

Abstract
Pancreas ductal adenocarcinoma (PDAC) is the third most common cause of cancer death in the USA. While other cancers with historically poor prognoses have benefited from new immunotherapies and targeted agents, the 5-year survival rate for PDAC patients has remained static. The accessibility to genomic testing has improved in recent years and it is now clear that PDAC is a heterogenous disease, with a subset of patients harboring actionable mutations. There are several targeted therapies approved by the Food and Drug administration (FDA) in PDAC: EGFR inhibitor erlotinib (combined with gemcitabine) in unselected patients, TRK inhibitors larotrectinib and entrectinib for patients with NTRK fusion mutation, the PD-L1 inhibitor pembrolizumab for mismatch repair deficient patients and the Poly-ADP-ribose polymerase (PARP) inhibitor olaparib in patients with germline BRCA mutation as a maintenance therapy. DNA damage repair (DDR) is paramount to genomic integrity and cell survival. The defective repair of DNA damage is one of the hallmarks of cancer and abnormalities in DDR pathways are closely linked with the development of malignancies and upregulation of these pathways linked with resistance to treatment. The prevalence of somatic and germline mutations in DDR pathways in metastatic PDAC is reported to be approximately 15–25%. Patients with DDR gene alterations benefit from a personalized approach to treatment. Recently, the POLO trial demonstrated a progression-free survival (PFS) benefit in metastatic PDAC patients with a germline BRCA1/2 mutation treated with maintenance olaparib following platinum-based induction chemotherapy. This was the first phase 3 randomized trial to establish a biomarker-driven approach in the treatment of PDAC and establishes a precedent for maintenance therapy in PDAC. The review herein aims to outline the current treatment landscape for PDAC patients with DDR gene-mutated tumors, highlight novel therapeutic approaches focused on surmounting tumor resistance and exploring new strategies which may lead to an expansion in the number of patients who benefit from these targeted treatments.
Keywords: Pancreas, BRCA, DNA-damage repair, homologous repair, ATM, BRCAness
Background
The discovery of immunotherapies and a variety of targeted agents in recent years has resulted in a considerable shift in survival data for a number of cancers with historically poor prognoses. In terms of survival, however, pancreas ductal adenocarcinoma (PDAC) remains with the lowest 5 year survival of 10%[1]. In 2021, 48,220 people in the US are expected to die from this disease, placing it third in leading causes of cancer death following lung and colon cancer[1].
In front-line metastatic PDAC, the combination of oxaliplatin, irinotecan, fluorouracil and leucovorin (FOLFIRINOX) and the combination of nanoparticle albumin bound paclitaxel (Nab-P)/gemcitabine are the standard-of-care therapies following the results of two landmark trials. [2,3]. The overall survival for patients included in these studies was 8.5–11.1 months, highlighting the dire need for additional treatment options for these patients[2,3]. A number of databases are publicly available which define actionability for genomic aberrancies[4]. Actionability is determined by a molecular alteration for which there is clinical or strong preclinical evidence of a predictive benefit from a specific therapy (in any malignancy type)[5]. PDAC is largely defined by somatic mutations in KRAS, TP53, CDKN2A and SMAD4[6,7] and with mutations in BRCA1, BRCA2, ATM, CHEK2 being the most commonly seen pathogenic germline variants[8,9]. The prevalence of somatic and germline variants in DDR pathways in metastatic PDAC is reported to be approximately 15–25%[10–12,8,9]. Thus, a significant minority of PDAC patients may benefit from tailored targeted DDR therapies. These targeted DDR therapies form the focus of this review.
DNA Damage Repair Pathways
Cells are estimated to experience over 20,000 DNA damaging events each day[13]. A finely tuned response of DNA repair and activation of cell cycle checkpoints has evolved to address these ubiquitous events[14]. The defective repair of DNA damage is a common hallmark of cancer with aberrations in DDR pathways being closely linked with the development of malignancies and upregulation of these pathways linked with resistance to DNA damaging chemotherapy and radiotherapy[14]. The main pathways involved in these processes are summarized in Figure 1. A number of these pathways are therapeutically targetable. There are three major mechanisms that identify and repair single-strand DNA damage: mismatch repair, nucleotide excision repair, and base excision repair. The mismatch repair (MMR) pathway corrects for inappropriate nucleotide insertions, deletions, and single nucleotide mismatched incorporations[15]. Defective MMR increases mutation rates up to 1,000-fold, leading to microsatellite instability (MSI) which is associated with cancer development[16]. MSI high tumors can be targeted with immune checkpoint inhibitors[17,18]. Therapeutically targeting the MMR pathway has been described extensively elsewhere and will not be covered in this review[19–22]. The nucleotide excision repair (NER) senses mostly larger nucleotide DNA adducts or ultraviolet (UV) induced damage[23]. The base excision repair (BER) pathway is responsible for sensing and repairing single-strand breaks (SSBs), the most common type of DNA damage. BER is also responsible for repairing damage that can be therapeutically induced with radiation and some chemotherapies[24], with polymerase 1 (PARP1) and PARP2 key facilitators in the process[14]. These PARP proteins can be targeted with a number of PARP inhibitors.
Figure 1:
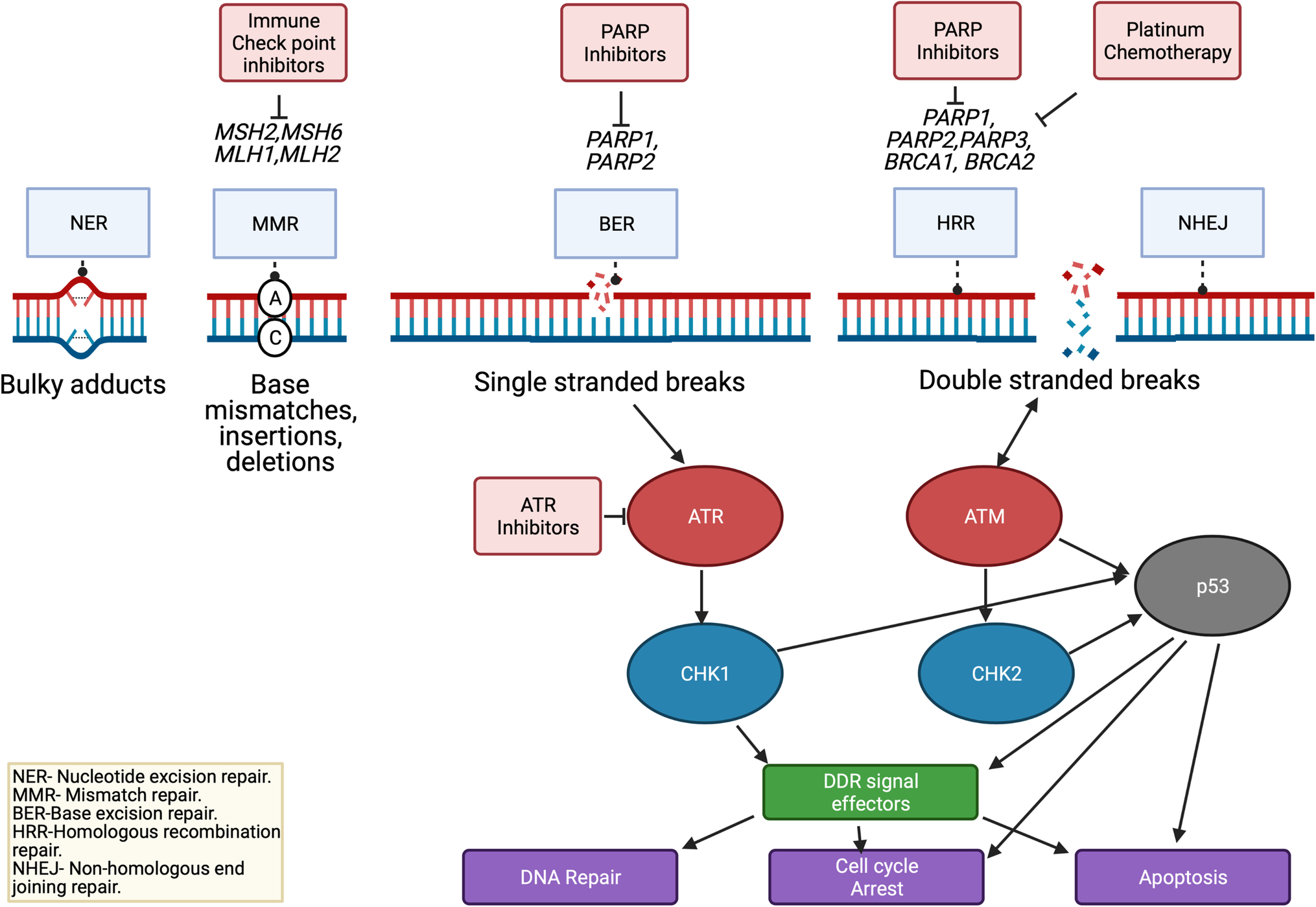
Targeting DDR Pathways in Pancreas Cancer
Made with Biorender.com
Unrepaired SSBs lead to collapse of the replication fork and double stranded breaks (DSBs), which are repaired by the homologous recombination (HR) or non-homologous end joining (NHEJ). DSBs are detrimental to cells and if left unrepaired, they can drive apoptosis or senescence. If mis-repaired, these breaks lead to the generation of chromosomal aberrations, resulting in genomic instability associated with carcinogenesis[25]. NHEJ directs re-ligation of the broken DNA molecule in a template-independent manner, and is active in all phases of the cell cycle and is more error-prone[26]. In contrast, homologous recombination repair (HRR) utilizes homology of a sister chromatid; therefore, with high fidelity and is largely restricted to the S and G2 phases of the cell cycle[27]. DNA damage sensing leads to recruitment of DDR proteins to site of damage, cell cycle activation and ultimately DNA repair[28]. The MRN (MRE11, RAD50 and NBN protein) complex is heavily involved in the initial steps of DSB damage repair. It activates ATM and ATR to allow formation of protruding 3’ ends at both side of the DSB and recruits BRCA1 to the site of damage[28]. BRCA1 in turn recruits BRCA2 and PALB2[29]. The DSB is resected to create a single-stranded 3′-overhang, which becomes rapidly coated with the ssDNA-binding protein RPA[30,31] and this overhang ultimately invades the sister chromatid[32]. Once an RPA-coated ssDNA filament is generated, RPA is replaced by Rad51 in an ATM/ CHK2/ BRCA1/ BRCA2/ PALB2-dependent fashion[33]. A number of these interactions are represented in Figure 1 and a number of these genes are directly or indirectly targetable. The nature of alterations of these genes across 33 different cancers was examined by Knijnenburg et al using TCGA PanCanAtlas[34]. Somatic mutations with accompanying loss of heterozygosity was observed in over 1/3 of DDR genes. Epigenetic silencing was also a frequent event, with silencing of direct repair genes (eg: EXO5, MGMT, ALBH3) occurring in 20% of samples[34].
