

Abstract
The nutrient sensors peroxisome proliferator-activated receptor γ (PPARγ) and mechanistic target of rapamycin complex 1 (mTORC1) closely interact in the regulation of adipocyte lipid storage. The precise mechanisms underlying this interaction and whether this extends to other metabolic processes and the endocrine function of adipocytes are still unknown. We investigated herein the involvement of mTORC1 as a mediator of the actions of the PPARγ ligand rosiglitazone in subcutaneous inguinal white adipose tissue (iWAT) mass, endocrine function, lipidome, transcriptome and branched-chain amino acid (BCAA) metabolism. Mice bearing regulatory associated protein of mTOR (Raptor) deletion and therefore mTORC1 deficiency exclusively in adipocytes and littermate controls were fed a high-fat diet supplemented or not with the PPARγ agonist rosiglitazone (30 mg/kg/day) for 8 weeks and evaluated for iWAT mass, lipidome, transcriptome (Rnaseq), respiration and BCAA metabolism. Adipocyte mTORC1 deficiency not only impaired iWAT adiponectin transcription, synthesis and secretion, PEPCK mRNA levels, triacylglycerol synthesis and BCAA oxidation and mRNA levels of related proteins but also completely blocked the upregulation in these processes induced by pharmacological PPARγ activation with rosiglitazone. Mechanistically, adipocyte mTORC1 deficiency impairs PPARγ transcriptional activity by reducing PPARγ protein content, as well as by downregulating C/EBPα, a co-partner and facilitator of PPARγ. In conclusion, mTORC1 and PPARγ are essential partners involved in the regulation of subcutaneous adipose tissue adiponectin production and secretion and BCAA oxidative metabolism.
Keywords: mTORC1, PPARγ, BCAA oxidation, adiponectin secretion, glyceroneogenesis, subcutaneous adipose tissue, C/EBPα
1. Introduction
Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear receptor that acts as an essential regulator of adipocyte formation, metabolic and endocrine functions and survival [1]. Indeed, full PPARγ inactivation is associated with adipocyte death and severe lipoatrophy in humans and mice [2–5]. PPARγ can be weakly activated by a variety of naturally occurring lipids such as polyunsaturated long-chain fatty acids, phosphatidylcholine, prostaglandin J2, among others, playing therefore an important role as a cellular lipid sensor [1]. PPARγ is also potently activated by the synthetic thiazolidinediones (TZDs), rosiglitazone and pioglitazone, which are used in the treatment of insulin resistance and Type 2 Diabetes [6]. Mechanistically, PPARγ activation and modulation of gene transcription involves its interaction with a ligand, followed by heterodimerization with the retinoic acid receptor (RXR), and recruitment of several coregulators [1].
Although at expense of few side effects, pharmacological PPARγ activation consistently improves insulin sensitivity and glucose homeostasis by promoting: fat redistribution from dangerous visceral to safer subcutaneous white and brown fat depots reducing therefore ectopic lipid deposition and lipotoxicity [7–11]; adipocyte secretion of adiponectin and other beneficial adipokines [12; 13]; adipocyte branched-chain amino acid (BCAA) oxidation and alleviation of insulin receptor substrate (IRS) inhibition by ribosomal protein S6 kinase 1 (S6K1) [14]; and a reduction in adipose tissue inflammation and proinflammatory cytokine secretion [15]. Specifically regarding lipid storage, PPARγ enhances all enzymatic steps involved in adipocyte fatty acid uptake and esterification in triacylglycerol including glycerol 3-phosphate generation [16; 17].
Along with PPARγ, another important regulator of adipocyte lipid storage is the nutrient and growth factor sensor mechanistic target of rapamycin complex 1 (mTORC1). mTORC1 is a multiprotein complex composed of regulatory-associated protein of mTOR (Raptor), mammalian lethal with Sec13 protein 8 (mLST8), DEP domain-containing mTOR-interacting protein (Deptor), proline-rich Akt substrate 40 (Pras40) and Tti1/Tel2 built around the serine/threonine kinase mTOR [18]. Similarly to PPARγ, mTORC1 also plays a central role in the regulation of key processes in adipocyte biology including adipogenesis and lipogenesis [19; 20]. Indeed, adipocyte deletion of mTORC1 essential component Raptor promotes a lipoatrophic state characterized by adipose tissue reduced triacylglycerol synthesis and lipid storage, increased oxidative metabolism, uncoupling protein-1 (UCP-1) content, inflammation and unrestrained lipolysis [21–24].
Evidence gathered over the years indicates that PPARγ and mTORC1 closely interact in the regulation of adipocyte lipid storage. The precise mechanisms underlying this interaction, however, and whether this extends to other metabolic processes in adipocytes are still unknown. Supporting adipocyte PPARγ and mTORC1 crosstalk, pharmacological mTORC1 inhibition with rapamycin was shown to impair PPARγ transcriptional activity and therefore preadipocyte differentiation into mature adipocytes in vitro [25]. Similarly, in vivo rapamycin administration impaired adipose tissue lipid storage by reducing tissue expression of a subset of PPARγ-regulated genes involved in triacylglycerol synthesis [26; 27]. Furthermore, combination of pharmacological PPARγ activation (rosiglitazone) and mTORC1 inhibition (rapamycin) in vivo revealed that mTORC1 is not only activated by rosiglitazone, but also acts as a critical mediator of the upregulation of lipoprotein lipase activity, lipid clearance/uptake, and subcutaneous and brown fat accretion induced by this ligand [27]. Altogether, these findings suggest the existence of a positive feedfoward interaction between PPARγ and mTORC1, through which these nutrient sensors potentiate each other activities. One limitation of those studies, however, relates to the use of rapamycin to inhibit mTORC1, as this macrolide displays partial inhibitory actions towards mTORC1, blocking the phosphorylation of some, but not all complex substrates [28] and, depending on the cell type, dose, route and period of administration, also inhibits mTORC2 [29], which was recently shown to modulate PPARγ activity [30].
In an attempt therefore to overcome those limitations and to investigate the full spectrum of biological processes in adipocytes regulated by the mTORC1/PPARγ crosstalk, mice with Raptor deletion and therefore mTORC1 deficiency exclusively in adipocytes were treated with the PPARγ ligand rosiglitazone and evaluated for adiponectin production and secretion, lipidome, transcriptome, respiration and BCAA metabolism in inguinal adipose tissue. Our main findings indicate that mTORC1 is an important regulator of adipose tissue adiponectin production and secretion, PEPCK mRNA levels and glyceroneogenesis, triacylglycerol synthesis and BCAA catabolism and a required mediator of the upregulation in these processes induced by PPARγ activation.
2. Methods
2.1. Mice and treatment
Animal experimental procedures were approved by the Animal Care Committee of the Institute of Biomedical Sciences, University of Sao Paulo, Brazil (#098/2010 and 093/2012, CEUA). Mice from The Jackson Laboratory (Bar Harbor, Maine, USA) and with a C57BL6/J background were kept at 23 ± 1°C, 12:12 h light-dark cycle, with free access to water and food ad libitum. Adipocyte Raptor deletion was produced by crossing RaptorLox/Lox (B6.Cg-Rptortm1.1Dmsa/J) mice with adiponectin-cre mice (B6;FVB-Tg(Adipoq-cre)1Evdr/J) as previously described [22]. Male RaptorLox/Lox; adiponectin-cre (referred henceforth as RapKO) and their littermates RaptorLox/Lox (referred henceforth as RapWT) mice were used in the experiments. RapWT and RapKO mice were fed a high-fat diet (HFD, 20% carbohydrates, 20% protein, 60% fat, %Kcal) supplemented or not with rosiglitazone (30 mg/kg/day) for 8 weeks and evaluated for body weight and food intake weekly. After 8 weeks, 6-h fasted mice were euthanized for tissue and blood harvesting. This rosiglitazone dose was previously shown to reduce visceral, improve glucose homeostasis and increase BCAA oxidation in mice [11; 14].
2.2. Indirect calorimetry
Mice were adapted to the metabolic cages for 2 consecutive days and evaluated for oxygen consumption (VO2), carbon dioxide production (CO2), spontaneous motor activity (SMA) and respiratory exchange ratio (RER, VCO2/VO2 ratio) during 24 h in a Comprehensive Laboratory Monitoring System calorimeter (Columbus Instruments).
2.3. Serum parameters
Serum adiponectin and leptin were measured with ELISA kits (Millipore, MA, USA) and serum BCAA was measured with a colorimetric kit (Biovision, CA, USA), following supplier recommendations.
2.4. RNAseq
Total RNA was extracted from 50 mg of inguinal white adipose tissue (iWAT) using trizol and the RNAeasy Mini Kit (Qiagen, USA). Ribosomal RNA (rRNA) was removed with the NEBNext depletion kit (E6310X) according to manufacturer’s instructions. After cDNA synthesis by Maximal Reverse Transcriptase (Thermo Fisher cat no. EP0742) and NEBNext Ultra II Non-directional RNA Second Strand Synthesis Module (cat no E6111L), paired-end sequencing libraries were generated using the Illumina Nextera XT DNA Library Preparation Kit (FC-131-1096) according to manufacturer instructions. Quantity and quality of RNA-Seq libraries were analyzed by Qubit and Agilent Bioanalyzer, respectively. Libraries were pooled at a final concentration of 2.22 pM and sequenced by a NextSeq 500 system (36 bp, paired-end reads).
