,000 known genes) (63). Also, the changes in abundance (64) and the ratio of posttranslational modifications of proteins in skeletal muscle can be detected (e.g., ~10,000 phosphorylation sites) (43). The incorporation of next-generation sequencing (i.e., RNA sequencing) to correctly interrogate the breadth and the complexity of the mammalian transcription is limited (65); for example, the top 1% of most highly expressed protein-coding genes commonly encompass up to 40% of sequencing reads (66). Furthermore, grouping differentially expressed genes into functional categories (e.g., Gene Ontology, Ingenuity pathway analysis) without utilizing a robust statistical approach to account for sampling biases can make any analysis unreliable (67). However, one can reliably quantify 30–40,000 RNA species with microarray technology (63), and when modeled properly, the variation in RNA could explain up to 73% of protein abundance changes (68).
Pillon et al. (69) used published transcriptomic profiling data sets (n = 66) with more than 1000 individuals and demonstrated 2000 genes affected by RET. Also, Gene Ontology analysis characterized that RET mainly upregulated the mRNA genes involved in extracellular matrix remodeling. However, despite the large sample size, Pillon et al. (69) assessed changes in gene expression in response to a RET regime without incorporating physiological changes, including, critically, skeletal muscle hypertrophy. Thus, whether these changes in gene abundance play a role in hypertrophy is unknown. Indeed, Raue et al. (70) identified that over 600 genes correlated with muscle growth and strength changes after 12 wk of RET. However, many of the growth-related genes were generic features of exercise adaptation(s) (70) and not specific to RET-induced skeletal muscle hypertrophy, per se (56). Rather than averaging transcriptional responses across a cohort of individuals, we propose that if we leverage individual responses (i.e., skeletal muscle hypertrophy) to a RET regime, one can determine the transcriptional signature specific to skeletal muscle hypertrophy.
We recently discovered a set of 141 genes correlated with the muscle growth response to chronic muscle loading in humans (n = 100) (63). The signature showed that muscle loading regulated the untranslated regions (UTR) of mRNA (length of their 3′ or 5′UTR), and this regulated-UTR length was closely correlated with muscle growth, despite levels of mRNA remaining unchanged (>1000 genes) (63). For example, the increase in length of BCAT2 3′UTR or EXT1 5′UTR was strongly related to gain in muscle mass after RET. Ours was the first study linking UTR regulatory events to skeletal muscle hypertrophy via RET; thus, it provided potential clues to the reported discordance between mRNA and corresponding protein levels (64). Also, performing within-individual paired muscle tissue analysis in this study strengthened the reliability of the obtained results by reducing the response heterogeneity by ~40% (71). Our study identified that RET activated the genes associated with extracellular matrix remodeling, angiogenesis, and mitochondria (e.g., FKBP1A, BCAT2, NID2) as central pathways for muscle growth (63). Collectively, utilizing transcriptome technology and leveraging individual heterogeneity in response to RET may help determine molecular regulators for RET-induced skeletal muscle hypertrophy. Nonetheless, the best approach to determine the molecular responses to RET will be to perform reliable gene expression profiling that is complementary to reliable high-throughput protein expression methods.
RE-INDUCED ACUTE CHANGES IN SYSTEMIC ANABOLIC HORMONES
Canonical ostensibly anabolic hormones (e.g., testosterone, growth hormone (GH) and its various isoforms, and IGF-1), the concentrations of which are moderately (usually well within the diurnal variation of the hormone) and transiently increased for 15–30 min after RE, have been proposed to be internal stimuli having causative roles in RET-induced skeletal muscle hypertrophy (72). However, despite numerous studies designed to probe this question directly, our group and others have found no support for the thesis that acute changes in serum anabolic hormones induced by RE are mechanistically responsible for skeletal muscle hypertrophy (28,73) or increments in MPS (74). Notably, serum cortisol (i.e., a catabolic hormone for skeletal muscle) was the only hormone shown to be associated with the change in type II fiber CSA resulting from RET (75).
Hypotheses for the potential role of acute changes in anabolic hormones mediating skeletal muscle hypertrophy originate from the observation that RE is an effective physiological stimulus for GH release (76). During an RE session, serum GH levels increase 10–20 min after initiation and peak at the end of the RE, returning to baseline values about 30 min post-RE (76). A relevant question is whether GH is a mediator of muscular growth at all? For example, GH infusion studies mimicking the response to RE show stimulation of MPS and decrement in MPB (77). However, RE-induced increase in serum GH was not associated with MPS (78). Furthermore, there was no additional effect in quadriceps protein synthesis and circumference during a RET program even when young men administered 40 μg·kg−1 of GH for 12 wk (79). Similarly, one bout of RE increased serum IGF-1 levels from 40–50 (resting levels) to 60–70 nM (74). However, the increased IGF-1 levels returned to the basal level within 30 min of post-RE (76), and there was no correlation between systemic changes in the IGF-1 level and MPS (74) or skeletal muscle hypertrophy over time (29,74). In addition, IGF-1 (15 μg·kg−1·d−1) administered for 1 yr in older women, who usually have lower basal GH and IGF-1 than young adults, did not change body composition (or any other measured outcome variable) compared with a placebo group (80). Therefore, changes in serum GH or IGF-1 in response to RE or exogenous administration do not seem to influence skeletal muscle hypertrophy.
Sex steroids are anabolic hormones that have been repeatedly investigated in skeletal muscle hypertrophy studies. Testosterone is an androgenic hormone, and previous studies have been shown that exogenous administrations of testosterone in supraphysiological doses to healthy eugonadal men (81,82) and replacement doses to hypogonadal men (83,84) significantly increase muscle mass and lean body mass. Also, administration of testosterone adjuvant to RET promoted muscle mass increase in older adults who have lower baseline levels of endogenous testosterone (85). RE endogenously increases the systemic concentration of testosterone by 2–4 times above baseline for ~15–30 min in healthy young men (86). Contrary to exogenous testosterone administration, this transient ~30 min spike in serum testosterone has a minimum impact on daily testosterone physiological fluctuation, and it is far lower (4- to 6-fold) than the concentrations reached by exogenous administrations of testosterone (Fig. 4) (86). Furthermore, we have repeatedly shown no association between changes in systemic testosterone concentrations and skeletal muscle hypertrophy response to RET (73,75). Instead, we have shown that androgen receptor (AR) content in skeletal muscle seems more relevant as a variable to predict the hypertrophic response to RET (73). In addition, it has been shown that RE increases AR binding to DNA, improving anabolic signaling (89). It is also worth noting that healthy eugonadal women, with ~10-fold lower circulating testosterone than men, show similar relative hypertrophy and strength gains in response to RET (90), which is an observation that is difficult to reconcile with testosterone being a mechanistically important, rather than a possibly permissive hormone in RET-induced skeletal muscle hypertrophy.
FIGURE 4.