DNA Damage Repair and PDAC
DDR Germline Mutations
Up to 10% of PDAC arise in the setting of an underlying pathogenic germline mutation. One study of 854 patients with PDAC, reported that up to 40% of those with germline HR-DDR–mutated PDAC did not have a significant family history of cancer[35]. From 2019, the National Comprehensive Cancer Network (NCCN) guidelines have been updated to recommend germline testing for all patients with confirmed pancreas cancer, using comprehensive gene panels for hereditary cancer syndromes[36]. Genes implicated in predisposition to PDAC include BRCA1, BRCA2, ATM, PALB2, STK11, MLH1, MSH2, MSH2, MSH6, PMS2 and CDKN2A with other genes such as BARD1, ATR, RAD51 and CHEK2 are considered as candidate risk genes[37–43,8]. Notably, many of these genes are involved in DDR. In a study of 159 patients with PDAC, a pathogenic germline alteration was discovered in 15%, with BRCA1 and BRCA2 mutations the most predominant, at rates of 2.5% and 8.2%, respectively[9]. Another study, using a larger gene panel and 615 patients reported a 19.8% rate of germline mutations, with BRCA1 mutations in 2.4%, BRCA2 mutations in 5.7%, PALB2 mutations in 0.2% and ATM (1.8%)[8]. Another study looking specifically at HRR genes and their impact on OS in PDAC, analyzed samples from 3078 patients[44]. One hundred and seventy-five (5.7%) were identified to carry a mutation in one of eight HRR genes: 67 (2.2%) BRCA2, 65 (2.1%) ATM, 20 (0.6%) BRCA1, 12 (0.4%) PALB2, 4 (0.1%) BRIP1, 4 (0.1%) RAD51C, 2 (0.1%) BARD1 and 1 (0.03%) in RAD51D[44]. As had been previously reported, mutation carriers were observed to be diagnosed with PDAC at a younger age (62.8 vs. 65.8 years, P < 0.001) and were more likely to present with metastatic disease at diagnosis (46.2% vs. 35.6%, P < 0.001) compared with noncarriers[44]. This rate of germline BRCA mutations is enriched in certain populations: 5–16% in Ashkenazi Jewish community, 5–19% in familial PDAC and 5–10% in those with a history of familial breast or ovarian cancer[45–49,9]. The rates of mutations in MMR genes (MLH1, MSH2, MSH6) are low, comprising of only 1–7 patents in a number of studies, which signified under 1% of patients in larger studies[9,8,50,51]. However, this is less explored in certain ethnicities and more global investigation is required and is underway[52].
Sporadic DDR Mutations
A review of pancreas tumor specimens within the Catalogue of Somatic Mutations in Cancer (COSMIC) database revealed that most pancreas cancers harbor somatic mutations, with the five most frequent aberrations occurring in KRAS, TP53, CDKN2A, SMAD4, and ARID1A genes[7]. Three percent of patients harbored a somatic ATM mutation[7]. A study examining whole genome sequencing and copy number variations of 100 PDAC patients identified 3 with a somatic BRCA2 mutation and 2 with a somatic BRCA1 mutation[39].
Next generation sequencing and advanced genome panels has enabled identification of alterations and variants in DDR genes. However, it is often unclear if these low frequency mutated genes or variants are truly driver mutations of tumorigenesis, especially when they occur alongside other frequent cancer driving genes like KRAS and TP53. It is also unclear, what events must occur for these genes to become pathogenic and if they fit the classical tumor suppressor rules of requiring loss of the second allele in the tumor[53].
BRCAness and beyond DDR-gene mutations
BRCAness is defined as being a phenocopy of BRCA1/2 mutation. It describes the situation in which an HRR defect exists in a tumor in the absence of a BRCA1/2 mutation [54]. Other non-core HR (non-BRCA1/2) gene somatic and germline mutations are candidates for displaying BRCAness. Tumors with HRR defects are highly sensitive to crosslinking agents (such as cisplatin) and DSBs that are induced by radiation and anti-topoisomerase I agents[55,14].
Genetic signatures beyond known HR gene mutations and scars may be more inclusive in identifying a DDR alteration and may enrich for response to sensitivity to DNA damaging agents e.g., signature 3 and HRDetect[56,57]. A number of assays that detect DDR deficient molecular signatures and various scores have been designed in an attempt to identify tumors which truly have a defective DDR pathway and thus may benefit from DDR targeted agents. For HR deficient tumors, breast and ovarian cancer fields have lead the way in development of HRD scores[58,59]. One commonly used HRD score comprises of the sum of three independent DNA-based measures of genomic instability: loss of heterozygosity, telomeric imbalance and large scale state transitions[60–62]. Other HRD classifiers have also been investigated in PDAC with a number being associated with platinum response and superior survival.[63,64]. In our group’s work of 262 advanced PDAC patients with both germline and somatic MSK-IMPACT available, comprehensive evaluation of methodologies for platinum sensitivity was performed and the strongest correlation was in the allelic status of the DDR gene and patients with bilallelic loss of both core (BRCA1/2, PALB2) and non-core HR genes derived great benefit from platinum[65]. In one study HRD genomic tumor classifiers suggested that 7% to 10% of PDACs that do not harbor germline BRCA/PALB2 have features of HRD[63]. In the same study pathogenic variants in germline ATM (n = 6) or germline CHEK2 (n = 2) did not result in HRD-PDAC by any of the classifiers[66]. Defining HRD using validated scores is crucial to broaden the indication for DDR-targeted agents. However, the resolution of genetic signature evaluation from targeted gene sequencing methods is limited in PDAC in view of low number of mutations and further investigation is needed[65]. Platinum sensitivity itself or a strong familial history of pancreatic cancer without the presence of germline HR mutations have also been studied in this context[67,68].
Targeting the DDR Pathway in PDAC Therapy
Results published from the “Know Your Tumor” program identified that patients with pancreas cancer with an actionable molecular alteration who received a matched therapy (n=46), had significantly longer median OS compared to those patients who received unmatched therapies (n=143; 2·58 years [95% CI 2·39 to not reached] vs 1·51 years [1·33–1·87]; HR 0·42 [95% CI 0·26–0·68], P=0·0004)[5]. Ninety-four (13%) of 189 patients with actionable mutations in this analysis had mutations in the DNA damage repair (DDR) pathway[5].
Treatment with Platinum Agents
Tumors with HRR defects are highly sensitive to crosslinking agents. Platinum agents are known for their ability to crosslink with purine bases in DNA. Most of these crosslinks are intrastrand and are usually repaired by NER but interstrand DNA crosslinks can cause catastrophic replicative and transcriptional stress, which leads to apoptosis [69]. In various malignancies, anti-tumor responses are enhanced in the presence of underlying double-strand DNA repair alterations in the tumor[70]. A recent systematic review of platinum use in HRR deficiency (HRD) in retrospective studies found the average weighted median OS in patients with HRD (n=137) treated with platinum based chemotherapy was 23.8 and was 17.1 months in patients without HRD (n=293)[71]. Without platinum-based chemotherapy, the average weighted median OS was 8.3 months in patients with HRD (n=69) and 12.0 months in patients without HRD (n=144)[71]. NCCN currently recommends FOLFIRINOX or cisplatin plus gemcitabine as first line treatment for individuals who have advanced PDAC, a good performance status, and pathogenic germline mutation in BRCA1/2 and PALB2[36].
To date, most data pertaining to platinum treatment in PDAC patients with DDR alterations has focused on alterations that are germline in origin. In 2011 a small study of 15 patients with germline BRCA mutations reported that of six patients who received platinum based chemotherapy as first line, five achieved a partial response (PR) to treatment and one a complete response(CR)[72]. This finding has been further supported by further studies. In 2014 a study of 71 BRCA germline patients found a superior OS for the 22 patients with stage 3/ 4 PDAC treated with platinum-based treatment versus those treated with non-platinum-based treatment. The majority of these patients were treated with cisplatin and three were treated with FOLFIRONOX[73]. The median OS was 22 months for the platinum exposed (n=22) compared with 9 months for the non-platinum (n=21) treatment groups (P<0.039)[73]. Later a further study reported that twenty-nine (76.3%) of 38 germline mutated BRCA1/2 patients who received platinum, achieved a treatment response in addition to five of nine patients with ATM alterations(55.6%)[8].