2.5. RNA-Seq analysis.
For RNA-Seq analysis, transcripts from the GCRm38/mm10 mouse transcriptome were quantified by quasi-mapping sequencing reads using Salmon [31]. Transcript counts were merged into gene counts using the tximport library [32]. Normalization and analysis of variance (ANOVA) were performed using EdgeR [33]. For ANOVA, we only tested genes that were detected in at least two samples with log2CPM ≥ 1. Genes were considered significant if they passed a false discovery rate (FDR) cutoff of FDR ≤ 0.05. Gene set enrichment analysis was performed using ShinyGO [34]. RNA-Seq raw and processed data are available at the Gene Expression Omnibus (GEO) repository accession number GSE166988.
2.6. RNA extraction and RT-qPCR
iWAT total RNA was extracted with Trizol (Invitrogen Life Technologies), reverse transcribed to cDNA, and evaluated by quantitative real-time PCR using a Rotor Gene (Qiagen) and SYBR Green as fluorescent dye as previously described [35]. Data are expressed as the ratio of target and reference gene (36B4), which was not significantly affected by either raptor deletion or rosiglitazone treatment. Primers sequences are detailed in Table 1.
Table 1.
Primers used in qPCR
| Gene | Nm | Forward | Reverse |
|---|---|---|---|
|
| |||
| 36b4 | NM_007475.5 | AGATTCGGGATATGCTGTTGGC | TCGGGTCCTAGACCAGTGTTC |
| Adiponectin | NM_009605.5 | GATGCAGGTCTTCTTGGTCCT | AGGTGAAGAGAACGGCCTTG |
| BCAT2 | NM_009737.3 | GGCTCCTACTTCCCTGGAGA | CAGCCACAGTGGGTCCATAG |
| BCKDHA | NM_007533.5 | TGGCTAGATCTCACCCCAGC | ATGCCGGAGATGACATTGGG |
| BCKDHB | NM_001305935.1 | AGTCCCTGCAGTATGGGCAAA | CCTGAGCGGTAGCGATACTT |
| BCKDK | NM_009739.3 | TGAGGATTGCTCACCGCATC | GGTCCTTGATCGGAGGGAAG |
| C/EBPα | NM_007678.3 | CAAGAACAGCAACGAGTACCG | GTCACTGGTCAACTCCAGCAC |
| DBT | NM_010022.4 | CAGACTGACCTGTGTTCGCT | GTGACGTGGCTGACTGTACT |
| DLD | NM_007861.5 | CCAGGTGCTGGAGAAATGGT | GCCTCTGATAAGGTCGGATGC |
| DsbA-L | NM_029555.2 | GGCTGGGCTTTGAGGTCCTA | ACTGGCCTTTTCGGGGAAC |
| ERO1-Lα | NM_015774.3 | AAGCGGACCAAGTTATGAGTTC | GTGCCTGAAGTTCTCTAGTTCTC |
| PEPCK | NM_011044.3 | CGATGACATCGCCTGGATGA | TCTTGCCCTTGTGTTCTGCA |
| PPARγ1 | NM_001127330.2 | CGGGCTGAGAAGTCACGT | TGTGTCAACCATGGTAATTTCAGT |
| PPARγ2 | NM_011146.3 | GGAATGCGAGTGGTCTTCCA | GCCTATGAGCACTTCACAAGAAAT |
| SLC25A44 | NM_178696.5 | ATGGAGGACAAACGGAACATC | GTGAACGGGTACACGCTGA |
| UCP-1 | NM_009463.3 | AGGCTTCCAGTACCATTAGGT | CTGAGTGAGGCAAAGCTGATTT |
2.7. Western blotting
Protein extracted from iWAT were resolved on polyacrylamide gels, transferred to PVDF membranes, blocked with 5% milk and incubated with primary and secondary antibodies, as previously described [35]. Primary antibodies were detailed in Table 2. Membranes were developed using the ECL enhanced chemiluminescence substrate (GE Healthcare Life Sciences). Densitometric analyses were performed with ImageJ (NIH).
Table 2.
Primary antibodies used in Western Blotting
| Primary antibody | Catalog number | Manufacturer |
|---|---|---|
|
| ||
| Adiponectin | ##2789 | Cell Signaling |
| C/EBPα | #2295 | Cell Signaling |
| Catalase | #14097 | Cell Signaling |
| Raptor | #2280 | Cell Signaling |
| SOD-1 | #AB13498 | Abcam |
| SOD-2 | #AB13533 | Abcam |
| Tubulin | #2125 | Cell Signaling |
| UCP-1 | #14670 | Cell Signaling |
2.8. iWAT valine oxidation
Valine oxidation was measured as previously described [14] by incubating iWAT explants (15–20 mg) in Krebs Ringer bicarbonate buffer [in mM]: 118 NaCl, 4.8 KCl, 1.25 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 HEPES (pH 7.4) containing 2% essentially fatty acid-free albumin, 5 mM glucose, 1 mM valine and 0.2 μCi/mL [U-14C] valine (PerkinElmer) in a hermetically closed vial. At the end of incubation, reaction was stopped with 6N H2SO4 and labeled 14CO2 was trapped in a piece of paper containing phenylethylamine:methanol (1:1) and counted.
2.9. iWAT pyruvate incorporation into triacylglycerol-glycerol.
Glyceroneogenesis was evaluated by rates of pyruvate incorporation into triacylglycerol-glycerol as previously described [36]. Briefly, iWAT explants (15–20 mg) were incubated in Krebs Ringer bicarbonate buffer [in mM]: 118 NaCl, 4.8 KCl, 1.25 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 HEPES (pH 7.4) containing 2% essentially fatty acid-free albumin, 5 mM glucose, 1 mM pyruvate and 0.2 μCi/mL [1-14C] pyruvate (PerkinElmer). At the end of incubation, reaction was stopped with 6N H2SO4 and the explants were washed, lipid extracted with chloroform:methanol (2:1) and counted as described [36].
2.10. iWAT oxygen consumption
iWAT explants were incubated in DMEM-4% BSA (w/v) under continuous stirring at 37°C and O2 consumption was measured with an Oroboros Oxygraph-2K essentially as previously described [35].
2.11. iWAT citrate synthase activity
iWAT was homogenized in 50 mM phosphate buffer and centrifuged at 13,000 rpm for 10 min. The supernatant was incubated with 0.1 M Tris-HCl, 500 μM oxaloacetate, 200 μM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), 100 μM acetyl-CoA and 0.1% Triton-X for 5 min at 30°C. DTNB reduction was followed at 412 nm [35].
2.12. iWAT lipidomics
iWAT lipidomics was performed essentially as previously described [37]. Briefly, iWAT (50 mg) was homogenized in 1 mL of 10 mM sodium phosphate buffer (pH 7.4) containing 100 μM deferoxamine mesylate. Briefly, samples aliquots (50 μL) were mixed with a cocktail of internal standards (Table 3) and lipids were extracted using methyl tert-butyl ether (MTBE) [37]. The organic phase containing lipids was dried under nitrogen stream, suspended in isopropanol and analyzed in an ultra-high-performance liquid chromatography (UHPLC Nexera, Shimadzu, Kyoto, Japan) coupled to electrospray ionization time-of-flight mass spectrometer (Triple TOF 6600, Sciex, Framingham, US). Samples (2 μl) were loaded into a CORTECS (UPLC C18 column, 1.6 μm, 2.1 mm i.d. × 100 mm) with a flow rate of 0.2 mL min−1 and the oven temperature maintained at 35°C. Mobile phases A (water:acetonitrile, 60:40) and B (isopropanol:acetonitrile:water, 88:10:2) contained ammonium acetate or ammonium formate (10 mM final concentration) for negative or positive ionization mode analyses, respectively. Lipids were separated using gradient elution: 40 to 100% B over the first 10 min, 100% B from 10–12 min, 100 to 40% B from 12–13 min, and 40% B from 13–20 min. The MS was operated in both positive and negative ionization modes, and the scan range was set at a mass-to-charge ratio of 200–2000 Da. MS/MS data were obtained by Information Dependent Acquisition (IDA®) mode with a period cycle time of 1.05 s with 50 ms acquisition time for MS1 scan and 20 ms acquisition time to obtain the top 20 precursor ions. Data acquisition was performed using Analyst® 1.7.1 with an ion spray voltage of −4.5 kV and 5.5 kV (for negative and positive modes, respectively) and a cone voltage at +/− 80 V. The curtain gas was set at 25 psi, nebulizer and heater gases at 45 psi, and interface heater of 450°C. The MS/MS data were analyzed with PeakView®. Lipid molecular species were manually identified based on their exact masses, specific fragments and/or neutral losses [38] with the help of an in-house manufactured Excel-based macro. Also, a maximum error of 5 mDa was defined for the attribution of the precursor ion. Lipid quantification was performed with MultiQuant®, where peak areas of each identified lipid precursor ions were normalized to those of the corresponding internal standards. Lipidomic processed data are available at 10.6084/m9.figshare.14229785.
Table 3.