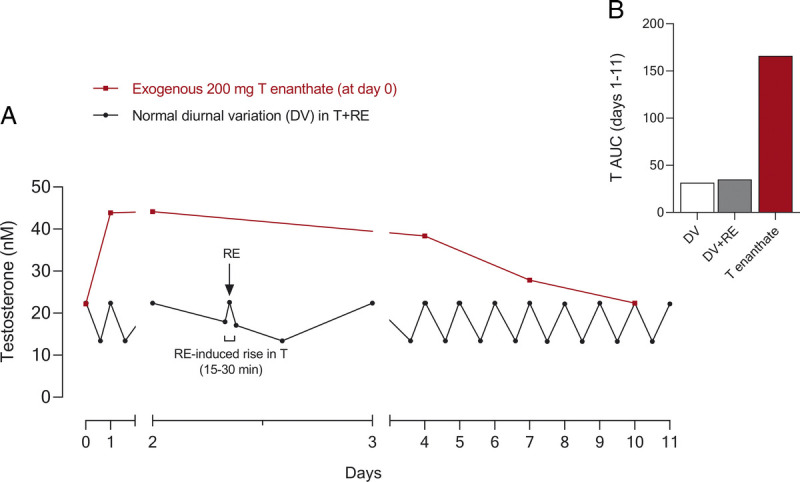
A comparison of the changes in exogenous vs endogenous testosterone in serum. A, Schematic of the circadian changes of serum testosterone throughout 11 d showing the effect of one bout of RE in healthy young men (RE-induced ~30 min testosterone peak; based on data from Willoughby and Taylor [87]). In addition, a representation of the effect of an intramuscular testosterone injection (200 mg testosterone enanthate) vs a schematic of the normal diurnal variation in testosterone concentrations throughout a given week (based on data from Dobs et al. (88)). B, Cumulative AUC of serum testosterone over the first 7 d comparing 1) DV changes, 2) DV + 1 bouts of RE, and 3) 200 mg T enanthate. The schematic is adapted (with permission) from Schroeder et al. (86). AUC, area under the curve; DV, diurnal variation; T, testosterone.
Estrogen may also be a relevant hormone acting to augment hypertrophy by decreasing muscle damage caused by exercise and upregulating anabolic signaling pathways relevant to muscle anabolism (e.g., insulin/IGF-1 and PI3K/Akt signaling) (72). However, there is no consensus on the role of estrogen in RET-induced skeletal muscle hypertrophy. Variables like the menstrual cycle phase and the testing of subjects before or after menopause are certain variables to control and investigate in future studies (91). We speculate, however, that the role of estrogen in skeletal muscle hypertrophy will follow the pattern of other androgenic hormones and be related to intrinsic muscle variables (receptor density and postreceptor signaling) as we and others have observed with testosterone and AR content (28,92).
RE-INDUCED MUSCLE DAMAGE
Muscle damage can significantly increase inflammatory mediators in skeletal muscle and induce satellite cell (SC) activation (93), affecting muscle regenerative processes. The gold standard method for assessing RE-induced muscle damage is via examination of ultrastructural changes, including z-band streaming or muscle swelling (edema) (94). Many, however, rely on indirect measures of proxy markers such as elevation in muscle soreness and creatine kinase (CK) activity in the blood, which is not a measure of damage per se. There is controversy regarding the validity of raised serum CK levels after RET as a relevant marker of myofiber damage and its relationship to MPS and hypertrophy (95). Damas et al. (95) demonstrated that MPS, in addition to markers of muscle damage (serum CK activity, indirect) and Z-disk streaming (direct), was highest after RE in untrained persons early in a RET program; however, neither measure was well correlated with MPS or RET-induced skeletal muscle hypertrophy. Nevertheless, after 10 wk of RET, the acute MPS was correlated with the degree of muscular hypertrophy observed, despite significantly lower muscle damage (95). Thus, muscle damage, which is progressively mitigated with chronic RE, is a poor proxy for MPS and skeletal muscle hypertrophy (95).
After RE, SC responds by activating the myogenic program to proliferate and either return to quiescence or differentiate, donating their nuclei to the existing myofibers (96). Damas et al. (97) reported increased SC content during the first week of RET, showing the more significant RET-induced muscle damage. However, there was no correlation between the SC content and MPS throughout RET (97), suggesting that SC may serve a more prominent role in myofiber repair during the initial stages of RET than the latter stages of RET showing muscle hypertrophy, which is contrary to previous dogma that muscle damage is concomitant and a prerequisite for muscle hypertrophy (98). Recent work from Roman et al. (99) demonstrated that local muscle damage can be repaired independent of SC through a mechanism related to nuclear migration. This alternative myofiber-autonomous repair mechanism challenges the role that SC may play in acute local muscle damage as myonuclear migration was sufficient for the local delivery of mRNAs necessary for efficient repair of the damaged sarcomeres (99).
Muscle damage induced by RE triggers an inflammatory response characterized by the release of several mediators (93) and proinflammatory cytokines (e.g., tumor necrosis factor alpha (TNF-α) and interleukin (IL) 1β)) that are known regulators of proproteolytic activity in skeletal muscle. In comparison, preclinical studies have shown that IL-1β and TNF-α have proproliferative effects in myoblast cells through mechanisms involving IL-6 and prostaglandins (100), and myotubes treated with IL-6 upregulate mTORC1 signaling and myotube protein synthesis (101). Nevertheless, when tested in humans, daily ingestion of anti-inflammatory medication during RET was reported to have no effects on muscle thickness in young (102) and hypertrophy in older adults (103). Our group found a correlation between the concentration of the IL-6 post-RE and changes in myofiber CSA in subjects submitted to RE (28). Therefore, inflammatory mediators might play a role in skeletal muscle hypertrophy, but this field demands further research exploring intrinsic and local mechanisms.
METABOLITES
Metabolites produced during muscular contractions have been posited to be potential internal determinants of RET-induced skeletal muscle hypertrophy (104). Because marked changes in metabolite concentrations always accompany RE (or any other form of muscle contraction), several different molecules are proposed to be involved in gene expression (105) and distinct protein signaling pathways (104). However, no causative research shows that any metabolite is a viable signaling candidate for triggering skeletal muscle anabolism in humans.
Elevated lactate, hydrogen ion, inorganic phosphate and reduced phosphocreatine are all elevated with muscle contraction (106). Based on this mechanism, it has been suggested that the reduced blood pH may promote muscle growth by potentiating GH release and increasing motor unit (MU) recruitment to maintain force output (107). Nonetheless, as pointed out in a preceding section of this review (see RE-Induced Acute Changes in Systemic Anabolic Hormones), changes in serum levels of GH (and its various isoforms) after RE are not correlated, mechanistically incongruent, and with stimulation of MPS (we note that collagen-predominant tissues like bone are markedly sensitive to GH) or hypertrophy (73,74). Furthermore, no additional muscle hypertrophy was observed after RET with blood flow restriction—the RET model used to elevate metabolites production by limiting blood flow—compared with traditional RET (108). Rather, RET with blood flow restriction showed weaker higher threshold MU recruitment (109). In addition, increased lactate concentration in plasma does not induce an additional increase in MPS (74) or CSA by MRI (76) after RE and RET, respectively. Overall, little-to-no evidence exists to suggest that any single metabolite, or even a plausible combination, influences RET-induced anabolic signaling or hypertrophy.