The data supporting increased efficacy of platinum agents in PDAC patients with somatic DDR mutations is less abundant. In 2017, Lowery et al observed an ORR to platinum of 34% among 50 PDAC patients who had somatic alterations in one or more genes associated with DDR, including BRCA2, FANCA, ATM, and ATR. In this dataset, somatic mutations in DDR genes failed to enrich for patients responding to platinum-based chemotherapy[74]. Thirteen patients in this dataset had somatic BRCA1 /2 mutations. They also observed a variance in clinical outcomes and response to platinum chemotherapy[74]. Most recently in 2020, Park et al presented data on the platinum treatment outcomes of 50 PDAC patients with HR deficiency (HRD) (15% germline and 4% somatic); again, reporting a significantly improved median PFS when treated with first line platinum compared to non-platinum treatment (12.6 [95%CI: 9.6–24.9] vs 4.4 [3.0–10.0] months) in HRD patients[65]. This study also reported the lack of difference in genomic instability or clinical outcome between germline and somatic HRD patients, which suggests that both may predict for benefit to platinum-based therapies[65].
Poly ADP Ribose Polymerase (PARP) Inhibitors
PARP enzymes are primarily involved in BER of SSB[75]. Inhibition of PARP enzymes arrests SSB repair and when these SSBs accumulate this leads to replication fork collapse and DSBs. As explored earlier, DSBs are repaired by one of two processes HRR and NHEJ with inability to repair DDBs leading cell cycle arrest and subsequent apoptosis[76]. In patients with mutations in HRR genes, for example in BRCA1/2 where tumors have defective DSB repair mechanisms, PARP inhibition can be used to create synthetic lethality in tumors, which can cause selective toxicity to malignant cells that lack HRR capacity whereas normal cells repair damage via their HRR mechanism[76].
The Pancreas OLaparib Ongoing (POLO) trial reported in 2019 was the first phase 3 randomized study to establish a biomarker-driven approach in treatment of PDAC, opening the door to a new era in personalized care for a subset of patients. In ovarian cancer the phase 3 SOLO-1 study demonstrated the benefit of maintenance olaparib in BRCA1/2 patients[77]. The POLO study took the same approach, aiming to extend PFS. The phase 3 results reported on 154 patients with PDAC who had a germline BRCA1/2 mutation, whose disease was stable or responding on front-line platinum-based therapy, and included 92 patients randomized to receive maintenance olaparib and 62 to placebo[78]. At 68% data maturity, the median PFS in olaparib group was 7.4 months vs. 3.8 months in placebo group (HR 0.53, 95% CI 0.35 to 0.92, P=0.004). At data cutoff after 108 deaths in July 2020, 13 patients remained on olaparib vs 2 on placebo[66]. More recently in 2021, mature OS data were reported and were similar for both groups: 19.0 and 19.2 months, respectively (HR 0.83 favoring olaparib; 95% CI 0.56–1.22; P= 0.3487)[66]. OS at 36 months was 33.9% for olaparib and 17.8% for placebo[66]. Median time from randomization to second disease progression or death was 16.9 months vs 9.3 months, respectively (HR, 0.66; 95% CI 0.43–1.02; P= 0.0613)[66]. The latter, showed a clear trend for treatment benefit of olaparib beyond disease progression but this was not statistically significant[66]. Importantly, there was no difference in quality of life scores between olaparib and placebo arms[78,79]. Nevertheless, this study is a landmark proof-of-concept study for maintenance PARP inhibitor in germline BRCA mutated metastatic PDAC and led to the Food and Drug Administration (FDA) approval of olaparib in this setting in PDAC in late 2019.
The use of this strategy in PDAC patients who harbor somatic DDR gene mutations is less clear; although reports from maintenance setting studies in ovarian cancer have demonstrated that patients with BRCA somatic mutations had a similar reduction in the risk of disease progression as those with germline BRCA variants (median PFS, 20.9 months vs. 11.0 months; HR 0.27; 95% CI, 0.08 to 0.90; P=0.02)[80]. A phase 2 study evaluating maintenance rucaparib in PDAC patients (n=24) whose disease has not progressed on first line platinum included 13 patients with germline mutations in BRCA2, three patients with germline mutations in BRCA1, two patients with germline mutations in PALB2 and one patient with a somatic mutation in BRCA2[81]. The median PFS was 9.1 months with an ORR of 36.8% (6 PR’s: 1 CR). Disease control rate (DCR) was 89.5% for at least eight weeks[81]. The sole patient with a somatic mutation was one of the seven responders with, the remaining six patients having mutations that were germline in origin (BRCA2 [n=4], and PALB2 [n=2])[81]
The use of PARP inhibitors as monotherapy in patients with PDAC whose disease has progressed on prior systemic chemotherapy is limited, with some conflicting results among different agents. The first phase 2 trial was a basket study for germline BRCA1/2 solid tumors which included 23 patients with advanced PDAC. The mean number of prior therapies was two and 65% of patients had received prior platinum[82]. The tumor response rate was 21.7% (95% CI, 7.5 to 43.7)[82]. Stable disease that persisted ≥ 8 weeks was observed in 34.8% of those with PDAC (n = 8; 95% CI, 16.4 to 57.3)[82]. There was no apparent difference in response rates in those treated with platinum (20%; 95% CI, 4.3 to 48.1) or without (25%; 95% CI, 3.2 to 65.1)[82]. 30.4% experienced serious AEs[82]. Of specific note, it is unclear whether these patients had experienced disease progression to prior platinum therapy. Rucaparib monotherapy following treatment with chemotherapy has also been evaluated in patients with PDAC who had a somatic or germline mutation in BRCA (16 of 19 enrolled had germline BRCA mutations)[83]. The ORR was 15.8% (3 of 19), two PR’s and 1 CR were confirmed with an additional CR unconfirmed[83]. Of further note, 2 of 3 patients with a somatic BRCA2 mutation had a PR[83]. In 2018 results of veliparib in pre-treated stage III/IV PDAC and known BRCA1/2 or PALB2 mutations were reported. Fourteen (88%) patients with PDAC had received and experienced disease progression on prior platinum therapy. In contrast to the above studies no confirmed responses were seen. Four (25%) had SD, whereas 11 (69%) had POD as best response. Survival data was modest with a median PFS of 1.7 months (95% CI 1.57–1.83) and median OS of 3.1 months (95% CI 1.9–4.1)[84]. The key reasons for these differences in outcomes between the several studies include the potency differences among the PARP inhibitors and the impact of acquired resistance to platinum agents which render PARP inhibitors significantly less effective[84].
Javle et al recently reported the first results of PARP inhibitor monotherapy in a BRCAness population, with 48 patients treated with olaparib following at least one systemic therapy[68]. BRCAness in these studies was defined as previously known DDR genetic alterations (DDR-GAs), personal or family history of BRCA-associated cancers (without DDR-GAs), or evidence of ATM loss on immunohistochemistry with twenty-four, seventeen and five patients respectively, recruited with these traits[68]. DDR-GAs included ATM (n = 14), PALB2 (n = 2), ARID1A (n = 3), BRCA somatic (n = 1), PTEN (n = 1), RAD51 (n = 1), CCNE (n = 1), and FANCB (n = 2)[68]. The median duration of treatment was 3 months. Two patients had a PR (4%; exact 95% CI, 0.53%–14.8%), 1 of which was not confirmed (2%); these PRs occurred in 1 patient with PALB2 and 1 with somatic ATM variant with the confirmed PR 3.9 months in duration[68]. Thirty-three patients experienced stable disease (72%; 95% CI, 59%–85%), of whom 11 (24%) experienced disease stability longer than 4 months[68]. Median PFS was 3.7 months (95% CI, 2.9–5.7) and was significantly higher for patients with DDR-GAs (5.7 months; 95% CI, 3.6–8.8 months; P= .008) and platinum-sensitive PDAC (4.1 months; 95% CI, 3.6–7.8 months; P = .01). The overall PFS is comparable to second line chemotherapy regimens but is lower than that noted among patients with BRCA variants who received olaparib in the POLO trial[68,66]. The estimated median OS was 9.9 months (95% CI, 7.6–16.1 months) and 13.6 months (95% CI, 9.69 to not reached) in the DDR-GA cohort[68]. There are a number of PARP monotherapy trials ongoing. NCT03601923 and NCT03553004 are evaluating the use of niraparib, a selective PARP1 and PARP2 inhibitor, in PDAC patients with mutations in HRR genes. A phase 2 study assessing rucaparib in patients with solid HRR deficient tumors is also still recruiting (NCT04171700).
Strategies to overcome resistance to PARP inhibition has been investigated and combining a platinum with a PARP inhibitor was hypothesized to lead to improved response and survival in patients with DDR gene mutations based on some promising phase 1 results[85]. A phase 1 dose escalation study of 18 unresectable PDAC patients treated with olaparib with cisplatin, irinotecan, and mitomycin C observed that the combination caused substantial toxicity with 16 patients (89%) experiencing grade 3 or greater adverse events[86]. One patient with a germline BRCA2 mutation had a durable clinical response lasting more than four years, but died from complications of treatment-related MDS[86]. A phase 2 study of 108 patients treated with veliparib and folinic acid, fluorouracil, and irinotecan (FOLFIRI) or FOLFIRI alone included 11 PDAC patients with stage IV disease (9%) with HRD, including 4 germline (BRCA1, BRCA2, ATM) and 7 somatic mutations (BRCA2, PALB2, ATM, CDK12). Additionally, 24 cancers (20%) had germline (n = 11, e.g., FANC, BLM, SLX4, CHEK2) or somatic mutations (n= 13, e.g., FANC, BLM, POLD1, RIF1, MSH2, MSH6) in other genes, not classified as HRD. Improvements in OS (11.9 vs 5.7 months) and PFS were observed in patients who had either germline or somatic BRCA-mutated or with non–BRCA-mutated HRD in comparison with the no-HRD group[87,88]. A phase 1/2 study of veliparib in combination with 5-fluorouracil and oxaliplatin (FOLFOX) in metastatic PDAC observed that the combination had greater activity in platinum-naïve patients and demonstrated again greater activity in those with HR mutations[89]. The ORR in HRD platinum naïve patients was 57%[89]. Activity of a combination platinum and PARP in a platinum naïve HRD population was then assessed in a randomized phase 2 trial of gemcitabine and cisplatin with or without veliparib in untreated patients with PDAC and a germline BRCA/PALB2 mutation. Results published in 2020 noted that the addition of veliparib did not improve ORR [90]. The ORR for veliparib arm (arm A) was 74.1% and 65.2% for standard chemotherapy arm (arm B) (P= .55). Disease control rates (DCR) was 100% for arm A and 78.3% for arm B (P= .02). The median PFS was 10.1 months for arm A (95% CI, 6.7 to 11.5 months) and 9.7 months for arm B (95% CI, 4.2 to 13.6 months; P= .73)[90]. Median OS for arm A was 15.5 months (95% CI, 12.2 to 24.3 months) and 16.4 months for arm B (95% CI, 11.7 to 23.4 months; P= .6). Grade 3 to 4 hematologic toxicities for arm A versus arm B were 13 (48%) versus seven (30%) for neutropenia, 15 (55%) versus two (9%) for thrombocytopenia and 14 (52%) versus eight (35%) for anemia[90]. The major take home points from this study are that cisplatin and gemcitabine is an active and reference standard for this patient population and that the addition of the PARP inhibitor added toxicity and did not improve outcome. All completed PARP inhibitor trials, in PDAC population are summarized in table 1.