Internal standards used in the untargeted lipidomics.
| Internal Standard | nmol in 50 L | Normalized lipid subclasses |
|---|---|---|
|
| ||
| FFA (13:0) | 2.333 | FFA |
| CL (14:0/14:0/14:0/14:0) | 0.403 | CL |
| PC (17:0/17:0) | 0.656 | PC; *PI; **CoQ |
| LPC (17:0) | 0.981 | LPC; **AC |
| PE (17:0/17:0) | 0.694 | PE |
| LPE (17:1) | 1.074 | LPE |
| PG (17:0/17:0) | 0.666 | PG |
| PS (17:0/17:0) | 0.654 | PS |
| Cer (d18:1/17:0) | 0.906 | Cer; **HexCer; **Sulfatide |
| SM (d18:1/17:0) | 0.697 | SM |
| DG (17:0/17:0) | 0.838 | DG |
| TG (17:0/17:0/17:0) | 0.589 | TG |
| CE (22:0) | 0.705 | CE; Cholesterol |
An external calibration curve relative to PC (17:0/17:0) was used for PI to determine class specific response factors. This calibration was performed using five different concentrations (from 0.03 ng to 1 ng per injection) of PI (14:1/17:0). The external calibration yielded a correction factor or response factor of 0.65 (PI) relative to PC (17:0/17:0).
No external calibration was performed for HexCer, Sulfatide, CoQ and AC, and so the concentration values are not absolute. The values need to be taken with caution, since they can only be compared within samples and not with other lipid compounds.
2.13. Statistical Analysis.
Differences between genotypes, treatments and their combination were evaluated by multifactorial ANOVA followed by Newman-Keuls test. Data were assessed for sphericity using Mauchly’s test, and whenever the test was violated, technical correction through the Greenhouse-Geisser test was performed. The statistical level of significance was set at P < 0.05. Data were analyzed using Graph Prism® (GraphPad Software Inc, San Diego, CA, USA). Results are presented as mean ± SEM.
3. Results
As illustrated in Figure 1, RapKO mice displayed reduced iWAT Raptor protein content (Fig. 1A), body weight gain (Fig. 1B–C), and masses of brown adipose tissue (BAT, Fig. 1D), visceral fat (epididymal + retroperitoneal, Fig. 1E) and iWAT (Fig. 1F), but no changes in food intake (Fig. 1G), rates of oxygen consumption expressed by mouse (Fig. 1H) and spontaneous motor activity (SMA, Fig. 1I). RapKO mice featured, on the other hand, increased respiratory exchange ratio (RER) indicating higher oxidation of carbohydrates (Fig. 1J). Along with adiposity, RapKO mice had reduced serum levels of leptin (Fig. 1K) and adiponectin (Fig. 1L), as well as reduced iWAT adiponectin mRNA and protein contents (Fig. 1M–N). Rosiglitazone administration to RapWT did not affect iWAT Raptor content (Fig. 1A), body weight gain (Fig. 1B–C), food intake (Fig. 1G), oxygen consumption (Fig. 1H) and SMA (Fig. 1I), but, as expected, it significantly increased BAT mass (Fig. 1D), reduced visceral adiposity (Fig. 1E), and increased RER (Fig. 1J) and serum and iWAT adiponectin contents (Fig. 1L and N). Interestingly, rosiglitazone did not show any of these well-established actions in RapKO mice, suggesting that adipocyte mTORC1 is a required mediator of those phenotypes associated with PPARγ agonism. In an attempt to investigate the mechanism by which mTORC1 deficiency and rosiglitazone affect adiponectin secretion, we evaluated iWAT mRNA levels of disulfide-bond A oxidoreductase like protein (DsbA-L) and endoplasmic reticulum oxidoreductase (Ero1-Lα), proteins involved in endoplasmic reticulum adiponectin multimerization. As illustrated in Figure 1O–P, adipocyte Raptor deletion significantly decreased DsbA-L and increased Ero1-Lα in iWAT. Rosiglitazone, on the other hand, did not affect DsbA-L and Ero1-Lα mRNA levels in both RapWT and RapKO iWAT.
Figure 1.
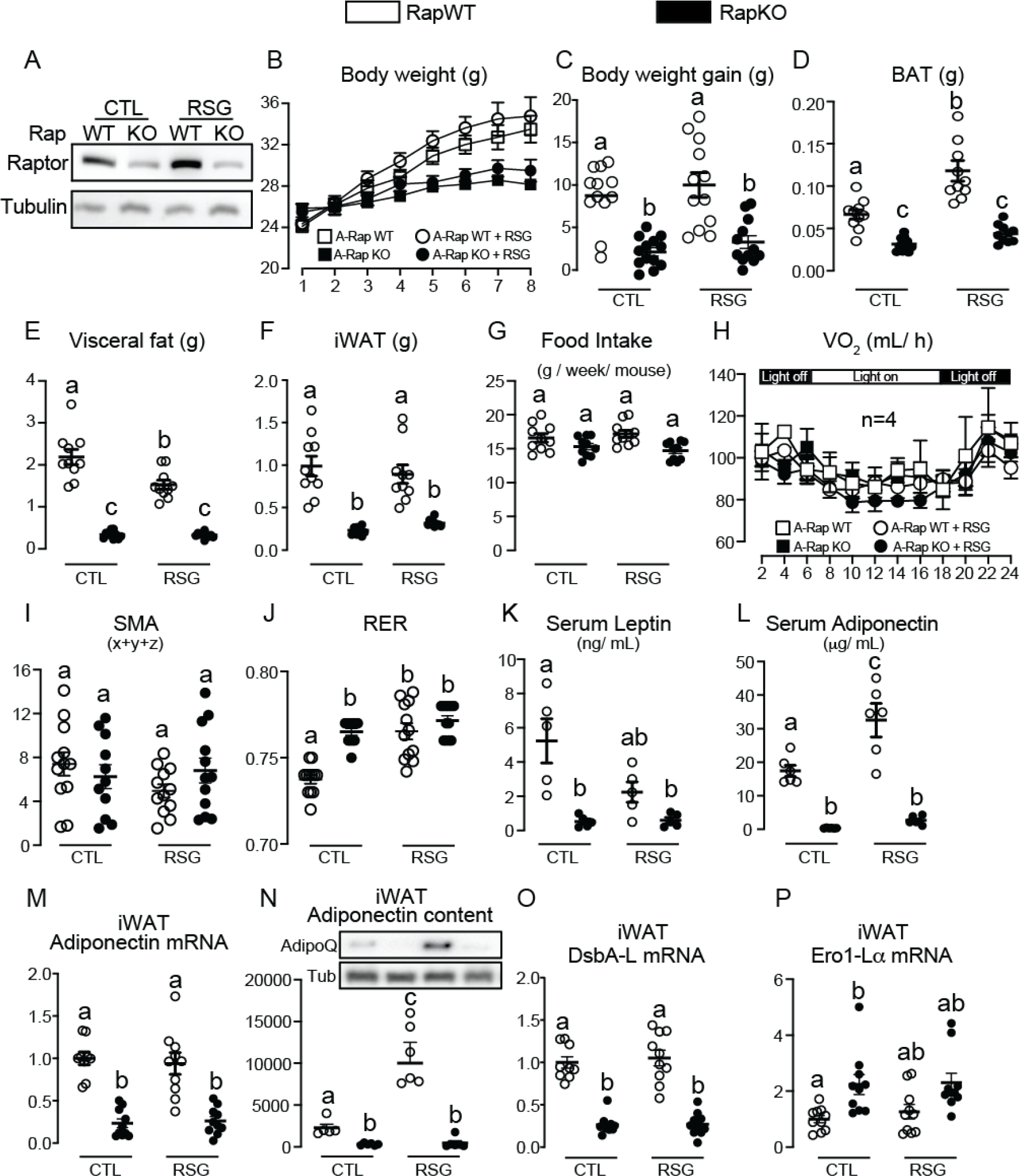
Adipocyte mTORC1 deficiency impairs iWAT adiponectin and blocks the activation of its synthesis and secretion by rosiglitazone. Inguinal white adipose tissue (iWAT) Raptor content (A), body weight (B), body weight gain (C), brown adipose tissue (BAT, D), visceral fat (E), and iWAT (F) masses, food intake (G), whole-body oxygen consumption (H), spontaneous motor activity (SMA, I), respiratory exchange ratio (RER, J), serum leptin (K) and adiponectin (L) and iWAT adiponectin mRNA (M) and protein (N) contents, and DsbA-L (O) and Ero1-La (P) mRNA levels in RapWT and RapKO mice fed with high-fat diet (HFD) containing or not rosiglitazone (RSG, 30 mg/kg/day) for 8 weeks. Values are expressed as mean ± SEM. n= 4–12 mice per group. Means not sharing a common superscript are significantly different from each other, p ≤ 0.05.