Reactive oxygen species and nitric oxide have been mentioned as potential mediators of skeletal muscle hypertrophy by activating MAPK signaling pathways and SC, respectively (110). In addition to recognizing that evidence supporting this claim is scarce, it is critical to consider the vast regulatory networks involved with RE-induced activation of MAPK (111) and SC (112) activated by mechanotransduction. Based on existing evidence, MAPK and SC activation should be recognized for their anabolic effect rather than reactive oxygen species and nitric oxide production per se.
Given that there are over 4200 metabolites in human serum, any metabolite may be directly/indirectly associated with anabolic signaling for muscle growth. However, the exercises that result in a lesser degree of skeletal muscle hypertrophy relative to RE (e.g., endurance or higher-intensity interval or sprint exercise) also result in significant increments in several metabolite concentrations similar to, or greater than, RE (113,114), further suggesting that metabolites are not the primary drivers of muscle hypertrophy.
CLINICAL ILLNESS AND AGING AND THE MECHANISMS INVOLVED IN MUSCLE HYPERTROPHY
As opposed to being the exclusive domain of athletes and bodybuilders, it is abundantly clear that RET is a useful therapeutic modality in clinical care. Importantly, we are beginning to gain critical mechanistic insight into how RET can affect diseased muscle to impart a less catabolic phenotype and greatly improve clinical outcomes. We highlight here some relevant and newer advances in these exciting areas.
Muscle loss in clinical illness(es) (e.g., cancer, COPD, cardiovascular disease, sepsis, and burns) and aging is, in part, a result of rates of MPB chronically exceeding rates of MPS. Specifically, proteolysis through the ubiquitin–proteasome system (UPS) has been considered a primary mechanism of muscle loss during clinical illness (115). Concomitantly, reduced PI3K–AKT/mTORC1 pathway activity has been considered the main mechanism underpinning an attenuated MPS response (115). Our understanding of mechanistic processes underlying muscle loss during illness is mostly derived from animal studies. Thus, much remains to be discovered about these complex mechanisms, particularly in humans, and there are currently no successful pharmacological treatments to prevent muscle wasting. However, previous studies have reported that RET can increase lean body mass, or prevent further losses, in several clinical populations, including cancer patients and survivors (1), patients with renal disease (116), and patients with Parkinson’s disease (117). Notably, RET counteracts skeletal muscle wasting and thus may be characterized as mitigation of muscle loss rather than true hypertrophy in several clinical populations. Also, previous studies highlighted the association between low muscle mass and poor clinical outcomes, such as treatment tolerability and survival in cancer patients (118,119). Understanding the mechanisms driving RET-induced skeletal muscle hypertrophy could improve therapeutics to improve clinical outcomes during clinical illness.
In many diseases (cancer, sepsis, diabetes, COPD, heart failure, and burns), increased systemic concentrations of inflammatory markers (e.g., TNF-α, IL-1β, and IL-6) have been shown to coordinate the changes in different mechanisms regulating muscle protein turnover and muscle regeneration (4). The increase in proinflammatory cytokines may promote proteolysis by stimulating the UPS and decreasing MPS (120). Also, high systemic levels of proinflammatory cytokines can negatively impact ribosomal biogenesis in skeletal muscle and myogenesis (121). Although RE-induced inflammation seems trivial in regulating the hypertrophic response in healthy young (102) and older adults (103), exposure to substantially higher or chronically elevated concentrations of systemic inflammatory markers during illness is associated with lower muscle mass or blunted MPS response (122). Thus, reducing the resting concentration of proinflammatory agents and elevating circulating levels of anti-inflammatory cytokines (e.g., IL-1 receptor antagonist, soluble TNF-receptor, IL-10), such as with regular RET, may attenuate muscle loss in clinical illness.
Elevated systemic levels of glucocorticoids (e.g., cortisol) have been observed in many diseases (cancer, sepsis, diabetes, renal disease, COPD, and heart failure), and this increment can happen because of exogenous therapeutic administration or endogenous cortisol secretion as part of the stress response to the disease state (4). The excessive glucocorticoid level in systemic circulation activates protein breakdown signaling, including FOXO1, FOXO3, NF-κB, and reduces PI3K–AKT/mTORC1 signaling pathway activity, thereby inducing muscle atrophy (123). However, 7-wk RET increased thigh CSA measured by computer tomography in renal transplants patients receiving prednisone therapy (124). Also, because a diminished capillary number was shown in such patients, the previous study suggested the reduced muscle perfusion (i.e., delivery of amino acids and oxygen) as a reason for atrophy during clinical illness (124). Our laboratory also found the lower capillary number and reduced angiogenesis-related markers protein expression in coronary artery disease patients with reduced SC number and abnormal muscle fiber–type shifting (125). However, 4 and 12 wk of stair climbing-based high-intensity interval training improved the compromised muscle characteristics (125).
In addition to altered metabolism, clinical illness patients experience significantly reduced physical activity. Muscle disuse can negatively modulate skeletal muscle remodeling leading to muscle atrophy by decreasing the anabolic signals activated by mechanical stimuli (i.e., mechanotransduction), such as the mTOR signaling pathway (3). Disuse atrophy has been linked to anabolic resistance in response to hyperaminoacidemia (i.e., feeding) (3). Furthermore, in vitro and animal model data (126) indicate that ceramide accumulated during inactivity may inhibit factors downstream of PA (see the previous section on mechanosensors). However, regular exercise training improved MPS response and reduced ceramide in obese patients with higher muscle ceramide content (127). Reduced physical activity during clinically illness attenuates the activation of anabolic pathway downstream (e.g., HIPPO and mTOR) that can be promoted by mechanical stimuli (i.e., loss of mechanotransduction). Also, the increased level of inflammatory markers and glucocorticoids decrease the activation of anabolic signaling pathway (PI3K–AKT/mTOR signaling) and inactivate FOXO transcription factors, thereby promoting gene expression–associated protein degradation. Although more complex mechanisms are involved in the muscle loss during clinical illness, improving the deteriorated variables, explained previously, that may be able to be improved by RET could be an effective strategy to maintain muscle mass in disease patients.
Although aging is not an illness per se, there are undoubtedly factors common in aging and certain disease states that are likely playing a role in the age-related sarcopenic loss of muscle mass such as increased inflammation factors (128) and loss of proteostasis (129). Besides that, although multifactorial in origin, reduced number and regenerative capacity of SC (130,131), fiber denervation (132), and deregulated intracellular communications (e.g., GH/IGF-1, testosterone, and myostatin) (72,133) have been considered as a cause of muscle loss in aging. Although a deep exploration of the mechanisms that underpin sarcopenic muscle loss cannot be undertaken here, we refer the reader to a recent review on the topic (134). A common finding that occurs with aging and in many muscle wasting disease states is that the response of MPS to normally robust anabolic stimuli is attenuated. This so-called anabolic resistance of MPS, noted in response to RE and protein ingestion (i.e., hyperaminoacidemia), likely relates to cellular mechanisms and signaling responses being attenuated. In addition, a persistent but low-grade, sterile inflammatory state (inflammaging) is likely also playing a role in suppressing MPS and possibly increasing proteolysis by activating UPS (135). Previous studies have shown that these age-related negative adaptations in skeletal muscle could be alleviated by performing RET, resulting in an increase in SC number (130), innervating MU (136), and reduction of inflammation (137). Also, despite the presence of anabolic resistance and low-grade inflammation, older persons almost invariably experience gains in strength with RET but a lesser degree of hypertrophy relative to their younger-age counterparts (138). Nonetheless, RET is a powerful consistent stimulus that should be a primary form of exercise prescribed to counteract age- and disease-related muscle loss. Figure 5 outlines several factors that are likely to play a role in age-related sarcopenic muscle loss, including, in our view, a primary contributor, which is periodic disuse events (2).