Table 1:
Selected Completed PARPi Trials in Pancreas Cancer
| Drugs | N | Phase | Cohort | Study design | Outcome | Reference |
|---|---|---|---|---|---|---|
| Olaparib | 154 | 2 | gBRCA1/BRCA2 mutation and had not progressed during first line platinum | Olaparib 200mg bid vs placebo | mPFS 7.4 vs 3.8 mos (HR 0.53; 95% CI, 0.35 to 0.82; P=0.004) mOS 19.0 vs 19.2 mos (HR 0.83 favoring olaparib; 95% CI 0.56–1.22; P= 0.3487. |
Golan et al[78,66] |
| Rucaparib | 24 | 2 | Platinum sensitive PDAC with BRCA1/BRCA2 or PALB2 mutations | Rucaparib 600mg bid | ORR 36.8% DCR for >8 weeks 89.5% mPFS 9.1 mos (no HR given) |
Binder et al[81] |
| Olaparib | 23 | 2 | gBRCA1/2 solid tumors | Olaparib 400mg bid | RR 21.7% SD 34.8% |
Kaufman et al[82] |
| Rucaparib | 19 | 2 | gBRCA1/2 and somatic BRCA1/2 | Rucaparib 600mg bid | ORR 15.9% DCR 31.6% or 44.4% in those with prior chemotherapy |
Shroff et al[83] |
| Veliparib | 16 | 2 | gBRCA1/2, PALB2 mutated PDAC | Veliparib 300 mg bid n=3), 400 mg bid (n=15) | RR 0% SD 25% mPFS 1.7mos (95% CI 1.57–1.83) mOS 3.1mos ((95% CI 1.9–4.1) |
Lowery et al[84] |
| Olaparib | 48 | 2 | Platinum-sensitive, metastatic PDAC, without gBRCA mutation and with BRCAness, previous chemotherapy | Olaparib 400mg bid | (n=46) RR 2% SD 72% mPFS 3.7mos (95% CI, 2.9–5.7) mOS 9.9mos (95% CI, 7.6–16.1 months) |
Javle et al[68] |
| Olaparib | 18 | 1 | Unresectable PDAC | MTD of olaparib and irinotecan, cisplatin, mitomycin C | Significant toxicity One 4-year response in germline BRCA mutated patient |
Yarchoan et al[86] |
| Veliparib | 108 | 2 | Metastatic PDAC | Veliparib plus modified FOLFIRI (no 5FU bolus) vs FOLFIRI alone | mOS was 5.1 vs 5.9 mos (HR 1.3, 95%CI 0.9–2.0, P = 0.21), and mPFS was 2.1 vs 2.9 mos (HR 1.5, 95%CI 1.0–2.2, P = 0.05) | Chiorean et al[87] |
| Veliparib | 31 | 1/2 | Metastatic PDAC | Veliparib (40 mg to 250 mg twice a day, days 1–7 of each 14-day cycle) and FOLFOX vs FOLFOX alone | ORR in HRR mutated platinum naïve patients of 57% | Pishvaian et al[89] |
| Veliparib | 50 | 2 | Stage 3/ 4 PDAC with gBRCA/PALB2 mutation | Veliparib 80mg bid 1–12 cycled every 3 weeks plus cisplatin and gemcitabine vs cisplatin and gemcitabine alone | RR 74.1% DCR 100% mPFS 10.1 (95% CI, 6.7 to 11.5 months) vs 9.7 (95% CI, 4.2 to 13.6 months; P = .73) mOS 15.5 (95% CI, 12.2 to 24.3 months) vs 16.4 (95% CI, 11.7 to 23.4 months; P = .6) |
O’Reilly et al[90] |
| Veliparib | 30 | 2 | Locally advanced PDAC | Veliparib 40mg bid, weekly gemcitabine, daily IMRT | OS for DDR mutated patients 19 mos 95% CI: 6.2–27.2 vs 14mos in DDR intact patients. | Tuli et al[121] |
RR: Response rate; SD: Stable disease; POD: Progression of disease; BID: twice daily, mPFS: Median progression free survival, mOS: Median overall survival; gBRCA: germline BRCA mutated, mos: months; wks: weeks); MTD Maximum tolerated dose; IMRT: Intensity modulated radiotherapy
Future Directions
In the POLO trial the median PFS was 9.1 months, demonstrating that many patients acquire PARP inhibitor resistance with exposure to PARP inhibitors[91,78]. Tumor cells can develop ways to restore HR repair (HRR repair restoration) or retain its HR defect but find an alternative mechanism to protect the replication fork[92,91]. The latter is often achieved by upregulation of the ATR/ ChK1 pathway which stabilizes the replication fork[92]. In BRCA1/2 mutated tumors, the most common mechanism of resistance to PARP inhibitors is achieved through intragenic secondary mutations, which restore BRCA1 and BRCA2 protein function[92]. Various other factors, such as reversion mutations, epigenetic modification, loss of PAR glycohydrolase (PARG) and restoration of ADP-ribosylation (PARylation) can also lead to PARP inhibitor resistance[91]. Deletion of shieldin, a key protein complex in NHEJ repair has also been found to confer resistance to PARP inhibitors in BRCA1 deficient cells[93]. To date combining chemotherapy with PARP inhibitors has not led to improved survival for platinum naive patients with HRD, although there is interest in continuing to explore this strategy given the theoretic advantage of delaying the emergence of resistance. A number of other strategies to combat resistance are being investigated in other cancers with prevalent DDR mutations. Table 2 summarizes ongoing trials targeting DDR in PDAC.