To further unveil processes regulated by the PPARγ-mTORC1 crosstalk, we performed a non-target lipidomic analysis of iWAT. Lipidomic analysis identified and quantified approximately 338 lipids in iWAT, among which 133 were significantly altered by either Raptor deficiency, or rosiglitazone, or their combination (Supplementary File 1 - 10.6084/m9.figshare.14229785). The most striking alterations found in lipidomics were the marked increase in RapKO iWAT content of the fatty acids palmitic (16:0) and oleic (18:1) acids either at the free form or conjugated to carnitine (Fig. 2A and B). Importantly, a similar response pattern was seen for other long-chain fatty acids such as palmitoleic (16:1), stearic (18:0) and linoleic (18:2) acids (Supplemental File 1 - 10.6084/m9.figshare.14229785). Interestingly, this accumulation of free fatty acids in RapKO mice was associated with a marked reduction in iWAT generation of glycerol 3-phosphate via glyceroneogenesis as evidenced by the reduced pyruvate incorporation into triacylglycerol-glycerol (Fig. 2C) and mRNA levels of its key enzyme phosphoenolpyruvate carboxykinase (PEPCK, Fig. 2D). The mRNA content of other enzymes involved in triacylglycerol synthesis, namely, glycerol 3-phosphate acyltransferase (GPAT) 1, 3 and 4 and diacylglycerol acyltransferase 1 (DGAT 1) was not significantly affected by mTORC1 deficiency (data not shown). In addition to free fatty acids and their respective acylcarnitines, another important lipidomic finding was a robust increase in RapKO iWAT content of some cardiolipins (Fig. 2E), which are exclusively found at mitochondrial inner membrane. Along with those changes in iWAT lipid profile, RapKO mice also displayed a significant increase in iWAT oxygen consumption (Fig. 2F), citrate synthase (CS) activity (Fig. 2G), a surrogate estimative of mitochondrial mass, and mRNA levels of the following proteins: PPAR coactivator 1α (PGC1α) (Fig. 2H), a coregulator previously shown to promote mitochondrial biogenesis and oxidative metabolism by modulating the activity of several transcriptional factors; carnitine palmitoyltransferase 1a (CPT1a) (Fig. 2I), the rate limiting enzyme that catalyzes long-chain fatty acid conjugation to carnitine required for mitochondrial entry and β–oxidation; and the uncoupling protein 1 (UCP-1) (Fig. 2J), a mitochondrial protein that dissipate proton gradient from ATP synthesis to heat production at mitochondrial inner membrane. Along with mRNA levels, adipocyte Raptor deletion significantly increased iWAT UCP-1 protein content (Fig. 2K and O); reduced the antioxidant enzyme catalase (Fig. 2L and O) that decomposes hydrogen peroxide to water and oxygen; did not affect cytosolic superoxide dismutase 1 (SOD-1, Fig. 2M and O), and significantly increased mitochondrial SOD-2 (Fig. 2N and O), antioxidant enzymes that convert superoxide into hydrogen peroxide. Rosiglitazone administration to RapWT mice did not affect iWAT free and carnitine-conjugated fatty acid contents (Fig. 2A–B), pyruvate incorporation into triacylglycerol-glycerol (Fig. 2C) and cardiolipin content (Fig. 2E), but it increased iWAT PEPCK mRNA levels (Fig. 2D), oxygen consumption (Fig. 2F), citrate synthase activity (Fig. 2G) and PGC1α mRNA levels (Fig. 2H), without significantly affecting CPT1a mRNA (Fig. 2I) and UCP-1 mRNA and protein contents (Fig. 2J–K). Rosiglitazone also increased iWAT protein content of the antioxidant enzymes catalase, SOD-1 and SOD-2 in RapWT mice (Fig. 2L–O). Rosiglitazone administration to RapKO mice, on the other hand, did not affect free and carnitine-conjugated palmitic and oleic acids contents (Fig. 2A and B, respectively), pyruvate incorporation into triacylglycerol-glycerol (Fig. 2C) and PEPCK mRNA levels (Fig. 2D), and partially reduced cardiolipin content (Fig. 2E) without significantly affecting iWAT respiration (Fig. 2F), citrate synthase activity (Fig. 2G), PGC1α and CPT1a mRNA levels (Fig. 2H and I), UCP-1 mRNA and protein content (Fig. 2J–K) and catalase, SOD-1 and SOD-2 protein contents (Fig. 2L–O).
Figure 2.
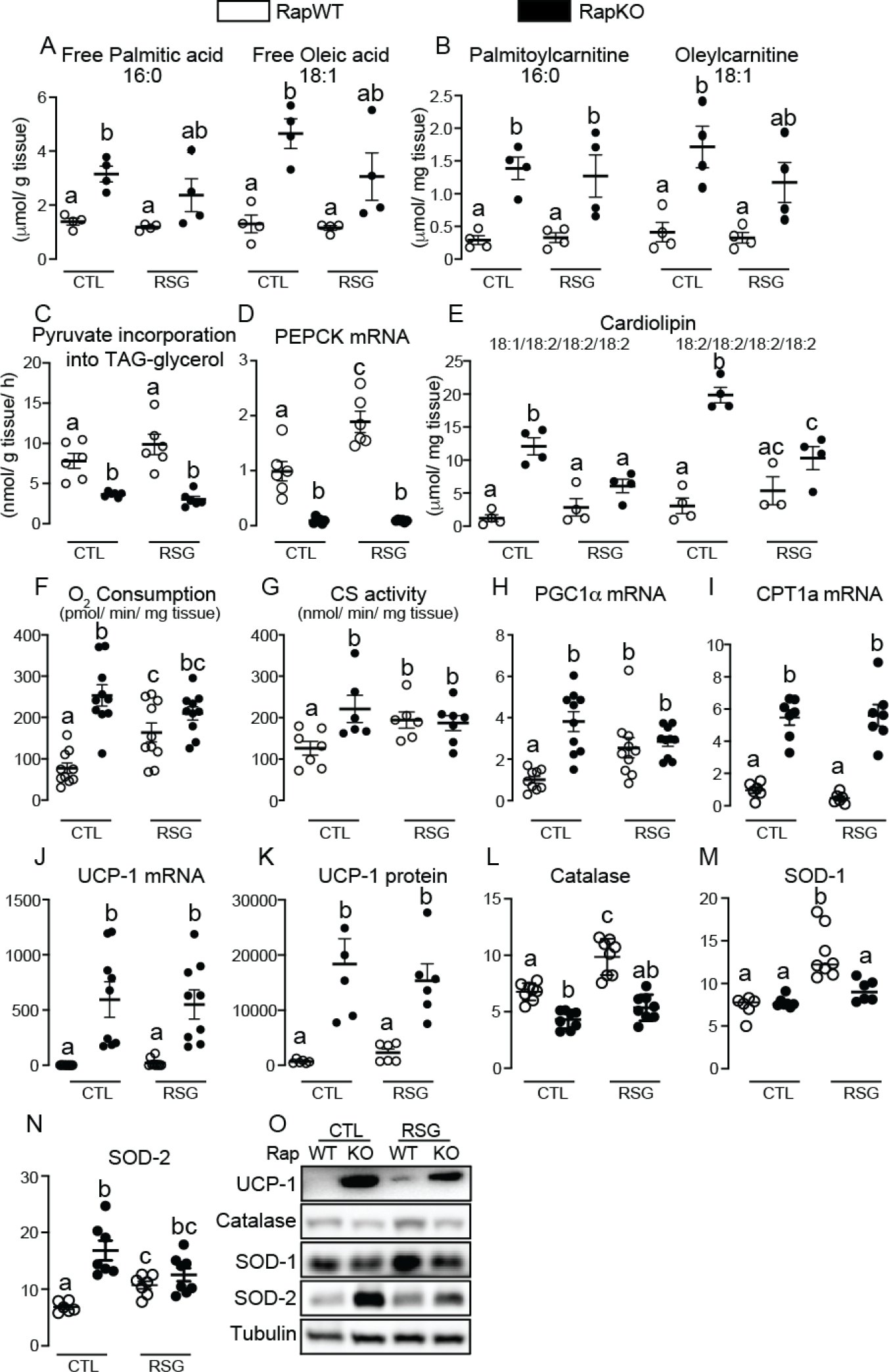
Adipocyte mTORC1 deficiency increases iWAT content of free fatty acids and acylcarnitines and oxidative metabolism. Inguinal white adipose tissue (iWAT) contents of the free fatty acids palmitate and oleate (A) and the acylcarnitines palmitoylcarnitine and oleylcarnitine (B), pyruvate incorporation into triacylglycerol-glycerol (C), PEPCK mRNA levels (D), cardiolipin content (E), oxygen consumption (F), citrate synthase (CS) activity (G), PGC1α (H), CPT1a (I), UCP-1 mRNA levels (J) and protein content of UCP-1 (K and O), catalase (L and O), SOD-1 (M and O) and SOD-2 (N and O) in RapWT and RapKO mice fed with high-fat diet (HFD) containing or not rosiglitazone (RSG, 30 mg/kg/day) for 8 weeks. Values are expressed as mean ± SEM. n= 4–10 mice per group. Means not sharing a common superscript are significantly different from each other, p ≤ 0.05.
To expand our knowledge about processes in adipocytes regulated by the crosstalk mTORC1-PPARγ, we next performed RNAseq analysis of iWAT. As illustrated in Figure 3, either adipocyte Raptor deficiency or rosiglitazone administration or their combination significantly altered mRNA levels of approximately 2425 genes. More specifically, adipocyte Raptor deletion altered the mRNA content of proteins mainly related to processes such as mitochondrial function and inflammation as revealed by Gene ontology enrichment analysis and KEGG (complete list at Supplemental File 2 - 10.6084/m9.figshare.14229785). To unveil clusters of genes in iWAT whose modulation by rosiglitazone requires mTORC1, we searched for genes that were significantly altered by rosiglitazone in RapWT, but not RapKO mice. We found that 408 genes met this condition (Figure 3A and B and Supplemental File 3 - 10.6084/m9.figshare.14229785). Gene ontology enrichment analysis and KEGG analysis of these genes revealed several metabolic processes such as tricarboxylic acid (TCA) cycle, malate-aspartate shuttle, mitochondrial respiration and electron transport, fatty acid β-oxidation, and catabolism of amino acids such as glutamate, tryptophan and the branched-chain amino acids (BCAA) leucine, isoleucine and valine (Fig. 3B). Next, we validated and extended RNAseq findings by evaluating iWAT BCAA metabolism. As depicted in Figure 4, RapKO mice displayed increased serum BCAA levels (Fig. 4A) in association with a reduced iWAT valine oxidation (Fig. 4B) and mRNA levels of key mitochondrial enzymes involved in BCAA oxidation such as branched-chain amino acid aminotransferase 2 (BCAT2, Fig. 4D), that catalyzes the first step in BCAA catabolism namely the transamination of BCAA to their respective alpha-keto acids, and dihydrolipoamide branched-chain transacylase E2 (DBT, Fig. 4G), a component of the branched-chain alpha-keto acid dehydrogenase complex (BCKDH) that catalyzes the conversion of alpha-keto acids to acyl-CoA and carbon dioxide. Interestingly, rosiglitazone administration reduced serum BCAA (Fig. 4A) and increased iWAT valine oxidation (Fig. 4B) and mRNA levels of SLC25A44 (Fig. 4C), a mitochondrial BCAA transporter; BCKDH subunits A (BCKDHA, Fig. 4E) and B (BCKDHB, Fig. 4F), DBT (Fig. 4G) and dihydrolipoamide dehydrogenase (DLD, Fig. 4H); and branched-chain keto acid dehydrogenase kinase (BCKDK, Fig. 4I). All these rosiglitazone actions were completely abolished in iWAT of RapKO mice.