FIGURE 5.
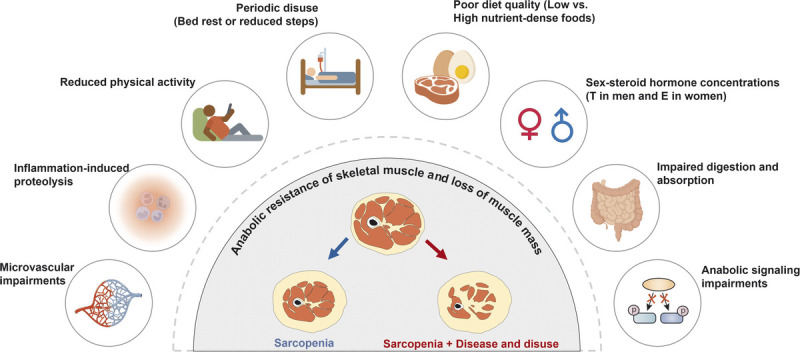
Representative factors that contribute to the anabolic resistance of skeletal muscle with aging and disease. This schematic is adapted (with permission) from McKendry et al. (2). E, estrogen; T, testosterone.
CONCLUSIONS AND FUTURE DIRECTIONS
Skeletal muscle hypertrophy is a complex process resulting from an intricate interplay between external and internal variables (Fig. 1), and RET is the most potent external variable that initiates a cascade of events that induce muscle hypertrophy (Fig. 6). Thus, understanding the internal variables activated by RE could provide valuable insight to induce skeletal muscle hypertrophy. However, compared with preclinical models, determining mechanisms from human studies is more challenging because of various factors (e.g., limitation of muscle tissue volume, experimental technical difficulty, ethics, and more). Nevertheless, mechanotransduction, translational capacity, and transcription seem to be very promising in identifying key mechanisms for RET-induced skeletal muscle hypertrophy in humans. Also, the anabolic mechanisms regulated by RET could be an important target to maintain muscle mass during disease and aging in general, although there are more complex mechanisms interfering with muscle homeostasis.
FIGURE 6.
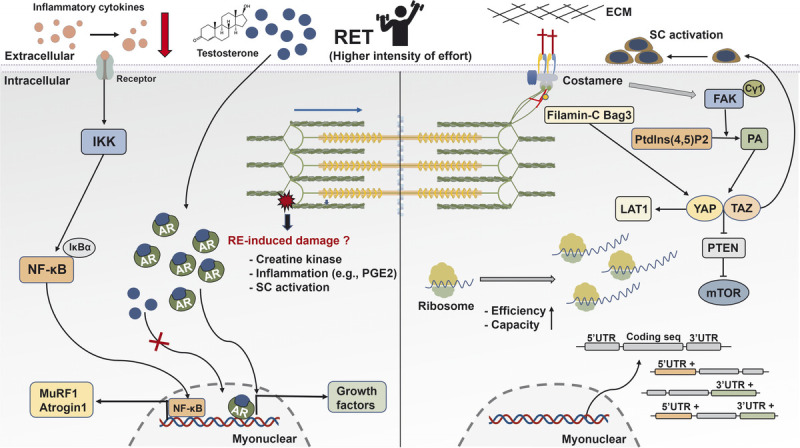
Schematic illustration of the mechanisms underpinning skeletal muscle hypertrophy response to RET. After mechanical stimuli, phospholipase Cγ1 colocalizes around FAK and catalyzes the conversion of Ptdlns(4,5)P2 to PA that activates the HIPPO pathway (YAP and TAZ). YAP and TAZ promote the mTOR signaling pathway via the suppression of PTEN, regulate SC activation, and mediate the expression of L-type amino acid transporter 1 (LAT1). Interaction between Filamin-C and Bag3 increases the activation of the HIPPO pathway. Translational capacity and efficacy via ribosome are essential determinants for muscle hypertrophy. Besides increased gene expression during RE, the UTR of mRNA is regulated and closely correlated with muscle growth. The AR content is associated with RET-induced hypertrophy, whereas RE-induced acute changes in serum testosterone show no association with muscle growth. In human trials, RE-induced increased CK and inflammation factors have no effects on muscle hypertrophy. However, RET reduces resting concentration of inflammation cytokines in aging and clinical illness. AR, androgen receptor; ECM, extracellular matrix; FAK, focal adhesion kinase; IκBα, NF-κB inhibitor alpha; IKK, IκB kinase; LAT1, L-type amino acid transporter 1; mTOR, mechanistic target of rapamycin; MuRF 1, muscle RING-finger protein 1; NF-κB, nuclear factor kappa light-chain enhancer of activated B; PGE2, prostaglandin E2; PTEN, phosphatase and tensin homolog; RE, resistance exercise; RET, resistance exercise training; SC, satellite cells; TAZ, transcriptional coactivator with PDZ-binding motif; UTR, untranslated region; YAP, Yes-associated protein 1.
The search for molecular signatures identifying the transcripts involved in skeletal muscle hypertrophy, particularly in clinical populations, creates several avenues for future investigation. Activation of mTOR is clearly a key component of muscle anabolism, but additional factors also seem to contribute to skeletal muscle hypertrophy beyond this protein kinase. Ribosomal biogenesis (i.e., translational capacity) and translational efficiency seem relevant and associated with acute RET responses in training-naive subjects, although the role of such variables in the long-term skeletal muscle hypertrophic response requires further research. Studies applying new methods, using in vivo measurement of rRNA synthesis, might bring additional input to assess the role of translational capacity in skeletal muscle hypertrophy, and endocrine-related factors, such as AR content in skeletal muscle and female sex hormones, may yet help us understand the role of these variables in skeletal muscle physiology and hypertrophy. More work is still required, but with the rapid development of technology, the up-to-date techniques in skeletal muscle, such as single-cell isolation and single-cell RNA-seq (139,140), could be considered to accelerate to uncover the mechanisms underpinning RET-induced skeletal muscle hypertrophy in humans.
Acknowledgments
S. M. P. thanks the Canada Research Chairs Program, the Canadian Institutes of Health Research, and Natural Science and Engineering Research Council of Canada for their support during the development of this work. E. A. N. is a tier 2 research productivity fellow supported by the Brazilian National Council for Scientific and Technological Development (CNPq; grant number 308584/2019-8). B. S. C. and A. C. Q. T. are supported by Alexander Graham Bell Doctoral Canada Graduate Scholarships. J. C. M. is supported by an Ontario Graduate Scholarship.