Table 2:
Novel Therapy Directions in DDR/HRD-Related Pancreas Cancer
| Drug | N | Cancer type | Phase | Design | Status | Reference |
|---|---|---|---|---|---|---|
| Niraparib | 32 | PDAC harboring deficiencies in HRR | 2 | Niraparib 200mg-300mg QD, palliative radiation therapy to a small field >1 week prior to day 1 of treatment | Recruiting | NCT03601923 |
| Niraparib | 18 | PDAC after previous chemotherapy | 2 | Niraparib 300mg daily for 28 days | Recruiting | NCT03553004 |
| Rucaparib | 220 | Solid tumors with HRR deleterious mutations | 2 | Rucaparib 600mg BID | Recruiting | NCT04171700 |
| Olaparib and pembrolizumab | 63 | Metastatic PDAC with HRR deficiency or exceptional treatment response to platinum | 2 | Arm A: Patients with mutations in core HR genes Arm B: Patients with mutations in non-core HR genes Arm C: Patients with previous PR/CR to platinum Pembrolizumab 200 mg Q3W. After the first 6 months (8 cycles), on C9D1 Pembrolizumab 400 mg Q6W. Olaparib 300 mg BID continuously (as a maintenance therapy). |
Recruiting | NCT04666740 |
| Dostarlimab and niraparib | 20 | gBRCA or somatic BRCA mutated pancreatic cancer | 2 | Niraparib QD on days 1–21. Dostarlimab on day 1 Q3W for cycles 1–4 and Q6W for subsequent cycles. Cycles repeat every 21 days for up to 2 years in the absence of POD or unacceptable toxicity | Recruiting | NCT04493060 |
| Niraparib and dostarlimab | 18 | BRCA mutated metastatic breast, pancreas, ovarian, fallopian tube, primary peritoneal. | 1 | Niraparib QD on days 1–28 of cycle 1. Cycle 2: patients receive niraparib QD on days 1–21 and dostarlimab on day 1. Repeats every 21 days for 4 cycles in the absence of POD or unacceptable toxicity. Beginning cycle 6, patients receive niraparib )D on days 1–42 and dostarlimab on day 1. Cycles repeat every 42 days for up to 24 months in the absence of POD or unacceptable toxicity | Not yet recruiting | NCT04673448 |
| Olaparib and pembrolizumab vs olaparib alone | 88 | Metastatic pancreatic cancer with gBRCA mutation SWOG S2001 |
2 | Arm A: Olaparib BID on days 1–21 and pembrolizumab IV on day 1. Repeats every 21 days for up to 18 cycles in the absence of POD or unacceptable toxicity. Cycle 19, patients receive Olaparib BID on days 1–42 and pembrolizumab day 1. Repeat every 42 days in the absence of POD or unacceptable toxicity. Arm B: olaparib BID on days 1–21. Cycles repeat every 21 days |
Recruiting | NCT04548752 |
| AZD6738 vs AZD6738 and olaparib | 68 | Solid tumors with ATM loss or ARID1A positivity in solid tumors | 2 | Arm A: BAF250a negative or ATM-Mutant receive AZD6738 PO twice a day on days 1–14. Treatment repeats every 28 days in the absence of POD or unacceptable toxicity. Arm B: BAF250a positive receive AZD6738 PO every day on days 1–7 and Olaparib twice a day on days 1–28. Treatment repeats every 28 days in the absence of disease POD or toxicity. |
Recruiting | NCT03682289 |
| Olaparib and durvalumab vs olaparib and durvalumab | 90 | Advanced solid tumors who have failed previous standard therapy | 2 | Arm A: Olaparib mg BID in combination with Durvalumab,1500 mg Q4W Arm B: Cediranib, 20 mg QD(5 days on 2 days off) in combination with Durvalumab 1500 mg Q4W. |
Recruiting | NCT03851614 |
| Niraparib and nivolumab and niraparib and ipilimumab | 84 | PDAC who have not progressed on platinum | 1/2 | Arm A: Niraparib 200mg QD on days 1–21 of each 21-day cycle. Nivolumab 240mg days 1 and 15 of odd-numbered cycles; day 8 of even numbered cycles Arm B: Niraparib 200mg QD on days 1–21 of each 21-day cycle Ipilimumab 3mg/kg day 1 of each cycle, for the first 4 cycles only |
Recruiting | NCT03404960 |
| Avelumab, binimetinib and talazoparib | 122 | RAS- mutant solid tumors | 1/2 | Arm A: Avelumab and binimetinib Arm B: Avelumab, binimetinib and talazoparib Arm C: Binimetinib and talazoparib |
Recruiting | NCT03637491 |
| Olaparib and cediranib | 126 | Advanced solid tumors | 2 | Cediranib maleate QD on day 1. Patients undergoing FMISO scan also receive Olaparib BID beginning the day after the second FMISO scan and the rest of the patients receive Olaparib BID beginning day 4 of cycle 1. Cycles repeat every 28 days (35 days for cycle 1) in the absence of POD or unacceptable toxicity | Recruiting | NCT02498613 |
| Olaparib and durvalumab vs cediranib and durvalumab | 90 | Advanced solid tumors | 2 | Arm A: Olaparib 300mg QD in combination with Durvalumab 1500 mg Q4W. Arm B: Cediranib20mg QD, 5 days on 2 days off in combination with Durvalumab 1500 mg Q4W |
Recruiting | NCT03851614 |
| Veliparib and dinaciclib | 118 | Advanced solid tumors | 1 | PART 1A: Veliparib BID on days 1–28 and dinaciclib, days 8 and 22. Cycles repeat every 28 days in the absence of POD or unacceptable toxicity PART 1B: Veliparib and dinaciclib as patients in Part 1A. PART 1C: Veliparib BID on days 1–7 of cycle 0. Veliparib BID on days 1–21 and dinaciclib on days 1, 4, 8, and 11 or days 1 and 8. Cycles repeat every 21 days in the absence of POD or unacceptable toxicity |
Recruiting | NCT01434316 |
| BAY1895344 usual chemotherapy | 54 | Advanced solid tumors | 1 | Gemcitabine on days 1 and 8, and BAY 1895344 BID on days 1–3 and 8–10. Cycles repeat every 21 days in the absence of POD or unacceptable toxicity | Not yet recruiting | NCT04616534 |
| BAY1895344 to usual chemotherapy | 87 | Advanced solid tumors, special focus on PDAC, SCLC, poorly differentiated neuroendocrine cancer. | 1 | Arm A: BAY 1895344 PO BID on days 1 and 2 and irinotecan liposome day 1. Cycles repeat every 14 days in the absence of POD or unacceptable toxicity. Arm B: Topotecan on days 1–5 and BAY 1895344 PO BID on days 2 and 5. Cycles repeat every 21 days in the absence of POD or unacceptable toxicity |
Not yet recruiting | NCT04514497 |
| AZD1775 and olaparib | 54 | Solid tumors who have PARP resistance | 1 | Olaparib BID on days 1–5 and 15–19 of each cycle and adavosertib PO QD on days 8–12 and 22–26 of each cycle. Cycles repeat every 28 days in the absence of POD or unacceptable toxicity | Recruiting | NCT04197713 |
| Copanlisib, olaparib and durvalumab | 102 | Solid tumors with mutations in HRR genes, PTEN, hotspot mutation in PIK3CA | 1 | Copanlisib hydrochloride on days 1, 8, and 15 and olaparib BID. Cycle 2, patients receive durvalumab on day 1. Cycles repeat every 28 days for 24 months in the absence POD or unacceptable toxicity | Recruiting | NCT03842228 |
QD: Once daily; BID: twice daily; HR: Homologous repair; PR: Partial response; CR: Complete response; QxW: Every x week; CxDy: Cycle x Day y; POD: Progression of disease; PO: orally.
PARP inhibition and Anti-Angiogenic Therapy
Anti-angiogenic therapy results in targeted inhibition of the vascular epidermal growth factor (VEGF) families and can induce a hypoxic tumor microenvironment which leads to downregulation of HR gene expression[94]. It has been suggested that this hypoxic state causes functional inactivation of BRCA1 and RAD51 in the absence of genetic mutations of these genes, thus inducing BRCAness[95,96]. In ovary cancer, bevacizumab, a humanized monoclonal antibody against VEGF-A, was added to olaparib as maintenance therapy in platinum responders in the phase 3 PAOLA-1/ENGOT-ov25 trial, demonstrating a PFS survival benefit for both BRCA mutated and HRD-positive patients[97]. Cediranib is an anti-angiogenic targeted kinase inhibitor against VEGFR1, VEGFR2, and VEGFR3. A phase 2 study of olaparib with or without cediranib in men with metastatic castration resistant prostate cancer reported a median PFS of 11.1 versus 4.0 months in combination arm and monotherapy arm, respectively (HR 0.54, 95% CI 0.317, 0.928, P=0.026) with HR mutation status pending. In a PARP resistance setting, cediranib plus olaparib for ovary cancer patients following disease progression on PARP inhibition, reported a sixteen-week PFS rate of 55% in platinum sensitive patients and 50% in platinum resistant group. This study also identified genomic mechanisms of resistance to PARP inhibitors including reversion mutations in BRCA1/2 and RAD51B (19%); CCNE1amplification (16%); ABCB1 upregulation (15%); and SLFN11 downregulation (7%). Patients with reversion mutations in HR genes and/or ABCB1 upregulation had poor outcomes[98]. A phase 3 study comparing single olaparib and olaparib plus cediranib combination against standard of care (SOC) chemotherapy in relapsed, platinum sensitive ovarian cancer found the combination had similar activity to SOC with ORR of 71.3% (SOC), 52.4% (olaparib), and 69.4% (combination)[99]. The combination failed to meet the primary endpoint of improved PFS[91]. In germline BRCA patients, HR for PFS was 0.55 (95% CI 0.73–1.30) for combination vs SOC[94]. Although the combination of PARP inhibition and anti-angiogenic therapy is novel, and significant activity has been observed in ovarian cancer. The theoretical role of anti-angiogenesis in perturbation of HR-gene transcription in non-HRD patients has not been proven in a clinical trial setting to date and evaluation of monoclonal antibody versus a tyrosine kinase inhibitor requires further investigation in ongoing trials. A phase 2 study combining olaparib and cediranib in advanced solid tumors, including PDAC is currently recruiting (NCT02498613).
PARP Inhibitors and Immune Checkpoint Blockade
PARP inhibition leads to an accumulation of DNA damage. Dying cells release endogenous molecules referred to as danger/damage associated molecular patterns (DAMPs)[100]. DAMPs act as neoantigens for anti-tumor immune response, involving activation of both innate and adaptive immune responses with the process being dependent on stimulator of interferon genes (STING) pathway activation[101–104]. Treatment with PARP inhibitors has also been found to increase PD-L1 expression [105,106]. The combination of PARP inhibitors and immune check point inhibitors has been studied in a number of malignancies, showing promising anti-tumor activity. The phase 1/2 MEDIOLA study was a basket study investigating combination olaparib with durvalumab (AstraZeneca), a PD-L1 inhibitor in patients with germline BRCA mutated ovarian and breast cancer, metastatic gastric cancer, and relapsed small cell lung cancer. In the metastatic breast cohort (n=34), the median duration of response was 9·2 months (95% CI 5·5–13·1) with ORR of 53%. At a median follow-up of 6·7 months (IQR 4·6–13·8), median PFS was 8·2 months (95% CI 4·6–11·8: 80% maturity). At a median follow-up of 19·8 months (IQR 14·4–25·5), median OS was 21·5 months (95% CI 16.2–25.763% maturity)[107]. In the platinum sensitive relapsed ovarian cohort (n=32), the ORR was 71.9% (95% CI: 53.25%, 86.25%) with a total of seven CRs. The median PFS was 11.1 months (95% CI: 8.2, 15.9) and OS for all patients was not reached with 87% of patients still alive at 24 months[108]. In PDAC, the combination of immunotherapy and PARP inhibitors is still in its infancy with a number of trials ongoing. A phase 2 study of olaparib and durvalumab vs cediranib and durvalumab in PDAC and other solid tumors is currently recruiting as part of the DAPPER trial (NCT03851614). Dostarlimab (ANB011, GSK) is an anti PD-1 monoclonal antibody which is currently being evaluated in combination with niraparib in patients with germline or somatic BRCA1/2 PDAC (NCT04493060, NCT04673448). A phase 2 study combining well known anti PD-L1 inhibitor pembrolizumab with olaparib in patients with metastatic PDAC and germline BRCA mutations is also recruiting (NCT04548752). Expanding beyond the BRCA mutation setting, the phase 2 POLAR trial evaluating olaparib and pembrolizumab combination in PDAC patients with HR-gene mutations or exceptional response to platinum-based therapy without HR-gene mutations, is also currently recruiting (NCT04666740).