Figure 3.
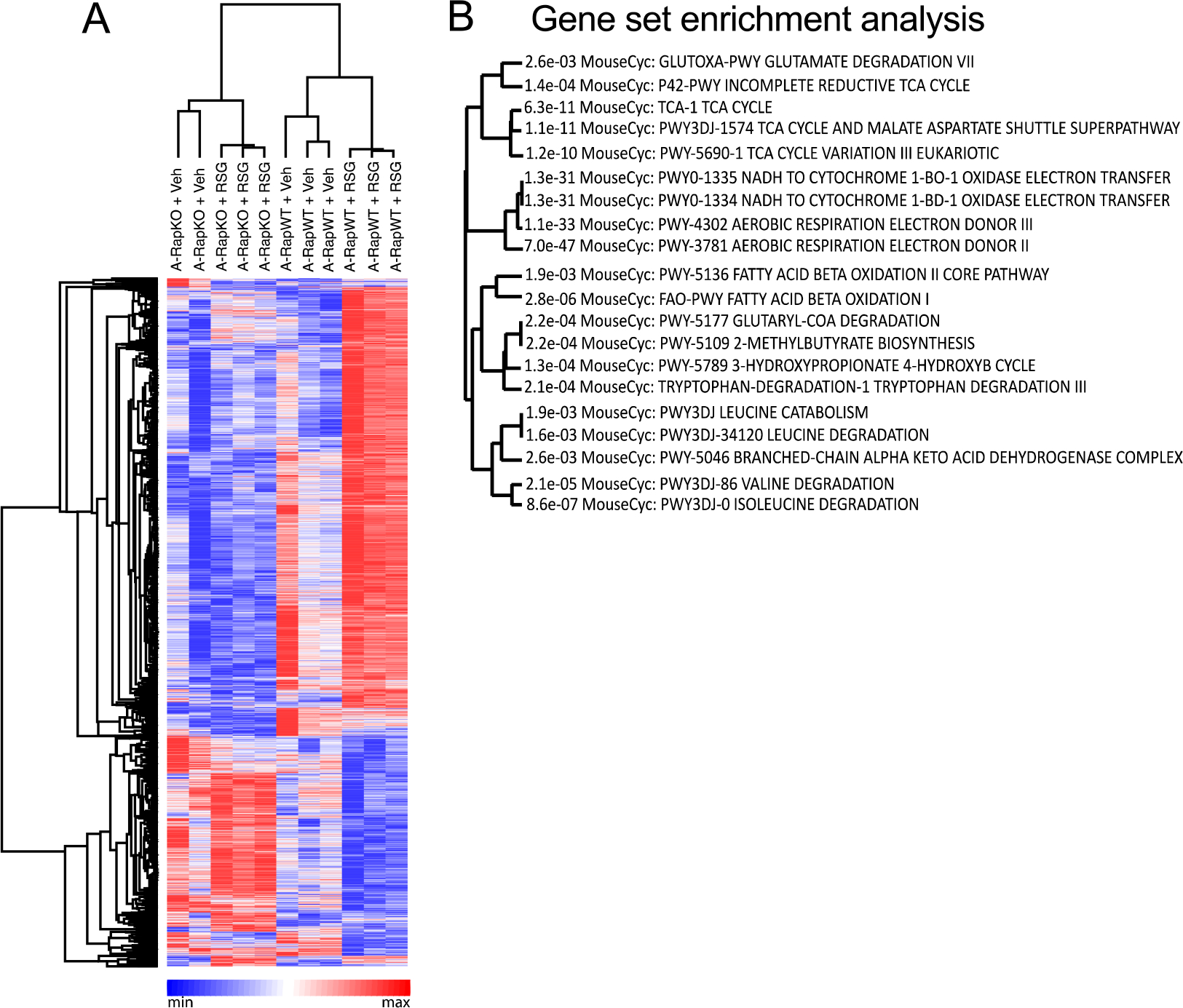
Adipocyte mTORC1 deficiency blocks rosiglitazone modulation of a subset of genes in iWAT. Heap map (A) and gene set enrichment analysis (B, ShinyGo) of genes modulated by rosiglitazone in inguinal white adipose tissue (iWAT) of RapWT, but not RapKO mice (A) fed with high-fat diet (HFD) containing or not rosiglitazone (RSG, 30 mg/kg/day) for 8 weeks. n= 2–3 mice per group.
Figure 4.
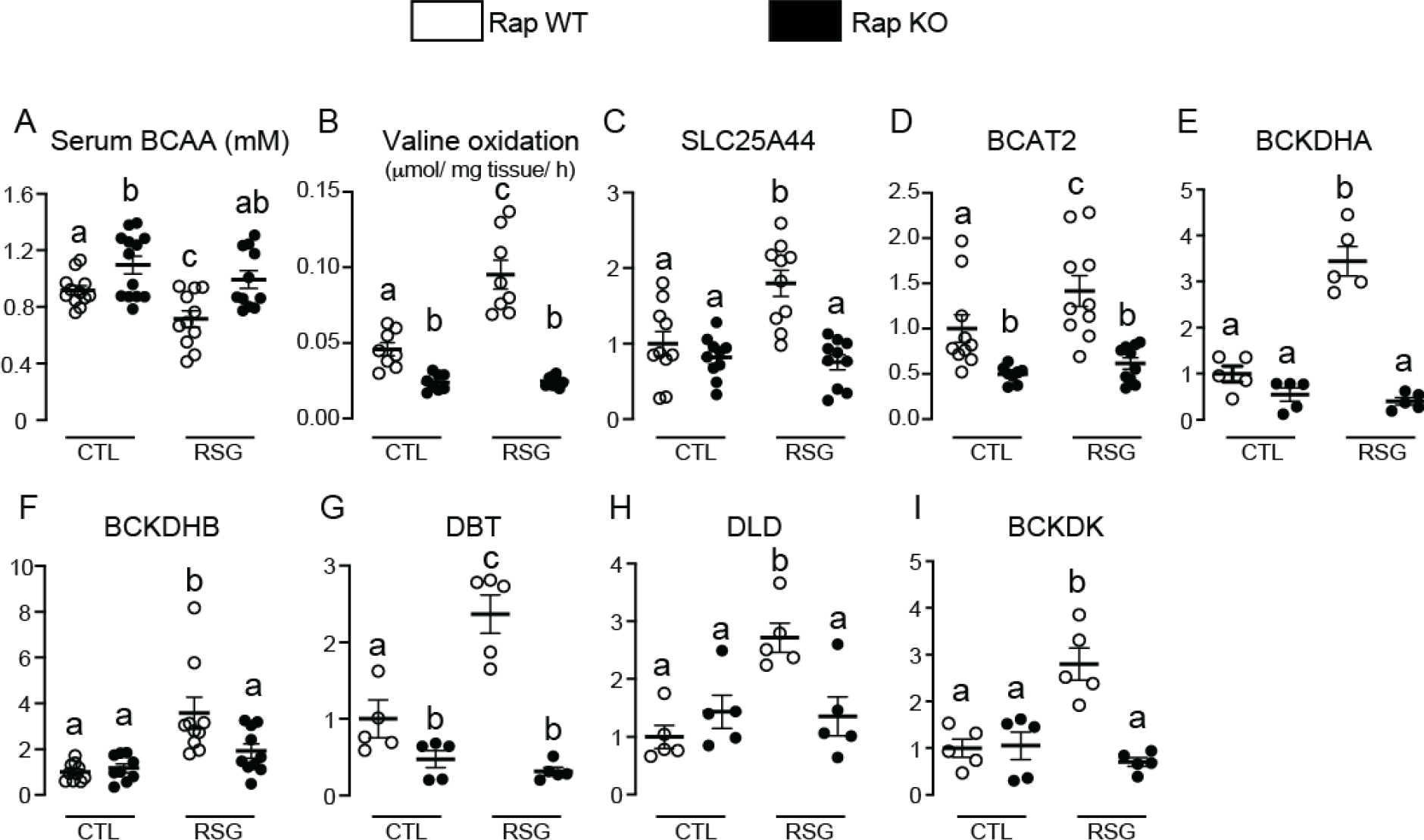
Adipocyte mTORC1 deficiency reduces iWAT BCAA oxidation and blocks the upregulation in this process induced by rosiglitazone. Serum branched-chain amino acids (BCAA) (A) and inguinal white adipose tissue (iWAT) valine oxidation (B), and mRNA levels of SLC25A44 (C), BCAT2 (D), BCKDHA (E), BCKDHB (F), DBT (G), DLD (H) and BCKDK (I) in RapWT and RapKO mice fed with high-fat diet (HFD) containing or not rosiglitazone (RSG, 30 mg/kg/day) for 8 weeks. Values are expressed as mean ± SEM. n= 5–10 mice per group. Means not sharing a common superscript are significantly different from each other, p ≤ 0.05.