S. M. P. reports grants from US National Dairy Council and a research contract with Roquette, during the conduct of the study; personal fees from US National Dairy Council; and nonfinancial support from Enhanced Recovery, outside the submitted work. In addition, S. M. P. has a patent Canadian 3052324 issued to Exerkine and a patent US 20200230197 pending to Exerkine, but reports no financial gains from these patents. C. L., E. A. N., B. S. C., J. C. M., and A. C. Q. T. declare no conflicts of interest. The results of the present study do not constitute endorsement by the American College of Sports Medicine. The authors declare that the study results are presented clearly, honestly, and without fabrication, falsification, or inappropriate data manipulation.
Contributor Information
CHANGHYUN LIM, Email: limc16@mcmaster.ca.
EVERSON A. NUNES, Email: nunese1@mcmaster.ca.
BRAD S. CURRIER, Email: currierb@mcmaster.ca.
JONATHAN C. MCLEOD, Email: mcleoj2@mcmaster.ca.
AARON C. Q. THOMAS, Email: thomasac@mcmaster.ca.
REFERENCES
- 1.Koeppel M, Mathis K, Schmitz KH, Wiskemann J. Muscle hypertrophy in cancer patients and survivors via strength training. A meta-analysis and meta-regression. Crit Rev Oncol Hematol. 2021;163:103371. [DOI] [PubMed] [Google Scholar]
- 2.McKendry J, Stokes T, McLeod JC, Phillips SM. Resistance exercise, aging, disuse, and muscle protein metabolism. Compr Physiol. 2021;11(3):2249–78. [DOI] [PubMed] [Google Scholar]
- 3.Glover EI Phillips SM Oates BR, et al. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol. 2008;586(24):6049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14(1):58–74. [DOI] [PubMed] [Google Scholar]
- 5.Wackerhage H, Schoenfeld BJ, Hamilton DL, Lehti M, Hulmi JJ. Stimuli and sensors that initiate skeletal muscle hypertrophy following resistance exercise. J Appl Physiol (1985). 2019;126(1):30–43. [DOI] [PubMed] [Google Scholar]
- 6.Gloerich M ten Klooster JP Vliem MJ, et al. Rap2A links intestinal cell polarity to brush border formation. Nat Cell Biol. 2012;14(8):793–801. [DOI] [PubMed] [Google Scholar]
- 7.Meng Z Qiu Y Lin KC, et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature. 2018;560(7720):655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harvey K, Tapon N. The Salvador–Warts–Hippo pathway—an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7(3):182–91. [DOI] [PubMed] [Google Scholar]
- 9.Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886–97. [DOI] [PubMed] [Google Scholar]
- 10.Watt KI Judson R Medlow P, et al. Yap is a novel regulator of C2C12 myogenesis. Biochem Biophys Res Commun. 2010;393(4):619–24. [DOI] [PubMed] [Google Scholar]
- 11.Jeong H Bae S An SY, et al. TAZ as a novel enhancer of MyoD-mediated myogenic differentiation. FASEB J. 2010;24(9):3310–20. [DOI] [PubMed] [Google Scholar]
- 12.Watt KI Turner BJ Hagg A, et al. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat Commun. 2015;6:6048. [DOI] [PubMed] [Google Scholar]
- 13.Goodman CA, Dietz JM, Jacobs BL, McNally RM, You JS, Hornberger TA. Yes-associated protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015;589(13):1491–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen CG, Ng YL, Lam WL, Plouffe SW, Guan KL. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015;25(12):1299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A. 2006;103(12):4741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furst DO, Osborn M, Nave R, Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol. 1988;106(5):1563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powers K, Schappacher-Tilp G, Jinha A, Leonard T, Nishikawa K, Herzog W. Titin force is enhanced in actively stretched skeletal muscle. J Exp Biol. 2014;217(Pt 20):3629–36. [DOI] [PubMed] [Google Scholar]
- 18.Puchner EM Alexandrovich A Kho AL, et al. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci U S A. 2008;105(36):13385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruger M, Kotter S. Titin, a central mediator for hypertrophic signaling, exercise-induced mechanosignaling and skeletal muscle remodeling. Front Physiol. 2016;7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishikawa K, Lindstedt SL, Hessel A, Mishra D. N2A Titin: signaling hub and mechanical switch in skeletal muscle. Int J Mol Sci. 2020;21(11):3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Centner T Yano J Kimura E, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306(4):717–26. [DOI] [PubMed] [Google Scholar]
- 22.Gregorio CC, Perry CN, McElhinny AS. Functional properties of the titin/connectin-associated proteins, the muscle-specific RING finger proteins (MURFs), in striated muscle. J Muscle Res Cell Motil. 2005;26(6–8):389–400. [DOI] [PubMed] [Google Scholar]
- 23.Ulbricht A Eppler FJ Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013;23(5):430–5. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman NJ Parker BL Chaudhuri R, et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015;22(5):922–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. 2021;101(3):1371–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reidy PT Borack MS Markofski MM, et al. Post-absorptive muscle protein turnover affects resistance training hypertrophy. Eur J Appl Physiol. 2017;117(5):853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchell CJ, Churchward-Venne TA, Bellamy L, Parise G, Baker SK, Phillips SM. Muscular and systemic correlates of resistance training–induced muscle hypertrophy. PLoS One. 2013;8(10):e78636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell CJ Churchward-Venne TA Parise G, et al. Acute post-exercise myofibrillar protein synthesis is not correlated with resistance training–induced muscle hypertrophy in young men. PLoS One. 2014;9(2):e89431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damas F Phillips SM Libardi CA, et al. Resistance training–induced changes in integrated myofibrillar protein synthesis are related to hypertrophy only after attenuation of muscle damage. J Physiol. 2016;594(18):5209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator–Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodson N, Philp A. The importance of mTOR trafficking for human skeletal muscle translational control. Exerc Sport Sci Rev. 2019;47(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Z Moore DR Hodson N, et al. Resistance exercise initiates mechanistic target of rapamycin (mTOR) translocation and protein complex co-localisation in human skeletal muscle. Sci Rep. 2017;7(1):5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abou Sawan S van Vliet S Parel JT, et al. Translocation and protein complex co-localization of mTOR is associated with postprandial myofibrillar protein synthesis at rest and after endurance exercise. Physiol Rep. 2018;6(5):e13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hannaian SJ Hodson N Abou Sawan S, et al. Leucine-enriched amino acids maintain peripheral mTOR–Rheb localization independent of myofibrillar protein synthesis and mTORC1 signaling postexercise. J Appl Physiol (1985). 2020;129(1):133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodson N McGlory C Oikawa SY, et al. Differential localization and anabolic responsiveness of mTOR complexes in human skeletal muscle in response to feeding and exercise. Am J Physiol Cell Physiol. 