PARP and other Targeted Agents
A number of other strategies to address PARP inhibitor resistance are in early drug development. ATR plays a significant role in resistance to PARP inhibition. In pre-clinical studies using ovarian models, PARP inhibitor-resistance was accompanied by increased ATR-Chk1 activity and sensitivity to ATR inhibition[109]. It is noted that PARP inhibitor resistant cells are significantly more sensitive to ATR inhibitors when combined with PARP inhibitors[109]. A phase 2 trial combining olaparib with AZD6738 (AstraZeneca), an ATR kinase inhibitor is currently recruiting. Patients with ATM loss/mutation or BAF250a (ARID1A) expression are eligible to participate, with only the ARID1A positive group eligible to receive the combination (NCT03682289). Another pre-clinical study observed that PARP inhibitor resistant cells had elevated RAS/MAPK signaling independent of mutations in KRAS and that resistance was at least in part reversed by MEK inhibition[110]. A phase 1 /2 study of combinations of avelumab (Pfizer) a PD-L1 inhibitor, binimetinib (Pfizer) a MEK1/2 inhibitor and the PARP inhibitor talazoparib (Pfizer) is currently recruiting (NCT03637491). Finally, pre-clinical studies have also identified that inhibition of cycling dependent kinase 12 (CDK12), a transcriptional regulator of the HR process, can reverse PARP inhibitor resistance in BRCA mutated triple negative breast cancer cell lines[111]. A phase 1 study combining veliparib and dinaciclib (Merck) in advanced solid tumors is currently recruiting (NCT01434316).
Targeting ATR
ATR plays an important role in maintaining genome integrity during DNA replication, through the phosphorylation and activation of CHK1 upon binding of replication protein A (RPA) to single-strand DNA. ATR binds to RPA and is activated through TOPBP1 complex and phosphorylated ATR activates downstream CHK1 kinase[112]. This CHK1 activation ultimately slows the cell cycle by activating the G2/M negative checkpoint regulators CDC25C and WEE1. Accumulating evidence suggests that targeting ATR can selectively sensitize cancer cells but not normal cells to DNA damage and this strategy can cause synthetic lethality in ATM-mutant cancer cells [113]. Gemcitabine activates the ATR/Chk1 pathway which affects tumor survival. Downregulation of this pathway has been shown to sensitize cancer cells to gemcitabine[114,115]. Two trials evaluating the addition of ATR inhibitor BAY1895344 (Bayer) to usual chemotherapy for advanced solid tumors, with special focus on PDAC has recently been registered but is not yet recruiting. (NCT04616534, NCT04514497). Evaluating HR status is a secondary outcome of these trials.
Inducing HRD in HR Proficient Tumors
A number of strategies to induce HRD in HR proficient tumors, thus expanding the use of DDR pathway targeting to more patients are currently underway. WEE1 is a tyrosine kinase that inhibits the activation of CDK1 and CDK2 and thus, acts as a cell cycle regulator in the G2/M and S phases of the cell cycle. AZD1775(AstraZeneca) is a small molecular inhibitor of WEE1 and has been shown to cause cell cycle acceleration and apoptosis when applied with DNA damaging agents[116]. In a pre-clinical study, WEE1 inhibition decreased the level of HR related proteins, suggesting WEE1 inhibition induces HRD by decreasing the ability of HR to repair cells. Co-treatment with AZD1775 and olaparib was found to be highly effective in BRCA proficient triple negative breast cancer cells. It was also observed that AZD1775 enhanced sensitivity to cisplatin and an ATR inhibitor[116]. A dose escalation study of AZD 1775 in combination with gemcitabine and radiation therapy in patients with locally advanced PDAC, demonstrated that the combination was well tolerated[117]. The median OS for all patients was 21.7 months (90% CI, 16.7 to 24.8 months), and the median PFS was 9.4 months (90% CI, 8.0 to 9.9 months)[117]. However, disappointingly no analysis related to HRD was reported. A phase 1 clinical trial of AZD1775 in patients who have BRCA mutated/ HRD solid tumors and whose disease has progressed on PARP inhibitor therapy is currently recruiting (NCT04197713).
ATM and ATR are PI3K-related protein kinase in DNA response pathways. In a pre-clinical study using triple negative breast cancer cells, it was observed that the suppression of PI3K resulted in HR impairment by BRCA1 and RAD51 downregulation and apoptotic cell death by the induction of DNA damage[118]. Further pre-clinical studies demonstrated that mTOR/PI3K inhibition enhanced the anti-tumor efficacy of PARP inhibitors, again in breast cancers[119,120]. A phase 1 study combining niraparib and copanlisib (Bayer), a PI3K inhibitor in a variety of gynecological cancers is currently recruiting(NCT03586661). A phase 1 study of copanlisib, olaparib and durvalumab in advanced solid tumors is also recruiting (NCT03842228).
Conclusion
New therapies are urgently needed for PDAC. Germline and somatic genomic testing are standard of care for all patients with PDAC. PDAC patients with mutations in core HR-genes (BRCA1/2, PALB2) involved in DDR benefit from a personalized approach to treatment, irrespective of whether the mutations are germline or somatic. Treatment with platinum chemotherapy in the first line setting for patients with HR-deficient PDAC has been demonstrated to impact survival. Maintenance therapy with olaparib following induction chemotherapy in patients with germline BRCA1/2 mutations has yielded promising results but resistance mechanisms to PARP inhibition remain a challenge. Many trials are underway to build on the results of the POLO trial including evaluating the combination of PARP inhibitor therapy and immunotherapy. While PARP inhibitor/chemotherapy combination trials have failed to demonstrate a survival benefit, a number of additional combination strategies to counteract PARP resistance are currently being investigated. Defining and identifying HRD tumors will be paramount going forward, with a number of promising HRD scores evaluated in recent years. Of high priority are strategies to induce HRD and overcome resistance to HRD and thus expand the number of patients who may benefit from DDR targeted therapy.
Funding Sources
Cancer Center Support Grant P30 CA0008748
David M. Rubinstein Center for Pancreas Research
Paul Calabresi Career Development Award K12 CA184746
Parker Institute for Immunotherapy Pilot Grant
Society of Immunotherapy for Cancer (SITC) - TimIOs
Footnotes
Disclosures
FC: No conflicts of interest to declare.
WP: Research funding to institution: Merck, Astellas, Gossamerbio.
EOR: Research Funding to MSK: Genentech/Roche, Celgene/BMS, BioNTech, BioAtla, AstraZeneca, Arcus, Elicio. Consulting Role: Cytomx Therapeutics (DSMB), Rafael Therapeutics (DSMB), Sobi, Silenseed, Molecular Templates, Boehringer Ingelheim, BioNTech, Ipsen, Polaris, Tyme, Seagen, Merck, AstraZeneca, Noxxon, BioSapien, Bayer (spouse), Genentech-Roche (spouse), Celgene-BMS (spouse), Eisai (spouse)
References
- 1.Siegel RL, Miller KD, Fuchs HE, & Jemal A (2021). Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians, 71(1), 7–33, doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med, 364(19), 1817–1825, doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med, 369(18), 1691–1703, doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prawira A, Pugh TJ, Stockley TL, & Siu LL (2017). Data resources for the identification and interpretation of actionable mutations by clinicians. Annals of Oncology, 28(5), 946–957, doi: 10.1093/annonc/mdx023. [DOI] [PubMed] [Google Scholar]
- 5.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. (2020). Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. The Lancet Oncology, 21(4), 508–518, doi: 10.1016/S1470-2045(20)30074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, et al. (2019). Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology, 156(8), 2242–2253.e2244, doi: 10.1053/j.gastro.2019.02.037. [DOI] [PubMed] [Google Scholar]
- 7.Heestand GM, & Kurzrock R (2015). Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget, 6(7), 4553–4561, doi: 10.18632/oncotarget.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. (2018). Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J Natl Cancer Inst, 110(10), 1067–1074, doi: 10.1093/jnci/djy024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salo-Mullen EE, O’Reilly EM, Kelsen DP, Ashraf AM, Lowery MA, Yu KH, et al. (2015). Identification of germline genetic mutations in patients with pancreatic cancer. Cancer, 121(24), 4382–4388, doi: 10.1002/cncr.29664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S, & Cardin D (2020). Targeting DNA Damage Repair Pathways in Pancreatic Adenocarcinoma. Current Treatment Options in Oncology, 21(8), 62, doi: 10.1007/s11864-020-00763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyer S, Paulus-Hock V, Upstill-Goddard R, Lampraki E, Jamieson N, Cooke S, et al. (2019). Defining DNA damage repair deficiency and replication stress in pancreatic cancer. Journal of Clinical Oncology, 37(4_suppl), 285–285, doi: 10.1200/JCO.2019.37.4_suppl.285. [DOI] [Google Scholar]
- 12.Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen W-J, et al. (2018). Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precision Oncology(2), 1–13, doi: 10.1200/PO.17.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindahl T, & Wood RD (1999). Quality control by DNA repair. Science, 286(5446), 1897–1905, doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 14.Curtin NJ (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer, 12(12), 801–817, doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 15.Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, et al. (2016). Genomic landscape of DNA repair genes in cancer. Oncotarget, 7(17), 23312–23321, doi: 10.18632/oncotarget.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. (2004). Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst, 96(4), 261–268, doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 357(6349), 409, doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. (2020). Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol, 38(1), 1–10, doi: 10.1200/jco.19.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beatty GL, Eghbali S, & Kim R (2017). Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. American Society of Clinical Oncology Educational Book(37), 267–278, doi: 10.1200/EDBK_175232. [DOI] [PubMed] [Google Scholar]