In an attempt to investigate the mechanisms underlying rosiglitazone failure to promote BCAA metabolism and a subset of PPARγ-regulated genes in RapKO mice, we evaluated iWAT mRNA and protein content of PPARγ and C/EBPα, a transcription factor that regulates PPARγ transcriptional activity. As illustrated in Figure 5, adipocyte Raptor deletion did not affect iWAT mRNA levels of PPARγ isoforms 1 and 2 (Fig. 5A and B, respectively), but it significantly reduced C/EBPα mRNA levels (Fig. 5C). Interestingly, despite not affecting mRNA levels, adipocyte Raptor deletion significantly reduced iWAT protein content of the PPARγ isoforms 1 and 2 (Fig. 5D–E and H). This reduction in iWAT C/EBPα mRNA induced by adipocyte Raptor deletion, on the other hand, was associated with a reduction in C/EBPα protein content (Fig. 5F–G and H). Rosiglitazone administration did not affect iWAT PPARγ1 and 2 and C/EBPα mRNA and protein contents in both RapWT and RapKO mice (Fig. 5A–D).
Figure 5.
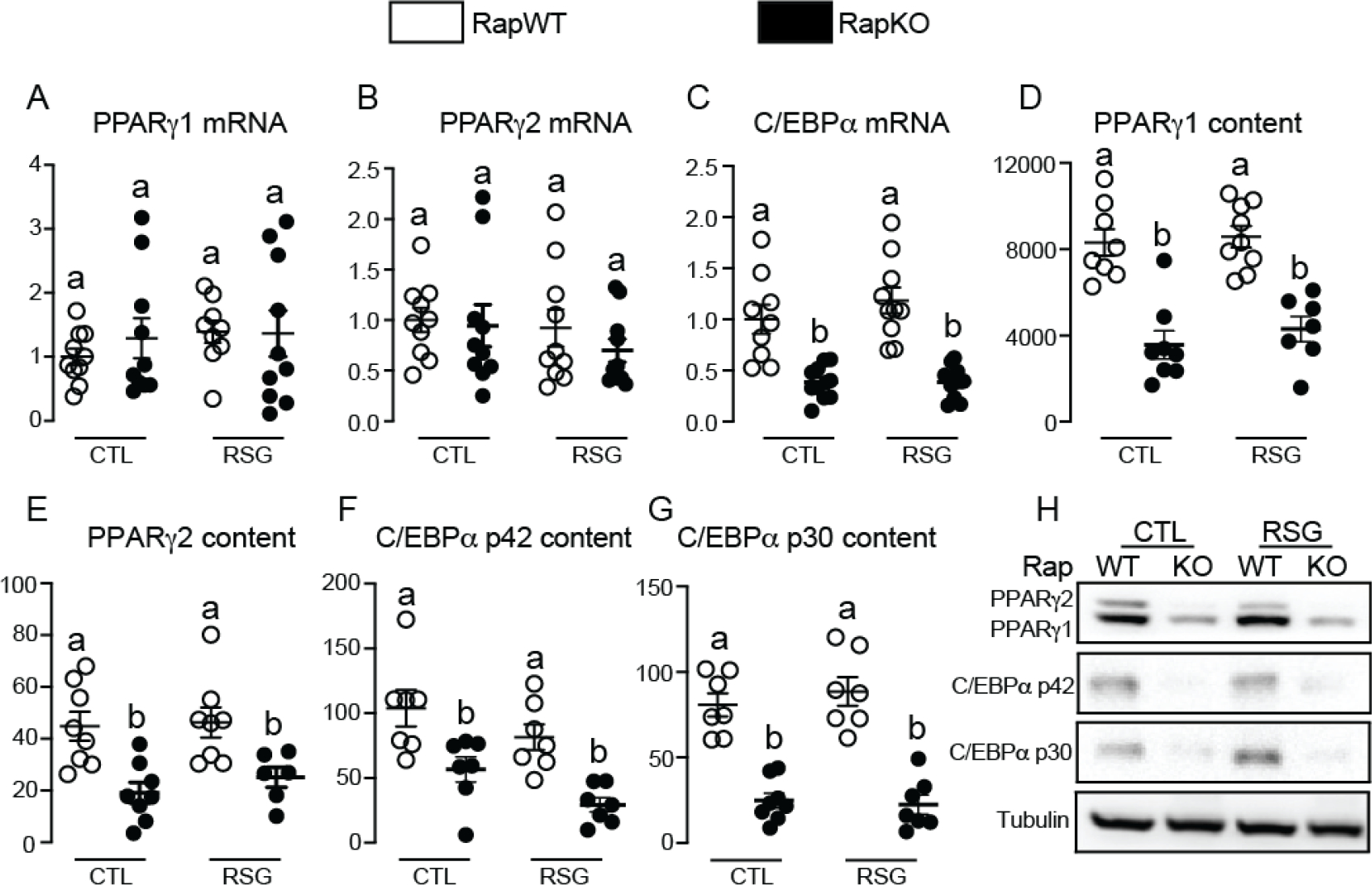
Adipocyte mTORC1 deficiency reduces iWAT PPARγ and C/EBPα protein contents. Inguinal white adipose tissue (iWAT) mRNA levels of PPARγ1 (A), PPARγ2 (B) and C/EBPα (C), and protein content of PPARγ1 (D and H), PPARγ2 (E and H), C/EBPα p42 (F and H) and C/EBPα p30 (G and H) in RapWT and RapKO mice fed with high-fat diet (HFD) containing or not rosiglitazone (RSG, 30 mg/kg/day) for 8 weeks. Values are expressed as mean ± SEM. n= 6–10 mice per group. Means not sharing a common superscript are significantly different from each other, p ≤ 0.05.
4. Discussion
We combined herein pharmacological PPARγ activation and adipocyte Raptor deletion to investigate the crosstalk between the nutrient sensors PPARγ and mTORC1 in the regulation of adipose tissue metabolism and secretory function. Our main findings indicate that adipocyte mTORC1 is a positive regulator of subcutaneous adipose tissue adiponectin production and secretion, glyceroneogenesis and PEPCK mRNA levels, triacylglycerol synthesis and BCAA oxidation and a required mediator of the upregulation in these processes induced by pharmacological PPARγ activation. Mechanistically, mTORC1 deficiency seems to impair PPARγ transcriptional activity by reducing adipose tissue PPARγ protein content, as well as by downregulating C/EBPα, a transcriptional factor previously shown to facilitate PPARγ binding to chromatin and to cooperate in the recruitment of coregulators and transcription initiation induced by this nuclear receptor [39].
A previous study has shown that mice bearing adipocyte mTORC1 deficiency display upon intake of a HFD reduced body weight gain and adiposity, increased food intake and carbohydrate utilization as evaluated by RER, but no major changes in spontaneous motor activity and energy expenditure evaluated by mouse oxygen consumption [21]. With exception of hyperphagia, these phenotypes were fully reproduced herein and were not significantly affected by rosiglitazone treatment. The precise mechanisms by which adipocyte mTORC1 deficiency reduces mice body weight gain are still unknown, but the absence of changes in food intake and energy expenditure indicates a possible impairment in food absorption. Supporting this notion, RapKO mice displayed increased fecal lipid content upon the intake of a HFD [21]. How adipocyte Raptor deletion impairs intestinal lipid absorption is still unknown.
We have previously shown that rosiglitazone administration attenuates the adipose tissue inflammation and systemic glucose intolerance associated with either pharmacological (rapamycin) or genetic (adipocyte Raptor deletion) mTORC1 inhibition in rodents [22; 40]. Despite those beneficial actions, we found here that rosiglitazone completely lost its ability to upregulate BAT mass and adiponectin production and secretion in mice bearing adipocyte mTORC1 deficiency. Specifically regarding adiponectin, our findings indicate that mTORC1 regulates all steps involved in the production and secretion of this adipokine. Indeed, adipocyte Raptor deletion not only markedly reduced iWAT adiponectin mRNA levels suggesting a role for mTORC1 in the regulation of adiponectin transcription, but also completely blocked the increase in adiponectin mRNA translation (protein content) and secretion (serum levels) induced by pharmacological PPARγ activation. Mechanistically, adipocyte mTORC1 deficiency seems to decrease iWAT adiponectin mRNA levels by reducing C/EBPα, a well-known inducer of adiponectin gene transcription [41], while it impairs adiponectin secretion by reducing DsbA-L, an oxidoreductase that mediates the adiponectin multimerization and secretion induced by rosiglitazone [42]. Noteworthy, in addition to adiponectin, adipocyte mTORC1 deficiency and rosiglitazone administration individually reduced serum levels of the anorexigenic adipokine leptin, such an effect that was not additive between treatments and, surprisingly, had no major impact on mice food intake. Why such low levels of leptin do not result in mouse hyperphagia upon either adipocyte mTORC1 deficiency or rosiglitazone treatment or their combination as it does in other conditions (lipoatrophy, for instance) remains to be investigated.