2017;313(6):C604–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abou Sawan S, Mazzulla M, Moore DR, Hodson N. More than just a garbage can: emerging roles of the lysosome as an anabolic organelle in skeletal muscle. Am J Physiol Cell Physiol. 2020;319(3):C561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogasawara R, Jensen TE, Goodman CA, Hornberger TA. Resistance exercise–induced hypertrophy: a potential role for rapamycin-insensitive mTOR. Exerc Sport Sci Rev. 2019;47(3):188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West DW Baehr LM Marcotte GR, et al. Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiol. 2016;594(2):453–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogasawara R Fujita S Hornberger TA, et al. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci Rep. 2016;6:31142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drummond MJ Fry CS Glynn EL, et al. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol. 2009;587(Pt 7):1535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogasawara R, Suginohara T. Rapamycin-insensitive mechanistic target of rapamycin regulates basal and resistance exercise–induced muscle protein synthesis. FASEB J. 2018;fj201701422R. [DOI] [PubMed] [Google Scholar]
- 43.Steinert ND Potts GK Wilson GM, et al. Mapping of the contraction-induced phosphoproteome identifies TRIM28 as a significant regulator of skeletal muscle size and function. Cell Rep. 2021;34(9):108796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henras AK, Plisson-Chastang C, O’Donohue MF, Chakraborty A, Gleizes PE. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6(2):225–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25(48):6384–91. [DOI] [PubMed] [Google Scholar]
- 46.Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA, Smith K. Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol (1985). 1992;73(4):1383–8. [DOI] [PubMed] [Google Scholar]
- 47.Kirby TJ, Lee JD, England JH, Chaillou T, Esser KA, McCarthy JJ. Blunted hypertrophic response in age skeletal muscle is associated with decreased ribosome biogenesis. J Appl Physiol (1985). 2015;119(4):321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Neil D, Glowatz H, Schlumpberger M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Curr Protoc Mol Biol. 2013; Chapter 4:Unit 4.19. [DOI] [PubMed] [Google Scholar]
- 49.Goldberg AL, Etlinger JD, Goldspink DF, Jablecki C. Mechanism of work-induced hypertrophy of skeletal muscle. Med Sci Sports. 1975;7(3):185–98. [PubMed] [Google Scholar]
- 50.Kotani T, Takegaki J, Tamura Y, Kouzaki K, Nakazato K, Ishii N. Repeated bouts of resistance exercise in rats alter mechanistic target of rapamycin complex 1 activity and ribosomal capacity but not muscle protein synthesis. Exp Physiol. 2021;106(9):1950–60. [DOI] [PubMed] [Google Scholar]
- 51.Hammarstrom D Ofsteng S Koll L, et al. Benefits of higher resistance-training volume are related to ribosome biogenesis. J Physiol. 2020;598(3):543–65. [DOI] [PubMed] [Google Scholar]
- 52.Nader GA von Walden F Liu C, et al. Resistance exercise training modulates acute gene expression during human skeletal muscle hypertrophy. J Appl Physiol (1985). 2014;116(6):693–702. [DOI] [PubMed] [Google Scholar]
- 53.McGlory C, Devries MC, Phillips SM. Skeletal muscle and resistance exercise training; the role of protein synthesis in recovery and remodeling. J Appl Physiol (1985). 2017;122(3):541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brook MS Wilkinson DJ Mitchell WK, et al. A novel D2O tracer method to quantify RNA turnover as a biomarker of de novo ribosomal biogenesis, in vitro, in animal models, and in human skeletal muscle. Am J Physiol Endocrinol Metab. 2017;313(6):E681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brook MS Wilkinson DJ Mitchell WK, et al. Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age-related anabolic resistance to exercise in humans. J Physiol. 2016;594(24):7399–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phillips BE Williams JP Gustafsson T, et al. Molecular networks of human muscle adaptation to exercise and age. PLoS Genet. 2013;9(3):e1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haun CT Vann CG Mobley CB, et al. Pre-training skeletal muscle fiber size and predominant fiber type best predict hypertrophic responses to 6 weeks of resistance training in previously trained young men. Front Physiol. 2019;10:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mobley CB Haun CT Roberson PA, et al. Biomarkers associated with low, moderate, and high vastus lateralis muscle hypertrophy following 12 weeks of resistance training. PLoS One. 2018;13(4):e0195203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gibbons JG, Branco AT, Yu S, Lemos B. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat Commun. 2014;5:4850. [DOI] [PubMed] [Google Scholar]
- 60.Figueiredo VC Wen Y Alkner B, et al. Genetic and epigenetic regulation of skeletal muscle ribosome biogenesis with exercise. J Physiol. 2021;599(13):3363–84. [DOI] [PubMed] [Google Scholar]
- 61.Atkinson G, Batterham AM. True and false interindividual differences in the physiological response to an intervention. Exp Physiol. 2015;100(6):577–88. [DOI] [PubMed] [Google Scholar]
- 62.Snapinn SM, Jiang Q. Responder analyses and the assessment of a clinically relevant treatment effect. Trials. 2007;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stokes T Timmons JA Crossland H, et al. Molecular transducers of human skeletal muscle remodeling under different loading states. Cell Rep. 2020;32(5):107980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson MM Dasari S Konopka AR, et al. Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 2017;25(3):581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deveson IW, Hardwick SA, Mercer TR, Mattick JS. The dimensions, dynamics, and relevance of the mammalian noncoding transcriptome. Trends Genet. 2017;33(7):464–78. [DOI] [PubMed] [Google Scholar]
- 66.Jiang L Schlesinger F Davis CA, et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21(9):1543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Timmons JA, Szkop KJ, Gallagher IJ. Multiple sources of bias confound functional enrichment analysis of global -omics data. Genome Biol. 2015;16(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li JJ, Biggin MD. Gene expression. Statistics requantitates the central dogma. Science. 2015;347(6226):1066–7. [DOI] [PubMed] [Google Scholar]
- 69.Pillon NJ Gabriel BM Dollet L, et al. Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat Commun. 2020;11(1):470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raue U Trappe TA Estrem ST, et al. Transcriptome signature of resistance exercise adaptations: mixed muscle and fiber type specific profiles in young and old adults. J Appl Physiol (1985). 2012;112(10):1625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacInnis MJ, McGlory C, Gibala MJ, Phillips SM. Investigating human skeletal muscle physiology with unilateral exercise models: when one limb is more powerful than two. Appl Physiol Nutr Metab. 2017;42(6):563–70. [DOI] [PubMed] [Google Scholar]