- 20.Balachandran VP, Beatty GL, & Dougan SK (2019). Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology, 156(7), 2056–2072, doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarantis P, Koustas E, Papadimitropoulou A, Papavassiliou AG, & Karamouzis MV (2020). Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World journal of gastrointestinal oncology, 12(2), 173–181, doi: 10.4251/wjgo.v12.i2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahin IH, Askan G, Hu ZI, & O’Reilly EM (2017). Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Annals of Oncology, 28(12), 2950–2961, doi: 10.1093/annonc/mdx503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yimit A, Adebali O, Sancar A, & Jiang Y (2019). Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nature Communications, 10(1), 309, doi: 10.1038/s41467-019-08290-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plo I, Liao ZY, Barceló JM, Kohlhagen G, Caldecott KW, Weinfeld M, et al. (2003). Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst), 2(10), 1087–1100, doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 25.Hoeijmakers JHJ (2001). Genome maintenance mechanisms for preventing cancer. Nature, 411(6835), 366–374, doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 26.Sishc BJ, & Davis AJ (2017). The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers, 9(7), 81, doi: 10.3390/cancers9070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapman JR, Taylor Martin R. G., & Boulton Simon J. (2012). Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Molecular Cell, 47(4), 497–510, doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 28.Perkhofer L, Gout J, Roger E, Kude de Almeida F, Baptista Simões C, Wiesmüller L, et al. (2021). DNA damage repair as a target in pancreatic cancer: state-of-the-art and future perspectives. Gut, 70(3), 606–617, doi: 10.1136/gutjnl-2019-319984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown JS, O’Carrigan B, Jackson SP, & Yap TA (2017). Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov, 7(1), 20–37, doi: 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Pearlman AH, & Hsieh P (2016). DNA mismatch repair and the DNA damage response. DNA Repair, 38, 94–101, doi: 10.1016/j.dnarep.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cimprich KA, & Cortez D (2008). ATR: an essential regulator of genome integrity. Nature Reviews Molecular Cell Biology, 9(8), 616–627, doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krejci L, Altmannova V, Spirek M, & Zhao X (2012). Homologous recombination and its regulation. Nucleic Acids Research, 40(13), 5795–5818, doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dietlein F, Thelen L, & Reinhardt HC (2014). Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends in Genetics, 30(8), 326–339, doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Reports, 23(1), 239–254.e236, doi: 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. (2017). Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 35(30), 3382–3390, doi: 10.1200/JCO.2017.72.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margaret AT (2019). NCCN Guidelines Updates: Pancreatic Cancer. Journal of the National Comprehensive Cancer Network J Natl Compr Canc Netw, 17(5.5), 603–605, doi: 10.6004/jnccn.2019.5007. [DOI] [PubMed] [Google Scholar]
- 37.Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, et al. (1996). Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res, 56(23), 5360–5364. [PubMed] [Google Scholar]
- 38.Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. (2016). Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov, 6(2), 166–175, doi: 10.1158/2159-8290.CD-15-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature, 518(7540), 495–501, doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knudsen ES, O’Reilly EM, Brody JR, & Witkiewicz AK (2016). Genetic Diversity of Pancreatic Ductal Adenocarcinoma and Opportunities for Precision Medicine. Gastroenterology, 150(1), 48–63, doi: 10.1053/j.gastro.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucas AL, Shakya R, Lipsyc MD, Mitchel EB, Kumar S, Hwang C, et al. (2013). High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clinical cancer research : an official journal of the American Association for Cancer Research, 19(13), 3396–3403, doi: 10.1158/1078-0432.CCR-12-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krantz BA, Yu KH, & O’Reilly EM (2017). Pancreas adenocarcinoma: novel therapeutics. Chin Clin Oncol, 6(3), 30, doi: 10.21037/cco.2017.06.14. [DOI] [PubMed] [Google Scholar]
- 43.Lee K, Yoo C, Kim KP, Park KJ, Chang HM, Kim TW, et al. (2018). Germline BRCA mutations in Asian patients with pancreatic adenocarcinoma: a prospective study evaluating risk category for genetic testing. Invest New Drugs, 36(1), 163–169, doi: 10.1007/s10637-017-0497-1. [DOI] [PubMed] [Google Scholar]
- 44.Yadav S, Kasi PM, Bamlet WR, Ho TP, Polley EC, Hu C, et al. (2020). Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clinical Cancer Research, 26(24), 6505, doi: 10.1158/1078-0432.CCR-20-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozçelik H, Schmocker B, Di Nicola N, Shi XH, Langer B, Moore M, et al. (1997). Germline BRCA2 6174delT mutations in Ashkenazi Jewish pancreatic cancer patients. Nat Genet, 16(1), 17–18, doi: 10.1038/ng0597-17. [DOI] [PubMed] [Google Scholar]
- 46.Lal G, Liu G, Schmocker B, Kaurah P, Ozcelik H, Narod SA, et al. (2000). Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res, 60(2), 409–416. [PubMed] [Google Scholar]
- 47.Ferrone CR, Levine DA, Tang LH, Allen PJ, Jarnagin W, Brennan MF, et al. (2009). BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol, 27(3), 433–438, doi: 10.1200/jco.2008.18.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stadler ZK, Salo-Mullen E, Patil SM, Pietanza MC, Vijai J, Saloustros E, et al. (2012). Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer, 118(2), 493–499, doi: 10.1002/cncr.26191. [DOI] [PubMed] [Google Scholar]
- 49.Chaffee KG, Oberg AL, McWilliams RR, Majithia N, Allen BA, Kidd J, et al. (2018). Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med, 20(1), 119–127, doi: 10.1038/gim.2017.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. (2015). Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology, 148(3), 556–564, doi: 10.1053/j.gastro.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, et al. (2018). Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clinical Cancer Research, 24(6), 1326, doi: 10.1158/1078-0432.CCR-17-3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golan T, Kindler HL, Park JO, Reni M, Macarulla T, Hammel P, et al. (2020). Geographic and Ethnic Heterogeneity of Germline BRCA1 or BRCA2 Mutation Prevalence Among Patients With Metastatic Pancreatic Cancer Screened for Entry Into the POLO Trial. Journal of Clinical Oncology, 38(13), 1442–1454, doi: 10.1200/JCO.19.01890. [DOI] [PubMed] [Google Scholar]
- 53.Golan T, & Brody JR (2020). Targeting homologous recombination addicted tumors: challenges and opportunities. Annals of Pancreatic Cancer, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lord CJ, & Ashworth A (2016). BRCAness revisited. Nature Reviews Cancer, 16(2), 110–120, doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 55.Kondo T, Kanai M, Kou T, Sakuma T, Mochizuki H, Kamada M, et al. (2018). Association between homologous recombination repair gene mutations and response to oxaliplatin in pancreatic cancer. Oncotarget, 9(28), 19817–19825, doi: 10.18632/oncotarget.24865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gulhan DC, Lee JJ, Melloni GEM, Cortés-Ciriano I, & Park PJ (2019). Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet, 51(5), 912–919, doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 57.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. (2017). HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med, 23(4), 517–525, doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. (2016). Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res, 22(15), 3764–3773, doi: 10.1158/1078-0432.Ccr-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takaya H, Nakai H, Takamatsu S, Mandai M, & Matsumura N (2020). Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Scientific Reports, 10(1), 2757, doi: 10.1038/s41598-020-59671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. (2012). Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. British journal of cancer, 107(10), 1776–1782, doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. (2012). Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with <em>BRCA1/2</em> Inactivation. Cancer Research, 72(21), 5454, doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 62.Birkbak NJ, Wang ZC, Kim J-Y, Eklund AC, Li Q, Tian R, et al. (2012). Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov, 2(4), 366, doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Golan T, O’Kane GM, Denroche RE, Raitses-Gurevich M, Grant RC, Holter S, et al. (2021). Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology, doi: 10.1053/j.gastro.2021.01.220. [DOI] [PubMed] [Google Scholar]
- 64.O’Kane GM, Denroche R, Picardo SL, Zhang A, Holter S, Grant RC, et al. (2020). Homologous recombination deficiency (HRD) scoring in pancreatic ductal adenocarcinoma (PDAC) and response to chemotherapy. Journal of Clinical Oncology, 38(4_suppl), 741–741, doi: 10.1200/JCO.2020.38.4_suppl.741. [DOI] [Google Scholar]
- 65.Park W, Chen J, Chou JF, Varghese AM, Yu KH, Wong W, et al. (2020). Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clinical cancer research : an official journal of the American Association for Cancer Research, 26(13), 3239–3247, doi: 10.1158/1078-0432.CCR-20-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. (2021). Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. Journal of Clinical Oncology, 39(3_suppl), 378–378, doi: 10.1200/JCO.2021.39.3_suppl.378. [DOI] [Google Scholar]
- 67.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. (2019). Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med, 21(1), 213–223, doi: 10.1038/s41436-018-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Javle M, Shacham-Shmueli E, Xiao L, Varadhachary G, Halpern N, Fogelman D, et al. (2021). Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA oncology, doi: 10.1001/jamaoncol.2021.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dasari S, & Tchounwou PB (2014). Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol, 740, 364–378, doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434(7035), 917–921, doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 71.Pokataev I, Fedyanin M, Polyanskaya E, Popova A, Agafonova J, Menshikova S, et al. (2020). Efficacy of platinum-based chemotherapy and prognosis of patients with pancreatic cancer with homologous recombination deficiency: comparative analysis of published clinical studies. ESMO Open, 5(1), e000578, doi: 10.1136/esmoopen-2019-000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowery MA, Kelsen DP, Stadler ZK, Yu KH, Janjigian YY, Ludwig E, et al. (2011). An Emerging Entity: Pancreatic Adenocarcinoma Associated with a Known BRCA Mutation: Clinical Descriptors, Treatment Implications, and Future Directions. [ 10.1634/theoncologist.2011-0185]. The Oncologist, 16(10), 1397–1402, doi: 10.1634/theoncologist.2011-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, et al. (2014). Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. British journal of cancer, 111(6), 1132–1138, doi: 10.1038/bjc.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. (2017). Real-Time Genomic Profiling of Pancreatic Ductal Adenocarcinoma: Potential Actionability and Correlation with Clinical Phenotype. Clin Cancer Res, 23(20), 6094–6100, doi: 10.1158/1078-0432.Ccr-17-0899. [DOI] [PubMed] [Google Scholar]