In addition to the endocrine function, adipocyte mTORC1 deficiency was also associated with important changes in iWAT lipidome, being the most striking phenotype a marked increase in tissue content of palmitic and oleic acids either at the free form or conjugated to carnitine. Mechanistically, this accumulation of free fatty acids in iWAT of RapKO mice seems to result from both an unrestrained lipolysis, as previously shown [24], and an impairment in fatty acid incorporation into triacylglycerol. Regarding the latter, analysis of mRNA levels of several enzymes involved in triacylglycerol synthesis revealed that adipocyte mTORC1 deficiency not only markedly reduced iWAT PEPCK mRNA, but also completely blocked the upregulation of this enzyme induced by rosiglitazone. PEPCK is a PPARγ-regulated key enzyme of glyceroneogenesis, a process that generates most of the glycerol 3-phosphate required for fatty acid esterification and triacylglycerol synthesis in adipose tissue of rodents [43]. Along with PEPCK mRNA, adipocyte mTORC1 deficiency markedly decreased glyceroneogenesis as evaluated by pyruvate incorporation in triacylglycerol-glycerol. Altogether these findings suggest that mTORC1 is an important regulator of glyceroneogenesis and a required mediator of the activation of this process induced by rosiglitazone in iWAT. Furthermore, these findings suggest the previously unrecognized notion that mTORC1 deficiency impairs triacylglycerol synthesis in iWAT at least in part by reducing glyceroneogenesis-mediated glycerol 3-phosphate generation.
Independently of the underlying mechanism, the build-up of toxic free fatty acids in iWAT of RapKO mice was associated with a shift in tissue metabolism towards oxidation as evidenced by the accumulation of the mitochondrial-related lipids acylcarnitine and cardiolipin along with the increases in tissue respiration, mitochondrial mass (citrate synthase activity) and mRNA levels of CPT1a, the enzyme that generates acylcarnitines, and UCP-1. Mechanistically, this increase in iWAT oxidative metabolism upon mTORC1 deficiency, which may be in part induced by PGC1α [44], seems to be a tissue attempt to minimize the harmful effects associated with tissue impaired triacylglycerol synthesis and free fatty acid accumulation. Two interesting aspects emerge from these data. Firstly, rosiglitazone completely failed in affecting tissue contents of free fatty acids and acylcarnitines in iWAT of RapKO mice, supporting the absolute mTORC1 requirement for PPARγ pro-lipid storage actions. Secondly, accumulation of free fatty acids was associated with a robust increase in iWAT UCP-1 content (browning/beiging) probably as an attempt to reduce the production of reactive oxygen species and protect mitochondrial against oxidative stress, which is an ancient, sometimes neglected function of uncoupling proteins [45]. Further supporting this notion, along with UCP-1, adipocyte mTORC1 deficiency significantly increased iWAT content of the mitochondrial antioxidant enzyme SOD-2.
In addition to lipid metabolism, mTORC1 and PPARγ interact in the regulation of other metabolic processes in adipose tissue. Indeed, by performing iWAT RNAseq analysis, we identified approximately 408 genes whose mRNA levels were significantly modulated by rosiglitazone in RapWT, but not RapKO mice, indicating that their regulation by PPARγ requires mTORC1. Functional clustering and KEGG analysis of these genes revealed the enrichment of proteins involved in several metabolic processes including the catabolism of amino acids, especially BCAA. Confirming and extending these findings, adipocyte mTORC1 deficiency not only reduced iWAT rates of valine oxidation and expression of BCAT2 and DBT, which resulted in increased serum BCAA levels, but also completely blocked the upregulation in BCAA oxidation and BCAT2 and DBT expressions, and the reduction in serum BCAA levels induced by rosiglitazone. Altogether, these findings raise the previously unrecognized notion that, in addition to adiponectin secretion, PEPCK mRNA levels and triacylglycerol synthesis, mTORC1 is also an important regulator of adipose tissue BCAA oxidation and a required mediator of the upregulation in this process induced by pharmacological PPARγ activation.
Considering the intense crosstalk between mTORC1 and PPARγ and their strong impact in adipocyte gene expression profile, several molecular mechanisms must control the interaction between these nutrient sensors. In this sense, we found herein strong evidence indicating that adipocyte mTORC1 deficiency impairs PPARγ transcriptional activity by at least two independent mechanisms. The first involves a post-transcriptional regulation of PPARγ by mTORC1 as evidenced by the marked reduction in PPARγ1 and 2 protein contents in the absence of changes in their respective mRNA levels in iWAT of RapKO mice. Such post-transcriptional regulation of PPARγ by mTORC1 may involve changes in PPARγ mRNA translation and/or protein stability and degradation. Indeed, a previous study has shown that mTORC1 inhibition in vitro impairs PPARγ translation through a mechanism that involves the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) [46]. Whether mTORC1 also regulates PPARγ protein stability and degradation is unknown. Additionally, adipocyte mTORC1 deficiency may impair PPARγ transcriptional activity by reducing iWAT expression and protein content of C/EBPα, a transcriptional factor previous shown to facilitate PPARγ binding to chromatin that cooperates in the cofactor recruitment and transcription initiation induced by this nuclear receptor [39]. Supporting this notion, a subset of genes whose modulation by PPARγ requires mTORC1 activity such as BCKDHB, for instance, was previously shown to be synergistically regulated by C/EBPα and PPARγ [39]. Taken together these findings strongly suggest the involvement of C/EBPα as one of the mechanisms by which adipocyte mTORC1 deficiency impairs PPARγ transcriptional activity. How mTORC1 impairs iWAT C/EBPα content is still unknown. Importantly, a large subset genes regulated by the mTORC1/PPARγ crosstalk do not have a C/EBPα response element in their promoters suggesting the existence of other mechanisms through which mTORC1 modulates PPARγ transcriptional activity. Possible candidate processes may involve mTORC1-mediated post-translational modifications of PPARγ and associated coregulators (phosphorylation, ubiquitination, sumoylation, acetylation, etc), as well as epigenetic changes (methylation, acetylation etc) of histones and promoter regions of target genes, among others.
A recent study has found that subcutaneous white preadipocytes bearing mTORC2 deficiency display reduced lipid deposition and PPARγ transcriptional activity after differentiation, suggesting a positive regulation of this nuclear receptor by mTORC2 [30]. Being mTORC2 an important upstream activator of mTORC1, it will be important in future studies to investigate whether those mTORC2 actions upon PPARγ involve mTORC1. Noteworthy, as previously reported by others and us [21; 22], adipocyte Raptor deletion and mTORC1 deficiency increases mTORC2 activity in adipose tissue due to the alleviation of the negative feedback catalyzed by mTORC1 target S6K1 on IRS-PI3K signaling, excluding therefore an involvement of mTORC2 in the impairment in PPARγ activity found in RapKO iWAT.
In conclusion, our findings indicate that mTORC1 and PPARγ are essential partners involved in the regulation of adipose tissue endocrine and metabolic functions. Adipocyte mTORC1 deficiency impairs adipose tissue adiponectin secretion, PEPCK mRNA levels and glyceroneogenesis, triacylglycerol synthesis and BCAA oxidation, and block the activation of these processes induced by the PPARγ ligand rosiglitazone, through a mechanism involving an impairment in protein content of PPARγ and its co-partner C/EBPα. Future studies are required to complete elucidate the mechanisms dictating PPARγ-mTORC1 crosstalk and regulation of adipocyte function.
Supplementary Material
Highlights.
Induction of adiponectin secretion by PPARγ agonism requires adipocyte mTORC1
Induction of iWAT glyceroneogenesis by PPARγ agonism requires adipocyte mTORC1
Induction of iWAT BCAA catabolism by PPARγ agonism requires adipocyte mTORC1
Adipocyte mTORC1 deficiency reduces iWAT PPARγ and C/EBPα protein contents
Funding
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP #15/19530-5 and 19/01763-4) and Brazilian National Council for Scientific and Technological Development (CNPq #303459/2016-6) to WTF. MLA, ASP, LAP, MFM, EC, TEO, TSV, MO-S, TB, ABC-F were recipients of fellowships from FAPESP (15/13508-8; 17/23040-9; 19/17660-0; 17/17582-3; 16/23169-9; 19/04271-5; 2017/17403-1; 17/12260-8; 15/22983-1; 18/11156-5). GRG and CAT were recipients of fellowships from CNPq (#142039/2017-9) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES #4193934). SM was supported by FAPESP #2013/07937-8.