- 72.Gharahdaghi N, Phillips BE, Szewczyk NJ, Smith K, Wilkinson DJ, Atherton PJ. Links between testosterone, oestrogen, and the growth hormone/insulin-like growth factor axis and resistance exercise muscle adaptations. Front Physiol. 2020;11:621226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morton RW Sato K Gallaugher MPB, et al. Muscle androgen receptor content but not systemic hormones is associated with resistance training–induced skeletal muscle hypertrophy in healthy, young men. Front Physiol. 2018;9:1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.West DW Kujbida GW Moore DR, et al. Resistance exercise–induced increases in putative anabolic hormones do not enhance muscle protein synthesis or intracellular signalling in young men. J Physiol. 2009;587(Pt 21):5239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morton RW Oikawa SY Wavell CG, et al. Neither load nor systemic hormones determine resistance training–mediated hypertrophy or strength gains in resistance-trained young men. J Appl Physiol (1985). 2016;121(1):129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.West DW Burd NA Tang JE, et al. Elevations in ostensibly anabolic hormones with resistance exercise enhance neither training-induced muscle hypertrophy nor strength of the elbow flexors. J Appl Physiol (1985). 2010;108(1):60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fryburg DA, Louard RJ, Gerow KE, Gelfand RA, Barrett EJ. Growth hormone stimulates skeletal muscle protein synthesis and antagonizes insulin’s antiproteolytic action in humans. Diabetes. 1992;41(4):424–9. [DOI] [PubMed] [Google Scholar]
- 78.West DW, Burd NA, Staples AW, Phillips SM. Human exercise-mediated skeletal muscle hypertrophy is an intrinsic process. Int J Biochem Cell Biol. 2010;42(9):1371–5. [DOI] [PubMed] [Google Scholar]
- 79.Yarasheski KE, Campbell JA, Smith K, Rennie MJ, Holloszy JO, Bier DM. Effect of growth hormone and resistance exercise on muscle growth in young men. Am J Physiol. 1992;262(3 Pt 1):E261–7. [DOI] [PubMed] [Google Scholar]
- 80.Friedlander AL Butterfield GE Moynihan S, et al. One year of insulin-like growth factor I treatment does not affect bone density, body composition, or psychological measures in postmenopausal women. J Clin Endocrinol Metab. 2001;86(4):1496–503. [DOI] [PubMed] [Google Scholar]
- 81.Bhasin S Storer TW Berman N, et al. The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med. 1996;335(1):1–7. [DOI] [PubMed] [Google Scholar]
- 82.Young NR, Baker HW, Liu G, Seeman E. Body composition and muscle strength in healthy men receiving testosterone enanthate for contraception. J Clin Endocrinol Metab. 1993;77(4):1028–32. [DOI] [PubMed] [Google Scholar]
- 83.Bhasin S Storer TW Berman N, et al. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J Clin Endocrinol Metab. 1997;82(2):407–13. [DOI] [PubMed] [Google Scholar]
- 84.Katznelson L, Finkelstein JS, Schoenfeld DA, Rosenthal DI, Anderson EJ, Klibanski A. Increase in bone density and lean body mass during testosterone administration in men with acquired hypogonadism. J Clin Endocrinol Metab. 1996;81(12):4358–65. [DOI] [PubMed] [Google Scholar]
- 85.Gharahdaghi N Rudrappa S Brook MS, et al. Testosterone therapy induces molecular programming augmenting physiological adaptations to resistance exercise in older men. J Cachexia Sarcopenia Muscle. 2019;10(6):1276–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schroeder ET, Villanueva M, West DD, Phillips SM. Are acute post-resistance exercise increases in testosterone, growth hormone, and IGF-1 necessary to stimulate skeletal muscle anabolism and hypertrophy? Med Sci Sports Exerc. 2013;45(11):2044–51. [DOI] [PubMed] [Google Scholar]
- 87.Willoughby DS, Taylor L. Effects of sequential bouts of resistance exercise on androgen receptor expression. Med Sci Sports Exerc. 2004;36(9):1499–506. [DOI] [PubMed] [Google Scholar]
- 88.Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab. 1999;84(10):3469–78. [DOI] [PubMed] [Google Scholar]
- 89.Cardaci TD, Machek SB, Wilburn DT, Heileson JL, Willoughby DS. High-load resistance exercise augments androgen receptor–DNA binding and Wnt/beta-catenin signaling without increases in serum/muscle androgens or androgen receptor content. Nutrients. 2020;12(12):3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roberts BM, Nuckols G, Krieger JW. Sex differences in resistance training: a systematic review and meta-analysis. J Strength Cond Res. 2020;34(5):1448–60. [DOI] [PubMed] [Google Scholar]
- 91.Hansen M. Female hormones: do they influence muscle and tendon protein metabolism? Proc Nutr Soc. 2018;77(1):32–41. [DOI] [PubMed] [Google Scholar]
- 92.Ahtiainen JP Hulmi JJ Kraemer WJ, et al. Heavy resistance exercise training and skeletal muscle androgen receptor expression in younger and older men. Steroids. 2011;76(1–2):183–92. [DOI] [PubMed] [Google Scholar]
- 93.Peake JM, Neubauer O, Della Gatta PA, Nosaka K. Muscle damage and inflammation during recovery from exercise. J Appl Physiol (1985). 2017;122(3):559–70. [DOI] [PubMed] [Google Scholar]
- 94.Beaton LJ, Tarnopolsky MA, Phillips SM. Variability in estimating eccentric contraction-induced muscle damage and inflammation in humans. Can J Appl Physiol. 2002;27(5):516–26. [DOI] [PubMed] [Google Scholar]
- 95.Damas F Phillips SM Lixandrao ME, et al. Early resistance training–induced increases in muscle cross-sectional area are concomitant with edema-induced muscle swelling. Eur J Appl Physiol. 2016;116(1):49–56. [DOI] [PubMed] [Google Scholar]
- 96.Snijders T Nederveen JP McKay BR, et al. Satellite cells in human skeletal muscle plasticity. Front Physiol. 2015;6:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Damas F Libardi CA Ugrinowitsch C, et al. Early- and later-phases satellite cell responses and myonuclear content with resistance training in young men. PLoS One. 2018;13(1):e0191039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moore DR, Phillips SM, Babraj JA, Smith K, Rennie MJ. Myofibrillar and collagen protein synthesis in human skeletal muscle in young men after maximal shortening and lengthening contractions. Am J Physiol Endocrinol Metab. 2005;288(6):E1153–9. [DOI] [PubMed] [Google Scholar]
- 99.Roman W Pinheiro H Pimentel MR, et al. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science. 2021;374(6565):355–9. [DOI] [PubMed] [Google Scholar]
- 100.Alvarez AM, DeOcesano-Pereira C, Teixeira C, Moreira V. IL-1beta and TNF-alpha modulation of proliferated and committed myoblasts: IL-6 and COX-2-derived prostaglandins as key actors in the mechanisms involved. Cell. 2020;9(9):2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao S, Durstine JL, Koh HJ, Carver WE, Frizzell N, Carson JA. Acute myotube protein synthesis regulation by IL-6-related cytokines. Am J Physiol Cell Physiol. 2017;313(5):C487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krentz JR, Quest B, Farthing JP, Quest DW, Chilibeck PD. The effects of ibuprofen on muscle hypertrophy, strength, and soreness during resistance training. Appl Physiol Nutr Metab. 2008;33(3):470–5. [DOI] [PubMed] [Google Scholar]
- 103.Trappe TA Carroll CC Dickinson JM, et al. Influence of acetaminophen and ibuprofen on skeletal muscle adaptations to resistance exercise in older adults. Am J Physiol Regul Integr Comp Physiol. 2011;300(3):R655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schoenfeld BJ. Potential mechanisms for a role of metabolic stress in hypertrophic adaptations to resistance training. Sports Med. 2013;43(3):179–94. [DOI] [PubMed] [Google Scholar]