- 75.Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. (2019). A decade of clinical development of PARP inhibitors in perspective. Annals of Oncology, 30(9), 1437–1447, doi: 10.1093/annonc/mdz192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ashworth A (2008). A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. Journal of Clinical Oncology, 26(22), 3785–3790, doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 77.Moore K, Colombo N, Scambia G, Kim B-G, Oaknin A, Friedlander M, et al. (2018). Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. New England Journal of Medicine, 379(26), 2495–2505, doi: 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 78.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. (2019). Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. New England Journal of Medicine, 381(4), 317–327, doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hall MJ, Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, et al. (2020). Pancreatic cancer (PaC)-specific health-related quality of life (HRQoL) with maintenance olaparib (O) in patients (pts) with metastatic (m) PaC and a germline BRCA mutation (gBRCAm): Phase III POLO trial. Journal of Clinical Oncology, 38(4_suppl), 648–648, doi: 10.1200/JCO.2020.38.4_suppl.648.31815583 [DOI] [Google Scholar]
- 80.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. (2016). Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. New England Journal of Medicine, 375(22), 2154–2164, doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 81.Binder KAR, Mick R, Hara M, Teitelbaum U, Karasic T, Schneider C, et al. (2019). Abstract CT234: A Phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in <em>BRCA1, BRCA2</em> or <em>PALB2</em>. Cancer Research, 79(13 Supplement), CT234, doi: 10.1158/1538-7445.AM2019-CT234. [DOI] [Google Scholar]
- 82.Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol, 33(3), 244–250, doi: 10.1200/jco.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. (2018). Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol, 2018, doi: 10.1200/po.17.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, et al. (2018). Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer, 89, 19–26, doi: 10.1016/j.ejca.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Reilly EM, Lowery MA, Segal MF, Smith SC, Moore MJ, Kindler HL, et al. (2014). Phase IB trial of cisplatin (C), gemcitabine (G), and veliparib (V) in patients with known or potential BRCA or PALB2-mutated pancreas adenocarcinoma (PC). Journal of Clinical Oncology, 32(15_suppl), 4023–4023, doi: 10.1200/jco.2014.32.15_suppl.4023.25332255 [DOI] [Google Scholar]
- 86.Yarchoan M, Myzak MC, Johnson BA 3rd, De Jesus-Acosta A, Le DT, Jaffee EM, et al. (2017). Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget, 8(27), 44073–44081, doi: 10.18632/oncotarget.17237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chiorean EG, Guthrie KA, Philip PA, Swisher EM, Jalikis F, Pishvaian MJ, et al. (2019). Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. Journal of Clinical Oncology, 37(15_suppl), 4014–4014, doi: 10.1200/JCO.2019.37.15_suppl.4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moffat GT, & O’Reilly EM (2020). The role of PARP inhibitors in germline BRCA-associated pancreatic ductal adenocarcinoma. Clin Adv Hematol Oncol, 18(3), 168–179. [PubMed] [Google Scholar]
- 89.Pishvaian MJ, Wang H, He AR, Hwang JJ, Smaglo BG, Kim SS, et al. (2020). A Phase I/II Study of Veliparib (ABT-888) in Combination with 5-Fluorouracil and Oxaliplatin in Patients with Metastatic Pancreatic Cancer. Clinical Cancer Research, 26(19), 5092, doi: 10.1158/1078-0432.CCR-20-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. (2020). Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J Clin Oncol, 38(13), 1378–1388, doi: 10.1200/jco.19.02931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li H, Liu Z-Y, Wu N, Chen Y-C, Cheng Q, & Wang J (2020). PARP inhibitor resistance: the underlying mechanisms and clinical implications. Molecular cancer, 19(1), 107–107, doi: 10.1186/s12943-020-01227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.D’Andrea AD (2018). Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair, 71, 172–176, doi: 10.1016/j.dnarep.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 93.Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, et al. (2018). DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell, 173(4), 972–988.e923, doi: 10.1016/j.cell.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. (2019). Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med, 11(492), doi: 10.1126/scitranslmed.aav4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. (2005). Hypoxia-Induced Down-regulation of <em>BRCA1</em> Expression by E2Fs. Cancer Research, 65(24), 11597, doi: 10.1158/0008-5472.CAN-05-2119. [DOI] [PubMed] [Google Scholar]
- 96.Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, & Glazer PM (2010). Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proceedings of the National Academy of Sciences, 107(5), 2201, doi: 10.1073/pnas.0904783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ray-Coquard IL, Pautier P, Pignata S, Pérol D, González-Martín A, Sevelda P, et al. (2019). LBA2_PR - Phase III PAOLA-1/ENGOT-ov25 trial: Olaparib plus bevacizumab (bev) as maintenance therapy in patients (pts) with newly diagnosed, advanced ovarian cancer (OC) treated with platinum-based chemotherapy (PCh) plus bev. Annals of Oncology, 30, v894–v895, doi: 10.1093/annonc/mdz394.053. [DOI] [Google Scholar]
- 98.Lheureux S, Oaknin A, Garg S, Bruce JP, Madariaga A, Dhani NC, et al. (2020). EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clinical Cancer Research, 26(16), 4206, doi: 10.1158/1078-0432.CCR-19-4121. [DOI] [PubMed] [Google Scholar]
- 99.Liu JF, Brady MF, Matulonis UA, Miller A, Kohn EC, Swisher EM, et al. (2020). A phase III study comparing single-agent olaparib or the combination of cediranib and olaparib to standard platinum-based chemotherapy in recurrent platinum-sensitive ovarian cancer. Journal of Clinical Oncology, 38(15_suppl), 6003–6003, doi: 10.1200/JCO.2020.38.15_suppl.6003. [DOI] [Google Scholar]
- 100.Vénéreau E, Ceriotti C, & Bianchi ME (2015). DAMPs from Cell Death to New Life. Frontiers in immunology, 6, 422–422, doi: 10.3389/fimmu.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gay CM, & Byers LA (2019). PARP Inhibition Combined with Immune Checkpoint Blockade in SCLC: Oasis in an Immune Desert or Mirage? J Thorac Oncol, 14(8), 1323–1326, doi: 10.1016/j.jtho.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 102.Vikas P, Borcherding N, Chennamadhavuni A, & Garje R (2020). Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Frontiers in Oncology, 10, 570–570, doi: 10.3389/fonc.2020.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, et al. (2016). The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity, 44(5), 1177–1189, doi: 10.1016/j.immuni.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 104.Mouw KW, Goldberg MS, Konstantinopoulos PA, & D’Andrea AD (2017). DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov, 7(7), 675–693, doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. (2017). PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res, 23(14), 3711–3720, doi: 10.1158/1078-0432.Ccr-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nature Communications, 8(1), 1751, doi: 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. (2020). Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol, 21(9), 1155–1164, doi: 10.1016/s1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 108.Drew Y, Kaufman B, Banerjee S, Lortholary A, Hong SH, Park YH, et al. (2019). 1190PD - Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Annals of Oncology, 30, v485–v486, doi: 10.1093/annonc/mdz253.016. [DOI] [Google Scholar]
- 109.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. (2020). Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nature Communications, 11(1), 3726, doi: 10.1038/s41467-020-17127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, et al. (2017). Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Science translational medicine, 9(392), eaal5148, doi: 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. (2016). CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Reports, 17(9), 2367–2381, doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rundle S, Bradbury A, Drew Y, & Curtin NJ (2017). Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers, 9(5), doi: 10.3390/cancers9050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fokas E, Prevo R, Hammond EM, Brunner TB, McKenna WG, & Muschel RJ (2014). Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treatment Reviews, 40(1), 109–117, doi: 10.1016/j.ctrv.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 114.Karnitz LM, Flatten KS, Wagner JM, Loegering D, Hackbarth JS, Arlander SJH, et al. (2005). Gemcitabine-Induced Activation of Checkpoint Signaling Pathways That Affect Tumor Cell Survival. Molecular Pharmacology, 68(6), 1636, doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 115.Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, et al. (2009). Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Molecular Cancer Therapeutics, 8(1), 45, doi: 10.1158/1535-7163.MCT-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ha D-H, Min A, Kim S, Jang H, Kim SH, Kim H-J, et al. (2020). Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Scientific Reports, 10(1), 9930, doi: 10.1038/s41598-020-66018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. (2019). Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. Journal of Clinical Oncology, 37(29), 2643–2650, doi: 10.1200/JCO.19.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guney Eskiler G (2019). The Interaction of PI3K Inhibition with Homologous Recombination Repair in Triple Negative Breast Cancer Cells. J Pharm Pharm Sci, 22(1), 599–611, doi: 10.18433/jpps30684. [DOI] [PubMed] [Google Scholar]
- 119.Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, et al. (2016). mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin Cancer Res, 22(7), 1699–1712, doi: 10.1158/1078-0432.Ccr-15-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, & Dey N (2014). Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia, 16(1), 43–72, doi: 10.1593/neo.131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tuli R, Shiao SL, Nissen N, Tighiouart M, Kim S, Osipov A, et al. (2019). A phase 1 study of veliparib, a PARP-1/2 inhibitor, with gemcitabine and radiotherapy in locally advanced pancreatic cancer. EBioMedicine, 40, 375–381, doi: 10.1016/j.ebiom.2018.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]