Abbreviations:
- AMPK
AMP dependent protein kinase
- BAT
brown adipose tissue
- BCAA
branched-chain amino acid
- BCAT2
branched-chain amino acid aminotransferase 2
- BCKDH
branched-chain alpha-keto acid dehydrogenase complex
- BCKDHA
branched-chain alpha-keto acid dehydrogenase complex subunit A
- BCKDHB
branched-chain alpha-keto acid dehydrogenase complex subunit B
- BCKDK
branched-chain keto acid dehydrogenase kinase
- CPT1a
carnitine palmitoyltransferase 1a
- DBT
dihydrolipoamide branched-chain transacylase E2
- Deptor
DEP domain-containing mTOR-interacting protein
- DLD
dihydrolipoamide dehydrogenase
- DsbA-L
disulfide-bond A oxidoreductase like protein
- Ero1-Lα
endoplasmic reticulum oxidoreductase
- H/E
hematoxylin and eosin
- HFD
high-fat diet containing 60% of calories from lard
- IRS
insulin receptor substrate
- iWAT
inguinal white adipose tissue
- mLST8
mammalian lethal with Sec13 protein 8
- mTORC1
mechanisitc target of rapamycin complex 1
- mTORC2
mechanisitc target of rapamycin complex 2
- PEPCK
phosphoenolpyruvate carboxykinase
- PGC1α
PPAR coactivator 1α
- PPARγ
peroxisome proliferator activated receptor γ
- Pras40
proline-rich Akt substrate 40
- RapKO
RaptorLox/Lox
- adiponectin-cre+/−
- RapWT
RaptorLox/Lox
- adiponectin-cre−/−
- Raptor
regulatory-associated protein of mTOR
- RXR
retinoic acid receptor
- TCA
tricarboxylic acid
- TZD
thiazolidinediones
- UCP-1
uncoupling protein 1
- WAT
white adipose tissue
Footnotes
Conflict of Interests
Authors do not have any conflict of interest relevant to this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. , 2013. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 19(5):557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. , 1999. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402(6764):880–883. [DOI] [PubMed] [Google Scholar]
- [3].Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, et al. , 2006. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab 4(4):303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA, 2013. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc Natl Acad Sci U S A 110(46):18656–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moreira RJ, Castro É, Oliveira TE, Belchior T, Peixoto AS, Chaves-Filho AB, et al. , 2020. Lipoatrophy-Associated Insulin Resistance and Hepatic Steatosis are Attenuated by Intake of Diet Rich in Omega 3 Fatty Acids. Mol Nutr Food Res:e1900833. [DOI] [PubMed] [Google Scholar]
- [6].Soccio RE, Chen ER, Lazar MA, 2014. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20(4):573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. , 2002. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 87(6):2784–2791. [DOI] [PubMed] [Google Scholar]
- [8].Laplante M, Festuccia WT, Soucy G, Gélinas Y, Lalonde J, Berger JP, et al. , 2006. Mechanisms of the depot specificity of peroxisome proliferator-activated receptor gamma action on adipose tissue metabolism. Diabetes 55(10):2771–2778. [DOI] [PubMed] [Google Scholar]
- [9].Laplante M, Sell H, MacNaul KL, Richard D, Berger JP, Deshaies Y, 2003. PPARgamma activation mediates adipose depot-specific effects on gene expression and lipoprotein lipase activity: mechanisms for modulation of postprandial lipemia and differential adipose accretion. Diabetes 52(2):291–299. [DOI] [PubMed] [Google Scholar]
- [10].Laplante M, Festuccia WT, Soucy G, Gélinas Y, Lalonde J, Deshaies Y, 2007. Involvement of adipose tissues in the early hypolipidemic action of PPARgamma agonism in the rat. Am J Physiol Regul Integr Comp Physiol 292(4):R1408–1417. [DOI] [PubMed] [Google Scholar]
- [11].Blanchard PG, Turcotte V, Côté M, Gélinas Y, Nilsson S, Olivecrona G, et al. , 2016. Peroxisome proliferator-activated receptor γ activation favours selective subcutaneous lipid deposition by coordinately regulating lipoprotein lipase modulators, fatty acid transporters and lipogenic enzymes. Acta Physiol (Oxf) 217(3):227–239. [DOI] [PubMed] [Google Scholar]
- [12].Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. , 2001. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50(9):2094–2099. [DOI] [PubMed] [Google Scholar]
- [13].Yang WS, Jeng CY, Wu TJ, Tanaka S, Funahashi T, Matsuzawa Y, et al. , 2002. Synthetic peroxisome proliferator-activated receptor-gamma agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes Care 25(2):376–380. [DOI] [PubMed] [Google Scholar]
- [14].Blanchard PG, Moreira RJ, Castro É, Caron A, Côté M, Andrade ML, et al. , 2018. PPARγ is a major regulator of branched-chain amino acid blood levels and catabolism in white and brown adipose tissues. Metabolism 89:27–38. [DOI] [PubMed] [Google Scholar]
- [15].Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. , 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112(12):1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Festuccia WT, Blanchard PG, Turcotte V, Laplante M, Sariahmetoglu M, Brindley DN, et al. , 2009. Depot-specific effects of the PPARgamma agonist rosiglitazone on adipose tissue glucose uptake and metabolism. J Lipid Res 50(6):1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Festuccia WT, Blanchard PG, Turcotte V, Laplante M, Sariahmetoglu M, Brindley DN, et al. , 2009. The PPARgamma agonist rosiglitazone enhances rat brown adipose tissue lipogenesis from glucose without altering glucose uptake. Am J Physiol Regul Integr Comp Physiol 296(5):R1327–1335. [DOI] [PubMed] [Google Scholar]
- [18].Saxton RA, Sabatini DM, 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell 169(2):361–371. [DOI] [PubMed] [Google Scholar]
- [19].Laplante M, Sabatini DM, 2009. An emerging role of mTOR in lipid biosynthesis. Curr Biol 19(22):R1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Festuccia WT, 2020. Regulation of Adipocyte and Macrophage Functions by mTORC1 and 2 in Metabolic Diseases. Mol Nutr Food Res:e1900768. [DOI] [PubMed] [Google Scholar]
- [21].Lee PL, Tang Y, Li H, Guertin DA, 2016. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol Metab 5(6):422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chimin P, Andrade ML, Belchior T, Paschoal VA, Magdalon J, Yamashita AS, et al. , 2017. Adipocyte mTORC1 deficiency promotes adipose tissue inflammation and NLRP3 inflammasome activation via oxidative stress and de novo ceramide synthesis. J Lipid Res 58(9):1797–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN, 2008. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8(5):399–410. [DOI] [PubMed] [Google Scholar]
- [24].Paolella LM, Mukherjee S, Tran CM, Bellaver B, Hugo M, Luongo TS, et al. , 2020. mTORC1 restrains adipocyte lipolysis to prevent systemic hyperlipidemia. Mol Metab 32:136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim JE, Chen J, 2004. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 53(11):2748–2756. [DOI] [PubMed] [Google Scholar]
- [26].Houde VP, Brûlé S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, et al. , 2010. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59(6):1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Blanchard PG, Festuccia WT, Houde VP, St-Pierre P, Brule S, Turcotte V, et al. , 2012. Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion. J Lipid Res 53(6):1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. , 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284(12):8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. , 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22(2):159–168. [DOI] [PubMed] [Google Scholar]
- [30].Hsiao WY, Jung SM, Tang Y, Haley JA, Li R, Li H, et al. , 2020. The Lipid Handling Capacity of Subcutaneous Fat Is Programmed by mTORC2 during Development. Cell Rep 33(1):108223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14(4):417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Soneson C, Love MI, Robinson MD, 2015. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ge SX, Jung D, Yao R, 2020. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8):2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Magdalon J, Chimin P, Belchior T, Neves RX, Vieira-Lara MA, Andrade ML, et al. , 2016. Constitutive adipocyte mTORC1 activation enhances mitochondrial activity and reduces visceral adiposity in mice. Biochim Biophys Acta 1861(5):430–438. [DOI] [PubMed] [Google Scholar]
- [36].Festuccia WT, Kawashita NH, Garofalo MA, Moura MA, Brito SR, Kettelhut IC, et al. , 2003. Control of glyceroneogenic activity in rat brown adipose tissue. Am J Physiol Regul Integr Comp Physiol 285(1):R177–182. [DOI] [PubMed] [Google Scholar]
- [37].Oliveira TE, Castro É, Belchior T, Andrade ML, Chaves-Filho AB, Peixoto AS, et al. , 2019. Fish Oil Protects Wild Type and Uncoupling Protein 1-Deficient Mice from Obesity and Glucose Intolerance by Increasing Energy Expenditure. Mol Nutr Food Res:e1800813. [DOI] [PubMed] [Google Scholar]
- [38].Han X, 2016. Part II: Characterization of lipids. Lipidomics : Comprehensive Mass Spectrometry of Lipids. John Wiley & Sons, Inc, p. 459. [Google Scholar]
- [39].Madsen MS, Siersbæk R, Boergesen M, Nielsen R, Mandrup S, 2014. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol 34(6):939–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Festuccia WT, Blanchard PG, Belchior T, Chimin P, Paschoal VA, Magdalon J, et al. , 2014. PPARgamma activation attenuates glucose intolerance induced by mTOR inhibition with rapamycin in rats. Am J Physiol Endocrinol Metab 306(9):E1046–1054. [DOI] [PubMed] [Google Scholar]
- [41].Qiao L, Maclean PS, Schaack J, Orlicky DJ, Darimont C, Pagliassotti M, et al. , 2005. C/EBPalpha regulates human adiponectin gene transcription through an intronic enhancer. Diabetes 54(6):1744–1754. [DOI] [PubMed] [Google Scholar]
- [42].Liu M, Zhou L, Xu A, Lam KS, Wetzel MD, Xiang R, et al. , 2008. A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc Natl Acad Sci U S A 105(47):18302–18307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nye CK, Hanson RW, Kalhan SC, 2008. Glyceroneogenesis is the dominant pathway for triglyceride glycerol synthesis in vivo in the rat. J Biol Chem 283(41):27565–27574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Feige JN, Auwerx J, 2007. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol 17(6):292–301. [DOI] [PubMed] [Google Scholar]
- [45].Rial E, Rodríguez-Sánchez L, Gallardo-Vara E, Zaragoza P, Moyano E, González-Barroso MM, 2010. Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim Biophys Acta 1797(6–7):800–806. [DOI] [PubMed] [Google Scholar]
- [46].Guntur KV, Guilherme A, Xue L, Chawla A, Czech MP, 2010. Map4k4 negatively regulates peroxisome proliferator-activated receptor (PPAR) gamma protein translation by suppressing the mammalian target of rapamycin (mTOR) signaling pathway in cultured adipocytes. J Biol Chem 285(9):6595–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.