- 105.van der Knaap JA, Verrijzer CP. Undercover: gene control by metabolites and metabolic enzymes. Genes Dev. 2016;30(21):2345–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.MacDougall JD, Ray S, Sale DG, McCartney N, Lee P, Garner S. Muscle substrate utilization and lactate production. Can J Appl Physiol. 1999;24(3):209–15. [DOI] [PubMed] [Google Scholar]
- 107.Dankel SJ, Mattocks KT, Jessee MB, Buckner SL, Mouser JG, Loenneke JP. Do metabolites that are produced during resistance exercise enhance muscle hypertrophy? Eur J Appl Physiol. 2017;117(11):2125–35. [DOI] [PubMed] [Google Scholar]
- 108.Ampomah K Amano S Wages NP, et al. Blood flow–restricted exercise does not induce a cross-transfer of effect: a randomized controlled trial. Med Sci Sports Exerc. 2019;51(9):1817–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bjornsen T Wernbom M Lovstad A, et al. Delayed myonuclear addition, myofiber hypertrophy, and increases in strength with high-frequency low-load blood flow restricted training to volitional failure. J Appl Physiol (1985). 2019;126(3):578–92. [DOI] [PubMed] [Google Scholar]
- 110.Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18(12):2238–51. [DOI] [PubMed] [Google Scholar]
- 111.Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4(11):a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fu X, Wang H, Hu P. Stem cell activation in skeletal muscle regeneration. Cell Mol Life Sci. 2015;72(9):1663–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirvonen J, Nummela A, Rusko H, Rehunen S, Harkonen M. Fatigue and changes of ATP, creatine phosphate, and lactate during the 400-m sprint. Can J Sport Sci. 1992;17(2):141–4. [PubMed] [Google Scholar]
- 114.Tesch PA, Colliander EB, Kaiser P. Muscle metabolism during intense, heavy-resistance exercise. Eur J Appl Physiol Occup Physiol. 1986;55(4):362–6. [DOI] [PubMed] [Google Scholar]
- 115.Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin–proteasome pathway in normal and disease states. J Nutr. 1999;129(1S Suppl):227S–37. [DOI] [PubMed] [Google Scholar]
- 116.Cheema BS, Chan D, Fahey P, Atlantis E. Effect of progressive resistance training on measures of skeletal muscle hypertrophy, muscular strength and health-related quality of life in patients with chronic kidney disease: a systematic review and meta-analysis. Sports Med. 2014;44(8):1125–38. [DOI] [PubMed] [Google Scholar]
- 117.Brienesse LA, Emerson MN. Effects of resistance training for people with Parkinson's disease: a systematic review. J Am Med Dir Assoc. 2013;14(4):236–41. [DOI] [PubMed] [Google Scholar]
- 118.Bland KA, Kouw IWK, van Loon LJC, Zopf EM, Fairman CM. Exercise-based interventions to counteract skeletal muscle mass loss in people with cancer: can we overcome the odds? Sports Med. 2022;52(5):1009–27. [DOI] [PubMed] [Google Scholar]
- 119.Pamoukdjian F, Bouillet T, Levy V, Soussan M, Zelek L, Paillaud E. Prevalence and predictive value of pre-therapeutic sarcopenia in cancer patients: a systematic review. Clin Nutr. 2018;37(4):1101–13. [DOI] [PubMed] [Google Scholar]
- 120.Bhatnagar S, Panguluri SK, Gupta SK, Dahiya S, Lundy RF, Kumar A. Tumor necrosis factor-alpha regulates distinct molecular pathways and gene networks in cultured skeletal muscle cells. PLoS One. 2010;5(10):e13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Costamagna D, Costelli P, Sampaolesi M, Penna F. Role of inflammation in muscle homeostasis and myogenesis. Mediators Inflamm. 2015;2015:805172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tuttle CSL, Thang LAN, Maier AB. Markers of inflammation and their association with muscle strength and mass: a systematic review and meta-analysis. Ageing Res Rev. 2020;64:101185. [DOI] [PubMed] [Google Scholar]
- 123.Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest. 2009;119(10):3059–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Horber FF, Hoopeler H, Scheidegger JR, Grunig BE, Howald H, Frey FJ. Impact of physical training on the ultrastructure of midthigh muscle in normal subjects and in patients treated with glucocorticoids. J Clin Invest. 1987;79(4):1181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lim C Dunford EC Valentino SE, et al. Both traditional and stair climbing-based HIIT cardiac rehabilitation induce beneficial muscle adaptations. Med Sci Sports Exerc. 2021;53(6):1114–24. [DOI] [PubMed] [Google Scholar]
- 126.Mebarek S Komati H Naro F, et al. Inhibition of de novo ceramide synthesis upregulates phospholipase D and enhances myogenic differentiation. J Cell Sci. 2007;120(Pt 3):407–16. [DOI] [PubMed] [Google Scholar]
- 127.Dube JJ Amati F Toledo FG, et al. Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia. 2011;54(5):1147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schaap LA, Pluijm SM, Deeg DJ, Visser M. Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am J Med. 2006;119(6):526 e9–17. [DOI] [PubMed] [Google Scholar]
- 129.Jiao J, Demontis F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr Opin Pharmacol. 2017;34:1–6. [DOI] [PubMed] [Google Scholar]
- 130.Verdijk LB, Snijders T, Drost M, Delhaas T, Kadi F, van Loon LJ. Satellite cells in human skeletal muscle; from birth to old age. Age (Dordr). 2014;36(2):545–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Domingues-Faria C, Vasson MP, Goncalves-Mendes N, Boirie Y, Walrand S. Skeletal muscle regeneration and impact of aging and nutrition. Ageing Res Rev. 2016;26:22–36. [DOI] [PubMed] [Google Scholar]
- 132.Cade WT, Yarasheski KE. Metabolic and molecular aspects of sarcopenia. In: Runge MS, Patterson C, editors. Principles of Molecular Medicine. Totowa (NJ): Humana Press; 2006. pp. 529–34. [Google Scholar]
- 133.McKay BR, Ogborn DI, Bellamy LM, Tarnopolsky MA, Parise G. Myostatin is associated with age-related human muscle stem cell dysfunction. FASEB J. 2012;26(6):2509–21. [DOI] [PubMed] [Google Scholar]
- 134.Wiedmer P Jung T Castro JP, et al. Sarcopenia—molecular mechanisms and open questions. Ageing Res Rev. 2021;65:101200. [DOI] [PubMed] [Google Scholar]
- 135.Rom O, Reznick AZ. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic Biol Med. 2016;98:218–30. [DOI] [PubMed] [Google Scholar]
- 136.Tam SL, Gordon T. Mechanisms controlling axonal sprouting at the neuromuscular junction. J Neurocytol. 2003;32(5–8):961–74. [DOI] [PubMed] [Google Scholar]
- 137.Liberman K, Forti LN, Beyer I, Bautmans I. The effects of exercise on muscle strength, body composition, physical functioning and the inflammatory profile of older adults: a systematic review. Curr Opin Clin Nutr Metab Care. 2017;20(1):30–53. [DOI] [PubMed] [Google Scholar]
- 138.Grgic J, Garofolini A, Orazem J, Sabol F, Schoenfeld BJ, Pedisic Z. Effects of resistance training on muscle size and strength in very elderly adults: a systematic review and meta-analysis of randomized controlled trials. Sports Med. 2020;50(11):1983–99. [DOI] [PubMed] [Google Scholar]
- 139.Orchard P Manickam N Ventresca C, et al. Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits. Genome Res. 2021;31(12):2258–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Murach KA Dimet-Wiley AL Wen Y, et al. Late-life exercise mitigates skeletal muscle epigenetic aging. Aging Cell. 2022;21(1):e13527. [DOI] [PMC free article] [PubMed] [Google Scholar]